
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDNA-inspired oligomers: from oligophosphates to functional materials
Mykhailo
Vybornyi
 *a,
Yuliia
Vyborna
b and
Robert
Häner
*c
*a,
Yuliia
Vyborna
b and
Robert
Häner
*c
aLaboratoire de Biochimie (LBC), ESPCI Paris, PSL Research University, CNRS UMR8231 Chimie Biologie Innovation, 10 Rue Vauquelin, 75005 Paris, France. E-mail: mykhailo.vybornyi@espci.fr
bSorbonne Université, Laboratoire Jean Perrin, 75005 Paris, France. E-mail: yuliia.vyborna@sorbonne-universite.fr
cDepartment of Chemistry and Biochemistry, University of Bern, Freiestrasse 3, CH-3012, Switzerland. E-mail: robert.haener@dcb.unibe.ch
First published on 2nd July 2019
Abstract
To exert its role of a functional polymer, DNA relies on a molecular self-assembly process that is driven by the interactions of only four units placed in a defined order. Extending the structural diversity of recognition motifs in DNA, to and beyond analogues of the nucleobases, will open doors to self-assembled materials with advanced programmable properties. DNA-inspired systems are becoming useful for numerous applications unachievable by the nucleic acids in their native composition. Potential applications of rationally designed oligo- and polyphosphodiesters reside in the areas of drug delivery, diagnostic signalling and imaging, in systems for efficient energy transfer or the precise ordering on the nanoscale. The field of DNA-inspired phosphodiesters highlights the general value and utility of precision in the composition of oligomers and polymers. In this tutorial review, we will summarize the approaches of directing the self-assembly of DNA-inspired, sequence-specific polyphosphodiesters into soft materials with unique properties. These data expose the so far underexploited potential of DNA-derived systems in solving some of the key issues in various technological areas, such as advanced biomaterials, morphologically defined soft matter or the controlled polymer folding and assembly. Moreover, precise positioning of structurally diverse molecules within a polymer chain creates unmatched opportunities for encoding information on the molecular level and transmitting it further to the microscopic and even macroscopic level via noncovalent interactions.
 Mykhailo Vybornyi | Mykhailo (Misha) Vybornyi got his BSc from the Kyiv National University (Ukraine) and MSc from the University of Angers (France) in nanotechnology and organic materials. In 2011, he joined the Häner group at the University of Bern (Switzerland) to work on the development of synthetic oligomers for 2D supramolecular polymerization. As an SNCF postdoc fellow, he conducted research with Prof. E. W. (Bert) Meijer at the Eindhoven University of Technology (the Netherlands). In 2018, Mykhailo moved to the industrial sector (Enamine, Kyiv, Ukraine). Since 2019, he has been a member of the LBC team (Prof. Andrew Griffiths and Dr. Philippe Nghe) at the ESPCI in Paris. |
 Yuliia Vyborna | Yuliia Vyborna graduated from the Kiev National University (Ukraine) with a MSc diploma in analytical chemistry in 2011. Later, she started PhD thesis under the supervision of Prof. Robert Häner at the University of Bern (Switzerland) with the major emphasis on elaborating methodologies toward DNA-grafted supramolecular polymers. Her doctoral work was awarded with a faculty prize for the best PhD thesis in chemistry and biochemistry in 2018. In 2019, Yuliia joined the group of Dr. Andre Estévez-Torres at the Sorbonne University to develop novel active matter systems. |
 Robert Häner | Robert Häner received his PhD in chemistry in 1987 from the ETH Zürich, Switzerland. After postdoctoral research at UC Berkeley and CalTech, he joined the Swiss pharmaceutical industry in 1990, where he held several research positions in the following years. In the year 2000 he was appointed full professor of organic chemistry at the University of Bern, Switzerland. His research interests focus on the understanding of fundamental aspects of the supramolecular organization of DNA-based nanomaterials and the application of modified nucleic acids in the fields of diagnostics and novel materials. |
Key learning points1. Solid-phase synthesis is the prevailing method for integration of diverse functional building blocks within an oligomer chain in a sequence-specific manner.2. The self-assembly behaviour of oligophosphodiesters can be encoded on a molecular level. 3. Structural mimicking of DNA becomes possible, functional mimicking in the area of intelligent materials is foreseeable. 4. DNA-inspired systems can lead to biocompatible soft materials with novel functions. 5. Oligophosphodiesters provide access to spherical objects, folded single-chain nanoparticles and to one and two-dimensional supramolecular polymers. |
Introduction
The self-assembly of nucleic acids is crucial to sustaining some of the central processes occurring in living organisms. Key features of this self-assembly process include the storage and programmable execution of instructions encoded in a DNA or RNA sequence.1 Strikingly, these instructions are based on non-covalent interactions between only four nucleobases (A, G, C, T/U). This limited set of recognition units has evolved over billions of years and has served its purpose as genetic material in an amazing way. Nevertheless, novel types of building blocks may result in DNA analogues with properties that are very different from the natural archetype. Inspired by the notion, numerous attempts have been made to further extend the functionality of nucleic acid based, self-assembled systems by introducing structural diversity to the smallest constitutional unit, the nucleotide.2 The most prominent examples of these alterations include the re-shaping of the pentose sugar or the replacement of the natural nucleobases. In the latter example, expanding the genetic alphabet to six letters has already entered the stage of applied technology and offers exciting opportunities for creating advanced biological tools.3 In the mentioned areas, formation of stable double strands via hybridization of complementary strands is central to the pursued applications. Nucleic acids are efficiently used in the shaping of complex, functional objects of predetermined topology, as elegantly shown in numerous DNA origami systems. Despite the high structural complexity of these sophisticated molecular architectures, their design largely relies on a single structural element, the double helix, which is driven by the dependable formation of stacked Watson–Crick base pairs. The arrangement of the nucleotides in a double helix is perfectly suited for storing genetic information. Among others, it helps preserving the chemical integrity of the nucleobases by placing them inside the hydrophobic core of the helix, the phosphate backbone ensures solubility in a biological environment and the sugar moieties support conformational preorganization for base pairing. However, these advantages for biological functions may represent limitations when it comes to other applications, such as the medical, diagnostic and technological areas. In man-made systems, nucleic acids have also gained considerable attention in nanotechnology and supramolecular chemistry as essential components of smart materials. Here, chemical alternatives to the nucleic acids are indispensable. Exploring the chemical space of potential alterations is highly desirable for acquiring a proper understanding of the rules that will enable molecular programming the self-assembly of DNA-inspired systems beyond natural and modified nucleosides (Fig. 1).4 While the hybridisation behaviour of chemically modified DNA structures have been extensively studied,5 information about their controlled folding into functional systems is still very limited.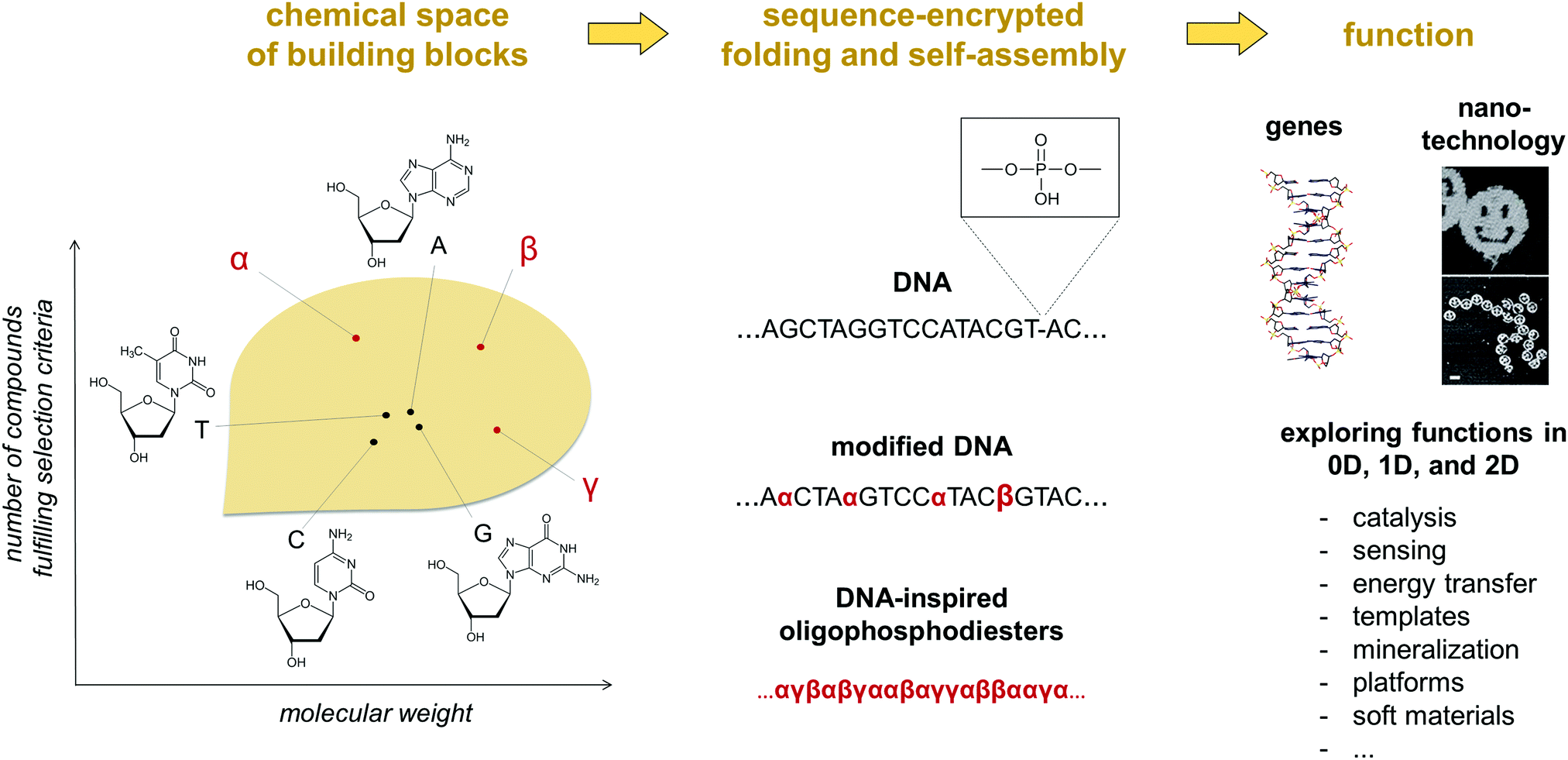 | ||
| Fig. 1 An illustration of the concept of chemical space applied to oligophosphodiesters. Within a selected set of parameters (e.g. chemical stability during solid phase synthesis, the number of hydrogen bond donor and acceptor sites, hydrophilicity/hydrophobicity, optical properties), there is a vast theoretical number of molecules, of which nucleobases occupy a tiny fraction. Integrating other compounds in oligophosphodiester chains can generate a diversity of dimensionally defined and functional, self-assembled or folded objects. For some of the reported examples of α, β, γ see Fig. 2. | ||
In this tutorial review, we will illustrate how the structure–function relation established for nucleic acids has inspired the creation of alternative functional systems. Over the past decade, this unapparent liaison has shaped an area of research in which the nucleic acids represent a part of a larger chemical space populated by sequence-specific oligo- or polyphosphodiesters.4 The notion will be supported by outlining some prospective areas of utilization of DNA-inspired systems and illustrating the potential of sequence-specific oligophosphodiesters for accessing functional self-assembled materials. To better demonstrate upcoming opportunities, the review also includes examples of self-assembling hybrid molecules as relevant intermediaries which facilitate a conceptual shift from DNA to entirely non-nucleoside oligophosphodiesters (Fig. 1). The term ‘oligophosphodiester’ implies the presence of negatively charged phosphates. It is self-explanatory that this chemical group is of central importance for structural considerations since it ensures water solubility and provides additional means to control the self-assembly (ionic strength, valency of counterions, pH).6 In the scope of this review, however, we will not explicitly address this part of the oligomers. The major focus will be put on the structural diversity of DNA-derived oligomers and the possibilities to modulate their properties by sequence control. Fig. 2 gives an overview on the monomeric building blocks (1–17) described in the following. Extensive research in monodisperse, sequence-specific oligophosphodiesters will help establishing general design principles based on the nature of their repetitive units. This knowledge is critical for the development of next-generation systems to address the demands of our society for smarter, safer, and eco-friendlier materials.
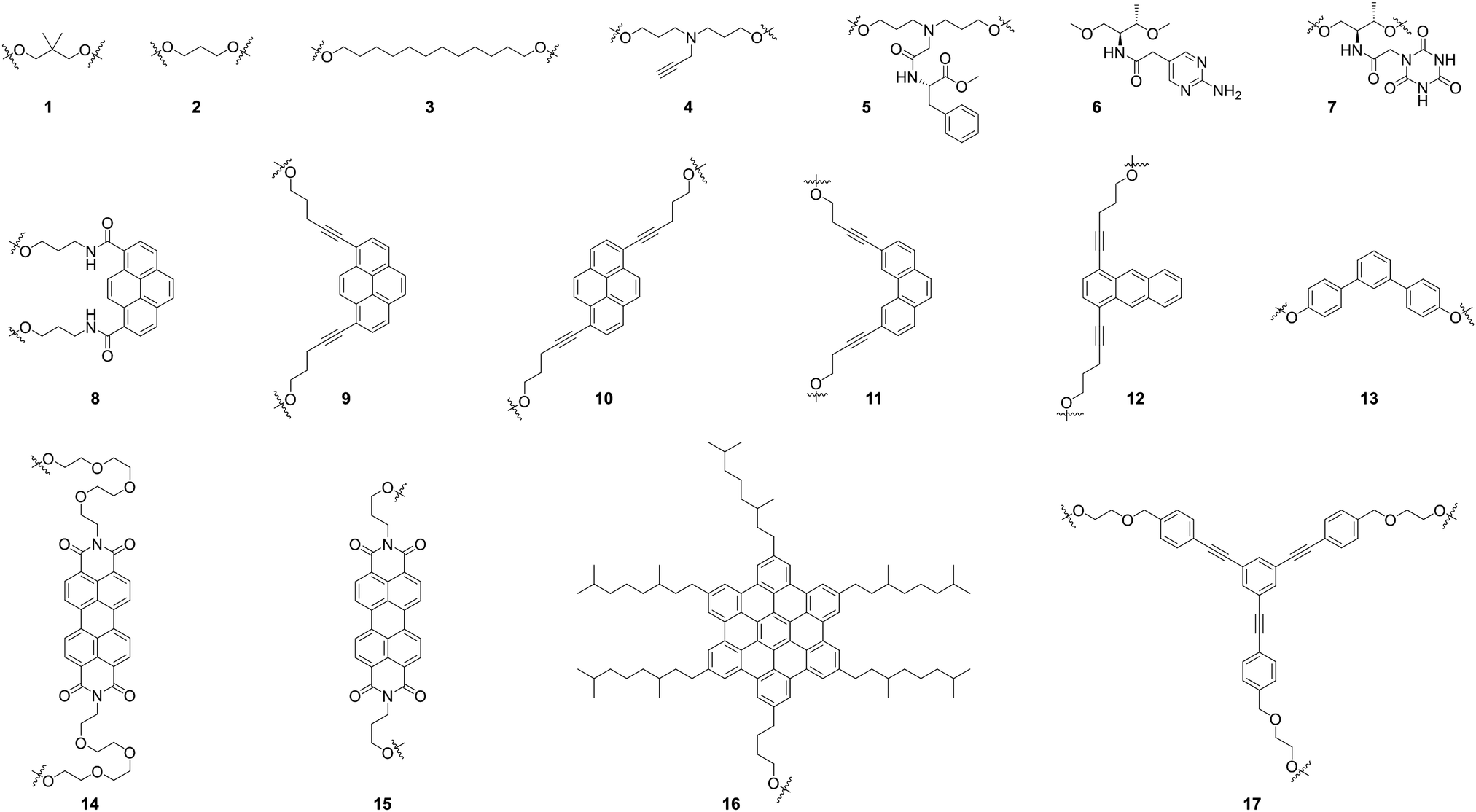 | ||
| Fig. 2 Selected monomeric building blocks described in this review. Building blocks 1–17 were introduced into oligomers via phosphoramidite chemistry. | ||
Self-assembling oligomers with a precise sequence will be presented and classified in accordance with the morphological shapes of the formed structures. The methods to analyze such systems will be provided on a case-to-case basis. The selection of works covered herein aims at presenting oligophosphodiesters as intriguing alternatives to nucleic acids in various fields so far exclusively reserved for DNA and RNA. These fields include, but are not limited to, modified nucleic acids for therapeutic applications, DNA as a scaffold for chromophore assembly, DNA/RNA origami, DNA-encoded libraries, self-assembly of DNA–polydisperse block copolymers and DNA–inorganic nanoparticles.
Synthesis of sequence-defined oligophosphodiesters
The dominant approach enabling the preparation of mono-disperse, sequence-defined oligophosphodiesters is given by automated solid-phase DNA synthesis.7Scheme 1 describes the key steps in a standard synthetic cycle – coupling of a solubilized phosphoramidite to a hydroxyl group attached to a solid phase, oxidation of P(III) to P(V) by a suitable oxidant, and finally acid-promoted deprotection of the newly introduced hydroxyl group. In general, commercial DNA/RNA synthesizers meet all the requirements for the stepwise integration of natural or modified nucleosides, as well as non-nucleosidic building blocks into an oligomeric chain. In the latter case, adjustments to the standard conditions may be required, including the identification of appropriate solvents for dissolving the phosphoramidites, adjusting of coupling times, preparation of functionalized solid supports and optimization of cleaving and deprotection conditions. The suitability of a molecule for solid-phase synthesis (SPS) is ultimately given by its chemical compatibility with the chemical reactions of the synthesis cycle. In general, the major obstacles encountered during incorporation of non-nucleosidic monomers reside in low solubility of the phosphoramidites, trace water content in solvents or acid lability of the building blocks. Release from the support is performed under basic conditions, often at elevated temperatures that may also be harmful to the oligomers. Thus, mild cleavage and deprotection procedures are required for sensitive building blocks. Low solubility and adsorption of the oligomeric product on the solid support may require several rounds of treatment of the latter with appropriate solvents. Fortunately, the standard protocols were developed for the rather sensitive natural nucleosides, thus, non-nucleosidic compounds are generally amenable to integration into oligomers by this approach. Alternatively, a solution phase synthesis may be envisaged, whereby the individual chemical steps of the synthesis cycle remain essentially the same as in SPS. The necessity of solution phase synthesis may arise in cases of poor solubility of the building blocks, precipitation due to mixing of reagents during the coupling step, inefficient coupling on solid phase or due to demand for quantities larger than typically obtained in SPS, which is in the sub-milligram range.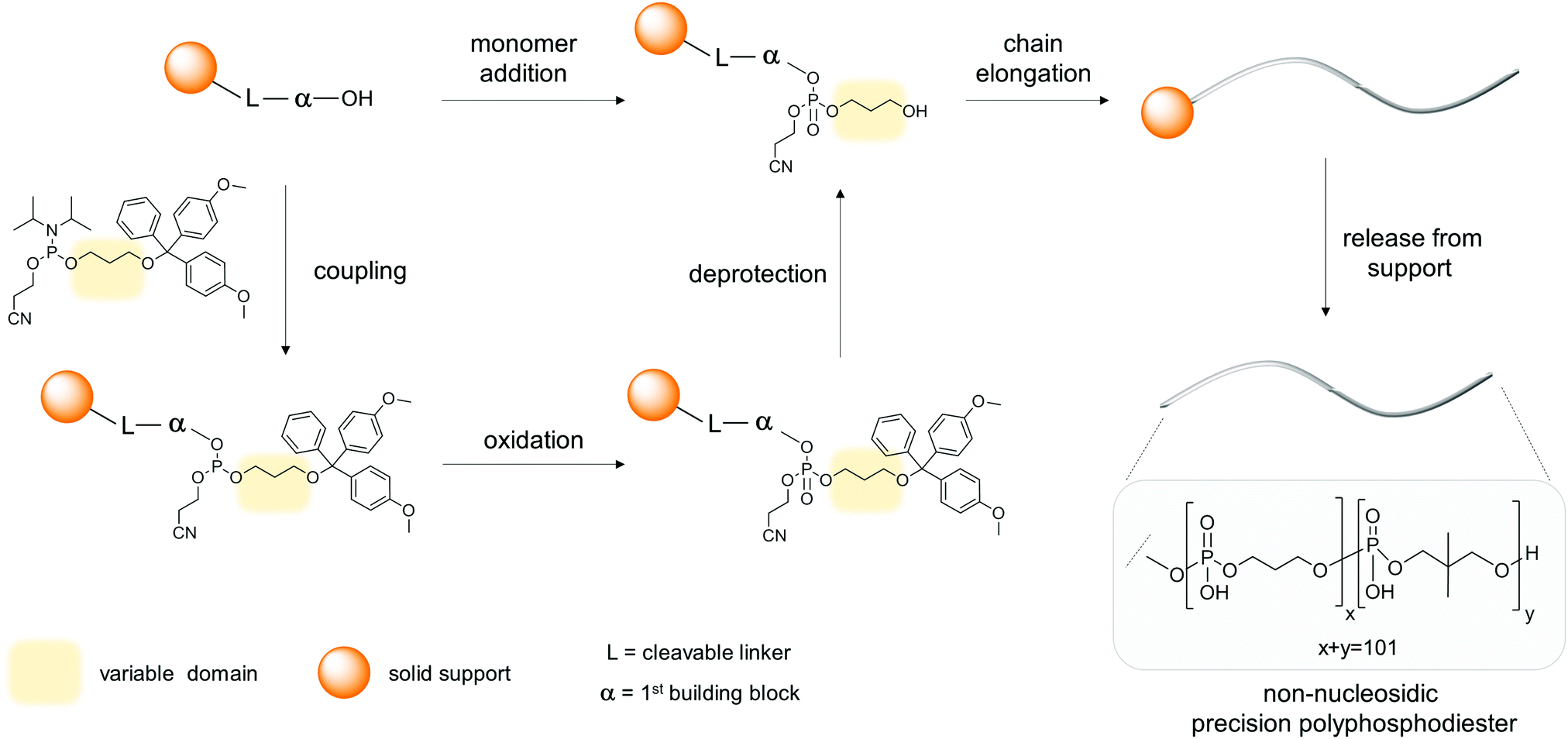 | ||
| Scheme 1 The solid phase synthesis used by Lutz to prepare polyphosphodiesters with up to 104 monomer units.10 Addition of one monomer, e.g. propane-1,3-diol, requires several synthetic steps (coupling, oxidation, deprotection). | ||
The first set of sequence-defined, monodisperse oligophosphodiesters (up to undecamers) completely devoid of nucleosides and solely based on perylene diimides 14 (PDIs) was presented in a seminal report by Li and co-workers in 2003.8 To prepare these oligomers, a demanding solution phase synthesis using standard phosphoramidite chemistry was required. Once synthesized, however, the PDI oligomers served as a rewarding model to explore molecular folding and self-assembly phenomena in multichromophore systems. In a series of papers, the Li group was able to consolidate and extend their findings on characteristic chromophore interactions. Several years later, in 2010, our group reported the first examples of entirely non-nucleoside precision oligophosphodiesters prepared by solid phase synthesis. The longest oligomers consisted of fourteen phosphodiester-linked pyrenes 8 (oligoarenotides) and one chiral diol.9 The work revealed surprising structural analogies between oligoarenotides and nucleic acids and prompted a series of further studies in the field of DNA-related systems. An inspiring example of using solid phase synthesis for the preparation of sequence-controlled oligophosphodiesters (SCP) containing over 100 units of 1 and 2 was reported by Lutz and co-workers in 2015 (Scheme 1).10 In a series of works, this group explored various synthetic approaches to derive precision oligophosphodiesters serving as binary-coded sequences in sufficient quantities and high purity. These characteristics are vital for the pursued application of coding and decoding of information embedded in sequence defined oligomers.
Increasing the structural diversity of molecular components for the solid phase synthesis is necessary to expand the functional potential of DNA-inspired systems. To this end, Sleiman and co-workers recently proposed a cost- and time-effective solution by preparing clickable tertiary amine monomers compatible with solid phase synthesis.11 Derivatization of alkynyl and carboxylate compounds (4 and 5, respectively) provided access to a variety of water-soluble sequence-defined oligomers.
With the reliable technology of solid-phase DNA synthesis in hands, various sequence-controlled oligophosphodiesters have become routinely accessible. Many of the reported DNA-inspired oligomers have already entered the field of supramolecular self-assembly, offering opportunities for creating advanced soft materials. The following chapters highlight the possibilities to control the dimensionality of self-assembled oligophosphodiesters, a crucial parameter for diverse applications. In parallel, interactions of self-assembled oligophosphodiesters with functional soft matter will be discussed.
Zero-dimensional supramolecular systems
An important subgroup is defined by quasi-zero-dimensional materials obtained from different precision oligophosphodiesters. The predominant representatives of such systems are self-assembled DNA-based micelles and vesicles. In this regard, a number of reports on synthetic polydisperse polyphosphodiesters illustrate their notable properties as biodegradable and biocompatible materials.12 At the end of the chapter, we will outline several lines of research in which precise molecular insertions into DNA chains can be used as a tool to control their folded structures and topologies. The topic of nucleic acids origami is largely excluded from the discussion to maintain the focus on the controlled folding of non-nucleosidic oligomer and polymer chains.Micelles belong to the most common type of aggregates which find numerous applications in molecular transfer, catalysis, and biomolecular sensing. Controlled self-assembly leading to micelles occurs in the case of amphiphilic oligomers with a low volume fraction of the hydrophobic segment. In monodisperse oligophosphodiesters, the hydrophilic fragment is mostly represented by DNA, whereas various types of lipophilic compounds play the role of the hydrophobic part.13 Often, such oligomers contain only one hydrophobic building block, which acts as a self-assembling unit leading to spherical aggregates. A relatively easy synthetic access to single lipophilic modifications of DNA ensured extensive research in the area with an emphasis on the biological behavior of the corresponding conjugates and less attention to their self-assembly behaviour. Filling this gap, Sleiman and co-workers conducted extensive testing of micelle formation from DNA–oligo(hexaethylene) conjugates.14 The oligonucleotide fragment was represented by a relatively bulky, pre-assembled DNA prism. This design puts certain restrictions on the number of DNA prisms which can reside on the outside of the sphere of segregated hydrophobic chains. Also the number of hexaethylene units 3 was used to modulate the formation of micelles. By carefully balancing these input parameters, the team managed to control formation and composition of the micelles by the molecular structure of the assembling units. Despite nonselective interactions of hydrophobic chains, the number of DNA conjugate units included in micelle was unique for each type of oligomers, which is by itself an unprecedented discovery.
Central to the properties of DNA micelles, the chemical composition of monomers results in a densely packed array of oligonucleotides (DNA corona) stretching out from the surface of a hydrophobic core. The DNA coverage on spherical surfaces can be modulated, yet with limited precision, by exploiting noncovalent interactions in multicomponent systems. In particular, DNA–lipid conjugates act as efficient anchors penetrating a lipid bilayer upon mixing with vesicles, forming so-called liposomal nucleic acids.15 Noteworthy, DNA liposomes are attractive substrates for mastering microscopic structural transformations in vesicular soft materials. For example, the group of Vogel successfully applied DNA encoding to cascades of liposome fusions, highlighting the potential of sequence-specific oligomers for the programming of complex interaction patterns and the mimicking of essential biological processes (Fig. 3A).16 This line of research has the potential to progressively evolve into more complex systems by exploiting the dynamic nature of soft materials. Recently, Varghese and co-workers demonstrated that the formation of DNA vesicles is possible by using host–guest interactions (Fig. 3B).17 To this end, a DNA strand was conjugated to a β-cyclodextrin (β-CD) via click chemistry. Introduction of a hydrophobic adamantane derivative triggers a cascade of noncovalent interactions. First, adamantane forms a stable host–guest complex with β-CD. Second, this complex self-assembles into vesicles due to the interactions between aliphatic and aromatic units covalently linked to adamantane. This work outlines a promising conceptual approach of introducing supramolecular interactions, which are dynamic and stimuli-responsive, for mastering the self-assembly of sequence-specific polymers into functional materials. The robustness of the β-CD–adamantane complex offers opportunities to explore a large chemical space of in situ generated dynamic oligophosphodiester amphiphiles.
 | ||
| Fig. 3 (A) DNA-coded liposome fusion. Figure adapted from ref. 16, with permission from John Wiley & Sons, Inc., copyright 2017. (B) Host–guest interactions resulting in DNA-grafted vesicles. Figure adapted from ref. 17, with permission from the Royal Chemical Society, copyright 2017. (C) Precision micelles forming monodisperse oligomers. Figure adapted from ref. 14, with permission from the American Chemical Society, copyright 2014. (D) Thermally induced aggregation of tetraethylene glycol-linked chromophores leading to the folding of DNA chains. Figure adapted from ref. 18, with permission from the American Chemical Society, copyright 2003. (E) Hydrophobic and stacking interactions modulating the spatial positioning of DNA strands. Figure adapted from ref. 19, with permission from the American Chemical Society, copyright 2012. (F) Triphenyl linkers guiding the assembly of DNA into cyclic structures. Figure adapted from ref. 20, with permission from the American Chemical Society, copyright 2007. | ||
Although significant advances in vesicular and micellar DNA systems illustrate the utility of liposomal nucleic acids, limited modularity of the biophysical properties of liposomes and their tendency towards fusion pose some drawbacks. For precision applications, a lack of dynamic control over the immersion of DNA–lipid conjugates into a bilayer and unknown surface densities of nucleic acids in individual self-assembled nanoparticles are issues that need to be addressed. The properties observed in experiments reflect a collective behavior of vesicles, which exist as a polydisperse population of supramolecular objects with theoretically different functional potential. The problems can be mitigated by several approaches relying on sequence precision. Thus, the Sleiman group focused on preparing compositionally defined oligomers with a precise number of hydrophobic units (Fig. 3C).21 These oligomers can be assembled into micelles with a controllable diameter and hydrophobicity. The possibility to synthesize such amphiphilic oligomers with a defined number of assembling units with desirable physicochemical properties provides access to programmable soft materials. A decisive parameter of these oligomers is the regular placement of phosphate groups in the hydrophobic region.
In general, low dimensional soft materials, such as micelles and vesicles, take a forefront position as biotechnology tools for efficient cargo delivery and compartmentalization.15 Nevertheless, these delivery systems are facing considerable challenges in several aspects, such as problems with loading, targeting and release of cargo, batch-to-batch reproducibility, or in the scale-up of materials. The community will surely benefit from higher precision in the composition of self-assembled oligomers. Extending the range of chemical designs of both, the hydrophobic and hydrophilic parts of oligophosphodiesters, are necessary steps to attain increased structural and functional diversity. Such supramolecular systems can find applications as advanced cargo compartments with stimuli-responsive and/or targeting properties.
Proper folding of oligomer and polymer chains occurs in nature to create catalytic macromolecules (e.g. proteins or ribozymes). Currently, the controlled folding of nucleic acids strongly concentrates on DNA origami – the art of folding long circular DNA into desired 3D shapes.22 Using noncovalent binders of nucleic acids was reported as an attractive, although considerably less modular, approach to change their 3D shapes. However, examples of point insertions of non-nucleoside molecules which modulate 3D arrangement of nucleic acids are quite limited. Due to lack of the required tools and the intrinsic problems involved with structure elucidation of nucleic acids, structures often remain scarcely analyzed in literature, not to mention the absence of detailed studies on controlled folding – not assembly – of oligophosphodiesters beyond the natural molecules. A further reason explaining this fact is associated with the limited resolution of available experimental techniques to analyze the 3D shape of synthetic nanoobjects. Additionally, most of the research in the area is focussed on chemical systems that tend to self-assemble rather than to fold. Developing reliable models to describe the folding process of sequence-defined synthetic polymers into desired shapes would be a significant step toward rational construction of functional systems. Toward this end, several groups provided conceptual foundations based on hybrid oligophosphodiesters. In 2003, Li and co-workers showed that the tertiary structure of a DNA can be controlled by site-specific incorporation of several tetraethylene glycol-perylene units (Fig. 3D).18 Monitoring the aggregation behaviour of the chromophores by changes in their absorption spectrum revealed an inverse temperature folding behaviour, i.e. the hydrophobic interactions between the chromophores and ethylene glycols (EGs) strengthen upon heating. In the latter case, the well documented property of EGs to aggregate upon heating due to lower critical solution temperature (LCST) was not explicitly analyzed in the paper, although it can contribute significantly to the thermal folding process. Importantly, the initial folding of the hydrophobic ethylene glycols perylenes at high temperature directed the subsequent folding of the single-stranded DNA parts. The groups of Nguyen and Schatz demonstrated that hydrophobic interactions between rigid small molecules, i.e. 1,3,5-trisalkynyl substituted benzenes 17, can impact the spatial positioning of DNA helices (Fig. 3E).19 The trivalent molecule was conjugated to three DNA strands. Depending on the choice of the DNA sequence, hybridisation leads to the formation of cage dimers or face-to-face dimers. The presence of single stranded T6 fragments which ensure conformational flexibility results in nanocages with two hydrophobic caps and three dsDNA pillars. Omitting the flexible T6 fragments, on the other hand, leads to the formation of highly stable face-to-face dimers. When exposure of the hydrophobic surfaces of the cores cannot be properly minimized in a dimer conformation, ill-defined networks are formed. This work further emphasizes the importance of hydrophobic interactions as an essential folding element for phosphodiester oligomers. Aromatic units are frequently used as spatial control elements in creating topologically defined systems, hence also in short cyclic oligonucleotides. Sleiman et al. proposed a method to incorporate a rigid triphenyl vertex in the middle of a DNA strand, facilitating the formation of otherwise restrained nanostructures.20 Conducting several hybridization–ligation–purification cycles allowed to access triangular, square, pentagon, and hexagon templates used for diverse modifications in functional systems.
It is noteworthy to mention that ill-ordered globular aggregates, either as products of self-assembly or folding, abound in supramolecular systems and often represent kinetically trapped states of low practical value. In such systems, vital properties including the dynamics of molecular exchange, conformational flexibility, and directionality of interactions can be significantly perturbed and most of the published data ignore these ill-defined formations. Devising reliable strategies for preparing and handling soft matter is one of the cornerstones in the field of DNA-inspired oligophosphodiesters. We believe that with the current pace of technological advances in instrumental and analytical techniques, the topic of controlled polymer folding will receive wider attention in the area of precision polymers. In this regard, oligophosphodiesters are valuable substrates due to the facile modulation of their precise composition and therefore help accessing a larger structural and functional diversity.
One-dimensional supramolecular systems
One-dimensional (1D) soft materials are indispensable for a broad range of technological applications. In particular, 1D assemblies of nucleic acids triggered widespread interest in the materials sciences for targeting functions beyond the obvious connection to biology.23 For example, DNA can act as a versatile template for mineralization of various inorganic materials, providing access to nanoscale structures unattainable by other approaches.24 The growth occurs via interactions of available negatively charged phosphate groups with suitable nucleation seeds followed by mineralization growth. This process is largely independent from the sequence and, thus, can be used for the conservation of the informational content of DNA. Another important aspect in this context is the fact that the long-range order of the stacked DNA base pairs is maintained during the mineralization process. This parameter is essential for using DNA wires in nanoelectronics and for constructing biosensing devices.25 Unfortunately, the limited scope of natural nucleobases, which are not optimal charge carriers, is an obstacle toward rapid advance of this area. To further develop these and other areas, the identification of diverse and economic structural DNA mimics with adaptable properties is a necessary step to approach future technological utilization.In 2011, our group reported the preparation and characterization of a 1D supramolecular polymer possessing astonishing structural similarities to long double-stranded DNA.26 The material was obtained by supramolecular polymerization of oligophosphodiester 18 (Fig. 4) composed of seven 1,8-disubstituted pyrene units. The oligomer forms fibroid assemblies due to strong non-covalent inter- and intramolecular interactions between pyrene bis-amides. The formation of fibers was unambiguously confirmed by a set of spectroscopic measurements and TEM and later by AFM analysis. Importantly, the supramolecular polymers can be rendered optically active, which is reflected by the emergence of a characteristic CD signature, by addition of small amounts of a nucleotide conjugate 19. The optical activity is explained by the interaction of cytosine with the pyrene stack. Earlier, we showed that cytosine can be replaced by another chiral auxiliary, even of a non-nucleoside nature.9 Introducing structural changes to the side chains of the 1,8-substituted pyrene core was investigated as the most straightforward strategy to modulate the properties of DNA-mimicking, one-dimensional supramolecular polymers. Thus, replacing the two amide groups of the pyrene core with triple bonds leads to the formation of micrometre long fibers even with very short oligomers, such as trimer 20 and at considerably lower ionic strength.27 The enhanced tendency to aggregate was ascribed to an increased hydrophobicity of the alkynyl linkers relative to carboxamides. Owing to the dynamic nature of supramolecular polymers, oligomers 21 and 22 are readily integrated into fibers by cooling a pre-heated mixture (above the temperature of disassembly) containing both types of oligomers.28 The trimer 21 contains 3,6-bisalkynyl substituted phenanthrene units, a close structural analog of pyrene, whereas 22 has an additional pyrene unit. Once mixed supramolecular polymers are formed, phenanthrene units act as efficient UV light collectors, transferring energy within a fiber over distances over 100 nm to fluorescent acceptor pyrenes. Thus, a remarkably effective light-harvesting antenna is created by the simple mixing of two short oligophosphodiesters. In a recent extension of this work, DNA photonic wires with differently arranged fluorophores were integrated into light-harvesting supramolecular phenanthrene polymers (Fig. 5). Optimal arrangement of the chromophores resulted in light-harvesting complexes with antenna effects ranging from 1.4 up to 23.29
 | ||
| Fig. 4 Structures of phosphodiester oligomers 18–22. | ||
 | ||
| Fig. 5 (A) Integration of DNA photonic wires into light-harvesting supramolecular polymers (SPs). Figure adapted from ref. 29, with permission from John Wiley & Sons, Inc., copyright 2019. (B) Writing of AuNPs onto DNA-grafted SPs. Figure adapted from ref. 30, with permission from John Wiley & Sons, Inc., copyright 2017. (C) Thermally induced supramolecular polymerization of diblock DNA oligophosphodiesters. Figure adapted from ref. 31, with permission from John Wiley & Sons, Inc., copyright 2015. (D) Accelerated DNA computing using DNA-grafted SPs. Figure adapted from ref. 32, with permission from the American Chemical Society, copyright 2018. (E) Incorporation of terminal cyanines leads to a change of the morphology from micelles to fibers. Figure adapted from ref. 33, with permission from the American Chemical Society, copyright 2018. (F) Control of the enzymatic activity in supramolecular dynamic systems. Figure adapted from ref. 34, with permission from the Springer Nature, copyright 2018. | ||
Another area of application of sequence-specific oligophosphodiesters is the preparation of DNA-grafted 1D supramolecular polymers. In several publications, Häner and co-workers demonstrated that the morphology of self-assembled diblock oligomers consisting of variable numbers of nucleotides and 1,6-bisalkynylsubstituted pyrenes 10 is highly dependent on their composition. Thus, oligomers containing 10 nucleotides and seven pyrenes self-assemble into 1D helical ribbons, whereas decreasing the number of pyrenes leads to the formation of micellar aggregates (Fig. 5C).31 The driving force of the 1D polymerization is the intermolecular stacking of folded pyrene oligomers, which is sufficiently strong to overcome the repulsion between neighboring DNA strands. In this case, the thermally controlled supramolecular polymerization occurs via a cooperative mechanism, which includes nucleation and elongation stages. Applying a slow cooling procedure (slower than 0.2 °C min−1) was crucial for obtaining morphologically uniform formations. Resolved by atomic force microscopy (AFM) and transmission electron microscopy (TEM), these assemblies appeared as several hundred nm long and 2–3 nm thick ribbons with a pronounced helical profile. Moreover, the oligonucleotides remain functional and selectively bind gold nanoparticles modified with a complementary strand.35 High surface density of DNA chains along the 1D platform can lead to unexpected consequences caused by noncovalent interactions.36 Upon introducing a 10 nt complementary strand to the assembled ribbons, the supramolecular polymers are transformed into large 3D networks due to blunt-end stacking, i.e. stacking interactions between terminal base pairs of the appended DNA duplexes. Apart from microscopic studies, the formation of 3D networks can be followed by changes in the UV-vis spectra of pyrenes, which serve as an extremely sensitive reporter of the relative orientation of the arrayed transition dipole moments of the chromophore. This system lends more weight to the opportunities of controlling spatial orientation of soft materials at the nanoscale by structurally remote events. Indeed, hybridization of edge-located oligonucleotides leads to distinct perturbations in the core of the 1D supramolecular polymer, a case resembling the mode of action exploited in naturally occurring riboswitches.37
The abovementioned observations demonstrate the feasibility of using precision oligomers for the controlled assembly by rational design. Recently, the Sleiman group reported an elaborate study on the molecular programming of self-assembly in sequence-defined oligomers (Fig. 5E).33 The molecules were prepared via solid-phase synthesis and included a 19 nt long DNA chain, 12 hexaethylene units (HE12), and one or two site-specific cyanine (Cy3) modifications. By introducing Cy3 monomers at the terminal positions of the HE12 block, self-assembly results in well-defined fibers. As confirmed by FRET experiments, these fibers are dynamic formations permanently exchanging their constituent oligomers. Placing a Cy3 unit in the middle of the HE12 chain or between DNA and HE12 fragments only yields spherical aggregates. The results showcase the potential of site-selective modifications in precision oligomers to guide the self-assembly into dimensionally variable architectures. The subtlety of noncovalent interactions, as demonstrated in this work, calls for a careful evaluation of small structural variations introduced into oligomers and further emphasizes the value of sequence precision in oligomer design. An example of shape-shifting pairs in hybrid DNA materials was discovered by Varghese and colleagues.38 A DNA conjugate of 23 forms vesicles which are transformed into cylindrical objects upon addition of a complementary oligonucleotide (Fig. 6). The switch to 1D morphology is probably caused by the increased charge density in double-stranded DNAs. The finding extends the scope of approaches to access shape-shifting objects in soft materials.

An attractive avenue for modulating the efficiency of oligonucleotide hybridization using supramolecular polymerization of DNA conjugates was proposed by Brunsveld and co-workers.39 The group prepared a series of bis-pyridine-based C3-symmetrical amphiphilic discotic molecules conjugated to oligonucleotides, in some cases labeled with fluorescent or quencher tags. FRET experiments were the major tool to analyse hybridization of complementary DNA strands; the self-assembly of discotic molecules into supramolecular wires was anticipated on the solid basis of previously reported systems using this scaffold. The data revealed increased stability of hybridization due to the high effective molarity of oligonucleotides arranged on the supramolecular wires. In this regard, this work opens intriguing opportunities to modulate the strength of binding between oligonucleotides by the means of a supramolecular polymer core. A superior level of control over the properties of self-assembled materials is offered by multicomponent assemblies. To this end, Kieltyka and co-workers applied the strategy of doping 1D supramolecular polymers with DNA conjugates (Fig. 5B).30 Chemical integrity of the self-assembling unit, in this study squaramide 24, is a prerequisite for the successful integration of oligonucleotide conjugates into the supramolecular stacks. The efficient incorporation of the targeted amounts of squaramide–DNA (1 mol%) oligomers was confirmed for the co-assembled materials by the sequence-specific binding to the protruding oligonucleotide strands. Thus, the programmability of DNA hybridization allowed the loading of the 1D scaffold with gold nanoparticles of different size, unambiguously proven by TEM. The process of loading is reversible, and the nanoscale objects can be easily removed from the 1D scaffold regenerating again an array of single-stranded oligonucleotides ready for loading with a different type of cargo. Such materials can find applications as stimuli-responsive, smart materials.
The dynamic nature of DNA-grafted supramolecular polymers was elegantly used by Meijer and co-workers to control the activity of enzymes via selective recruitment (Fig. 5F).34 The functional polymer was assembled from two monomers – 1,3,5-benzenetricarboxamide (BTA) and the corresponding DNA conjugate, prepared via click chemistry from azide 25. These molecules share a BTA self-assembling motif, ensuring efficient integration of variable amounts of the conjugate (0–100 mol%) in the stacks of supramolecular polymers via triple hydrogen bonding, stacking and hydrophobic interactions. The formation of micrometre long fibers was revealed by various techniques, including optical spectroscopy, fluorescence microscopy, cryo-TEM, and most recently AFM. Single-stranded DNAs protruding from the polymer core were used for selective and reversible recruitment of enzyme TEM1-β-lactamase and its inhibitor protein (BLIP). In the absence of the inhibitor, enzymatic catalysis leads to the hydrolysis of a substrate. Introducing BLIP leads to significant suppression of the enzymatic activity due to the association of the enzyme–inhibitor pair along the supramolecular scaffold. In a follow-up study, Merkx and colleagues used the same supramolecular polymer to provide a tool for accelerating DNA-based computing (Fig. 5D).32 It was shown that a 1D platform sufficiently improves the kinetic parameters of DNA strand displacement reactions due to the high local concentration of oligonucleotides.
The programmable self-assembly of sequence-specific oligophosphodiesters sets the stage for the preparation of covalent materials with diverse morphologies. Recently, Häner and Yu presented a strategy toward the synthesis of nanotubes via a preorganization–polymerization process.40 In this design, a trimer of anthracene 12 was chosen as a suitable unit for forming tubular objects via self-assembly. Light-induced covalent linkage of the anthracene units within the supramolecular polymer led to the formation of covalent nanotubes, as demonstrated by optical spectroscopy, TEM and AFM experiments.
A remarkable way of assembling tubular objects with a narrow pore from precision oligophosphodiesters was reported by Asanuma and co-workers.41 By using two D-threoninol nucleic acids, each consisting of decameric aminopyrimidine 6 and cyanuric acid 7, the formation of a hexameric circular complex was achieved via multiple in-plane oriented hydrogen bonds. Upon melting, the system displays substantial hysteresis of 55 °C due to the coordinated interactions of many H-donors and acceptors. This approach of constructing supramolecular assemblies can find further applications in preparation of soft materials with enhanced thermal stability.
Environmental conditions emerge as a predominant factor governing the formation of soft materials. In this regard, there is an illustrative example by Liu and co-workers who explored the fiber-micelle transformation in self-assembling dendron–DNA conjugates by varying the content of THF in water.42 In the absence of THF, micrometre-long fibrillary structures with a diameter of 20 nm can exclusively be detected in TEM experiments. The fibers consist of a hydrophobic core of the assembled dendritic entities with oligonucleotides stretched outside the 1D platform. Adding 10% of THF, followed by removal of the organic solvent via dialysis, yields undefined micellar aggregates. The transition was demonstrated by TEM and dynamic light scattering experiments. Importantly, re-annealing of the micellar aggregates in pure water restores the original fibrillary morphology.
Applying thermal stimuli is a suitable approach to avoid kinetically trapped states and obtain the desired set of properties.43 Isothermal mixing of two kinds of 1D DNA-grafted supramolecular polymer containing complementary oligonucleotide strands leads to the formation of cross-linked networks. Despite the dynamic nature of self-assembled materials, these networks remain unchanged for days. Upon heating, the 3D networks disintegrate because of the disassembly of both DNA crosslinks and stacked pyrenes forming the supramolecular polymer core. Controlled cooling of the multicomponent mixture produces only individual ribbons. This happens because pyrenes form stacked platforms at a higher temperature than DNA hybridization takes place, integrating two kinds of complementary strands within the same core. Later, high strand density prevents network formation.
An alternative design approach to arrive at 1D supramolecular polymers in sequence-specific oligomers is based on end-to-end stacking of chromophores.44 Thus, Lewis and Rybtchinski reported a double-stranded DNA capped at both sides with a perylenediimide 15. The strong stacking of the chromophores accounts for the formation of 1D fibers. The work reveals the importance of precise positioning of self-assembling units in driving the aggregation. The groups expanded the set of PDI–DNA conjugates by attaching variable numbers of self-complementary (GC)n or (AT)n strands where n ≤ 3 to the PDI unit. Self-assembly behavior revealed that stacking of PDIs prevails in most cases. However, the properties of assembled materials can be modulated by DNA hybridization, leading to a diversity of morphological shapes.
A valid strategy that received little attention so far is the use of non-aqueous solvents to prepare 1D assemblies from oligophosphodiesters. By exposing a double-stranded DNA, in which each strand is conjugated to a dendritic oligoethylene chain, to acetonitrile leads to the formation of micrometre-long fibers.45 It is tempting to assume that structural DNA mimics can undergo similar transformations and thus be used for controlled surface depositions for various applications.
Despite significant advances achieved over the past decade, the field of 1D materials obtained from DNA conjugates and DNA-inspired building blocks is still in an early phase. Currently, two main approaches toward their preparation include the self-assembly of individual, sequence-specific oligophosphodiesters and the supramolecular copolymerization of 1D fibers and DNA conjugates carrying anchoring entities. In the former case, the general design principles, unfortunately, remain poorly understood; however, several reports indicate that the presence of aromatic units is beneficial, if not necessary. Exploring additional chemical diversity is necessary to reach predictability of aggregation patterns for a wider set of molecular entities. Stronger integration of computational tools would be highly beneficial for getting deeper insights into self-assembly mechanisms.46 In the latter case, the general issues raised in the chapter of vesicular assemblies, such as a description of individual components and control over the dynamics of oligomer exchange, remain largely unexplored. Also, integrating non-nucleosidic oligomers into 1D soft matter emerges as an attractive method to enhance the functional potential of supramolecular polymers.
Two-dimensional supramolecular assemblies
Two-dimensional materials have received unprecedented attention over the past two decades. They raise high expectation with regard to their properties, quite distinct from one-dimensional systems, that may help solving persistent technological problems in the areas of energy, catalysis, molecular filtration or ultra-strong polymer production.47 The challenge to bring 2D materials closer to application consists in the limited synthetic accessibility of such materials, high costs of production, and restricted variability of their chemical composition. Furthermore, to address a broad range of practical demands requires access to 2D materials with diversified functional cores and also with surfaces that are amenable to controlled and precise modification. In this regard, separating individual sheets from multi-layered objects with nanoscale precision or devising synthetic strategies for direct production emerge as central issues. The following discussion reveals that oligophosphodiesters can be assembled into 2D systems via rational sequence design. The abundance of phosphates in these oligomers facilitates self-assembly into individual nanoscale sheets. The chapter again avoids the large number of reports utilizing the DNA origami approach to access 2D materials.The first example of a 2D supramolecular polymer assembled from DNA-inspired oligomers was reported in 2013 (Fig. 7A).48 It was the result of a serendipitous discovery that stimulated our interest in this morphologic shape. The assembly of a trimer of 1,6-bis-alkynyl substituted, phosphodiester linked pyrene 10 occurs via a temperature-controlled process and leads to the formation of supramolecular nanosheets with a thickness of 2 nm and up to ten micrometres in lateral dimensions. This 2D material is water soluble because both surfaces of the nanosheets are covered with a network of phosphates, whereas the hydrophobic pyrenes are sandwiched in between. The internal organization of pyrenes within 2D arrays corresponds to a stacked, ladder-folded trimer. The model is based on experimentally observed changes in the UV-vis spectra upon aggregation (emergence of J- and H-type bands for different electronic transitions) and theoretical calculations of the electronic interaction of the transition dipole moments of 1,6-substituted pyrenes.
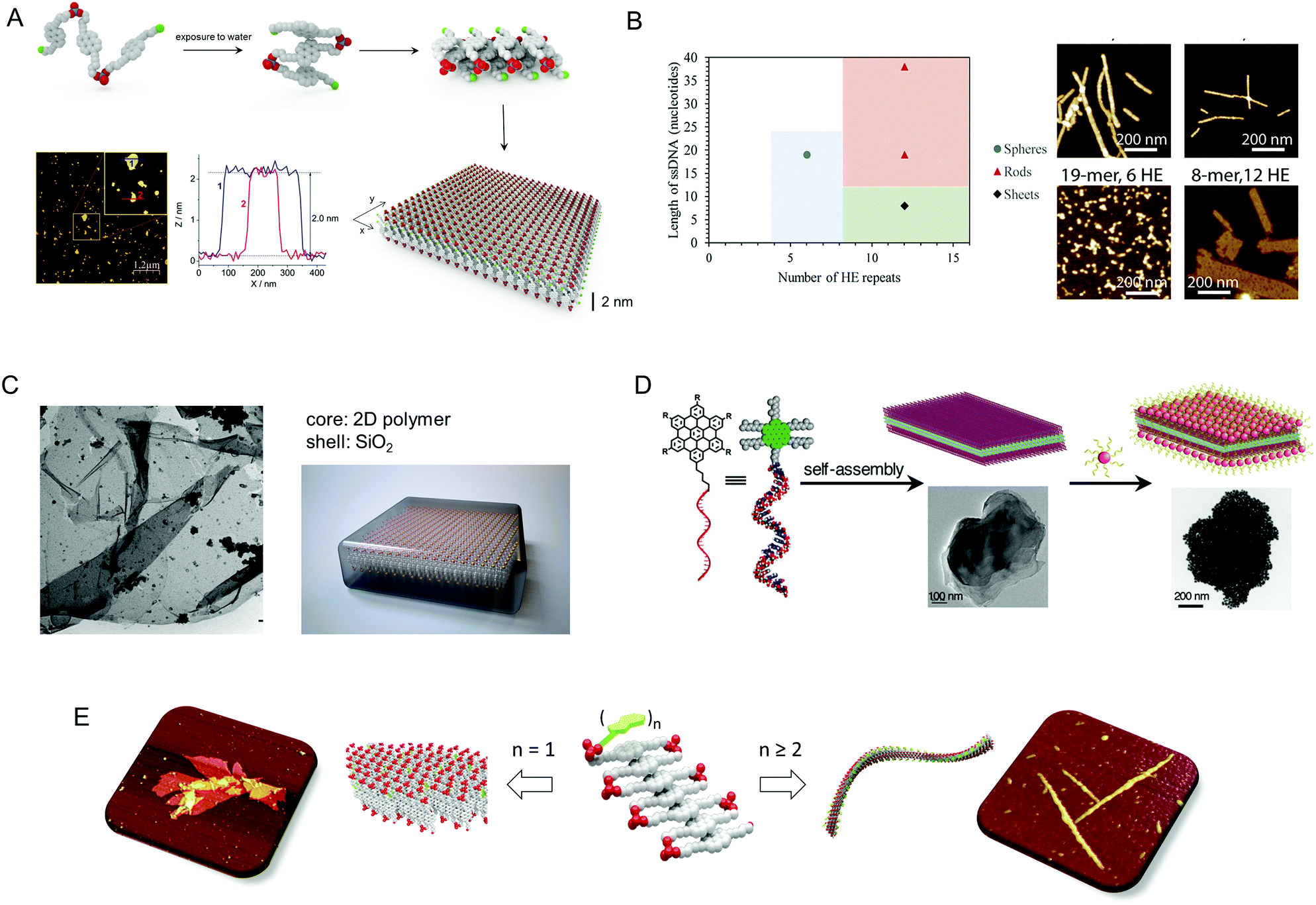 | ||
| Fig. 7 (A) Illustration of the cooperative self-assembly process of 1,6-substituted pyrene trimers leading to 2D SPs. Figure adapted from ref. 48, with permission from John Wiley & Sons, Inc., copyright 2019. (B) Assembly of SPs of different morphological shapes depending on the number of hydrophilic (DNA) and hydrophobic parts. Figure adapted from ref. 33, with permission from the American Chemical Society, copyright 2018. (C) 2D SPs as templates for the growth of a nanoscale SiO2 shell. Figure adapted from ref. 27, with permission from John Wiley & Sons, Inc., copyright 2017. (D) Self-assembly of small-molecule/DNA conjugates into 2D DNA-grafted supramolecular materials. Figure adapted from ref. 49, with permission from the American Chemical Society, copyright 2017. (E) The number of nucleobases attached to the chain of phosphodiester-linked pyrenes differentiates between self-assembly into 2D or 1D objects/structures. Figure adapted from ref. 50, with permission from the Royal Chemical Society, copyright 2017. | ||
The supramolecular nature of the nanosheets and, most importantly, their 2D morphology was found to respond in a highly sensitive manner to weak, uni-directional mechanical perturbations, such as the internal flow in a sample cuvette, as revealed by chiroptical methods.51 Upon stirring of a solution containing the assembled nanosheets, a strong exciton coupled signal emerges in the area of pyrene absorption. Stirring of the solution in opposite direction leads to the reversal of the sign of exciton coupling. The magnitude of the effect is dependent on the size of the 2D aggregates, with smaller sheets predictably resulting in a weaker response. This example of 2D supramolecular systems responding to weak mechanical perturbations illustrates the importance of internal ordering of monomers within ultrathin soft matter to act cooperatively for adapting to environmental stimuli. Due to the multi-charged nature of the nanosheets in an aqueous environment, it is possible to use them as nucleation templates for biomineralization.27 In this approach, a shell of various inorganic materials with a controlled thickness can be grown at the outside of a template. Once oligophosphodiester derived nanosheets are formed, 2D silica sheets can be prepared by adapting the protocols previously developed and successfully applied to long double-stranded DNAs (Fig. 7C).24 It is noteworthy, that chemically similar 1D platforms yield the corresponding 1D silica rods. This work obliterates the borders between natural nucleic acids and DNA-inspired self-assemblies for certain applications in the materials sciences by demonstrating the availability of close structural alternatives to nucleic acids.
To tackle the preparation of DNA-grafted 2D supramolecular polymers, a set of oligomers possessing self-assembling aromatic units with a propensity to form planar shapes was conceived in recent studies (Fig. 7E).50 Diblock oligophosphodiester containing seven 1,6-bis-substituted pyrenes and a variable number of nucleotides (from 1 to 20) were investigated. The data confirmed that noncovalent interactions of pyrenes in aqueous buffer drive the self-assembly in all cases, but the morphology of the formed architectures is determined by the number of nucleotides. In this design, 2D supramolecular polymers tolerate the presence of only one nucleotide, and only 1D helical ribbons were detected starting from two nucleotides. These data agree with a proposed mechanism of 2D self-assembly by stacking of folded units since conjugated dinucleotides efficiently obstruct lateral stacking thereby preventing growth in one of the dimensions.
In 2017, DNA-decorated 2D soft materials have been reported by Varghese and colleagues via a controlled self-assembly pathway (Fig. 7D).49 The group managed to design and conjugate the aromatic motif 16, a hexa-peri-benzocoronene, onto DNA to direct the self-assembly of nanosheets. Although nanosheets appeared as multilayer formations based on the results from the HR-TEM and AFM experiments, the surface DNA strands retained their ability to bind complementary oligonucleotides. This was confirmed by covering the sheet surface with gold nanoparticles via a DNA-mediated process. Importantly, the self-assembly process showed little dependence on the properties of the oligonucleotide segment, which demonstrates the robustness of the method. Furthermore, recent results from the Sleiman group support the accessibility of 2D self-assembled materials via rational sequence design. By decreasing the length of an oligonucleotide part and increasing the number of hexaethylene fragments, a morphological shift from rods, extensively analysed in the paper, to sheets can be followed by AFM.33 Although further understanding of the rules of molecular programming of the self-assembly of oligophosphodiesters is required, these reports provide a conclusive reply to the question whether 2D materials are accessible via self-assembly of amphiphilic sequence-specific oligomers.
Orthogonal surface functionalization is one of the fundamental challenges in the area of synthetic 2D materials. The possibility to modify free-floating nanosheets by differentiation of the two opposite surfaces (Janus nanosheets), can solicit new avenues for applications of soft matter. For example, Liu and co-workers utilized planar DNA origami to test the concept of a frame-guided assembly of hydrophobic dendritic chains.52 Importantly, a densely packed 2D array of hydrophobic molecules served as a template to bind and organize other hydrophobic units introduced in the system. Such systems can become valuable as membrane mimics with a tailored set of functions.
Synthetic oligophosphodiesters have occupied an important niche in research on 2D self-assembled materials. Such nanosheets can mimic or modulate certain properties inherently associated with different biomaterials – nucleic acids, structural proteins or lipid bilayers – and thus bear a high degree or functional potential. We believe that with the existing examples of oligophosphodiesters assembling in 2D presented above, the field has already matured to the point of addressing more fundamental questions. For example, exploring the dynamics of molecular exchange in noncovalent 2D materials is necessary to complement our current knowledge about their 1D counterparts. Or how can we create segregated domains in synthetic 2D systems, which could e.g. mimic lipid rafts? What are the properties of 2D copolymers? Can small molecules induce a controlled degree of curvature in otherwise planar sheets? What are the properties of chiral nanosheets? Addressing these and many other questions will help shaping 2D soft materials for future technological demands.
Conclusions
Recent advances in supramolecular chemistry, polymer science, and DNA/RNA nanotechnology have inspired the emergence of the new field of precision oligophosphodiesters. In a simplistic definition, these oligomers represent a universal version of single-stranded nucleic acids, in which different types of functions are encrypted in the sequence. By studying precision oligophosphodiesters, the major emphasis is placed on the informational content of each unit embedded in a polymer string as a means to attain the desired functions. The overall properties of such systems are manifested by the net outcome of noncovalent interactions, both intra- and intermolecular ones. Depending on their strength under given environmental conditions, oligophosphodiesters can self-assemble into morphologically defined architectures and, in some instances, are prone to folding into single polymer nanoparticles.The dominating role of nucleic acids as programmable building blocks in nanotechnology is comprehensible and justified by an overwhelming number of reports and applications. However, this bias inevitably narrows some lines of research to the selective DNA hybridization paradigm. To receive wider acceptance as a competitive alternative to the nucleic acids, research with oligophosphodiesters will need to pass a number of critical milestones. One of the biggest problems – and at the same time an exciting opportunity – lies in a low coverage of the relevant chemical space of non-nucleosidic monomers that have so far been incorporated in precision oligophosphodiesters. Once a sufficient diversity of scaffolds and the corresponding information on their properties will be available, molecular programming to direct assembly and folding patterns of a broad set of molecules, similar to base pairing in DNA nanotechnology, will become feasible. To be successful, future research in this direction will depend on a considerable expansion of the variability of synthetically accessible, information-rich building blocks of non-nucleosidic nature. A rational based access to different morphological shapes will complement the existing methods of molecular scaffolding. Devising strategies toward preventing the aggregation of folded single polymer chains and controlled biasing of folding and self-assembly processes are yet other poorly explored research directions.
Continuous progress in the field will help solving existing problems in the material sciences, such as biocompatible catalysis, information encryption and transmission, conducting molecular wires or template-based applications. Entirely non-nucleosidic oligophosphates will have their impact on existing technologies.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by the Swiss National Foundation (grant 200020_169030) is gratefully acknowledged.Notes and references
- V. A. Bloomfield, D. M. Crothers and I. Tinoco, Nucleic Acids – Structures, Properties, and Functions, University Science Books, Sausalito, 2000 Search PubMed.
- O. I. Wilner and I. Willner, Chem. Rev., 2012, 112, 2528–2556 CrossRef CAS PubMed.
- D. A. Malyshev and F. E. Romesberg, Angew. Chem., Int. Ed., 2015, 54, 11930–11944 CrossRef CAS PubMed.
- N. Appukutti and C. J. Serpell, Polym. Chem., 2018, 9, 2210–2226 RSC.
- Z. Zhao, T. Du, F. Liang and S. Liu, Int. J. Mol. Sci., 2018, 19, 2283 CrossRef PubMed.
- S. C. L. Kamerlin, P. K. Sharma, R. B. Prasad and A. Warshel, Q. Rev. Biophys., 2013, 46, 1–132 CrossRef CAS PubMed.
- M. H. Caruthers, Acc. Chem. Res., 1991, 24, 278–284 CrossRef CAS.
- W. Wang, L. S. Li, G. Helms, H. H. Zhou and A. D. Q. Li, J. Am. Chem. Soc., 2003, 125, 1120–1121 CrossRef CAS PubMed.
- R. Häner, F. Garo, D. Wenger and V. L. Malinovskii, J. Am. Chem. Soc., 2010, 132, 7466–7471 CrossRef PubMed.
- A. A. Ouahabi, M. Kotera, L. Charles and J.-F. Lutz, ACS Macro Lett., 2015, 4, 1077–1080 CrossRef.
- D. de Rochambeau, Y. Sun, M. Barlog, H. S. Bazzi and H. F. Sleiman, J. Org. Chem., 2018, 83, 9774–9786 CrossRef CAS PubMed.
- T. Steinbach and F. R. Wurm, Angew. Chem., Int. Ed., 2015, 54, 6098–6108 CrossRef CAS PubMed.
- M. Schade, D. Berti, D. Huster, A. Herrmann and A. Arbuzova, Adv. Colloid Interface Sci., 2014, 208, 235–251 CrossRef CAS PubMed.
- C. J. Serpell, T. G. Edwardson, P. Chidchob, K. M. Carneiro and H. F. Sleiman, J. Am. Chem. Soc., 2014, 136, 15767–15774 CrossRef CAS PubMed.
- A. Lopez and J. Liu, Langmuir, 2018, 34, 15000–15013 CrossRef CAS PubMed.
- P. M. G. Löffler, O. Ries, A. Rabe, A. H. Okholm, R. P. Thomsen, J. Kjems and S. Vogel, Angew. Chem., Int. Ed., 2017, 56, 13228–13231 CrossRef PubMed.
- S. K. Albert, H. V. P. Thelu, M. Golla, N. Krishnan and R. Varghese, Nanoscale, 2017, 9, 5425–5432 RSC.
- W. Wang, W. Wan, H. H. Zhou, S. Q. Niu and A. D. Q. Li, J. Am. Chem. Soc., 2003, 125, 5248–5249 CrossRef CAS PubMed.
- I. Eryazici, I. Yildirim, G. C. Schatz and S. T. Nguyen, J. Am. Chem. Soc., 2012, 134, 7450–7458 CrossRef CAS PubMed.
- F. A. Aldaye and H. F. Sleiman, J. Am. Chem. Soc., 2007, 129, 13376–13377 CrossRef CAS PubMed.
- T. G. W. Edwardson, K. M. M. Carneiro, C. J. Serpell and H. F. Sleiman, Angew. Chem., Int. Ed., 2014, 53, 4567–4571 CrossRef CAS PubMed.
- P. W. K. Rothemund, Nature, 2006, 440, 297–302 CrossRef CAS PubMed.
- S. P. W. Wijnands, E. W. Meijer and M. Merkx, Bioconjugate Chem., 2019 DOI:10.1021/acs.bioconjchem.9b00095.
- R. N. Grass, R. Heckel, M. Puddu, D. Paunescu and W. J. Stark, Angew. Chem., Int. Ed., 2015, 54, 2552–2555 CrossRef CAS PubMed.
- J. C. Genereux and J. K. Barton, Chem. Rev., 2010, 110, 1642–1662 CrossRef CAS PubMed.
- A. L. Nussbaumer, D. Studer, V. L. Malinovskii and R. Häner, Angew. Chem., Int. Ed., 2011, 50, 5490–5494 CrossRef CAS PubMed.
- M. Vybornyi, Y. Vyborna and R. Häner, ChemistryOpen, 2017, 6, 488–491 CrossRef CAS PubMed.
- C. B. Winiger, S. Li, G. R. Kumar, S. M. Langenegger and R. Häner, Angew. Chem., Int. Ed., 2014, 53, 13609–13613 CrossRef CAS PubMed.
- M. Kownacki, S. M. Langenegger, S. X. Liu and R. Häner, Angew. Chem., Int. Ed., 2019, 58, 751–755 CrossRef CAS PubMed.
- W. E. M. Noteborn, V. Saez-Talens and R. E. Kieltyka, ChemBioChem, 2017, 18, 1995–1999 CrossRef CAS PubMed.
- Y. Vyborna, M. Vybornyi, A. V. Rudnev and R. Häner, Angew. Chem., Int. Ed., 2015, 54, 7934–7938 CrossRef CAS PubMed.
- W. Engelen, S. P. W. Wijnands and M. Merkx, J. Am. Chem. Soc., 2018, 140, 9758–9767 CrossRef CAS PubMed.
- D. Bousmail, P. Chidchob and H. F. Sleiman, J. Am. Chem. Soc., 2018, 140, 9518–9530 CrossRef CAS PubMed.
- S. P. W. Wijnands, W. Engelen, R. P. M. Lafleur, E. W. Meijer and M. Merkx, Nat. Commun., 2018, 9, 65 CrossRef PubMed.
- Y. Vyborna, M. Vybornyi and R. Häner, Chem. Commun., 2017, 53, 5179–5181 RSC.
- Y. Vyborna, M. Vybornyi and R. Häner, J. Am. Chem. Soc., 2015, 137, 14051–14054 CrossRef CAS PubMed.
- A. Serganov and E. Nudler, Cell, 2013, 152, 17–24 CrossRef CAS PubMed.
- S. K. Albert, H. V. P. Thelu, M. Golla, N. Krishnan, S. Chaudhary and R. Varghese, Angew. Chem., Int. Ed., 2014, 53, 8352–8357 CrossRef CAS PubMed.
- M. Á. Alemán García, E. Magdalena Estirado, L.-G. Milroy and L. Brunsveld, Angew. Chem., Int. Ed., 2018, 57, 4976–4980 CrossRef PubMed.
- H. Yu and R. Häner, Chem. Commun., 2016, 52, 14396–14399 RSC.
- H. Kashida, Y. Hattori, K. Tazoe, T. Inoue, K. Nishikawa, K. Ishii, S. Uchiyama, H. Yamashita, M. Abe, Y. Kamiya and H. Asanuma, J. Am. Chem. Soc., 2018, 140, 8456–8462 CrossRef CAS PubMed.
- L. Wang, Y. Feng, Z. Yang, Y. M. He, Q. H. Fan and D. Liu, Chem. Commun., 2012, 48, 3715–3717 RSC.
- Y. Vyborna, M. Vybornyi and R. Häner, Bioconjugate Chem., 2016, 27, 2755–2761 CrossRef CAS PubMed.
- A. K. Mishra, H. Weissman, E. Krieg, K. A. Votaw, M. McCullagh, B. Rybtchinski and F. D. Lewis, Chem. – Eur. J., 2017, 23, 10328–10337 CrossRef CAS PubMed.
- K. M. M. Carneiro, F. A. Aldaye and H. F. Sleiman, J. Am. Chem. Soc., 2010, 132, 679–685 CrossRef CAS PubMed.
- D. Bochicchio and G. M. Pavan, ACS Nano, 2017, 11, 1000–1011 CrossRef CAS PubMed.
- P. Payamyar, B. T. King, H. C. Ottinger and A. D. Schlüter, Chem. Commun., 2016, 52, 18–34 RSC.
- M. Vybornyi, A. V. Rudnev, S. M. Langenegger, T. Wandlowski, G. Calzaferri and R. Häner, Angew. Chem., Int. Ed., 2013, 52, 11488–11493 CrossRef CAS PubMed.
- S. K. Albert, I. Sivakumar, M. Golla, H. V. P. Thelu, N. Krishnan, L. Joseph, Ashish and R. Varghese, J. Am. Chem. Soc., 2017, 139, 17799–17802 CrossRef CAS PubMed.
- Y. Vyborna, S. Altunbas, M. Vybornyi and R. Häner, Chem. Commun., 2017, 53, 12128–12131 RSC.
- N. Micali, M. Vybornyi, P. Mineo, O. Khorev, R. Häner and V. Villari, Chem. – Eur. J., 2015, 21, 9505–9513 CrossRef CAS PubMed.
- C. Zhou, Y. Zhang, Y. Dong, F. Wu, D. Wang, L. Xin and D. Liu, Adv. Mater., 2016, 28, 9819–9823 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2019 |