
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stimuli-responsive self-assembly of nanoparticles
Marek
Grzelczak
 *ab,
Luis M.
Liz-Marzán
*bc and
Rafal
Klajn
*d
*ab,
Luis M.
Liz-Marzán
*bc and
Rafal
Klajn
*d
aDonostia International Physics Center (DIPC), Manuel de Lardizabal 4, 20018 Donostia-San Sebastián, Spain. E-mail: marek.grzelczak@dipc.org
bIkerbasque, Basque Foundation for Science, 48013 Bilbao, Spain
cCIC biomaGUNE and CIBER-BBN, Paseo de Miramón 182, 20014 Donostia-San Sebastián, Spain. E-mail: llizmarzan@cicbiomagune.es
dDepartment of Organic Chemistry, Weizmann Institute of Science, Rehovot 76100, Israel. E-mail: rafal.klajn@weizmann.ac.il
First published on 28th January 2019
Abstract
The capacity to respond or adapt to environmental changes is an intrinsic property of living systems that comprise highly-connected subcomponents communicating through chemical networks. The development of responsive synthetic systems is a relatively new research area that covers different disciplines, among which nanochemistry brings conceptually new demonstrations. Especially attractive are ligand-protected gold nanoparticles, which have been extensively used over the last decade as building blocks in constructing superlattices or dynamic aggregates, under the effect of an applied stimulus. To reflect the importance of surface chemistry and nanoparticle core composition in the dynamic self-assembly of nanoparticles, we provide here an overview of various available stimuli, as tools for synthetic chemists to exploit. Along with this task, the review starts with the use of chemical stimuli such as solvent, pH, gases, metal ions or biomolecules. It then focuses on physical stimuli: temperature, magnetic and electric fields, as well as light. To reflect on the increasing complexity of current architectures, we discuss systems that are responsive to more than one stimulus, to finally encourage further research by proposing future challenges.
 Marek Grzelczak | Marek Grzelczak is an Ikerbasque Research Associate at Donostia International Physics Center, San Sebastian where he leads the Colloidal Systems Chemistry Group. His current research interests cover the synthesis and self-assembly of nanoparticles for artificial photosynthesis and biosensing. He was awarded with Young Researchers Award of Real Sociedad Española de Química in 2017. |
 Luis M. Liz-Marzán | Luis Liz-Marzán, is Ikerbasque Professor and Scientific Director of CIC biomaGUNE, in San Sebastián (Spain), since September 2012. He graduated in chemistry from the University of Santiago de Compostela, was postdoc at Utrecht University and Professor at the University of Vigo (1995–2012). He has been Invited Professor at several universities and research centers worldwide and has received numerous research awards. His major research activity is devoted to understand the growth mechanisms of metal nanocrystals, to tailor their surface chemistry and direct self-assembly. He also works on the design of biomedical applications based on the plasmonic properties of well-defined metal nanoparticles and nanostructures. |
 Rafal Klajn | Rafal Klajn completed his PhD in Chemical and Biological Engineering at Northwestern University in 2009. Since then, he is based at the Department of Organic Chemistry of the Weizmann Institute of Science (Israel), where he is currently an associate professor. The interests of his research group revolve around nanoscale self-assembly and reactivity, and the development of new stimuli-responsive nanomaterials. He is the recipient of the 2010 IUPAC Prize for Young Chemists, the 2013 ACS Victor K. LaMer Award, the 2016 Netherlands Scholar Award for Supramolecular Chemistry, and the 2019 New Horizons Solvay Lectureship in Chemistry. |
Introduction
Using a relatively simple repertoire of building blocks (such as amino acids and lipid surfactants) the natural systems have developed a rich variety of sophisticated functions, ultimately giving rise to life.1–4 These functions and behaviors include self-healing,5 signal amplification,6,7 homeostasis,8,9 and camouflage,10 and they are made possible by integrating simple building blocks within complex chemical reaction networks. Although research towards recreating these and other complex behaviors in a synthetic laboratory setting is still in its infancy,11–13 synthetic chemists have the advantage of accessing a virtually endless toolbox of building blocks, which can come with beneficial properties (catalytic activity, electrical conductivity, and so on). Among these, ligand-protected inorganic nanoparticles (NPs) are particularly attractive. NPs can be synthesized in the form of different sizes and shapes and, depending on the composition of the inorganic core, they can exhibit a variety of properties difficult or impossible to achieve with organic molecules (e.g., superparamagnetism,14 localized plasmon resonance,15 and photon upconversion16). Additional advantages of NPs are that they can readily be imaged (using methods such as transmission electron microscopy), interfacial effects can positively impact the properties of both the organic and the inorganic counterparts of these hybrid constructs, and many of their properties are dependent on and can be controlled by the degree of NP aggregation.Established protocols exist for functionalizing NPs with monolayers of organic ligands.17 Depending on the inorganic core of the NP, different anchoring groups are used. Arguably, the best known are sulfur-based ligands (mainly thiols), which have a high affinity to noble metal (mainly gold) NPs.18 Catechols, phosphates, and related ligands can form robust monolayers on the surfaces of metal oxide NPs; for silica, silanes are typically the ligands of choice. A variety of functional groups can be installed on the other end (i.e., at the ω position) of the ligand molecules, thus effectively decorating the outer surface of the NP. It is predominantly these terminal groups that determine the solubility of NPs and as such can be used to direct NP self-assembly. Importantly, NPs can simultaneously be functionalized with more than one type of ligand – in fact, it is possible to functionalize a single NP with a large library of molecules (e.g., a single 5 nm gold NP is capable of accommodating as many as 300 thiolate ligands17). This observation highlights the great number of possible materials that arise by integrating organic and nanosized inorganic components.
This review attempts to present stimuli-responsive NPs as the building blocks for constructing chemical reaction networks. To this end, we categorized the existing systems in terms of the external stimuli they respond to. Stimuli-responsiveness of NPs can originate either from the organic ligands adsorbed on NP surfaces or from the inorganic cores of the NPs. Diverse stimuli that have successfully been used to direct reversible NP self-assembly and which are discussed here, include chemical stimuli – solvents, acid/base signals, metal ions, gases, biomacromolecules, and redox signals – as well as physical stimuli – temperature, magnetic fields, and light. As a particularly attractive class of materials, we highlight NPs capable of simultaneously responding to more than one type of stimulus. The ultimate goal of this paper is to encourage synthetic chemists to use ligand-functionalized NPs as the building blocks of increasingly more complex chemical systems that will in the future rival the elegance, with which nature employs dynamic self-assembly for processes critical to life.
1. Solvents
Depending mostly on the ligand molecules bound to their surfaces, nanoparticles exhibit good solubility in certain solvents, and poor in others (non-solvents). Therefore, introducing a stimulus in the form of a non-solvent to a solution of NPs in a good solvent is a readily accessible and practical method for assembling NPs into higher-order structures. This approach typically relies on destabilizing (desolvating) ligand molecules covalently bound to the NP surfaces. For short ligands, the process results in decreasing steric repulsion between the ligands, giving rise to attractive van der Waals (vdW) interactions between the NP cores. With long ligands (e.g., polymer-based brushes), however, attractive vdW interactions do not overcome the steric brush repulsion, and self-assembly is driven predominantly by solvophobic interactions. In both cases, NPs within the aggregates are held together by noncovalent interactions, which can be broken either upon removing the stimulus (non-solvent) or upon adding a competing stimulus (a good solvent).In a classical example, attractive interactions between differently sized (18–62 nm) polystyrene (PS)-stabilized gold NPs in THF were induced upon the addition of water (which is a poor solvent for PS).19 The initially formed small spherical clusters of Au NPs gradually increased in size and eventually precipitated from the solution (Fig. 1a). The short interparticle distances within these clusters entailed strong coupling between the localized surface plasmon resonance (LSPR) modes of nearby NPs, which was manifested by a change in the color of the sample from red to blue. Importantly, the growth of these clusters could be arrested by adding a polymeric surfactant comprising a hydrophobic PS and a hydrophilic poly(acrylic acid) block; whereas the former block stabilized the hydrophobic NP clusters by intercalating between the NP-bound PS chains, the latter provided the clusters with solubility in aqueous solvents. The kinetically trapped clusters were stable in solution over long periods of time (months); however, treating this solution with a good solvent (THF) induced re-solvation of PS chains and consequently, partial disassembly of the clusters. Quantitative disassembly was achieved by removing water from the system, which brought the system back to the original state featuring non-interacting NPs in THF. A controlled partition of the clusters could also be achieved using another good solvent, dioxane, whose addition decreased the interfacial energy of the clusters.20 This process was investigated using kinetically trapped clusters containing PS-capped Au NPs of two sizes, 10 and 40 nm. Interestingly, the fission process occurred such that the ratio between differently sized NPs in the “daughter” clusters was preserved (Fig. 1b).
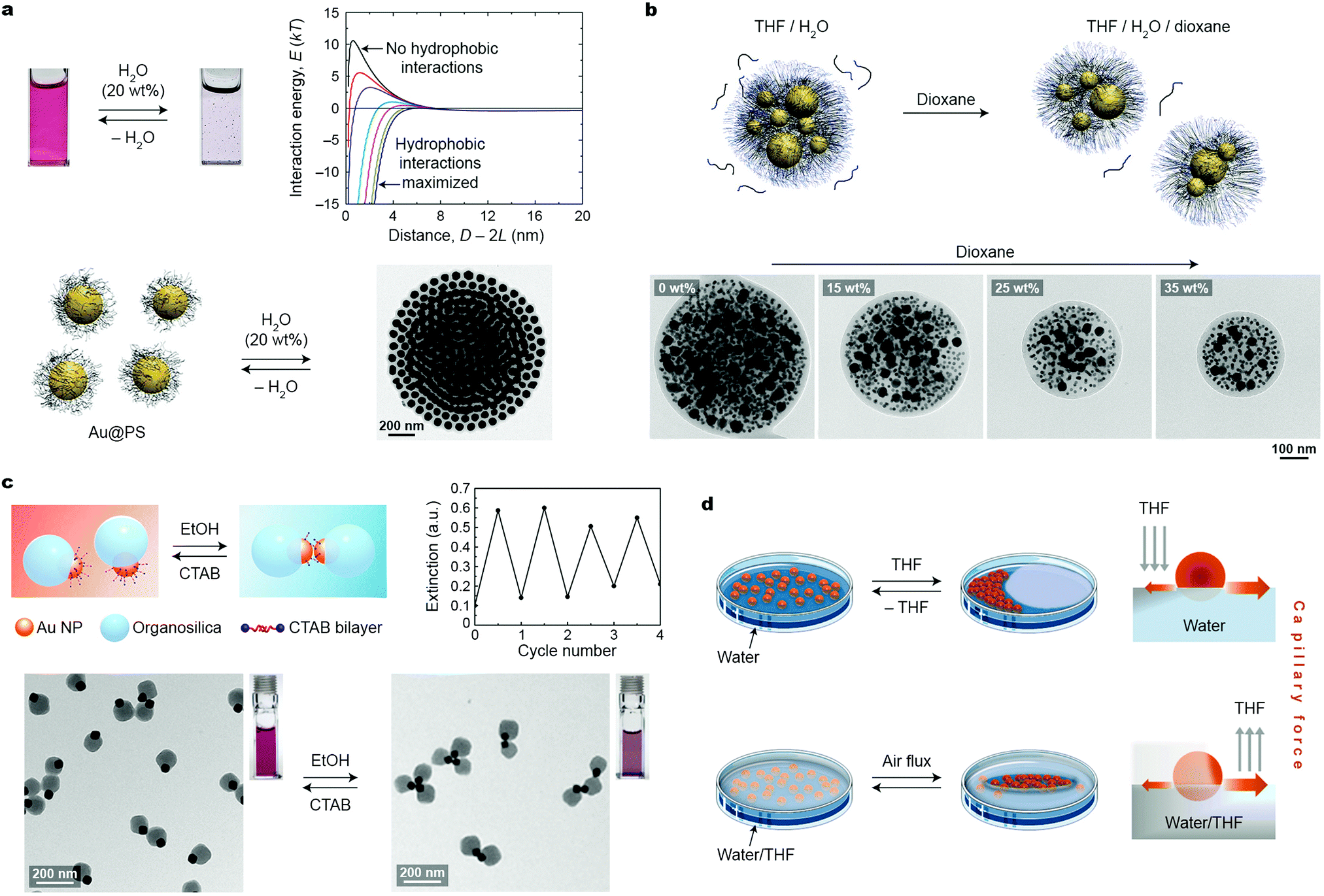 | ||
| Fig. 1 Solvent-induced self-assembly. (a) Reversible self-assembly of polystyrene-grafted Au NPs in THF using water. The transmission electron microscopy (TEM) image shows an NP cluster deposited on a TEM substrate. The plot in the top right corner shows calculated NP–NP interaction energies as a function of interparticle separation for different values of the hydrophilic–hydrophobic balance.19 Adapted with permission from ref. 19 (copyright 2012, American Chemical Society). (b) Top: NP cluster fission induced by a good solvent (dioxane). Bottom: TEM images of representative NP clusters in the presence of increasing amounts of dioxane. Adapted with permission from ref. 20 (copyright 2013, Royal Society of Chemistry). (c) Reversible self-assembly of Au–organosilica Janus NPs into discrete NP oligomers upon alternating additions of ethanol and CTAB stimuli. Adapted with permission from ref. 22 (copyright 2016, American Chemical Society). (d) Two approaches allowing for dynamic self-assembly of NPs at the liquid–air interface via capillary forces. Adapted with permission from ref. 23 (copyright 2013, American Chemical Society). | ||
Structural transitions in assemblies of PS-stabilized Au NPs have also been studied by exploiting the interplay between attractive hydrophobic forces and repulsive electrostatic forces. Kumacheva et al. worked with PS-functionalized 23 nm Au NPs prepared by the ligand-exchange reaction between thiolated PS chains and positively charged (decorated with cetyltrimethylammonium bromide; CTAB) NPs.21 When a DMF solution of these NPs was treated with water, two distinct types of assemblies were observed, depending on the length of PS and the amount of water in the final solution. For PS chains of <20 kDa, introducing 5–10 vol% of water afforded one-dimensional assemblies of Au NPs as a result of the competition between the hydrophobic attraction and electrostatic repulsion. With longer (>20 kDa) PS chains, self-assembly was initiated at lower (2–5 vol%) contents of water, and resulted in globular NP clusters instead. Only in the presence of relatively high (∼15 vol%) amounts of water was the transition to linear assemblies observed. Importantly, these transitions were reversible: for example, by gradually decreasing the amount of water in a solution of linear aggregates of NPs coated with PS of high molecular weight (50 kDa), a transition to the globular state was observed, followed by disassembly at ∼2 vol% of water.21 Taken together, these results indicate that solvents can serve as appealing stimuli to direct reversible transitions between different self-assembled states.
An important feature of solvent-induced self-assembly is that it is applicable to NPs having different morphologies. Discrete assemblies of NPs can be obtained by “protecting” a part of their surface with a bulky, solubilizing domain, while inducing attractive interactions through the exposed part. This strategy is exemplified by NPs comprising both a small, CTAB-coated Au domain and a large organosilica domain.22 Addition of an ethanol “stimulus” to an aqueous solution of these “Janus” NPs displaced the protective CTAB coating by the organic solvent molecules, and consequently, induced attractive vdW interactions between the Au domains. This process led to the formation of NP dimers or trimers, depending on the relative sizes of the Au and organosilica domains (Fig. 1c). Free CTAB used as a counter-stimulus effectively induced the disassembly process by providing extra stabilization to non-assembled Janus NPs. Subsequent reassembly–disassembly cycles could be performed upon alternately introducing ethanol and CTAB to the system.22
Dynamic self-assembly of 8 nm Au NPs functionalized with a mixture of a nonpolar (undecanethiol) and a polar (NMe3+-terminated) thiol was investigated by Sashuk, Fiałkowski, and co-workers.23 These amphiphilic NPs readily spread out at the water–air interface, forming a thin red film. Localized addition of an organic solvent (THF) onto the interface created a gradient of surface tension, inducing the migration of NPs towards the area of high tension (i.e., away from the place where THF was added) and formation of a densely packed NP monolayer (Fig. 1d, top). Notably, this process was associated with a pronounced color change in the NP film from red to purple, indicative of interparticle plasmon coupling. In addition to monitoring color changes, the assembly process was verified by scanning electron microscopy (SEM) and in situ small-angle X-ray scattering (SAXS) measurements. The resulting assemblies existed only as long as the surface tension gradient was maintained; the system gradually (within several days) retuned to equilibrium, regenerating the original film of NPs dispersed at the interface. The authors contend that the NPs temporarily existed in an ordered (i.e., high-entropy) state at the expense of entropy production associated with solvent mixing/evaporation.23 An alternative approach to form dynamically self-assembling NP films was based on the localized removal of THF from a water/THF mixture onto which a thin film of the same NPs was deposited (Fig. 1d, bottom). Directing a stream of air at a given location of the interface induced evaporation of THF, and consequently a local increase in surface tension, which triggered rapid NP self-assembly at this location. Interestingly, by delivering compressed air through an array of plastic nozzles, patterns of NP films at the liquid–air interface could be created. These patterns were “self-erasing” in that discontinuing the gas flow led to equilibration of the system and the disassembly of the dense NP films. Recently, the reversible, solvent-induced self-assembly was realized in the confinement provided by mesoporous silica capsules, which offered additional advantages related to reproducible plasmon shifts.24
2. Acid/base
It has long been recognized that the colloidal stability of nanoparticles is largely dependent on the NP surface charge, which typically originates from the terminal groups of the NP-bound ligands (e.g., ω-functionalized thiolates for noble metal NPs). NPs with high positive or negative charges are colloidally stable in polar solvents due to electrostatic double layer repulsions; conversely, decreasing the absolute value of the surface charge can induce NP self-assembly. These behaviors can conveniently be modulated using pH-responsive terminal groups (Fig. 2a). An important requirement, however, is that the (de)protonation of these groups occurs around neutral pH (ligand-protected NPs are unstable under highly acidic or basic conditions). A simple molecule meeting these criteria is 2-fluoro-4-mercaptophenol (FMP, pKa = 8.3), whereby the fluoro substituent renders the OH group rather acidic. Working with small (6–10 nm) Au NPs functionalized with FMP, Grzybowski et al. showed that decreasing the solution pH from ∼9 to ∼7 resulted in protonation of the terminal –O− groups, which induced NP assembly (Fig. 2a, top).25 By decorating NPs with basic ligands, the opposite behavior can be achieved. For example, Duan and co-workers prepared 14 nm Au NPs functionalized with a mixed ligand shell comprising poly(2-(diethylamino)ethyl methacrylate)26 (PDEA), with a pKa of ∼7.3. Below the pKa value, the NPs were readily soluble in water; self-assembly could be induced upon raising the pH and deprotonating the terminal –NR2H+ groups (Fig. 2a, bottom). The reversible, pH-controlled NP self-assembly was employed for the dynamic formation of hot-spots for surface-enhanced Raman scattering (SERS). To achieve this goal, Hervés, Pérez-Juste and co-workers worked with Au and Ag NPs functionalized with penicillamine (PEN), whose –COO− group can be protonated (and NP self-assembly induced) at pH < 3.7. The assembly process resulted in the formation of hot-spots, greatly enhancing the local electric field near the NP surfaces. Well-defined SERS spectra of PEN could be acquired at pH = 3; raising the pH to 7 resulted in the disappearance of the vibrational pattern characteristic for PEN. Interestingly, the process could be repeated for at least three cycles.27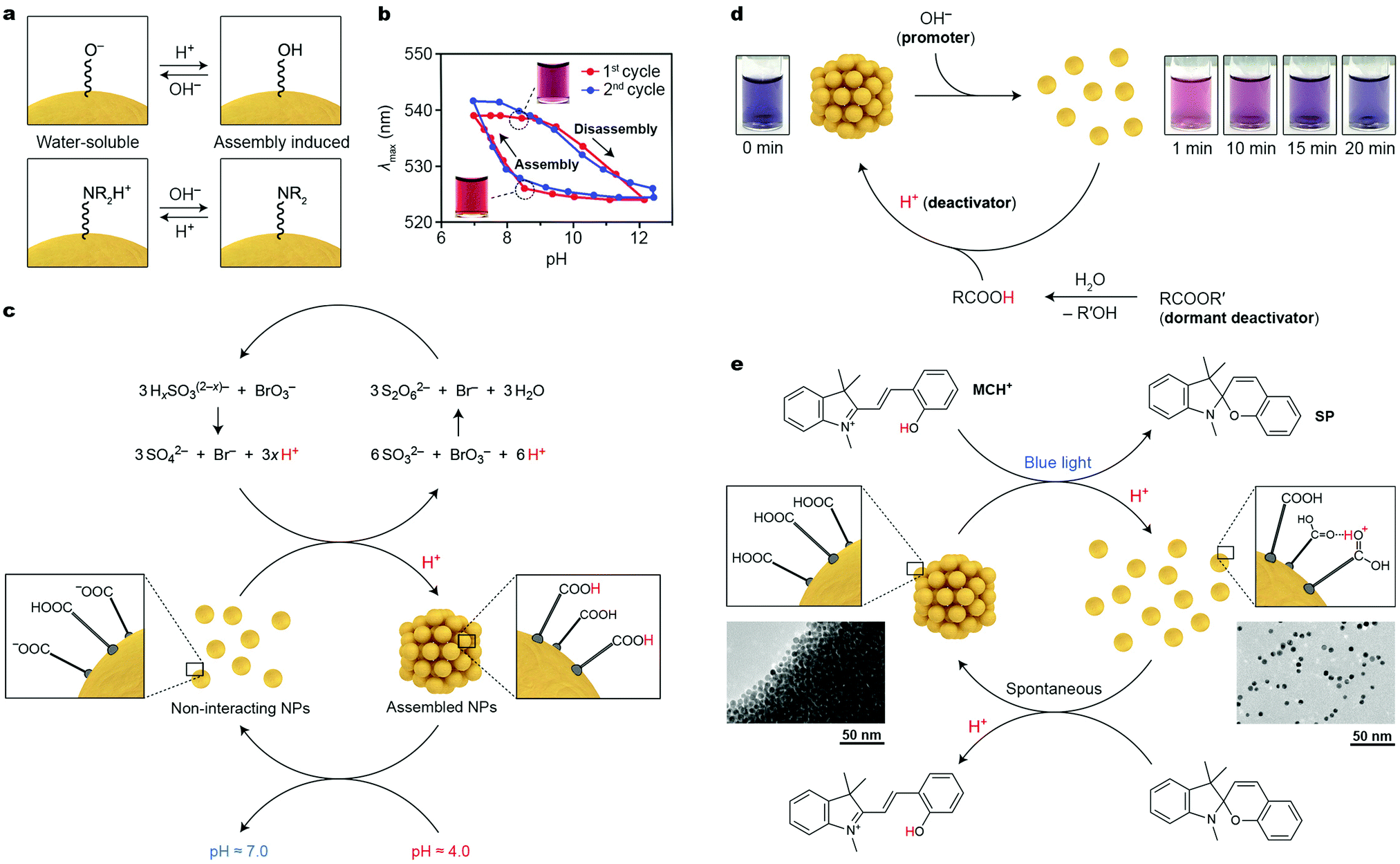 | ||
| Fig. 2 Acid/base-controlled self-assembly. (a) Modulating the interparticle interactions by (de)protonating the terminal groups of nanoparticle-bound ligands. (b) Hysteresis during reversible self-assembly of NPs induced by pH changes. Adapted with permission from ref. 25 (copyright 2010, American Chemical Society). (c) Coupling reversibly self-assembling NPs with the bromate/sulfite oscillator (x = 1, 2). (d) Controlling the lifetime of disaggregated NPs by using a mixture of a promoter and a dormant deactivator. Adapted with permission from ref. 30 (copyright 2015, American Chemical Society). (e) Reversible self-assembly of NPs controlled by a photoacid. Adapted with permission from ref. 31 (copyright 2015, Nature Publishing Group). | ||
A notable feature of many pH-responsive NPs is the ability to exist in two different states (i.e., free and assembled) at the same pH value, depending on the history of the sample.25,28 This bistability is manifested as a hysteresis in pH vs. degree-of-aggregation plots, as illustrated in Fig. 2b. Depending on whether the sample of FMP-protected Au NPs at pH = 8.3 was prepared by raising the pH from 7 or decreasing it from 12, the particles were aggregated or disassembled, respectively. It is remarkable that each of the two states at pH = 8.3 was stable for more than six months.25
Transient and out-of-equilibrium NP assemblies can be engineered by incorporating pH-responsive NPs within media featuring time-varying H+ concentration profiles. Again, an important consideration in designing such systems is that, for reversible self-assembly to take place, the pKa value of the NP-bound acid should fall within the pH window of the medium. In this context, much attention has been devoted to oscillating reactions, which often exhibit large-amplitude pH changes. Nabika and co-workers incorporated Au NPs functionalized with 12-mercaptododecanoic acid (MDA) within the so-called bromate–sulfite oscillator. In this oscillator, oxidation of protonated sulfites with bromate leads to the production of H+; however, once the concentration of H+ reaches a critical value, another redox reaction is initiated, in which protons are consumed (see Fig. 2c), resulting in pH oscillations between pH ∼ 7 and ∼4. At the high pH value, the –COOH groups of MDA are largely deprotonated, and the NPs do not interact with one another, whereas at pH = 4, self-assembly is induced. When the reaction was carried out in a continuously stirred tank reactor (CSTR), oscillations could be sustained indefinitely. Interestingly, the large period of oscillations (∼1 h) characteristic of the bromate–sulfite oscillator allows one to observe the pronounced hysteresis28 described in the previous paragraph. In a related study, FMP-functionalized Au and Ag NPs were integrated with the methylene-glycol/sulfite/gluconolactone (MGSG) oscillator, which exhibits reversible pH changes between ∼6.8 and ∼9.3 (recall that pKa of FMP = 8.3).29 Notably, the oscillation period of the MGSG oscillator is considerably shorter (∼100 s compared with ∼1 h for the bromate–sulfite system), allowing the reversible NP (dis)assembly rate to be modulated.
Walther and co-workers devised an alternative method to control the lifetimes of NP aggregates. Their method is based on simultaneously supplying the system with a “promoter” and a “dormant deactivator”.30 The promoter (typically, a base) triggers a fast self-assembly/disassembly process, whereas a deactivator (an acid), which is slowly generated from its dormant state (an ester), induces the opposite reaction. To demonstrate the proof-of-concept, these researchers worked with small, 4.0 nm Au NPs functionalized with FMP, which were originally in the assembled state. Injecting OH− resulted in the disassembly of NP aggregates (“1 min” in Fig. 2d). However, a dormant deactivator – methyl formamide – introduced at the same time gradually hydrolyzed and the concentration of H+ slowly increased, ultimately resulting in NP reassembly (Fig. 2d). An important feature of this strategy is that the choice of the dormant deactivator (e.g., between the rapidly hydrolyzing ε-caprolactone and the more stable gluconic acid δ-lactone) allows for controlling the lifetime of the out-of-equilibrium state (here, corresponding to free NPs) between minutes and days.
Unfortunately, introducing chemical stimuli (here, acids and bases) to a closed system leads to the accumulation of by-products (such as water and salt), which limits the number of assembly–disassembly cycles. A convenient way to overcome this limitation lies in the use of photoacids and photobases, which can transiently decrease or increase, respectively, the solution pH upon exposure to light. In this context, spiropyran-derived photoacids (e.g., MCH+ in Fig. 2e) are most frequently used. The pH decrease brought about by exposing a solution of MCH+ to blue light is large enough to partially protonate the mercaptoundecanoic acid (MUA) ligands on Au NP surfaces, and to solubilize the NPs in polar, non-aqueous solvents (Fig. 2e) (note that here, the –COOH groups act as bases). Although the pKa of the conjugated acid (i.e., –COOH2+) is very high, it is possible to protonate –COOH groups within dense layers on NP surfaces in solvents such as methanol (see Fig. 2e, right).31 Once light is turned off, the system relaxes to its original state: the closed form of the molecular switch (SP in Fig. 2e) removes the extra H+ from the NP surfaces, and NP reassembly is initiated. The disassembly–assembly sequence can be repeated more than 100 times without any appreciable fatigue.31 Notably, the process is associated with a pronounced color change: whereas in the ground state the NPs are quantitatively aggregated and do not contribute to the color of the sample, the light-exposed sample is deep-red due to free Au NPs. The reversible disassembly reaction readily proceeded, not only in methanolic solutions, but also in methanogels based on crosslinked poly(ethylene glycol). Irradiating these gels locally (through a mask) can be used to create images, which, similar to the metastable NPs in the disaggregated form, persisted only for a specific period of time (ca. three minutes), after which other images could be created in the same piece of gel.
It would arguably be more interesting to use light to induce the formation of transient NP aggregates, rather than to transiently disassemble NP aggregates. To accomplish this goal, the same MUA-protected Au NPs were placed in water. At a high pH and in the presence of a water-soluble version of the photoacid (MCH+ appended with a sulfonate group), a sufficient number of the terminal groups on NP surfaces existed in the –COO− form and the NPs exhibited high solubility.32 Irradiation with blue light, however, induced the release of protons from the sulfonated MCH+, protonation of the NPs, and consequently, their self-assembly. Unfortunately, spiropyran-derived photoacids are hydrolytically unstable, and the number of assembly–disassembly cycles in water was limited to ∼20.32
3. Metal ions
Early interest in assembling plasmonic nanoparticles using metal ions as the stimuli was motivated by the need to develop colorimetric systems for detecting these often-toxic species. The binding of metal ions is usually achieved using carboxylate groups, and the resulting complexes typically display a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 stoichiometry. Provided that the two COO− groups interacting with a metal ion reside on different NPs, the metal ions can be used to bring the particles together, which, for plasmonic NPs, is associated with pronounced color changes owing to coupling of the LSPR modes of neighboring NPs. Short peptides are well suited as NP ligands to achieve this goal: their N-termini can strongly bind to NP surfaces, whereas the exposed C-termini (i.e., carboxylate groups) can interact with metal ions present in the solution. Mandal and co-workers worked with Au NPs functionalized with a H2N-Leu-Aib-Tyr-COO− tripeptide.33 The relatively long chain of this ligand provided sufficient steric as well as electrostatic repulsions to prevent bulk precipitation of the NPs in the absence of metal ions. The balanced interplay between the attractive and repulsive forces also served to facilitate the disassembly of NP aggregates, which could be achieved using a strong chelator for metal ions, such as EDTA.
:2 stoichiometry. Provided that the two COO− groups interacting with a metal ion reside on different NPs, the metal ions can be used to bring the particles together, which, for plasmonic NPs, is associated with pronounced color changes owing to coupling of the LSPR modes of neighboring NPs. Short peptides are well suited as NP ligands to achieve this goal: their N-termini can strongly bind to NP surfaces, whereas the exposed C-termini (i.e., carboxylate groups) can interact with metal ions present in the solution. Mandal and co-workers worked with Au NPs functionalized with a H2N-Leu-Aib-Tyr-COO− tripeptide.33 The relatively long chain of this ligand provided sufficient steric as well as electrostatic repulsions to prevent bulk precipitation of the NPs in the absence of metal ions. The balanced interplay between the attractive and repulsive forces also served to facilitate the disassembly of NP aggregates, which could be achieved using a strong chelator for metal ions, such as EDTA.
Unfortunately, binding divalent metal ions by carboxylates (and generating COO−–M2+–−OOC moieties) neutralizes the charge on the NPs and typically leads to uncontrolled NP precipitation. To tackle this issue, Pillai et al. systematically studied how the colloidal stability of the NPs is affected by the incorporation of different amounts of thiols terminated with positively charged groups (NMe3+) within monolayers of COO−-terminated ligands.34 Upon addition of Pb2+, NPs co-functionalized with 80% of a COO−- and 20% of an NMe3+-terminated thiol formed small aggregates of ∼50 nm in size (demonstrated by dynamic light scattering (DLS), atomic force microscopy, and TEM), which exhibited maximum absorbance at ∼528 nm. Importantly, including the positively charged component within the protective coating was also essential for reversing the self-assembly. The subsequently added NaOH sequestered Pb2+ from NP aggregates, resulting in the disassembly. Within aggregates composed of NPs lacking the positively charged groups, however, Pb2+ ions were bound too tightly and could not be removed using NaOH.34
Self-assembly can be highly directional for NPs whose surfaces are functionalized with patches of metal-ion-responsive ligands. Such site-selective functionalization can be readily achieved in anisotropic NPs; for example, the side faces of CTAB-coated Au nanorods are protected more strongly than their tips, where the ligand exchange reaction occurs preferentially. This property was explored by Newcome et al., who organized rod-shaped gold NPs into one-dimensional assemblies using an Fe2+-based crosslinker (Fig. 3b).35 Because the tips of these nanorods could accommodate multiple crosslinker molecules, self-assembly into strictly one-dimensional assemblies was not possible, and a small population of branched structures was unavoidable (Fig. 3b). Disassembly could be achieved using NaOH, which scavenged the Fe2+ ions. Unfortunately, this process was not reversible: the subsequent addition of Fe2+ did not result in self-assembly, which was attributed to the chelation of Fe2+ by two terpyridine units on the same nanorod tip. Better success was achieved using Cd2+, whose complexes with terpyridine ligands are weaker and more labile. Here, reversible self-assembly of one-dimensional assemblies was possible by mixing equal amounts of Cd2+-free nanorods with ones saturated with the metal salt.
 | ||
| Fig. 3 Inducing nanoparticle self-assembly using metal ions. (a) Self-assembly of peptide-decorated Au NPs into three-dimensional NP networks using divalent metal ions. Disassembly can be induced using a strong chelator for metal ions, such as EDTA. Adapted with permission from ref. 33 (copyright 2008, Wiley-VCH). (b) Tip-to-tip reversible self-assembly of Au nanorods through the formation of metal ion–terpyridine complexes. Adapted with permission from ref. 35 (copyright 2010, Wiley-VCH). | ||
Additional examples of NP self-assembly induced by metal ions are discussed in Section 12, in the context of multi-stimuli-responsive nanoparticles.
4. Gases
Gaseous signals represent a unique type of external stimuli for inducing nanoparticle self-assembly. Gases can usually be removed from the system by mild heating, sonication, or purging with an inert gas; therefore, the assembly–disassembly cycle can be accomplished without accumulating any chemical byproducts (“waste”) in the system. Carbon dioxide is a particularly attractive gaseous stimulus because it is abundant, nontoxic, and biocompatible. Responsiveness to CO2 can be achieved by decorating NPs with amino groups, which can respond to CO2 either “indirectly” (by undergoing protonation; Fig. 4a) or by forming carbamates (Fig. 4d).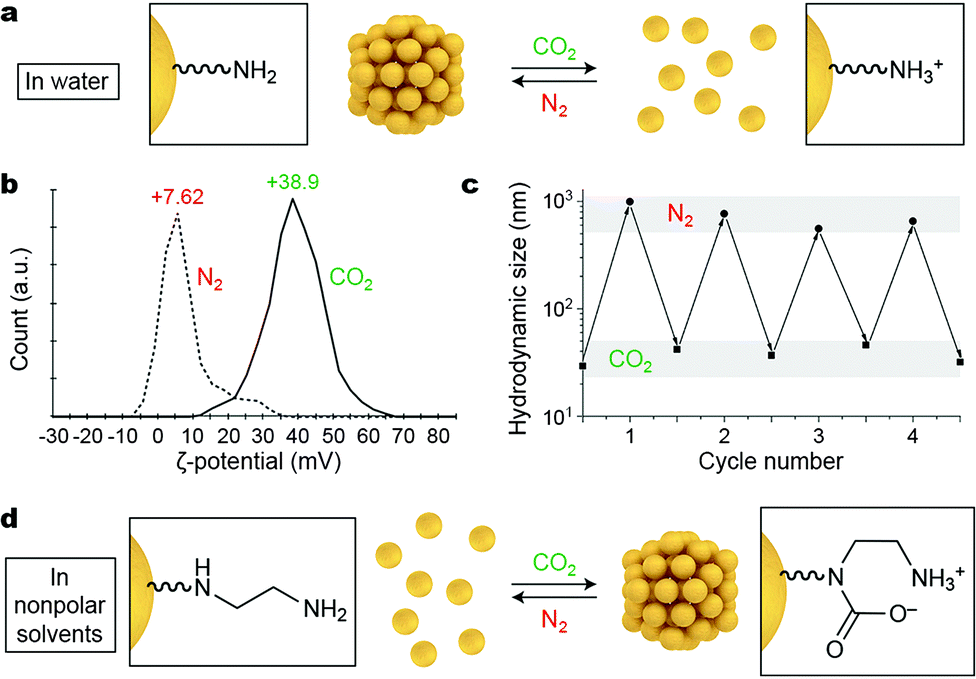 | ||
| Fig. 4 Reversible self-assembly of NPs induced by gaseous signals. (a) Affecting the assembly state of amine-decorated NPs in water by changing the solution pH. (b) Reversible changes in the ζ-potential of 1,8-diaminooctane-functionalized NPs upon purging the system with CO2 and N2. (c) Reversible changes in the hydrodynamic size of the same NPs upon alternately introducing CO2 and N2. Adapted with permission from ref. 36 (copyright 2014, Wiley-VCH). (d) Affecting the assembly state of diamine-decorated NPs in nonpolar solvents by direct binding of CO2. | ||
Lu et al. described an elegant, one-step method to prepare CO2-responsive NPs by reducing iron(III) acetylacetonate in the presence of 1,8-diaminooctane.36 This synthesis affords relatively monodisperse, ∼8 nm iron oxide NPs, which exhibit a low value of zeta (ζ) potential (∼7.6 mV) and are thus poorly soluble in water. Purging the system with CO2 results in partial protonation of the terminal NH2 groups on the NPs (Fig. 4a), leading to a large increase in the ζ-potential of the NPs to ∼+39 mV (Fig. 4b). The resulting NPs exhibit high colloidal stability in water owing to (i) the electrostatic double layer repulsion between like-charged NPs and (ii) efficient hydration of the terminal NH3+ groups. NP reassembly was triggered by purging with N2, which eliminates CO2 from the system and increases the solution pH to its initial value. The reversible disassembly/assembly process could be monitored by DLS, which showed that average particle size oscillated between ∼30 nm and ∼500 nm upon purging with CO2 and N2, respectively (Fig. 4c).36 In a related study, Zhao, Ma, and co-workers synthesized Au NPs functionalized with a thiolated polymer containing multiple tertiary amine groups.37 In the absence of CO2, this polymer was hydrophobic, and the NPs formed an opaque suspension in water. Bubbling CO2 for ca. 20 min produced enough NH3+ groups to render the NPs water-soluble. The subsequent introduction of N2 induced re-assembly of the NPs within a similar time scale.
The profound increase in NP surface polarity upon introduction of CO2 can be used not only to redisperse NPs in water, but can also trigger assembly in nonpolar solvents. This has been demonstrated using NPs decorated with diamine moieties, which have a strong affinity towards CO2 (Fig. 4d).38 Au NPs co-functionalized with an ethylenediamine-terminated thiol and an alkanethiol background ligand were readily soluble in degassed toluene. Interestingly, exposing these NPs to atmospheric CO2 was sufficient to initiate the self-assembly process. Disassembly was shown to proceed efficiently upon gentle heating, purging with inert gases (N2, Ar), or sonicating, and the assembly–disassembly cycle could be repeated multiple times.38
5. Biomacromolecules
Switchability at the molecular level is a central feature in sustaining the functionality of living systems. Virtually all biological processes are governed by chemical signals, rendering biomolecules attractive candidates for developing synthetic systems based on reversibly self-assembling nanoparticles. Great efforts have been devoted to organizing DNA-functionalized NPs of various sizes, shapes, and compositions into well-defined higher-order architectures; however, the use of DNA as input to control NP self-assembly in a reversible fashion has remained largely underexplored. In an early example, Niemeyer and co-workers demonstrated the reversible co-assembly of Au NPs functionalized with two distinct DNA sequences (A and B in Fig. 5a) using single-stranded oligonucleotides as the assembly and disassembly inputs (Fa and Fd in Fig. 5a).39 The Fa oligonucleotide contained a sequence complementary to A (i.e., A′), followed by one complementary to B (i.e., B′), and could therefore act as a crosslinker for the two types of NPs. In addition, Fa contained a short oligonucleotide sequence C′, which remained unhybridized. C′ was crucial in the disassembly step, which was induced by input of Fd with a sequence complementary to Fa (Fig. 5a). Importantly, the assembly–disassembly cycle was successfully repeated multiple times (Fig. 5a, bottom), with the Fa–Fd duplex as the only chemical waste.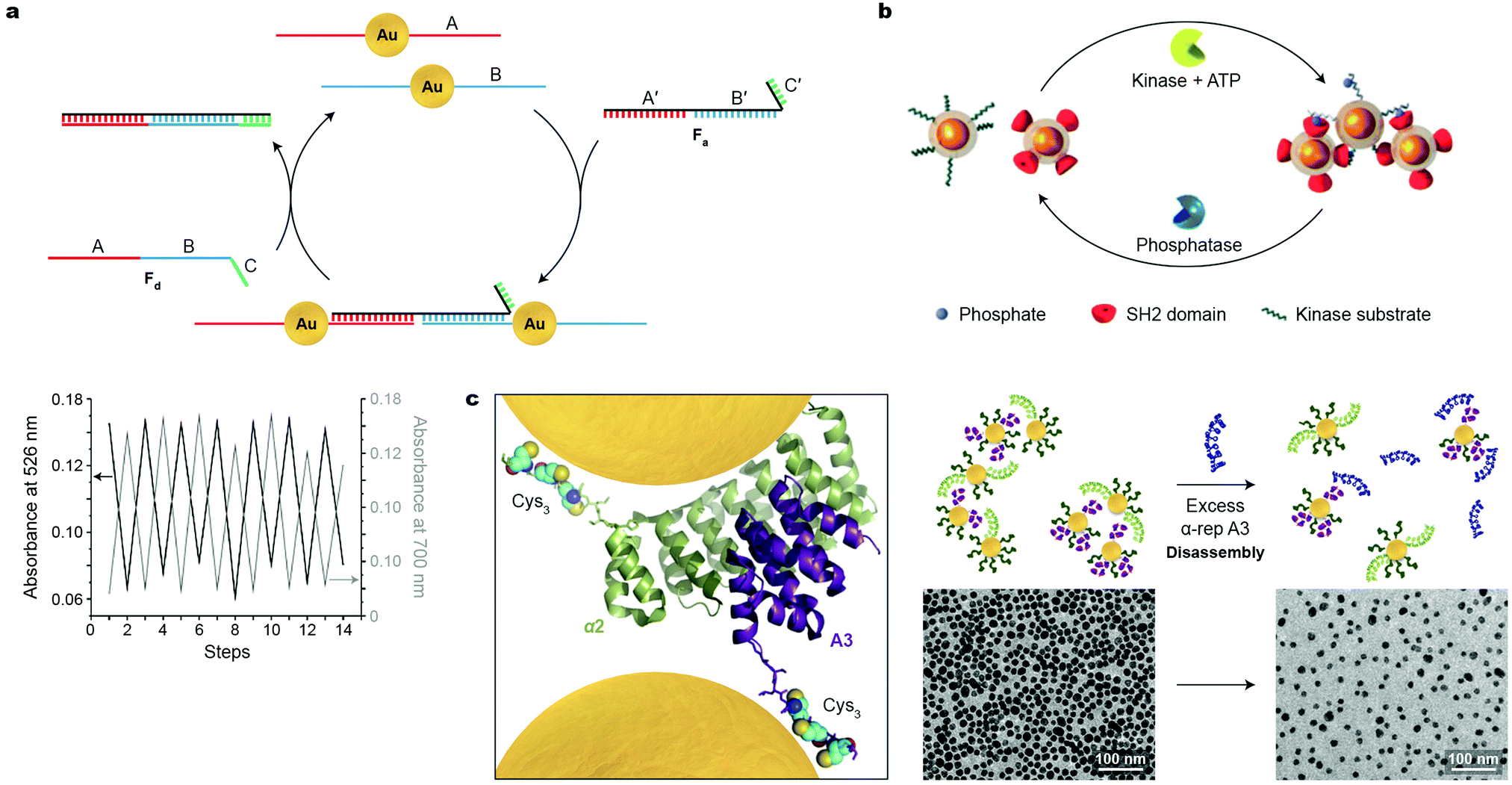 | ||
| Fig. 5 Reversible self-assembly of nanoparticles induced by biomacromolecules. (a) Top: Strategy conceived for reversible assembly of DNA-functionalized Au NPs using oligonucleotides with complementary sequences. Bottom: Reversible self-assembly followed by UV/Vis absorption spectroscopy. Adapted with permission from ref. 39 (copyright 2004, Wiley-VCH). (b) Reversible self-assembly of magnetic NPs controlled by competing enzymatic reactions. Adapted with permission from ref. 40 (copyright 2007, Wiley-VCH). (c) Left: X-ray crystal structure of a tightly interacting native A3–α2 protein pair (Cys3 tags and NPs added for illustration purposes). Right: Schematic illustration (top) and TEM images (bottom) of the disassembly process induced by a competing protein as the stimulus. Adapted with permission from ref. 41 (copyright 2016, American Chemical Society). | ||
Reversible NP self-assembly can also be achieved by employing enzyme-catalyzed reactions. Bhatia et al. devised a creative system based on the classic pair of antagonistic enzymes – tyrosine kinase and phosphatase – as the opposing stimuli to reversibly assemble and disassemble iron oxide NPs.40 This system incorporated two populations of NPs (Fig. 5b): one modified with a peptide chain containing a tyrosine residue prone to phosphorylation, and the other – with SH2 domains, that is, structurally conserved protein domains that recognize and bind to phosphorylated tyrosines. In the presence of kinase and ATP, phosphorylation of the former NPs was initiated, inducing their interactions with the latter type of NPs, which was manifested by a rapid increase in hydrodynamic size. Bringing the superparamagnetic NPs in close proximity to each other induced attractive interactions between their magnetic dipoles, markedly affecting the relaxation of nearby protons, and allowing the protein activity to be followed by magnetic resonance imaging (MRI). Subsequent introduction of phosphatase to the system triggered the removal of the phosphate groups, provided that its activity (a function of concentration) was higher than that of tyrosine kinase. This hydrolysis reaction regenerated the original tyrosine residues, causing the NP aggregates to disassemble.40 These results indicate that antagonistic biochemical processes can be coupled to, and dynamically control, self-assembly of inorganic NPs.
Similar to but to a lesser extent than DNA, various proteins have been used to assemble NPs, although examples of reversible assembly are scarce. An ideal candidate for inducing NP assembly should form strong and selective protein–NP as well as protein–protein interactions. To meet these criteria, Dujardin, Minard et al. employed the so-called α-repeat protein pairs, i.e., pairs of synthetic proteins exhibiting strong (nM) association constants.41 The X-ray crystal structure of one such pair composed of tightly interacting A3 and α2 partners is shown in Fig. 5c, left (in purple and green, respectively). In order to ensure strong attachment to gold NPs, both partners were equipped with a Cys3 handle—a thiol-rich oligopeptide sequence—on their C-termini. Co-assembly of A3-Cys3-functionalized Au NPs, 11 nm in diameter, with similarly sized α2-Cys3-functionalized NPs at a protein–NP molar ratio of 30:1 afforded large-area, freestanding, one-NP-thick films (Fig. 5c, center). Interestingly, decreasing the protein–NP ratio to 20:1 resulted in small, oligomeric raft-like NP assemblies.41 The formation of the anisotropic, 2D assemblies was attributed to the electrostatic repulsion between like-charged proteins grafted on the NPs. Importantly, this process was successfully reversed—and a solution of free NPs was regenerated—by adding a ten-fold excess of free A3 protein (Fig. 5c, right). Although this methodology can be extended to prepare assemblies of differently sized and shaped NPs, and to other artificial tightly interacting protein pairs, it remains a formidable challenge to increase the number of assembly–disassembly cycles.
6. Redox reactions
An important issue to consider when designing redox-responsive nanoparticles is the chemical sensitivity of the NP–ligand interface. For example, the thiolate–gold linkage can be broken in both oxidizing and reducing environments. Fortunately, a variety of redox-switchable organic moieties can be cycled between the reduced and the oxidized form within a narrow window of potentials, under which ligand-protected NPs remain unaffected. Stoddart, Grzybowski et al. synthesized metallic (gold, silver, palladium, and platinum) NPs functionalized with the electron-rich tetrathiafulvalene (TTF),42–44 which has a high affinity towards the electron-deficient macrocycle cyclobis(paraquat-p-phenylene) (CBPQT4+), forming a TTF⊂CBPQT4+ host–guest inclusion complex in polar aprotic solvents, such as MeCN (Fig. 6a). Additionally, TTF can be reversibly converted between neutral and dicationic (TTF2+) forms under mild conditions. Once oxidized, TTF loses its affinity to the tetracationic CBPQT4+, and the host–guest inclusion complex disassembles, but it can be reformed after adding a reducing agent (Fig. 6a).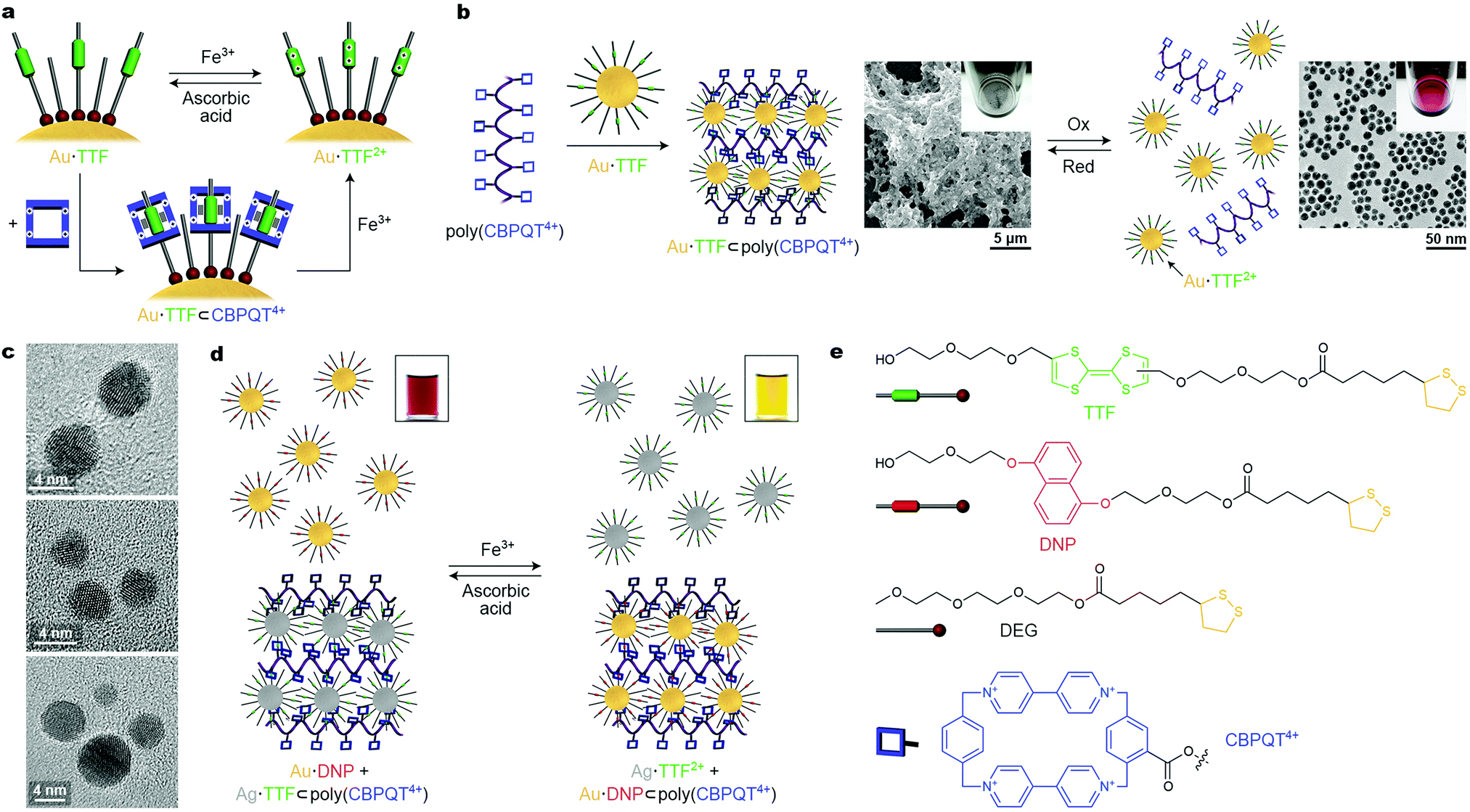 | ||
| Fig. 6 Controlling nanoparticle self-assembly by redox signaling. (a) Reversible modulation of the oxidation state of TTF (top) and reversible formation of a pseudorotaxane (bottom) on the NP surface. (b) Crosslinking TTF-functionalized Au NPs with a polymer containing pendant CBPQT4+ moieties (left) and redox-controlled disassembly and reformation of the crosslinked material (right). (c) TEM images of redox-responsive Au NP dimers, trimers, and tetramers obtained by reacting “monofunctionalized” (i.e., containing only one TTF ligand) NPs with a CBPQT4+ dimer, trimer, and tetramer, respectively. (d) Redox-controlled “sponge” for NPs allows for redox-controlled release of NPs of one type (here, TTF-functionalized Ag NPs), concomitant with sequestering other NPs (here, DNP-functionalized Au NPs). (e) Structural formulas of the key building blocks of redox-switchable NP assemblies. Adapted with permission from ref. 42 (copyright 2009, Nature Publishing Group) (a, b, d and e) and ref. 43 (copyright 2009, American Chemical Society) (c). | ||
To employ the redox-controlled inclusion complex formation for NP self-assembly, a linear polymer containing multiple pendant CBPQT4+ units, i.e., poly(CBPQT4+) was synthesized.42 Owing to its multiple TTF binding sites, poly(CBPQT4+) acted as a “glue” for TTF-functionalized gold NPs (Au·TTF in Fig. 6, whereby the number of TTF groups per NP could be controlled by dilution with a “dummy” ligand, DEG in Fig. 6e). Upon mixing Au·TTF with poly(CBPQT4+), hierarchical self-assembly of the NPs commenced: initially, short chains comprising 4–5 NPs were observed; interestingly, the length of these chains coincided with the length of an individual poly(CBPQT4+) molecule in its extended conformation. These NP chains further aggregated to afford elongated structures with a width of ∼50 nm. Eventually, this process led to a crosslinked NP network as a black precipitate (Fig. 6b, center). Upon addition of an oxidant (e.g., Fe3+), a near-instantaneous disassembly of the NPs was triggered as a result of the TTF → TTF2+ reaction, and a red solution of non-interacting NPs was recovered (Fig. 6b, right). This process was reversible: the addition of a reducing agent (e.g., ascorbic acid) induced the formation of the initial Au·TTF NPs and NP assembly process was re-initiated (and took about 20 min to complete). Although this procedure could be repeated only a few times due to accumulation of by-products, reversible disassembly/reassembly of NPs could also be achieved electrochemically at a potential of +0.9 V and 0 V (vs. Ag/AgCl), respectively, over multiple cycles.42 The reversible assembly of NPs driven by redox-switchable host–guest interactions was also achieved using “monofunctionalized” NPs, each containing only one molecule of the functional (i.e., TTF) ligand on its surface. By reacting these NPs with molecules containing two, three, and four CBPQT4+ groups, discrete redox-responsive NP dimers, trimers, and tetramers were obtained (Fig. 6c).43
The polymer-based system was extended to include NPs of another type, and to develop the concept of a redox-switchable “sponge” for NPs. Once the TTF moieties are oxidized to TTF2+ and their host Au NPs are dissociated from the polymer (see Fig. 6b, right), the CBPQT4+ groups on the polymer chain can be engaged in a different type of host–guest interaction, e.g., with another electron-rich guest, dialkoxynaphthalene (DNP). Although the affinity of CBPQT4+ to DNP is significantly lower than to TTF, DNP cannot be easily oxidized and in an oxidative environment, the DNP⊂CBPQT4+ complex prevails (i.e., the binding strength of CBPQT4+ follows the order TTF > DNP > TTF2+). Therefore, when a complex of TTF-functionalized Ag NPs with poly(CBPQT4+) was treated with an oxidizing agent in the presence of DNP-functionalized Au NPs, the release of Ag·TTF2+ was accompanied by the capture of Au·DNP, with the polymer remaining in the precipitated form where it indeed acted as a “sponge” for NPs (Fig. 6d). Upon the addition of a reducing agent, the opposite reaction was observed: poly(CBPQT4+) regained its high affinity to Ag·TTF, which displaced Au·DNP in the solid phase, and the solution assumed its original red color, characteristic of dispersed gold NPs.
7. Other small-molecule stimuli: dynamic covalent chemistry on nanoparticle surfaces
As we have seen so far, reversible, stimuli-responsive self-assembly of nanoparticles typically relies on the formation of noncovalent interactions between the NPs – either via direct NP–NP interactions, or, in the case of mediated self-assembly, using noncovalent crosslinkers. An alternative approach was proposed by Borsley and Kay, who demonstrated that Au NPs could be assembled covalently yet reversibly using boronate ester chemistry.45 As building blocks, these researchers used small, ∼3.8 nm Au NPs functionalized with thiols terminated with boronic acid moieties, which can form robust covalent linkages with 1,2-dihydroxybenzenes (catechols) (Fig. 7a). Upon the addition of different ditopic catechols, NP aggregation commenced (Fig. 7b), which eventually led to an insoluble material. Treating the resulting precipitate with a different chemical stimulus, namely, monofunctional catechol, broke the covalent bridges between the NPs, and a solution of free particles could be obtained (Fig. 7a, right). Notably, both assembly and disassembly proceeded quantitatively. Unfortunately, it took five days for the NPs to precipitate, and as many as 42 days to redissolve upon addition of catechol. Interestingly, when the disassembly process was quenched after 35 days, a colloidally stable solution of small NP aggregates, ca. 20 nm in diameter, was obtained. Other drawbacks of this method include limitations in the choice of solvent (a MeOH–CH2Cl2 mixture) and the necessity to use a large excess of base (∼1 M N-methylmorpholine). Nevertheless, the use of dynamic covalent chemistry in the context of inducing interparticle interactions remains largely unexplored and holds potential to greatly expand and enrich the existing toolbox of available stimuli.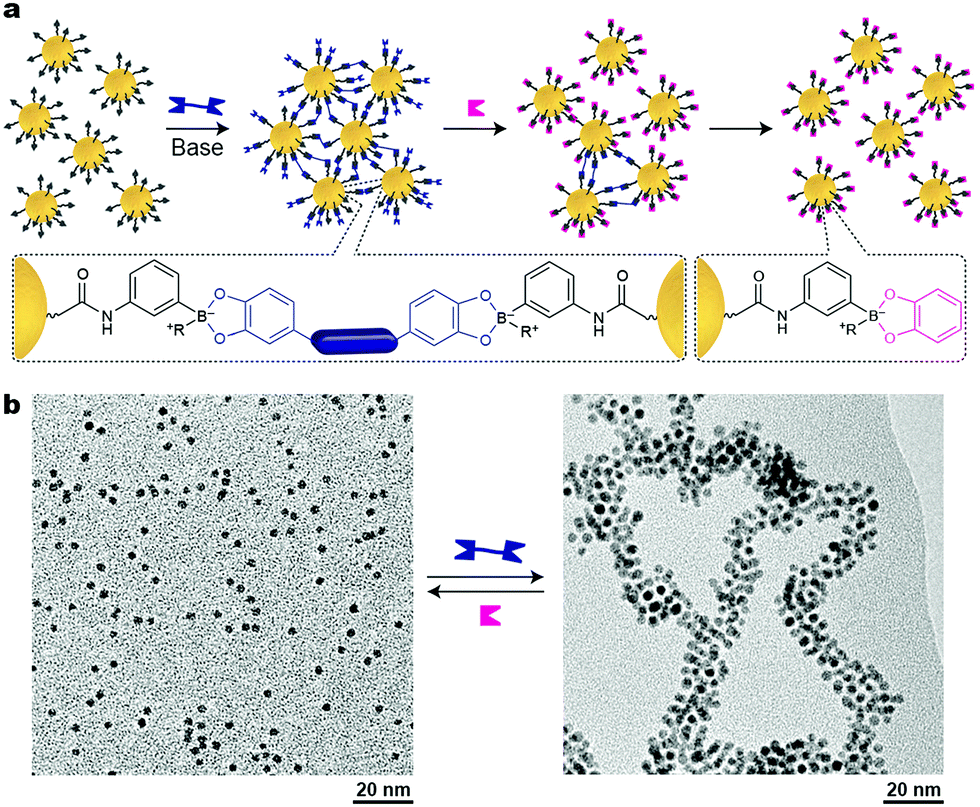 | ||
| Fig. 7 Nanoparticle aggregation driven by dynamic covalent chemistry. (a) NP assembly followed by disassembly induced by a ditopic and a monotopic boronic acid, respectively. (b) TEM images of free and covalently assembled Au NPs. Adapted with permission from ref. 45 (copyright 2016, Royal Society of Chemistry). | ||
8. Temperature
Temperature is a readily accessible stimulus that can be delivered and removed rapidly and often in a noninvasive manner. Thermally responsive nanomaterials are typically designed by functionalizing NPs with molecules exhibiting large changes in properties (e.g., conformation and intermolecular interactions) upon varying the temperature. Recently, Macfarlane et al. proposed the concept of “nanocomposite tectons”—gold NPs grafted with dense monolayers of polymer chains terminated with complementary recognition units—capable of self-assembling into colloidal crystals (Fig. 8a).46 The terminal groups on these NPs—diaminopyridine and thymine—interact with each other strongly by hydrogen bonding in nonpolar solvents and can guide the self-assembly process upon slowly decreasing the solution temperature. This strategy could be applied to Au NPs having different core sizes (10–40 nm) and polymer molecular weights (3.7–11 kDa). By systematically varying the NP core diameter as well as the length of the polymer chain, the authors gained interesting insights into the thermodynamics of self-assembly. For example, increasing the polymer molecular weight (on 10 nm NPs) from 3.7 to 11 kDa led to a drastic decrease (80 → 25 °C) in the disassembly temperature. These results were rationalized using both enthalpic and entropic arguments: first, increasing the polymer length decreases the density of the binding groups, weakening the interparticle interactions; second, a relatively high entropic penalty is associated with assembling NPs decorated with long polymer chains.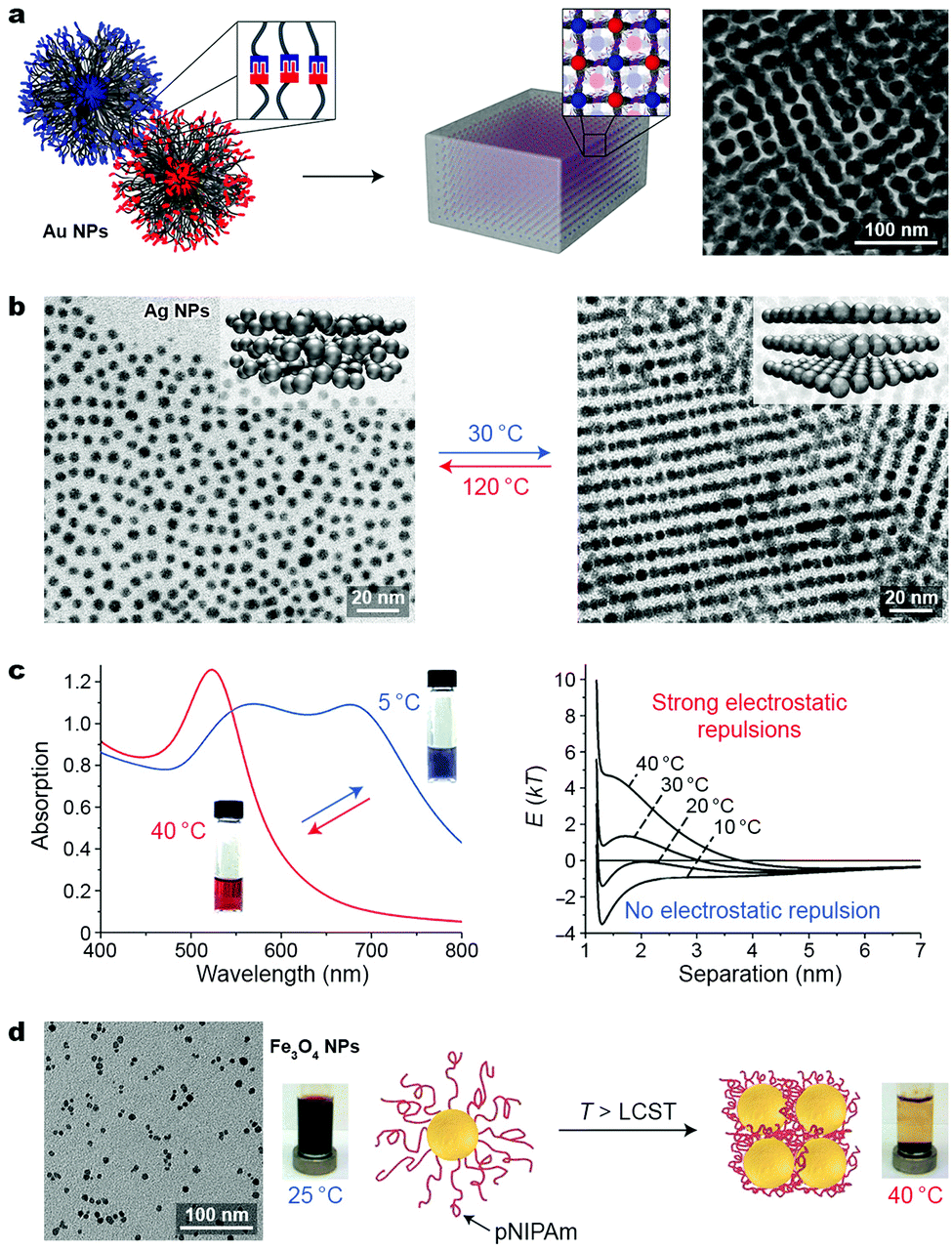 | ||
| Fig. 8 Temperature-controlled self-assembly of nanoparticles. (a) Three-dimensional arrangement of polymer-grafted Au NPs through reversible hydrogen bond formation between terminal functional groups: thymine and diaminopyridine. Adapted with permission from ref. 46 (copyright 2016, American Chemical Society). (b) Temperature-induced transition between isotropic (left) and lamellar (right) phases of Ag NPs grafted with liquid-crystalline ligands. Adapted with permission from ref. 47 (copyright 2015, Nature Publishing Group). (c) Temperature-induced change in the optical properties (left) and interaction potentials (right) between negatively charged gold NPs embedded in agarose gel. Adapted with permission from ref. 48 (copyright 2012, Wiley-VCH). (d) Self-assembly and magnetic separation of magnetite NPs grafted with thermoresponsive polymer chains. Adapted with permission from ref. 50 (copyright 2011, American Chemical Society). | ||
Taking advantage of the temperature-induced phase transition in liquid crystals (LCs), Lewandowski, Górecka et al. fabricated thermally responsive silver NPs by decorating their surfaces with LC-like molecules.47 Thin films of these NPs were studied using temperature-dependent SAXS. Above 100 °C, an isotropic distribution of the metallic cores in the films was observed (Fig. 8b, left), with a mean interparticle distance of 7.2 nm. Upon cooling to 30 °C, the NPs self-assembled into structures with long-range order, composed of stacked layers of NPs (see the inset in Fig. 8b, right), with an in-layer NP–NP distance of ∼6.1 nm and an interlayer distance of ∼8.4 nm, as evidenced by SAXS and TEM. This structural transformation translated into a relatively large difference in the optical properties of the materials: whereas the original film exhibited maximum absorption at 446 nm (approaching that for a suspension of non-interacting Ag NPs), a slow decrease in temperature (within tens of minutes) resulted in a significant (20 nm) red-shift of the absorption band, as a result of LSPR coupling. A smaller but still significant (14 nm) red-shift was observed when the temperature decreased more rapidly (tens of seconds). Importantly, these materials enable continuous tunability of plasmonic properties, as opposed to switching between two defined states (typically free and assembled NPs), which characterizes most switchable systems.
Yin and co-workers proposed a different mechanism to control the optical properties of NP-based materials. Specifically, they embedded negatively charged, 15 nm gold NPs inside agarose gels.48 At 40 °C, the gel was red, characteristic of non-interacting NPs. By decreasing the temperature to 5 °C, NPs self-assembled into linear chains as a result of the competition between attractive van der Waals forces and repulsive electrostatic interactions, giving rise to a blue gel (Fig. 8c, left). Although reversible self-assembly could also be achieved in solution, only inside the gel was uncontrolled aggregation and precipitation of the NPs prevented. Using theoretical modeling, the authors showed that the self-assembly process was governed by temperature-dependent changes in the ζ-potential of the NPs, whereas attractive vdW forces remained constant over the investigated temperature range (Fig. 8c, right).48
Increasing temperature can be used not only to induce disassembly of NP aggregates – it can also trigger NP self-assembly. For example, an equimolar mixture of positively and negatively charged Au NPs (6–7 nm in diameter) was stable in a high ionic strength solution (∼2 M salt) at 10 °C, but aggregation commenced upon warming the sample up to 40 °C.49 These particles were functionalized with thiols terminated with the NMe3+ and COO− groups, respectively, and their high colloidal stability at a low temperature was due to the stabilization of the charged groups by the dissolved salt. An increase in temperature resulted in desorption of the stabilizing ions from the NP ligands, which activated electrostatic interactions between the particles. The process was reversible and cooling the system led to the original solution of dispersed NPs.49
More attention has recently been devoted to NPs coated with thermoresponsive polymers, most notably poly(N-isopropylacrylamide) (pNIPAm). Thermoresponsive polymers are characterized by a lower critical solution temperature (LCST; ∼37 °C), above which they precipitate from aqueous solutions. Below the LCST, pNIPAm chains are readily solvated by water molecules. As temperature increases, hydrogen bonds between pNIPAm's amide groups and water molecules weaken and the relative contribution of hydrophobic interactions between the isopropyl groups increases. This results in the expulsion of water and collapse of pNIPAm chains into a globular structure. Davis and co-workers studied the behavior of ∼8 nm magnetite NPs coated with a poly(N-isopropylacrylamide-co-Nile Red) copolymer.50 At room temperature, the polymer chains were hydrated and the NPs were stable in solution, even in the presence of a magnet (Fig. 8d, left). When heated above the LCST, however, the particles lost their hydration layers and were rapidly removed from the solution (Fig. 8d, right). Owing to the magnetic nature of the NPs, this process could also be followed by NMR relaxometry, where the reduction of the T2 relaxation times was observed at increased temperatures, indicative of NP aggregation. Moreover, the fluorescent dye included in the NP coating could be used to follow the phase transition fluorometrically: collapse of the polymer chains resulted in dye aggregation, and consequently, a precipitous decrease in fluorescence intensity. In a related study, copolymers of N-isopropylacrylamide and acrylamide with different LCSTs (51 °C and 65 °C) were used to control the assembly of Au NPs on which they were grafted.51 Notably, this strategy was applicable to Au NPs with sizes ranging from ∼10 to ∼50 nm.
Unfortunately, temperature-induced phase transition of NP-grafted thermoresponsive polymers typically leads to uncontrolled NP precipitation.42 To address this shortcoming and to prepare colloidally stable aggregates, Lequeux, Sanson et al. studied the temperature-induced aggregation behavior of polymer-coated Au NPs in the presence of free polymer chains in solution.52 In this study, 6 nm NPs were functionalized with another thermoresponsive polymer, statistical poly(EOx–POy), where EO = ethylene oxide and PO = propylene oxide. Depending on the EO/PO molar ratio, the aggregation temperature of these NPs was controlled in the range 12–38 °C. To prevent uncontrolled aggregation of the NPs above the LCST, thermosensitive triblock copolymer Pluronic 123 (P123) was added to the solution. P123 exhibits a critical micellization temperature of 21 °C, above which solvated chains of P123 assembled into well-defined micelles, which stabilized the NPs if the NP-grafted polymers were also dehydrated. For example, switching the temperature of a solution of Au NPs coated with poly(EO75–PO25) containing 1% P123 between 15 °C and 35 °C changed its λmax between ∼521 nm and ∼538 nm in a highly reversible fashion. Interestingly, the sizes of the NP aggregates could be controlled by the amount of added P123; in the presence of a large excess of the polymer, individual NPs were stabilized even above the LCST and no assembly was observed. Yet another approach to produce colloidally stable NP aggregates was devised by Kaplan and co-workers, who functionalized 12 nm Au NPs with recombinant silk-elastin-like protein polymer (SELP) chains.53 These chains were composed of a repeating elastin-like pentapeptide sequence, GVGVP, and a silk-like hexapeptide sequence, GAGAGS, and provided the NPs with a 13 nm-thick coating. Upon heating to 50–60 °C, hydrogen bonds were formed between the valine residues within the elastin-like block, resulting in a conformational change from hydrophilic random coils to hydrophobic β-turns, inducing attractive interactions between the NPs. The silk-like sequences, on the other hand, served to stabilize the NP aggregates in solution. A solution of non-interacting NPs was obtained upon cooling the solution to room temperature.
9. Magnetic fields
A significant advantage of using an external magnetic field as the stimulus for NP self-assembly is that it can be delivered instantaneously and through boundaries that may be impermeable to chemical agents or light. Moreover, depending on the setup used, a magnetic field can be applied throughout the entire sample or locally, to a precise location. In order for NPs to respond to relatively weak external magnetic fields, their inorganic cores must contain atoms of ferromagnetic metals, such as iron, nickel, or cobalt. This is a notable difference compared with the systems described in the previous sections, whereby the NPs’ responsiveness to external stimuli stemmed from organic ligand monolayers on the NP surfaces, rather than being “encoded” in their inorganic cores. Therefore, functionalizing magnetic NPs with molecules that respond to other stimuli serves as a facile route toward multi-stimuli-responsive NPs; such systems are discussed in Section 12.In ferromagnetic materials, magnetic spins on neighboring atoms are coupled, giving rise to high values of magnetic susceptibility. If particles from these materials are small enough (e.g., <50 nm for magnetite, Fe3O4), all spins are oriented in the same direction, giving rise to one giant magnetic dipole moment (“superdipole”, see Fig. 9a). Depending on their size, these single-domain NPs can be either ferromagnetic or superparamagnetic (SPM)—in the former case, the particles readily attract one another and spontaneously precipitate from the solution. In the latter case, however, the dipole moments are free to rotate and the particles can be readily solubilized in various solvents.
 | ||
| Fig. 9 Magnetic-field-induced self-assembly of superparamagnetic NPs. (a) Scheme illustrating the reversible self-assembly of SPM NPs using an external magnetic field. (b) Self-assembly of linear chains of magnetite NPs into thick bundles and, (c) TEM images illustrating the reversible self-assembly process. Adapted with permission from ref. 55 (copyright 2015, American Chemical Society). (d) Magnetite NPs functionalized with monolayers of a first-generation MacMillan imidazolidinone organocatalyst as a magnetically recyclable catalyst. Adapted with permission from ref. 56 (copyright 2012, American Chemical Society). | ||
The behavior of SPM NPs in response to an external magnetic field is illustrated in Fig. 9a. Upon applying a field to a stable suspension on non-interacting SPM NPs (Fig. 9a, left), the dipole moments of these NPs tend to align in the direction of the applied field (Fig. 9a, center) and start interacting with one another by means of magnetic dipole–dipole interactions. Provided that the energy of these interactions overcomes the thermal energy, self-assembly of NPs into one-dimensional chains occurs (Fig. 9a, right). Importantly, this process is reversible: once the field is turned off, the NPs’ dipole moments randomize and the aggregates disassemble. The assembly–disassembly cycle can, in principle, be repeated indefinitely.
Whereas subjecting SPM NPs to a homogeneous magnetic field can lead to a stable suspension of linear NP aggregates, applying a field gradient can be used to remove the NPs from solution. This was demonstrated by Colvin et al., who studied the behavior of differently sized (4–20 nm) magnetite NPs in a commercial magnetic separator.54 Interestingly, they showed that the efficient removal of NPs from solution using a low-gradient separator could not be explained by a simple model of non-interacting particles, but that it was facilitated by the high-field gradients developed at NP surfaces, which triggered attractive interparticle interactions. In addition, this study highlighted the importance of NP size for efficient yet reversible aggregation. For particles smaller than 9 nm, NP removal was cumbersome because high-strength fields had to be applied to induce interparticle interactions and to achieve NP aggregation. When, on the other hand, the particles were larger than 12 nm, NP recovery from the separator column was incomplete even after removing the field. These findings were applied to remove toxic arsenic ions from water; the strategy could be applied to As(III) and As(V), both of which can strongly interact with nanocrystalline Fe3O4.54
Reversible, magnetic field-induced self-assembly can be applied not only to individual SPM NPs, but also to aggregates thereof. An interesting example of hierarchical self-assembly using magnetic forces was reported by Aida and co-workers, who encapsulated small (2–4 nm) Fe3O4 NPs within a mutant of chaperonin GroEL.55 By decorating the portal regions of GroEL with metal ion-binding moieties, supramolecular polymerization of the protein into one-dimensional, micron-long fibers could be achieved in the presence of Mg2+ (Fig. 9b). Once subjected to an external magnetic field, these fibers assembled into thick bundles, which spontaneously disassembled when the field was removed (Fig. 9b and c). Importantly, the protein jacketing was essential toward achieving reversible self-assembly: applying the same magnetic field to a solution of Fe3O4 NPs lacking the protein coating resulted in aggregates, within which the strong interparticle interactions prevented disassembly. An intriguing possibility of applying these reversibly self-assembling fibers for controlling biological events has been suggested.55
Another important application of SPM NPs lies in recyclable catalysis. In an illustrative example, Pericàs and co-workers immobilized the first-generation MacMillan imidazolidinone catalyst onto ∼5 nm Fe3O4 NPs using a silane anchoring group (Fig. 9d).56 The supported imidazolidinone efficiently catalyzed a model Friedel–Crafts alkylation of N-substituted pyrroles with a range of α,β-unsubstituted aldehydes, and the resulting products could be obtained with up to 98% yield and with up to a 91% enantiomeric excess. An important feature of this strategy is that the catalyst could be quantitatively removed from the reaction mixture by magnetic decantation. Notably, the catalytic NPs could be reused for six consecutive runs with only a small decrease in reaction yield and virtually no decrease in enantiomeric excess.56
10. Electric fields
The use of electric fields as external stimuli to control the dynamic self-assembly of nanoparticles remains highly unexplored. Only during the last couple of years two interesting experimental examples have been reported, where the authors demonstrated that an external electric field can maintain the nanoparticles in the assembled state. It was demonstrated that an electrochemical liquid–liquid two-phase system comprising immiscible electrolytes is an exciting playground to study nanoparticle self-assembly in the presence of an electric field.57 The interface between the liquids became permeable to ions once a sufficiently high voltage was applied across it. The direction of the electric field is therefore a trigger to form the array, since it can either push the nanoparticles toward the interface or move them away depending on the field direction.57 Montelongo et al. experimentally realized electro-tunable arrays based on the reversible self-assembly of 12-mercaptododecanoic acid-stabilized plasmonic gold nanoparticles at a liquid–liquid interface comprising water and 1,2-dichloroethane.58 Both the aqueous and the organic phase contained ions (10 mM) from NaCl and tetrabutylammonium tetraphenylborate, respectively. Upon the application of negative polarization (−0.2 V) for the aqueous phase relative to the organic phase, the NPs accumulated at the interface, which resulted in a redshift and enhancement of the reflectance maximum. The positive polarization (+0.2 V), on the other hand, pushed NPs back into the bulk, blueshifting and damping the maximum in the reflectance spectrum. An interesting feature of this system was the possibility of fully recovering the initial optical properties. The authors constructed a switchable window–mirror device that underwent a transition from transmissive to reflective states, at negative (mirror) and positive (window) potentials.Precise control over the use of electric fields to produce colloidal crystals has been demonstrated by Yu et al.59 The authors used silver nanocrystals of 6.6 nm diameter, coated with 1-dodecanethiol in toluene, to form superlattices in the presence of an applied electric field of 400 V cm−1 on gold-coated electrodes. Once the electric field was turned off, the superlattices dissolved and by turning it back on the superlattices could be regrown. The authors observed that the nanoparticles initially formed superlattices exclusively on the cathode. However, the presence of Br− ions (0.7 mM) neutralizing the surface charge resulted in no superlattice formation. Above 1 mM of Br− ions, the surface charge was reversed to negative, leading to the formation of superlattices on the positively charged electrode. At even higher concentration of Br− ions (1.6 mM), well-defined polyhedral colloidal crystals (∼10 μm) of FCC structure formed within 20 minutes. The mechanism behind the electric field-driven reversible crystal formation involved the following steps: generation of charged nanocrystals that migrated toward the oppositely charged electrode through electrophoretic forces followed by gradual neutralization by charge transfer from the particles to the electrode; accumulation of the neutralized nanocrystals close to the electrode surface until reaching a balance between their diffusion toward the bulk solution; and the electrophoretic force. Such a subtle balance between the electrophoretic force toward the electrode and the diffusion of the nanoparticles toward the solution required a threshold (of 40 V cm−1) to reach the saturation state and consequently crystal formation.
11. Light
Similar to a magnetic field, light is an external stimulus that can be delivered instantly, to precise locations, and to closed systems. An additional advantage of light is that it can be applied in the form of different wavelengths, which can potentially trigger different reactions. A simple way to render NPs photoresponsive is to functionalize their surfaces with monolayers of ligands terminated with light-switchable moieties. Most work to date has focused on thiolated azobenzenes, which can form robust monolayers on the surfaces of metallic NPs, making them soluble in hydrophobic solvents (such as toluene or hexane), owing to the nonpolar nature of the trans isomer of azobenzene (Fig. 10a, left). Upon exposure to low-intensity UV light, azobenzene rapidly isomerizes to its metastable cis configuration, which is significantly more polar than the trans form (Fig. 10a, right).50 As a result, the particles lose their colloidal stability and spontaneously assemble into aggregates comprising thousands to millions of individual NPs (e.g., Fig. 10b). Similar to cis-azobenzene, these aggregates are metastable and gradually disassemble once UV irradiation is discontinued. Owing to the chemical robustness of the azobenzene switch, the assembly–disassembly sequence can be repeated more than one hundred times without appreciable fatigue.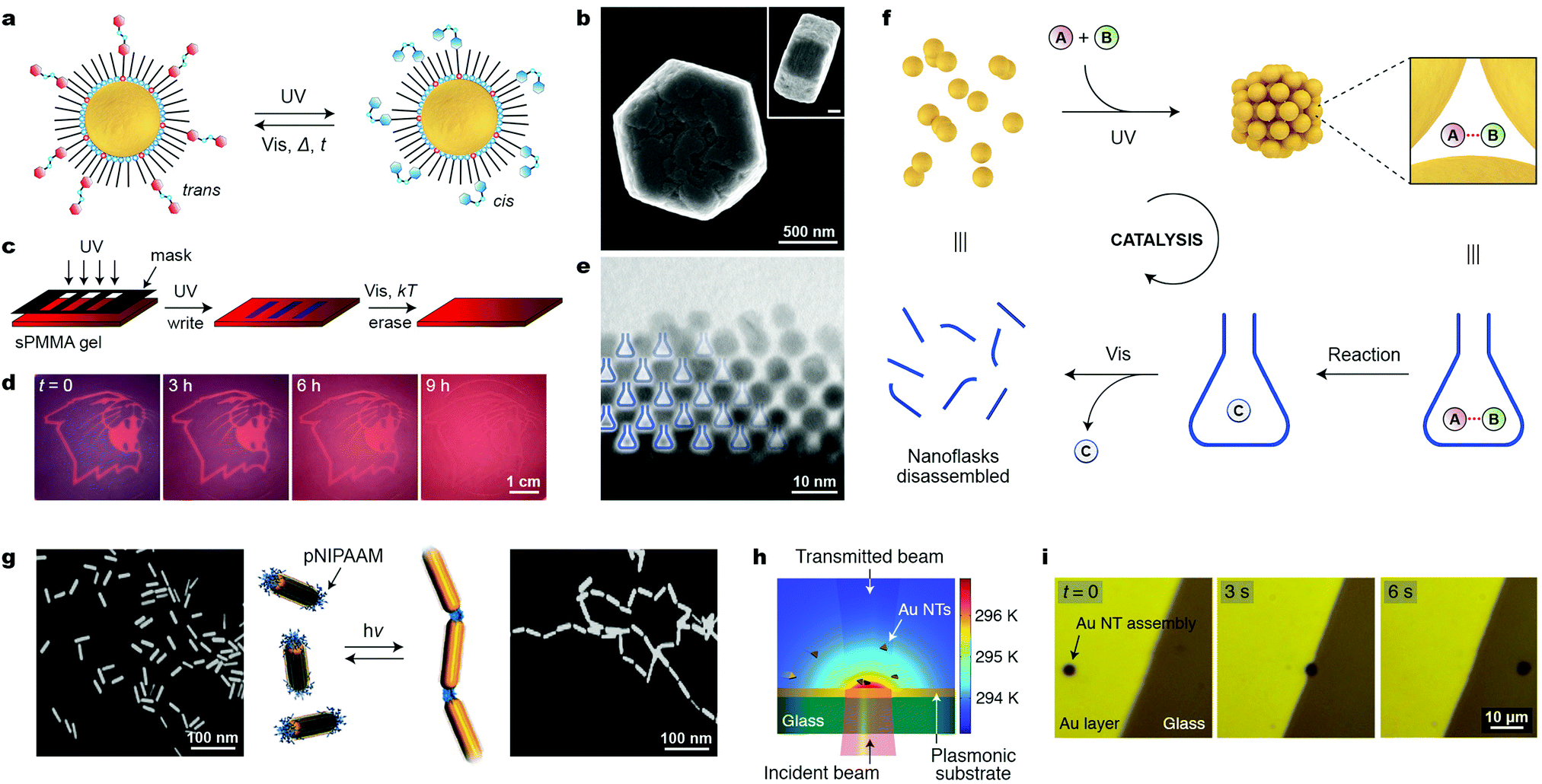 | ||
| Fig. 10 Controlling the assembly of nanoparticles using light. (a) Photoinduced isomerization of azobenzene on a nanoparticle surface. The trans → cis isomerization is accompanied by a large change in NP surface polarity and it can initiate the self-assembly process. (b) SEM image (top view) of a typical aggregate obtained from 5 nm Au NPs. The inset corresponds to a side view (scale bar, 200 nm). (c) Creating transient images using metastable NP aggregates as the “ink”. (d) Gradual (within 9 h) disappearance of an image accompanying the spontaneous disassembly of NP aggregates. (e) TEM image of an edge of a NP aggregate, whereby the nanopores between the NPs are represented as “nanoflasks”. (f) Schematic illustration of how the reversible formation of nanopores within NP aggregates can accelerate a chemical reaction. (g) Light-controlled collapse of thermoresponsive polymers at the tips of gold nanorods drives their self-assembly. (h) Self-assembly of gold nanotriangles (NTs) driven by light-induced thermophoresis. (i) Light-controlled stabilization and long-distance optical manipulation of an ensemble of Au NTs. Adapted with permission from ref. 60 (copyright 2016, Nature Publishing Group) (b, e and f), ref. 68 (copyright 2009, Wiley-VCH) (c and d), ref. 65 (copyright 2009, Royal Society of Chemistry) (g), and ref. 67 (copyright 2016, American Chemical Society) (h and i). | ||
For plasmonic NPs, the assembly process is accompanied by LSPR coupling, leading to color change. By exposing the sample to light locally (e.g., through a mask), it is possible to create high-contrast patterns, a finding that led to the development of “self-erasable” NP films (Fig. 10c).50 These films are based on an organogel – syndiotactic poly(methyl methacrylate) – soaked with Au or Ag NPs decorated with different amounts of thiolated azobenzene. Fig. 10d shows an example of an image created in a nanoparticle-doped gel, where the purple regions correspond to UV-irradiated areas. Importantly, the lifetimes of these images can be controlled by adjusting the fractional coverage of azobenzene on the NP surface: the higher the surface concentration of cis-azobenzene, the stronger the interparticle interactions, and the more persistent the images.50 Notably, no such control is possible in films based on small-molecule photochromic dyes.
Another attractive application of azobenzene-functionalized NPs is illustrated in Fig. 10f, where the nanopores between the NPs within densely packed aggregates have been used as “nanoreactors” capable of accelerating various bimolecular reactions (i.e., between A and B in Fig. 10f).51 Rich in cis-azobenzene residues, these “dynamically self-assembling nanoflasks” are polar, and can efficiently trap a variety of polar molecules present in the hydrophobic solvent, in which the UV-induced self-assembly takes place. Once concentrated in the nanoflasks, the polar molecules undergo chemical reactions faster and, more importantly, with altered stereoselectivities. It was demonstrated, for example, that whereas 9-(hydroxymethyl)anthracene dissolved in toluene dimerizes to afford exclusively the anti isomer of the [4+4] dimer, the same reaction occurring in the presence of the “dynamic nanoflasks” leads to as much as >80% of the syn isomer, thus strongly suggesting substrate preorganization in these nanosized flasks. Once the reaction has taken place, the product (C in Fig. 10f) can be conveniently released to the solution during the disassembly step. The assembly/disassembly sequence can be repeated multiple times, allowing for small amounts of NPs to convert a relatively large quantity of the substrate into the product.60
Spiropyran is another common photoswitch that has been used to control NP self-assembly.61 Huang, Chen, and co-workers worked with gold NPs co-functionalized with thiolated poly(ethylene glycol) (PEG) and polymethacrylate with pendant spiropyran groups.52 Owing to the presence of the hydrophilic PEG chains, these particles were soluble in polar solvents, such as water/DMF mixtures. Exposing the closed-ring isomer of spiropyran to UV light results in its transformation into a planar, conjugated open-ring form (merocyanine), which has a high propensity to self-aggregate. Aggregation between merocyanine units on different NPs initiated attractive interactions between the particles, which, interestingly, did not assemble in an uncontrolled fashion but rather afforded discrete aggregates comprising only three NPs, on average. This finding was rationalized by the reorganization of ligands within the mixed monolayers in a way that maximized the population (i) of the merocyanine groups between the NPs, and (ii) of the solubilizing PEG chains on the periphery of the aggregates.62 The assembly process entailed the formation of SERS-active hot-spots between the NPs, which translated into significantly higher (by a factor of ∼4) SERS activity of the aggregates compared with non-aggregated NPs. A limitation of this system, however, is the long irradiation times required to complete both the assembly (∼3 h of UV) and the disassembly (∼7 h of visible light) steps.
Ravoo and co-workers conceived a different way to control the self-assembly of NPs using light. Rather than covalently attaching photoresponsive molecules to NPs, they employed bis-arylazopyrazoles as non-covalent, light-responsive “glue” for Au NPs functionalized with β-cyclodextrin (β-CD).63 Arylazopyrazoles can be toggled between the trans and the cis form, with trans having a strong affinity to β-CD, with which it forms a host–guest inclusion complex. Similar to azobenzenes, the trans → cis isomerization in arylazopyrazoles can be induced by UV light, whereas the back-isomerization occurs upon exposure to visible light. There are, however, two significant advantages over azobenzenes: first, the photoisomerization reactions in both directions proceed with high yield, with photostationary states typically containing >90% of cis and trans isomer under UV and visible light, respectively. Second, most cis-arylazopyrazoles are far more stable than are typical cis-azobenzenes, with thermal half-lives as long as 1000 days. Therefore, arylazopyrazoles behave to a good approximation as truly bistable switches. With these considerations in mind, molecules containing two arylazopyrazole groups were used as crosslinkers for gold NPs functionalized with a heptathiolated β-CD. Mixing these two components afforded spherical assemblies of Au NPs, ∼200 nm in diameter, which could be disassembled upon exposure to UV light.63 Subsequent irradiation with visible light regenerated the assemblies, and at least four subsequent disassembly–reassembly cycles were performed without appreciable fatigue.
Reversible, light-controlled self-assembly of NPs can also be achieved using thermoresponsive polymers. Two approaches have been developed to induce phase transitions in thermoresponsive polymers using light. Taking advantage of the thermoresponsive behavior of N,N-dimethylacrylamide (DMAm)-based polymers, Zhao et al. synthesized a random copolymer of DMAm and an acrylamide derivatized with an azobenzene group (AzoAm).64 The resulting polymer, p(DMAm-co-AzoAm), exhibited significantly different values of LCST depending on the state of azobenzene; whereas the ground-state, p(DMAm-co-trans-AzoAm) had an LCST of ∼34 °C, UV irradiation decreased it to ∼20 °C. In other words, the trans-rich copolymer was highly water-soluble at room temperature, but exposure to UV light—as opposed to increasing temperature—induced the phase transition and the collapse of the polymer chains. Note that the solubility in water of the polymer containing trans isomers may appear contradictory to the examples discussed in previous paragraphs, where the cis isomers ensured solubility. Such a behavior is due to the presence of the amide groups linking the azobenzene moieties with chain backbone, an architecture that facilitates intramolecular hydrophobic interactions and stacking of azobenzenes selectively in the trans state.64 Interestingly, this polymer could act as a good stabilizer of Au NPs in aqueous solution – provided it was in the hydrated state. Exposure for 4 min to UV initiated NP self-assembly, which took ∼2 h to complete. Subsequent irradiation with visible light (also 4 min), followed by 30 min of stirring, regenerated the original solution of NPs stabilized with hydrated polymer chains, and the assembly/disassembly sequence could be repeated several times.64
The second approach for controlling the phase transition of thermoresponsive polymers using light does not require that photoswitchable molecules be present; rather, it takes advantage of the plasmonic properties of NPs. When irradiated with high-intensity visible light, plasmonic NPs can convert absorbed light energy into heat, thereby increasing temperature in the vicinity of the NP surface. Kumacheva et al. prepared gold nanorods whose tips were functionalized with a thiol-terminated pNIPAm (see Section 8).65 Exposing an aqueous solution of these nanorods to near-infrared (NIR) radiation (matching the rods’ longitudinal plasmon band) resulted in the collapse of the polymer chains, which initiated strong vdW interactions between the ends of the nanorods, affording linear nanorod assemblies (Fig. 10g). Analogous end-to-end assemblies were obtained upon heating the sample to 45 °C; however, the elegance of the light-induced approach lies in the fact that the temperature increase is confined to the immediate surroundings of the NP surface, and that no bulk heating is necessary. The linear chains of nanorods disassembled spontaneously within several minutes once the NIR irradiation was discontinued.65
The light-induced collapse of pNIPAm chains was also applied to control the self-assembly of pNIPAm-decorated 60 nm Au NPs.66 These particles were obtained by a place-exchange reaction on citrate-stabilized particles and displayed a partial negative charge. Upon exposure to 532 nm laser radiation (which coincides with the LSPR peak of these NPs) for 5 min, self-assembly into monodisperse, spherical clusters of ∼300 nm in diameter was obtained. The self-limiting growth of these aggregates was explained by the accumulation of negative surface charge as the aggregate size increased. Once irradiation ceased, the absorption at ∼670 nm, characteristic of Au NP aggregates, persisted for 10 s, after which it rapidly (within less than 1 s) shifted back to ∼539 nm (free NPs). This rapid blue-shift was rationalized by pNIPAm undergoing the phase transition and exerting strong elastic forces on the NPs. Notably, the same 532 nm laser had no effect on similarly sized, pNIPAm-decorated silver NPs which lack LSPR modes at this wavelength.66
A conceptually different strategy for assembling plasmonic NPs using light relies on thermophoretic effects.67 Zheng and co-workers used a flat substrate decorated with gold nanoislands as the template for self-assembly of positively charged Au nanotriangles (NTs) from a solution containing like-charged micelles at a high concentration. When the substrate was exposed to a 532 nm laser beam, the local temperature increase induced an electrostatic potential between the cold and the hot regions, and consequently, the migration of the NTs towards the irradiated substrate (Fig. 10h). Interestingly, whereas the underlying gold substrate was required to initiate the self-assembly process, once formed, the Au NT aggregate produced enough heat to maintain the potential in the system. As a result, the aggregate remained stable even after being transported to a glass substrate, as shown in Fig. 10i. Turning off the laser eliminated the concentration gradient of the charged species, and the aggregates disassembled. Importantly, this method was applicable to NPs of other sizes and compositions, such as gold and silver nanospheres.67
12. Multi-stimuli responsive nanoparticles
A very interesting feature of nanoparticles is that they can be designed to reversibly assemble in response to different (orthogonal) external stimuli, thereby giving rise to dual- or even multi-responsive NPs. This can be achieved by decorating the surfaces of NPs (i) with multiresponsive ligands, (ii) with mixed monolayers comprising different ligands, each responding to different stimuli, or (iii) by taking advantage of the stimuli-responsive nature of the NP core. The first approach is illustrated by a system developed by Wang, Jiang, and co-workers, who prepared Au NPs functionalized with a spiropyran-terminated thiol (Fig. 11a).69 Owing to multiple ethylene glycol units within the stabilizing layer, these NPs were readily soluble in water, both under ambient and UV light, where spiropyran exists in the closed and open form, respectively. Of these two isomers, only the latter can interact with certain metal cations, with which it forms complexes of 2:1 stoichiometry (cf.Fig. 11a, right). Therefore, only when both stimuli—UV and metal ions (here, Cu2+)—were present, did NP self-assembly commence and the system behaved like an AND logic gate. Because only a few Cu2+ ions were sufficient to induce the aggregation process, these NPs self-assembled in response to (and could be used to detect) Cu2+ concentrations as low as 300 nM. When either of the two stimuli was removed (in the case of Cu2+, by the addition of a strong complexing agent, EDTA), the NP aggregates disassembled, which could be followed conveniently by monitoring the color of the sample (Fig. 11a).
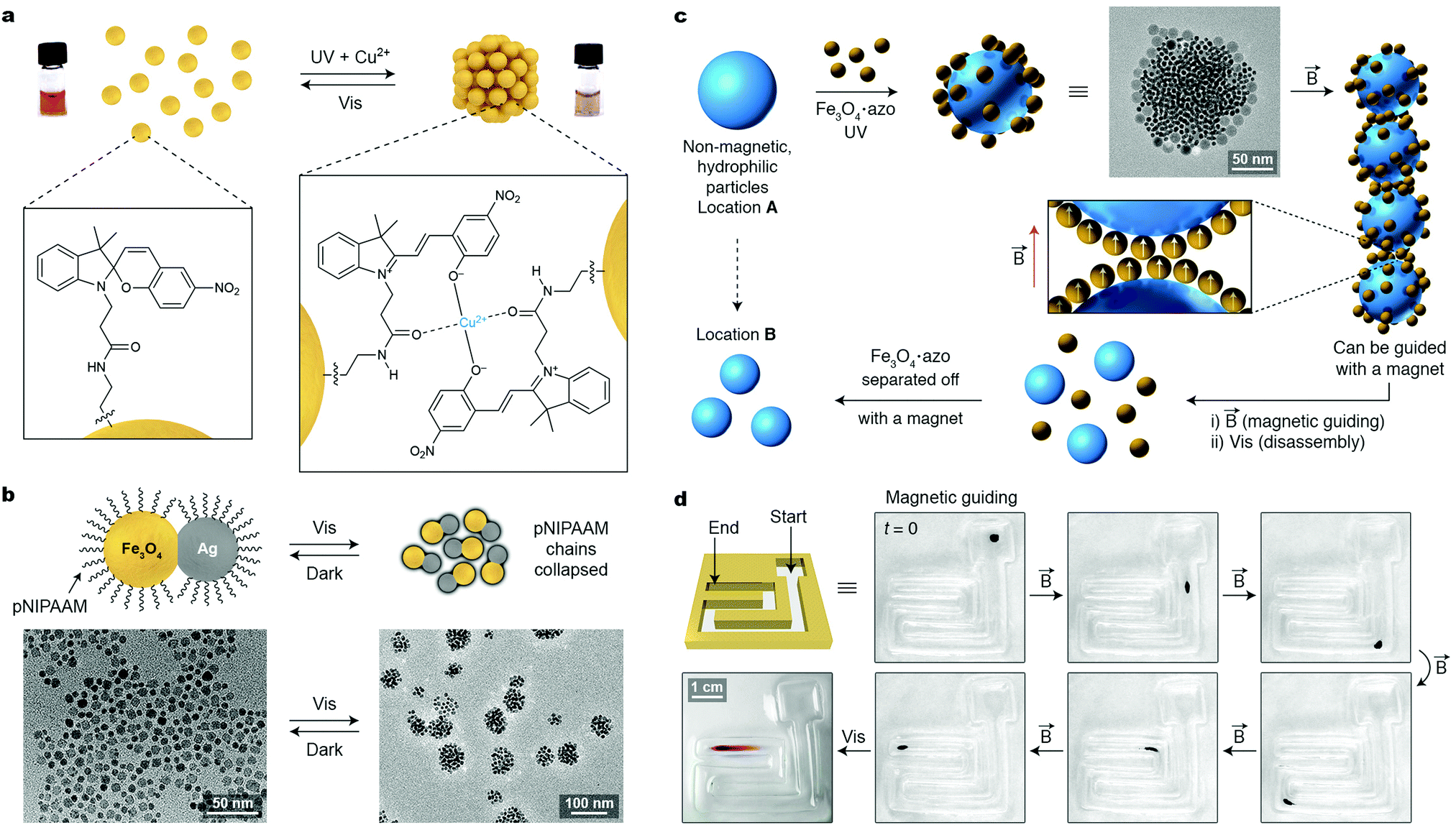 | ||
| Fig. 11 Multi-stimuli-responsive nanoparticles. (a) Spiropyran-functionalized gold NPs assembling upon simultaneous exposure to UV light and Cu2+ ions. Adapted with permission from ref. 69 (copyright 2011, Wiley-VCH). (b) pNIPAm-coated Ag–Fe3O4 heterodimeric NPs responding to three external stimuli: temperature, magnetic field, and light. Adapted with permission from ref. 72 (copyright 2013, Royal Society of Chemistry). (c) Schematic illustration of a strategy envisioned to magnetically manipulate diamagnetic particles with the help of dynamically self-assembling SPM coatings. (d) Magnetic guiding of gold NPs enclosed within SPM shells. Adapted with permission from ref. 73 (copyright 2012, American Chemical Society). | ||
The second approach to dual-responsive NPs can be exemplified by Au NPs co-functionalized with a mixture of azobenzene- and ethylenediamine-terminated thiols.38 Upon exposure to UV light or CO2, each of these two moieties becomes more polar, which can initiate NP self-assembly in nonpolar media (e.g., toluene). The composition of the mixed monolayer could be adjusted such that neither UV nor CO2 alone gave rise to a polarity increase that was large enough to induce NP assembly; only when both of these stimuli were applied simultaneously could self-assembly be achieved, which represents another example of an AND logic gate. Removal of either of the two stimuli was sufficient to recover the original solution of free NPs.38
Third, responsiveness to external stimuli can be “encoded” in the NPs’ inorganic cores. By functionalizing the surfaces of SPM iron oxide NPs with monolayers of azobenzene molecules, Klajn et al. obtained particles responsive to light and magnetic fields in a cooperative fashion. Only 11 nm in diameter, these NPs were too small to assemble in response to a weak external magnetic field alone. However, under the simultaneous exposure to UV light and magnetic field, these dual-responsive NPs initially formed spherical assemblies (triggered by UV light; see Section 11), which gradually increased in size. Interestingly, when these assemblies reached ∼100 nm in diameter, they became responsive to the applied magnetic field, which induced their rapid self-assembly driven by magnetic dipole–dipole interactions. The process resulted in long threads with a uniform thickness corresponding to the diameter of the smallest magnetically responsive NP clusters, i.e., ∼100 nm. At any stage of the process, visible light could be applied to cancel out all attractive interactions in this conceptually interesting system, where responsiveness to one type of external stimulus (magnetic field) can be activated by another (UV light).70
Another type of light/magnetically switchable system was prepared by incorporating hydrophobic magnetite NPs within the bilayer membranes of CD vesicles.71 These vesicles assembled spontaneously in water from an amphiphilic β-CD derivatized with seven long-alkyl chains, and they exposed multiple CD binding pockets to the outer solution. Owing to the SPM properties of the Fe3O4 NPs embedded within their membranes, the vesicles responded to external magnetic fields and could be assembled into linear aggregates of several micrometers in length. Subsequent addition of a bis-azobenzene crosslinker preserved these aggregates even after the removal of the field. This stabilization was achieved by strong supramolecular interactions between the trans-azobenzene “guests” and CD “hosts” within neighboring vesicles. However, exposure to UV light induced a transformation of trans- to cis-azobenzene, which has no affinity to β-CD—consequently, a solution of free vesicles could be regenerated.
A simple yet elegant example of NPs’ response to three types of external stimuli was reported by Lu and co-workers, who functionalized Ag–Fe3O4 heterodimeric NPs with pNIPAm shells (Fig. 11b).72 The thermoresponsive nature of pNIPAm was used to control the assembly state of these NPs: heating their aqueous solutions above ∼32 °C (i.e., the LCST of pNIPAm) induced the dehydration of the NP-bound polymer chains, destabilizing the particles in solution and inducing their aggregation. The same effect was achieved by exposing the NPs to sunlight illumination, which was absorbed by the Ag domains and converted into heat (Fig. 11b). However, no aggregation was observed upon illuminating pNIPAm-coated Fe3O4 NPs lacking the Ag counterpart. In addition, the Ag–Fe3O4 heterodimeric NPs have SPM properties and can be removed from the solution with the help of a magnet, but only following sunlight illumination, which activated the particles’ responsiveness to external magnetic field.
Multi-stimuli-responsive NPs offer novel and unique applications, which would be hard to achieve otherwise. One such example is illustrated in Fig. 11c and d, where light-/magnetically responsive NPs were used to form magnetic coatings around diamagnetic particles and to guide them in nonpolar solvents using weak external magnetic fields.73 As described in Section 11, azobenzene-coated NPs subjected to UV light in nonpolar solvents self-assemble into compact aggregates to minimize the contact with the hydrophobic environment. When the process takes place in the presence of highly polar particles in the system, however, the cis-azobenzene-coated NPs (Fe3O4·azo in Fig. 11c) preferentially attach to the surfaces of these particles rather than form aggregates on their own. Upon applying a magnetic field, the resulting core–shell objects interact with one another by multiple dipole–dipole interactions (see the inset in Fig. 11c), forming linear assemblies, which can be guided using weak magnets. Once delivered to a desired location, the diamagnetic “cargo” can be released by shining visible light, which induces the cis → trans azobenzene back-isomerization, stripping off the magnetic shells from the diamagnetic particles. The sequence of photographs in Fig. 11d demonstrates how 5 nm Au NPs can be rapidly transported from one end of a channel to the other end, with the help of only 4% of the Fe3O4·azo carriers. These gold particles were functionalized with a thiolated azobenzene at a relatively high surface concentration of azobenzene. Exposing a mixture of Au and Fe3O4 NPs to UV light afforded core–shell assemblies (see the TEM image in Fig. 11c) whereby the Au particles were localized in the cores to maximize the attractive interactions between the cis-azobenzene groups. The thin (submonolayer) shell of Fe3O4·azo was sufficient to render the assemblies responsive to a magnetic field, and to manipulate the non-magnetic entities over long distances. The red color seen in the last frame in Fig. 11d results from the disassembly of the core–shell assemblies and the release of free Au NPs following exposure to visible light.
Conclusions and outlook
The rich variety of examples discussed above makes us confident that ligand-protected nanoparticles have a great potential to be utilized as key components of complex chemical reaction networks and that the time is ripe to start utilizing them as the building blocks for constructing synthetic life-like systems and materials.74 Notwithstanding, much remains to be accomplished in the field of stimuli-responsive self-assembly of NPs. In the following, we outline several research directions and highlight opportunities for continued studies in this area.In the context of redox-switchable NPs, little efforts have been devoted to guiding their assembly under electrochemical control. In particular, we believe that the potential of redox-switchable radical-pi interactions remains largely unrealized. Furthermore, the use of photoredox catalysts (either added to the solution or co-adsorbed on NP surfaces) will make it possible to effectively control self-assembly of redox-responsive NPs using light.
Regarding light-induced self-assembly of azobenzene-functionalized NPs, an interesting challenge will be to selectively (using different colors of light) induce self-assembly in a mixture containing NPs of various sizes, each functionalized with a differently substituted azobenzene. An exciting question concerns the maximum number of NP types that can be addressed in an orthogonal fashion,75 with the help of azobenzenes responding to distinct wavelengths of light spanning the UV and visible regions. Additional opportunities—such as very long thermal half-lives76,77 and NIR-responsiveness78—are provided by recently developed photoswitches, such as donor–acceptor Stenhouse adducts79 and hydrazone-based switches,80 which are yet to be placed and operated reversibly81 on NP surfaces.
Another issue that deserves more attention, in the context of reversible NP self-assembly, is curvature-dependent properties of NP-adsorbed ligands. It has long been recognized that pH-,82 redox-,4 and other stimuli-responsive ligands can behave differently83,84 when deposited on differently sized NPs; for example, the pKa values of carboxylic acids at the terminal positions of thiolate ligands are largely dependent on the average distance between these groups, which is determined by NP curvature. Therefore, selective self-assembly of NPs of a particular size from mixtures comprising various NP sizes should be possible.
We foresee that much attention will be devoted during coming years to what has come to be known as dissipative self-assembly. In this mode of self-assembly, the addition of a chemical signal results in the formation of transient NP assemblies, which disassemble as the signal undergoes a chemical reaction.85–88 This concept can be exemplified by a hypothetical system, whereby carbon dioxide, in addition to rapidly inducing NP self-assembly (compare with Section 4), is slowly reduced to methanol, which is inert towards the NPs – in such a case, NP aggregates will persist only in the presence of a sustained supply of CO2. A more ambitious goal concerns coupling NP self-assembly to oxidation of toxic chemicals, or to controlled decomposition of energetic materials, such as nitrogen-rich compounds.
From a broader materials science perspective, dynamic systems and networks comprising nanoparticles and functional surface ligands represent an excellent starting point for designing and realization of dynamic assemblies that, apart from storing information, would perform programmed tasks related to, e.g., sensing or spatiotemporal catalysis.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
L. M. L.-M. and M. G. acknowledge funding from the Spanish MINECO (Grants MAT2013-46101R and MAT2017-86659-R). R. K. acknowledges support from the Minerva Foundation. L. M. L.-M. is recipient of an ERC Advanced Grant (787510, 4DBIOSERS). We thank Anton Hanopolskyi, Kristina Kucanda, and Michal Sawczyk for helpful discussions. In memory of Prof. Jacob Israelachvili (1944–2018).References
- J. H. van Esch, R. Klajn and S. Otto, Chem. Soc. Rev., 2017, 46, 5474–5475 RSC.
- S. Mann, Angew. Chem., Int. Ed., 2013, 52, 155–162 CrossRef CAS PubMed.
- B. A. Grzybowski and W. T. S. Huck, Nat. Nanotechnol., 2016, 11, 584–591 CrossRef PubMed.
- A. Coskun, P. J. Wesson, R. Klajn, A. Trabolsi, L. Fang, M. A. Olson, S. K. Dey, B. A. Grzybowski and J. F. Stoddart, J. Am. Chem. Soc., 2010, 132, 4310–4320 CrossRef CAS PubMed.
- J. C. Cremaldi and B. Bhushan, Beilstein J. Nanotechnol., 2018, 9, 907–935 CrossRef CAS PubMed.
- J. Shah and J. Zeier, Front. Plant Sci., 2013, 4, 30 Search PubMed.
- S. Soh, M. Byrska, K. Kandere-Grzybowska and B. A. Grzybowski, Angew. Chem., Int. Ed., 2010, 49, 4170–4198 CrossRef CAS PubMed.
- H. H. Zepik, E. Blochliger and P. L. Luisi, Angew. Chem., Int. Ed., 2001, 40, 199–202 CrossRef CAS PubMed.
- X. M. He, M. Aizenberg, O. Kuksenok, L. D. Zarzar, A. Shastri, A. C. Balazs and J. Aizenberg, Nature, 2012, 487, 214–218 CrossRef CAS PubMed.
- D. Stuart-Fox and A. Moussalli, Philos. Trans. R. Soc., B, 2009, 364, 463–470 CrossRef PubMed.
- R. Merindol and A. Walther, Chem. Soc. Rev., 2017, 46, 5588–5619 RSC.
- B. A. Grzybowski, K. Fitzner, J. Paczesny and S. Granick, Chem. Soc. Rev., 2017, 46, 5647–5678 RSC.
- S. De and R. Klajn, Adv. Mater., 2018, 30, 1706750 CrossRef PubMed.
- A. H. Lu, E. L. Salabas and F. Schuth, Angew. Chem., Int. Ed., 2007, 46, 1222–1244 CrossRef CAS PubMed.
- C. Zhan, X. J. Chen, J. Yi, J. F. Li, D. Y. Wu and Z. Q. Tian, Nat. Rev. Chem., 2018, 2, 216–230 CrossRef.
- M. Haase and H. Schafer, Angew. Chem., Int. Ed., 2011, 50, 5808–5829 CrossRef CAS PubMed.
- A. M. Smith, K. A. Johnston, S. E. Crawford, L. E. Marbella and J. E. Millstone, Analyst, 2017, 142, 11–29 RSC.
- I. Kanelidis and T. Kraus, Beilstein J. Nanotechnol., 2017, 8, 2625–2639 CrossRef PubMed.
- A. Sánchez-Iglesias, M. Grzelczak, T. Altantzis, B. Goris, J. Pérez-Juste, S. Bals, G. Van Tendeloo, S. H. Donaldson, B. F. Chmelka, J. N. Israelachvili and L. M. Liz-Marzán, ACS Nano, 2012, 6, 11059–11065 CrossRef PubMed.
- M. Grzelczak, A. Sánchez-Iglesias and L. M. Liz-Marzán, Soft Matter, 2013, 9, 9094–9098 RSC.
- R. M. Choueiri, A. Klinkova, H. Thérien-Aubin, M. Rubinstein and E. Kumacheva, J. Am. Chem. Soc., 2013, 135, 10262–10265 CrossRef CAS PubMed.
- H. C. Hu, F. Ji, Y. Xu, J. Q. Yu, Q. P. Liu, L. Chen, Q. Chen, P. Wen, Y. Lifshitz, Y. Wang, Q. Zhang and S. T. Lee, ACS Nano, 2016, 10, 7323–7330 CrossRef CAS PubMed.
- V. Sashuk, K. Winkler, A. Żywociński, T. Wojciechowski, E. Górecka and M. Fiałkowski, ACS Nano, 2013, 7, 8833–8839 CrossRef CAS PubMed.
- A. Sánchez-Iglesias, N. Claes, M. G. Solís, J. M. Taboada, S. Bals, L. M. Liz-Marzán and M. Grzelczak, Angew. Chem., Int. Ed., 2018, 57, 3183–3186 CrossRef PubMed.
- D. W. Wang, B. Kowalczyk, I. Lagzi and B. A. Grzybowski, J. Phys. Chem. Lett., 2010, 1, 1459–1462 CrossRef CAS.
- L. Cheng, A. P. Liu, S. Peng and H. W. Duan, ACS Nano, 2010, 4, 6098–6104 CrossRef CAS PubMed.
- P. Taladriz-Blanco, N. J. Buurma, L. Rodríguez-Lorenzo, J. Pérez-Juste, L. M. Liz-Marzán and P. Hervés, J. Mater. Chem., 2011, 21, 16880–16887 RSC.
- H. Nabika, T. Oikawa, K. Iwasaki, K. Murakoshi and K. Unoura, J. Phys. Chem. C, 2012, 116, 6153–6158 CrossRef CAS.
- I. Lagzi, B. Kowalczyk, D. W. Wang and B. A. Grzybowski, Angew. Chem., Int. Ed., 2010, 49, 8616–8619 CrossRef CAS PubMed.
- T. Heuser, A. K. Steppert, C. M. Lopez, B. L. Zhu and A. Walther, Nano Lett., 2015, 15, 2213–2219 CrossRef CAS PubMed.
- P. K. Kundu, D. Samanta, R. Leizrowice, B. Margulis, H. Zhao, M. Börner, T. Udayabhaskararao, D. Manna and R. Klajn, Nat. Chem., 2015, 7, 646–652 CrossRef CAS PubMed.
- D. Samanta and R. Klajn, Adv. Opt. Mater., 2016, 4, 1373–1377 CrossRef CAS.
- S. Si, M. Raula, T. K. Paira and T. K. Mandal, ChemPhysChem, 2008, 9, 1578–1584 CrossRef CAS PubMed.
- A. Rao, S. Roy, M. Unnikrishnan, S. S. Bhosale, G. Devatha and P. P. Pillai, Chem. Mater., 2016, 28, 2348–2355 CrossRef CAS.
- Y. T. Chan, S. N. Li, C. N. Moorefield, P. S. Wang, C. D. Shreiner and G. R. Newkome, Chem. – Eur. J., 2010, 16, 4164–4168 CrossRef CAS PubMed.
- S. C. Chen, C. X. Guo, Q. P. Zhao and X. M. Lu, Chem. – Eur. J., 2014, 20, 14057–14062 CrossRef CAS PubMed.
- J. M. Zhang, D. H. Han, H. J. Zhang, M. Chaker, Y. Zhao and D. L. Ma, Chem. Commun., 2012, 48, 11510–11512 RSC.
- J.-W. Lee and R. Klajn, Chem. Commun., 2015, 51, 2036–2039 RSC.
- P. Hazarika, B. Ceyhan and C. M. Niemeyer, Angew. Chem., Int. Ed., 2004, 43, 6469–6471 CrossRef CAS PubMed.
- G. von Maltzahn, D. H. Min, Y. X. Zhang, J. H. Park, T. J. Harris, M. Sailor and S. N. Bhatia, Adv. Mater., 2007, 19, 3579–3583 CrossRef CAS.
- K. L. Gurunatha, A. C. Fournier, A. Urvoas, M. Valerio-Lepiniec, V. Marchi, P. Minard and E. Dujardin, ACS Nano, 2016, 10, 3176–3185 CrossRef CAS PubMed.
- R. Klajn, M. A. Olson, P. J. Wesson, L. Fang, A. Coskun, A. Trabolsi, S. Soh, J. F. Stoddart and B. A. Grzybowski, Nat. Chem., 2009, 1, 733–738 CrossRef CAS PubMed.
- M. A. Olson, A. Coskun, R. Klajn, L. Fang, S. K. Dey, K. P. Browne, B. A. Grzybowski and J. F. Stoddart, Nano Lett., 2009, 9, 3185–3190 CrossRef CAS PubMed.
- R. Klajn, L. Fang, A. Coskun, M. A. Olson, P. J. Wesson, J. F. Stoddart and B. A. Grzybowski, J. Am. Chem. Soc., 2009, 131, 4233–4235 CrossRef CAS PubMed.
- S. Borsley and E. R. Kay, Chem. Commun., 2016, 52, 9117–9120 RSC.
- J. Y. Zhang, P. J. Santos, P. A. Gabrys, S. Lee, C. Liu and R. J. Macfarlane, J. Am. Chem. Soc., 2016, 138, 16228–16231 CrossRef CAS PubMed.
- W. Lewandowski, M. Fruhnert, J. Mieczkowski, C. Rockstuhl and E. Górecka, Nat. Commun., 2015, 6, 6590 CrossRef PubMed.
- Y. D. Liu, X. G. Han, L. He and Y. D. Yin, Angew. Chem., Int. Ed., 2012, 51, 6373–6377 CrossRef CAS PubMed.
- K. J. M. Bishop, B. Kowalczyk and B. A. Grzybowski, J. Phys. Chem. B, 2009, 113, 1413–1417 CrossRef CAS PubMed.
- S. Balasubramaniam, N. Pothayee, Y. N. Lin, M. House, R. C. Woodward, T. G. St. Pierre, R. M. Davis and J. S. Riffle, Chem. Mater., 2011, 23, 3348–3356 CrossRef CAS.
- K. L. Hamner and M. M. Maye, Langmuir, 2013, 29, 15217–15223 CrossRef CAS PubMed.
- C. Durand-Gasselin, N. Sanson and N. Lequeux, Langmuir, 2011, 27, 12329–12335 CrossRef CAS PubMed.
- Y. A. Lin, X. X. Xia, M. Wang, Q. R. Wang, B. An, H. Tao, Q. B. Xu, F. Omenetto and D. L. Kaplan, Langmuir, 2014, 30, 4406–4414 CrossRef CAS PubMed.
- C. T. Yavuz, J. T. Mayo, W. W. Yu, A. Prakash, J. C. Falkner, S. Yean, L. L. Cong, H. J. Shipley, A. Kan, M. Tomson, D. Natelson and V. L. Colvin, Science, 2006, 314, 964–967 CrossRef PubMed.
- S. Sim, D. Miyajima, T. Niwa, H. Taguchi and T. Aida, J. Am. Chem. Soc., 2015, 137, 4658–4661 CrossRef CAS PubMed.
- P. Riente, J. Yadav and M. A. Pericàs, Org. Lett., 2012, 14, 3668–3671 CrossRef CAS PubMed.
- M. E. Flatte, A. A. Kornyshev and M. Urbakh, J. Phys.: Condens. Matter, 2008, 20, 073102 CrossRef.
- Y. Montelongo, D. Sikdar, Y. Ma, A. J. S. McIntosh, L. Velleman, A. R. Kucernak, J. B. Edel and A. A. Kornyshev, Nat. Mater., 2017, 16, 1127–1135 CrossRef CAS PubMed.
- Y. X. Yu, D. Yu and C. A. Orme, Nano Lett., 2017, 17, 3862–3869 CrossRef CAS PubMed.
- H. Zhao, S. Sen, T. Udayabhaskararao, M. Sawczyk, K. Kučanda, D. Manna, P. K. Kundu, J.-W. Lee, P. Král and R. Klajn, Nat. Nanotechnol., 2016, 11, 82–88 CrossRef CAS PubMed.
- P. K. Kundu, S. Das, J. Ahrens and R. Klajn, Nanoscale, 2016, 8, 19280–19286 RSC.
- L. Zhang, L. W. Dai, Y. Rong, Z. Z. Liu, D. Y. Tong, Y. J. Huang and T. Chen, Langmuir, 2015, 31, 1164–1171 CrossRef CAS PubMed.
- L. Stricker, E. C. Fritz, M. Peterlechner, N. L. Doltsinis and B. J. Ravoo, J. Am. Chem. Soc., 2016, 138, 4547–4554 CrossRef CAS PubMed.
- A. Housni, Y. Zhao and Y. Zhao, Langmuir, 2010, 26, 12366–12370 CrossRef CAS PubMed.
- D. Fava, M. A. Winnik and E. Kumacheva, Chem. Commun., 2009, 2571–2573 RSC.
- T. Ding, V. K. Valev, A. R. Salmon, C. J. Forman, S. K. Smoukov, O. A. Scherman, D. Frenkel and J. J. Baumberg, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 5503–5507 CrossRef CAS PubMed.
- L. H. Lin, X. L. Peng, M. S. Wang, L. Scarabelli, Z. M. Mao, L. M. Liz-Marzán, M. F. Becker and Y. B. Zheng, ACS Nano, 2016, 10, 9659–9668 CrossRef CAS PubMed.
- R. Klajn, P. J. Wesson, K. J. M. Bishop and B. A. Grzybowski, Angew. Chem., Int. Ed., 2009, 48, 7035–7039 CrossRef CAS PubMed.
- D. B. Liu, W. W. Chen, K. Sun, K. Deng, W. Zhang, Z. Wang and X. Y. Jiang, Angew. Chem., Int. Ed., 2011, 50, 4103–4107 CrossRef CAS PubMed.
- S. Das, P. Ranjan, P. S. Maiti, G. Singh, G. Leitus and R. Klajn, Adv. Mater., 2013, 25, 422–426 CrossRef CAS PubMed.
- J. H. Schenkel, A. Samanta and B. J. Ravoo, Adv. Mater., 2014, 26, 1076–1080 CrossRef CAS PubMed.
- H. Han, J. Y. Lee and X. M. Lu, Chem. Commun., 2013, 49, 6122–6124 RSC.
- O. Chovnik, R. Balgley, J. R. Goldman and R. Klajn, J. Am. Chem. Soc., 2012, 134, 19564–19567 CrossRef CAS PubMed.
- M. Grzelczak, J. Colloid Interface Sci., 2019, 537, 269–279 CrossRef CAS PubMed.
- D. Manna, T. Udayabhaskararao, H. Zhao and R. Klajn, Angew. Chem., Int. Ed., 2015, 54, 12394–12397 CrossRef CAS PubMed.
- D. Bléger, J. Schwarz, A. M. Brouwer and S. Hecht, J. Am. Chem. Soc., 2012, 134, 20597–20600 CrossRef PubMed.
- H. Qian, S. Pramanik and I. Aprahamian, J. Am. Chem. Soc., 2017, 139, 9140–9143 CrossRef CAS PubMed.
- Y. Yang, R. P. Hughes and I. Aprahamian, J. Am. Chem. Soc., 2014, 136, 13190–13193 CrossRef CAS PubMed.
- N. Mallo, E. D. Foley, H. Iranmanesh, A. D. W. Kennedy, E. T. Luis, J. M. Ho, J. B. Harper and J. E. Beves, Chem. Sci., 2018, 9, 8242–8252 RSC.
- X. Su and I. Aprahamian, Chem. Soc. Rev., 2014, 43, 1963–1981 RSC.
- J. Ahrens, T. Bian, T. Vexler and R. Klajn, ChemPhotoChem, 2017, 1, 230–236 CrossRef CAS.
- D. W. Wang, R. J. Nap, I. Lagzi, B. Kowalczyk, S. B. Han, B. A. Grzybowski and I. Szleifer, J. Am. Chem. Soc., 2011, 133, 2192–2197 CrossRef CAS PubMed.
- T. Zdobinsky, P. S. Maiti and R. Klajn, J. Am. Chem. Soc., 2014, 136, 2711–2714 CrossRef PubMed.
- T. Moldt, D. Brete, D. Przyrembel, S. Das, J. R. Goldman, P. K. Kundu, C. Gahl, R. Klajn and M. Weinelt, Langmuir, 2015, 31, 1048–1057 CrossRef CAS PubMed.
- B. G. P. van Ravensteijn, W. E. Hendriksen, R. Eelkema, J. H. van Esch and W. K. Kegel, J. Am. Chem. Soc., 2017, 139, 9763–9766 CrossRef CAS PubMed.
- R. K. Grotsch, A. Angi, Y. G. Mideksa, C. Wanzke, M. Tena-Solsona, M. J. Feige, B. Rieger and J. Boekhoven, Angew. Chem., Int. Ed., 2018, 57, 14608–14612 CrossRef PubMed.
- M. Sawczyk and R. Klajn, J. Am. Chem. Soc., 2017, 139, 17973–17978 CrossRef CAS PubMed.
- M. P. Conte, J. K. Sahoo, Y. M. Abul-Haija, K. H. A. Lau and R. V. Ulijn, ACS Appl. Mater. Interfaces, 2018, 10, 3069–3075 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2019 |