
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cyclometallated tridentate platinum(II) arylacetylide complexes: old wine in new bottles
Ashanul
Haque†
a,
Linli
Xu†
 b,
Rayya A.
Al-Balushi
c,
Mohammed K.
Al-Suti
d,
Rashid
Ilmi
d,
Zeling
Guo
be,
Muhammad S.
Khan
*d,
Wai-Yeung
Wong
*be and
Paul R.
Raithby
*f
b,
Rayya A.
Al-Balushi
c,
Mohammed K.
Al-Suti
d,
Rashid
Ilmi
d,
Zeling
Guo
be,
Muhammad S.
Khan
*d,
Wai-Yeung
Wong
*be and
Paul R.
Raithby
*f
aDepartment of Chemistry, College of Science, University of Hail, Kingdom of Saudi Arabia
bDepartment of Applied Biology & Chemical Technology, The Hong Kong Polytechnic University, Hung Hom, Hong Kong, P. R. China. E-mail: wai-yeung.wong@polyu.edu.hk
cDepartment of Basic Sciences, College of Applied and Health Sciences, A’Sharqiyah University, P.O. Box 42, Ibra 400, Sultanate of Oman
dDepartment of Chemistry, Sultan Qaboos University, P.O. Box 36, Al-Khod 123, Sultanate of Oman. E-mail: msk@squ.edu.om
eThe Hong Kong Polytechnic University Shenzhen Research Institute, Shenzhen 518057, P. R. China
fDepartment of Chemistry, University of Bath, Claverton Down, Bath BA2 7AY, UK. E-mail: p.r.raithby@bath.ac.uk
First published on 13th November 2019
Abstract
Square planar platinum(II) complexes have been known for 150 years and pincer complexes, supported by a tridentate chelating ligand such as terpyridyl, have been known for more than 70 years. The development of cyclometallated platinum(II) pincer complexes, in which the tridentate ligand forms one or more platinum–carbon bonds, has been much more recent. Particularly, in terms of their solution and solid-state luminescence these cyclometallated complexes show substantial advantages over their terpyridyl analogues. This tutorial review introduces the reader to the area of platinum(II) cyclometallated pincer chemistry and shows the advantage of having an alkynyl group in the fourth coordination site on the metal. The basic design principles for the preparation of highly luminescent platinum(II) cyclometallated pincer complexes are outlined and the strategy to improve the luminescence further by chemical manipulation of the pincer ligand and of the auxiliary ligand in the fourth coordination site are illustrated with recent examples from the literature. Recent applications of these cyclometallated pincer complexes in the area of opto-electronics is described, with emphasis on their use in OLEDs, OFETs and as NLO materials as well as demonstrating their potential use as triplet photosensitizers and as metal ion sensors. The aim of this review is to show the recent advances in this rapidly developing research field and to highlight the future promise of these materials.
 Ashanul Haque | Dr A. Haque is an Assistant Professor of Chemistry at University of Hail, Saudi Arabia. He received his PhD Degree in Chemistry (2014) from Jamia Millia Islamia, India under the supervision of Prof. Imran Ali and Prof. Kishwar Saleem. In 2014 he joined Prof. Muhammad Khan's group at Sultan Qaboos University, Oman, where he stayed for ∼four years. His present research interests are centered on design and development of organic and organometallic scaffolds for biological and opto-electronics applications. Dr Haque has an h-index of 14, an i10-index of 19 and 876 citations to his 44 publications. |
 Linli Xu (right) and Zeling Guo (left) | Dr Linli Xu (right) obtained her PhD degree from Guangzhou Institute of Chemistry, Chinese Academy of Sciences (CAS) in 2010. She then carried out research at Technical Institute of Physics and Chemistry, CAS, from 2012 to 2017. She now works at the Hong Kong Polytechnic University as a research fellow in the group of Prof. Wai-Yeung Wong and her research focuses on polymer materials and carbon materials. Zeling Guo (left) received her BSc degree from the Department of Chemistry, Zhejiang University (ZJU) in 2016. She is currently a PhD student in the Department of Applied Biology and Chemical Technology, The Hong Kong Polytechnic University under the guidance of Prof. Wai-Yeung Wong. Her research interest focuses on luminescent transition metal complexes. |
 Rayya A. Al-Balushi | Dr Rayya A. Al Balushi received her MSc degree in Chemistry from Sultan Qaboos University, Oman (2006) and PhD from the same University in 2016 under the supervision of Professor Muhammad S. Khan. Her PhD project focused on the design, synthesis, and characterization of some novel metalla-yne and poly(metalla-yne) materials. She received the Best PhD Thesis award from Sultan Qaboos University in 2017. Al-Balushi is currently an Assistant Professor in the College of Applied and Health Sciences, A’Sharqiyah University (ASU), Oman. Her research interests include design and development of conjugated polymers for photoswitch, photovoltaic, and LED applications. |
 Rashid Ilmi (left), Muhammad S. Khan (centre) and Muhammad K. Al-Suti (right) | Dr Ilmi received his PhD (2013) form Jamia Millia Islamia, New Delhi, India. He joined (2013) Dr C. Pitris at the University of Cyprus as a postdoctoral fellow, and in 2017 Prof. Khan's group at Sultan Qaboos University, Oman. His current research interests include the synthesis of small heterocyclic organic molecules and their transition metal and lanthanide complexes. Prof. Khan was an 1851 Exhibition Scholar and received his PhD (1983) from the University of Cambridge, UK. Currently he is a Professor of Inorganic Chemistry at Sultan Qaboos University, Oman. He received many medals and awards, the recent ones include the Best Researcher Award, College of Science, SQU (2005, 2017), and the National Research Award, The Research Council, Oman (2018). His research interests include the design of novel sustainable energy materials incorporating conjugated organic, organometallic polymers, and lanthanide coordination complexes for opto-electronic applications. Dr. Al-Suti received his PhD (2006) from the University of Bath, UK. He is a Researcher (A) in the Department of Chemistry at SQU. His research interests include the synthesis and characterization of conjugated organic poly-ynes and poly-metalla-ynes and their applications in opto-electronics. |
 Wai-Yeung Wong | Professor Wai-Yeung Wong obtained his BSc (Hons.) and PhD degrees from the University of Hong Kong. After postdoctoral works at Texas A&M University and the University of Cambridge, he joined Hong Kong Baptist University from 1998 to 2016 and now works at the Hong Kong Polytechnic University as Chair Professor of Chemical Technology and Associate Dean of Faculty of Applied Science and Textiles. Among his awards are the RSC Chemistry of the Transition Metals Award, FACS Distinguished Young Chemist Award and State Natural Science Award from China. His research focuses on synthetic inorganic/organometallic chemistry, aiming at developing photofunctional metal–organic molecules and polymers. |
 Paul R. Raithby | Professor Paul Raithby is a Professor of Inorganic Chemistry at the University of Bath. He is a Fellow of the European Academy of Sciences (EURASC) and of the Royal Society of Chemistry. He was awarded the RSC Corday Morgan Medal in 1988 and the RSC Prize for Structural Chemistry in 2008. He has published over 860 refereed research papers. His current research focuses on coordination chemistry, the development of the chemistry of platinum poly-ynes as sensor materials, and he has pioneered the development of time-resolved crystallographic techniques for determining the structures of crystalline, excited state complexes with millisecond lifetimes. |
Key learning points(1) Photo-physical properties, structure–property relationships and applications of cyclometallated tridentate platinum(II) arylacetylide complexes.(2) Emphasis on C^N^N, N^C^N and C^N^C cyclometallated ligands. (3) Effect of varying cyclometallating and auxiliary ligands, and size of the metallacycle on excited states. (4) Comparison with bidentate analogues. (5) Key examples of complexes with intriguing applications. |
1. Introduction
Organometallic complexes and polymers are an important class of functional materials that display versatile and tunable properties both in solution and in the solid state.1 Among the most promising subsets of this class of materials are the luminescent square planar platinum(II) complexes that show a range of interesting optoelectronic properties and have found applications as sensor materials.2 Of particular significance are the platinum poly-ynes, that contain alkyne linker groups,3 and the platinum “pincer” complexes in which three of the co-ordination sites around the platinum(II) centre are occupied by a tridentate ligand, such as terpyridine.4 While the platinum terpyridine complexes have been studied extensively over the last two decades, and their physical and material properties established,5,6 related cyclometallated complexes, those with an aromatic polydentate ligand directly bound to platinum(II) centre through a covalent metal–carbon bond, have been studied in much less detail. However, from the studies undertaken these cyclometallated complexes have shown that they are particularly versatile and have some advantages over their terpyridine analogues.7 Their unique structural features coupled with tunable photoluminescence (PL) properties make these materials excellent candidates for a range of important applications including in OLEDs, OFETs and as non-linear optical (NLO) materials.In this tutorial review we will focus on platinum(II) complexes bearing tridentate cyclometallated ligands (C^N^N, N^C^N, C^N^C, Fig. 1a). They can also be classified on the basis of size and number of chelating rings (for example: 5-5-, 5-6-, and 6-6-membered metallacycle formed by the incorporation of an atom between ligating species and the proximal aromatic carbons). In the cases under discussion the fourth coordination site trans to the central ring of the tridentate ligand is occupied by an alkyne whose presence is particularly beneficial to the enhancement of the luminescent properties of the complexes. We wish to emphasise the advantages of these cyclometallated complexes over their N^N^N analogues, which have anyway been reviewed extensively in the recent past.8
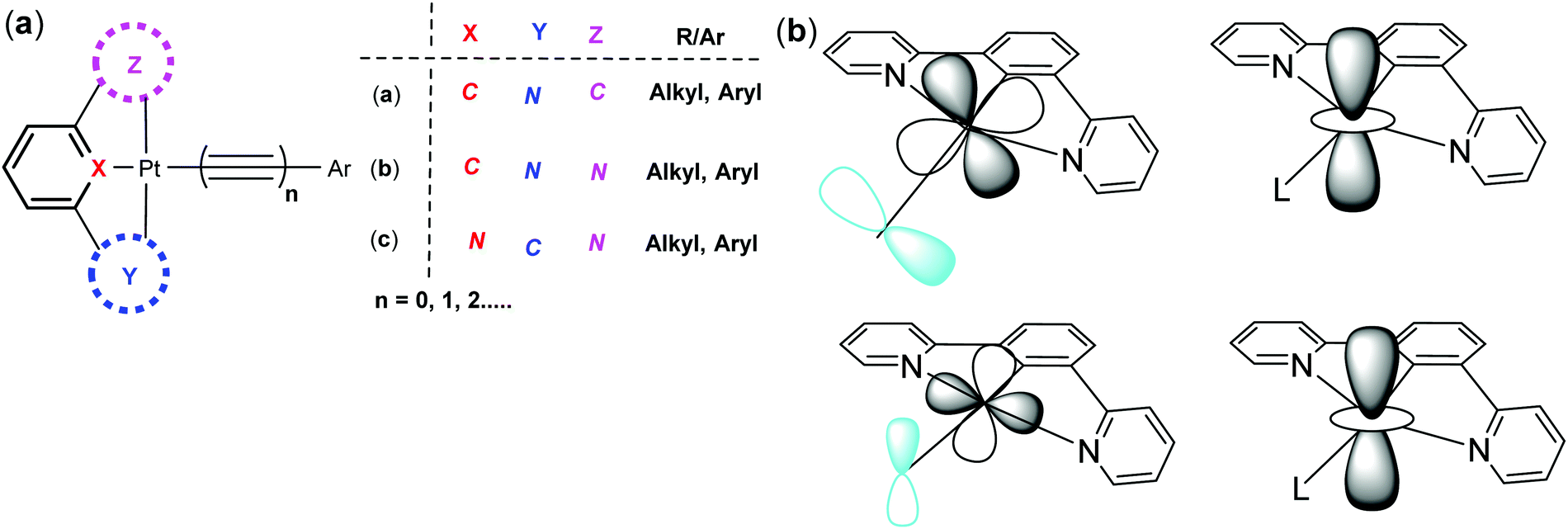 | ||
| Fig. 1 (a) Types of cyclometallated Pt(II) arylacetylide complexes focused upon in this review, and (b) the valence Pt dxy and dxz orbitals interact strongly with π-type molecular orbitals on the monodentate ancillary ligand (left), while the dz2 orbitals of adjacent molecules can interact in the stacked geometry typically adopted in the solid state (right). | ||
The first question to answer is why the cyclometallated platinum(II) alkynes display higher luminescence and are more versatile than their terpyridine analogues? To understand this we need to look at the metal and ligand energy levels within the complexes. The nature of the electronic properties of the cyclometallated species had been established by Williams et al. before 2010.9
Essentially, to maximise the luminescent efficiency for any Pt(II) complex the radiative rate constant for the emissive state must be maximised and the rates of non-radiative decay minimised. For square planar Pt(II) complexes, when there are no significant steric effects from the ligands, the metal orbitals make a significant contribution to the excited states. The high spin–orbital coupling constant associated with the heavy Pt atom facilitates rapid intersystem crossing from the singlet state to the triplet state. Since the rate of intersystem crossing vastly exceeds radiative decay rate constants for the singlet states it is reasonable to assume that the emission emanates from the triplet states. Thus, maximising the metal orbital contributions to the lowest energy triplet excited state will maximise the radiative decay.
Now, let us consider the factors that influence non-radiative decay. In the strong-field Pt(II) square planar complexes the unoccupied dx2−y2 orbital is strongly antibonding because of the ligand field stabilisation. If, however, this orbital is occupied there will be an elongation of the platinum–ligand bonds and a significant distortion of the metal geometry. This will facilitate non-radiative decay of the metal-centred (d–d) excited state to the ground state. Even if other excited states, such as charge-transfer states, lie at lower energies than the d–d states, these d–d states can still contribute to non-radiative decay if they can be accessed thermally.
Strategically, in order to design a platinum(II) complex with optimum luminescence we must produce a complex in which
• the Pt(II) orbitals must contribute substantially to the lowest-energy triplet state
• the lowest energy excited singlet state should not be a d–d state but a charge-transfer or ligand-based state
• the energy gap between the lowest-lying excited state and the higher lying d–d state should be as large as possible.
In platinum(II) complexes the energy gap between the lowest-lying excited state and the d–d state can be increased if strong-field ligands, such as cyanide or alkynyl ligands, are used.10 This is why having an alkynyl ligand in the fourth coordination site on a square planar Pt(II) pincer complex is particularly helpful,5 and why cyclometalled platinum(II) alkynyl complexes are the focus of this review. Additionally, the anionic nature of the alkynyl ligand means that for a mono-cyclometallated Pt(II) complex with the C^N^N and N^C^N-type pincer ligands the complex is neutral, which improves the solubility and stability of the complex in hydrocarbon solvents, and facilitates intermolecular interactions in the solid-state since there are no counterions to disrupt the packing arrangements of the molecules. More generally, the characteristics of the monodentate ancillary ligand (L = halides, cyanides, pseudo-halides, alkynyls, cyanides, etc.) that occupies the fourth coordination position on the Pt(II) centre also plays an important role in the solid-state aggregation and the formation of intermolecular interactions.11
A significant strategic advantage of having a cyclometallated pincer ligand co-ordinated to the Pt(II) centre over their N^N^N analogues is the formation of the Pt–C bond(s) that occurs through deprotonation of one or more of the aromatic C–H bonds, formally generating negatively charged “C” units. The “C−” bonded to the metal acts as a very strong σ-donor providing the metal with a very strong ligand-field while π-acceptor character is maintained through any co-ordinated pyridine rings. Thus, the energy of the high-lying d–d states is raised further by comparison to the N^N^N systems and the cyclometallated complexes display higher levels of luminescence. In this arrangement the d-orbitals are high lying and the dxy and dxz orbitals have the correct symmetry to interact with π-type orbitals of the alkynyl ligand. The dz2 orbital is also available to overlap with dz2 orbitals on the Pt(II) centres in adjacent molecules to assist in the formation of molecular stacks in the solid-state (Fig. 1b).11
Having established the basic design features for improving the luminescent properties of cyclometallated Pt(II) pincer complexes over the N^N^N-type analogues there are a number of chemical and structural features that can be manipulated in order to improve the solution and solid-state luminescence properties further. These include:
• changing the type of the pincer ligand used, from C^N^N through N^C^N to C^N^C, etc.
• changing the ring size of the Y and Z rings (Fig. 1a) from 6-membered to 5-membered and altering the nature of the atoms in the rings, e.g. from C to S, and by adding substituents to any or all of the pincer ring systems.
• changing the overall charge on the complex, in combination with changes in the nature of the pincer and the monodentate alkynyl or other ligands and the presence or absence of counterions.
• changing the steric and electronic properties of the monodentate alkynyl ligands.
• changing the geometry of the overall Pt(II) complex, e.g. changing the steric bulk or planarity of the complex.
An analysis of the effect of these changes, with examples taken from the recent literature, forms the next part of this review.
2. Design features of luminescent cyclometallated platinum(II) complexes
2.1. General features
From the structural viewpoint, rigidity and planarity of a Pt(II) square planar complex can promote high luminescence levels by reducing the non-radiative decay pathways caused by structural distortions, and the rigid tridentate pincer ligands are particularly effective at achieving this. An analysis of all the single-crystal X-ray structure determinations in the Cambridge Structural Database (CSD)12 (Version 5.40 (November 2018 + 2 updates)) for the structural fragments (N^N^N)Pt–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C, (C^N^N)Pt–CC and (N^C^N)Pt–CC allows the angles between the three six-membered arene rings in the pincer ligands to be measured and the deviations from planarity determined (Table 1).
C, (C^N^N)Pt–CC and (N^C^N)Pt–CC allows the angles between the three six-membered arene rings in the pincer ligands to be measured and the deviations from planarity determined (Table 1).
| No. hits in CSD | Range of interplanar angles between ring X and rings Y and Z (°) | Mean interplanar angle (°) | |
|---|---|---|---|
| N^N^N pincer | 60 | 0.0–11.06 | 4.11 |
| C^N^N pincer | 29 | 0.8–11.77 | 3.82 |
| N^C^N pincer | 7 | 0.0–7.97 | 3.56 |
The data from Table 1 for all three co-ordinated pincer ligands show that the three ligands show little deviation from planarity, and in the N^N^N and N^C^N there are some pincer ligands that are precisely planar as required by crystallographic symmetry. While it is appealing to suggest that the symmetric N^C^N ligand shows the least deviation from planarity, the range of values recorded in the data and the small differences between the data for the different pincers preclude firm assessments being made. However, it is certainly the case that the tridentate pincers show smaller deviations from planarity than related bidentate bipyridyl ligands in Pt(II) complexes.13
It may then be the features of the co-ordination environment around the metal centre that are more important in determining the differences in the luminescent properties of the cyclometallated platinum(II) alkynyl pincer complexes. This, indeed, seems to be the case where the symmetric N^C^N systems result in shorter Pt–C distances than the comparable Pt–C bonds in the asymmetric C^N^N systems. The average Pt–C and Pt–N bond lengths associated with the N^C^N and C^N^N ligands in the seven (N^C^N)Pt–CC and twenty-nine (N^C^N)Pt–CC complexes, where rings X, Y and Z (Fig. 1a) are all six-membered, obtained from a search of the CSD12 (Version 5.40 (November 2018 + 2 updates)) and are presented in Table 2, which confirms that the average Pt–C bond length is ca. 0.07 Å shorter in the N^C^N complexes than in the C^N^N complexes, which is likely to result in a stronger σ donor character in the former.
C and twenty-nine structures of the (C^N^N)Pt–CC complexes
| No. CSD hits | Pt–C (Å) | Pt–N1 (Å) | Pt–N2 (Å) | |
|---|---|---|---|---|
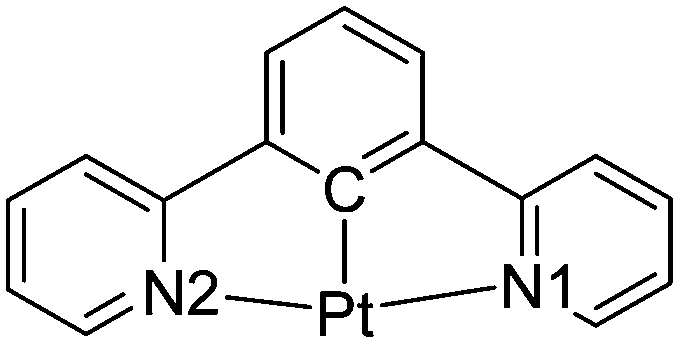
|
7 | 1.939 | 2.030 | 2.034 |
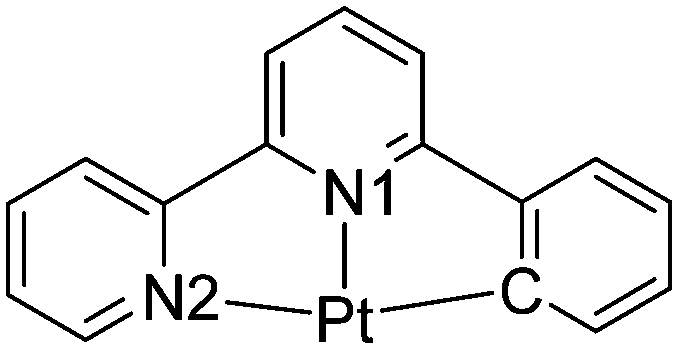
|
29 | 2.014 | 1.987 | 2.093 |
A search of the CSD for equivalent complexes where either Y and Z or both X and Z are five-membered rings results in a relatively small number of hits, and within these sets there is some diversity in the nature of the rings, so a meaningful statistical analysis is not possible. All that is clear is that when the X atom on the central six-membered ring in any of the complexes is a cyclometallated carbon atom, the Pt–C bond length is the shortest bond involving the pincer coordinating atoms in the structure. This, again, emphasises the significance of having a strong C σ-donor atom trans to the linear alkyne ligand and suggests that cyclometallation of the central ring in the pincer ligand is a key design feature for generating luminescent platinum(II) complexes.
In the solid-state and in solution at high concentrations aggregation of the planar Pt(II) complexes occurs with the interaction between the complexes often being a metallophilic Pt(II) (d8)⋯Pt(II) (d8) interaction. This results in a metal–metal-to-ligand charge transfer (MMLCT) process which is sensitive to the degree of Pt⋯Pt interaction.14 These metallophilic interactions coupled with π⋯π interactions make the solid-state luminescence properties of the materials significantly different to those in solution. Assuming an approximate van der Waals radius of Pt(II) to be about 1.75 Å, which is similar to the intermolecular π⋯π stacking interaction of 3.35 Å, it would be expected that there would be significant intermolecular Pt⋯Pt interactions at 3.5 Å and below. A CSD12 study of the Pt⋯Pt interactions in the twenty-nine (C^N^N)Pt–CC complexes, of which two were salts, with associated counterions and a further six had solvent of crystallisation in the lattice, and of the seven (N^C^N)Pt–CC complexes, of which two had solvent of crystallisation showed a range of Pt⋯Pt distances. For the symmetric (N^C^N)Pt–CC complexes four of the seven structures had intermolecular interactions between 3.229–4.886 Å, with two of the structures having distances below 3.5 Å. For the (C^N^N)Pt–CC complexes, twenty of the twenty-nine had Pt⋯Pt contacts in the range 3.145–5.501 Å, with six of those having Pt⋯Pt contacts of less than 3.5 Å. It might be thought that the presence of solvent in the lattice, or for the ionic systems, with counterions present, might reduce the possibility of Pt⋯Pt interactions, but this is not the case. The two salts for the C^N^N have among the shortest Pt⋯Pt contacts and a couple of the systems with solvent in the lattice also have short Pt⋯Pt interactions. So it is perfectly possible to have solvent present without losing the Pt⋯Pt interaction and its associated luminescence properties, but small alterations in the Pt⋯Pt separation, with a resulting change in colour, makes some of these complexes ideal sensors for specific gases and volatile organic compounds.11 A careful examination of the pincer and alkynyl ligand substituents in the two classes of cyclometallated systems suggests that short Pt⋯Pt interactions are less likely to occur when the ligands are bulky, preventing the close approach of the metal centres, but with the small number of crystal structures available to study this hypothesis is not conclusive. Similarly, for the cyclometallated systems with 5,6-5 and 5,6,6-type pincer ligands there are insufficient examples to draw firm conclusions about the factors that favour short Pt⋯Pt intermolecular contacts in these complexes in the solid-state.
In this section we have used the crystallographic data from the known cyclometallated platinum(II) alkynyl complexes to underpin the design rules to optimise the formation and fine tuning of highly luminescent complexes outlined in the previous section. In the following sections we highlight the application of these rules using examples from the recent literature where changing one or more features of the complexes alters their luminescent properties.
2.2. Platinum(II) alkynyl complexes with C^N^N pincer ligands
Following on from the first synthesis of the simplest tridentate pincer C^N^N ligand (6-phenyl-2,2′-bipyridine, PhBpy) and its corresponding Pt(II) complexes, a plethora of studies have been carried out to confirm the structure–property relationship in C^N^N-based systems.15 A systematic variation of the coordinating and ancillary ligands has helped to gain an understanding of the key factors for tuning the luminescent properties (Fig. 2a). For example, the replacement of chloride (Cl−) auxiliary from a cyclometallated Pt(II)–PhBpy complex (1a) by arylacetylide unit (1b–f) allows the tuning of the emission wavelength (up to 100 nm in visible region) as well as the quantum yield (Φ).16 Photophysical studies (in CH2Cl2 solution at room temperature) indicated that complex 1b exhibits characteristic transitions: a broad MLCT transition band at ∼430 nm (which was red-shifted and more intense compared to non-alkynylated auxiliaries) along with a high energy band (at ∼335 nm) attributed to intraligand (IL) charge-transfer transitions of the C^N^N ligand.17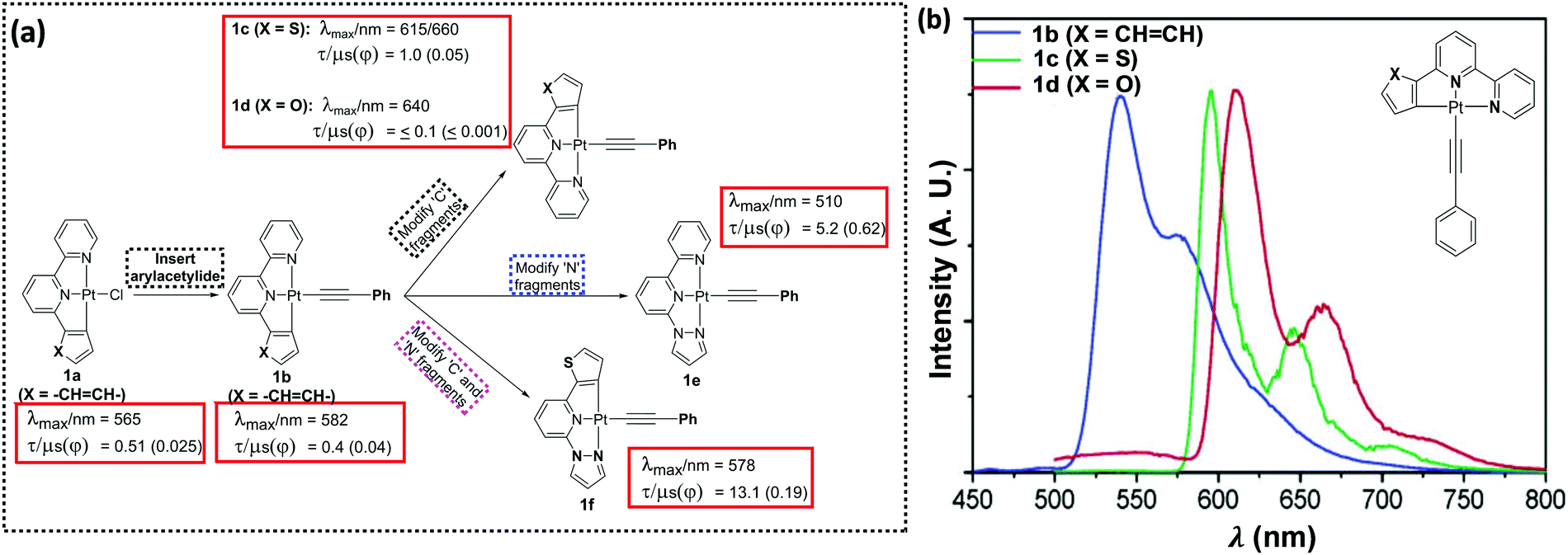 | ||
| Fig. 2 (a) Strategies to tune PL properties of a C^N^N-based Pt(II) complex, (b) emission spectra of 1b–d in alcoholic glass at 77 K (λex = 350 nm, concentration ∼1 × 10−5 mol dm−3). This figure has been reproduced from ref. 16 with permission from the American Chemical Society. | ||
Both MLCT and 1IL transitions were found to be sensitive to the nature of proximal C^ and N^-donors. For example, when the side-arm phenyl group is replaced by thienyl or furyl moieties (1c and 1d), 1IL transitions move to lower energy (higher wavelength). A noticeable red shift in the edge of the lower-energy absorption band also occurs. PL studies displayed an emission at 582 nm (in CH2Cl2 solution at 298 K) for 1b, which expectedly moved to the red in case of 1c (at λmax = 616 nm, shoulder at 660 nm) (Fig. 2b). A similar observation has also been made when one of the N-donating sites of bipyridyl (Bpy) core is replaced by other N-donors.18 Complexes bearing 2-phenyl-6-(1H pyrazol-1-yl)pyridine such as 1e showed blue shifted emission (λmax = 510 nm) compared to 1b, but high quantum yield (Φ = 0.62). The Huang–Rhys ratio (S), which indicates degree of molecular distortion in the excited state with respect to the ground state, decreases in sequence: 1b (∼0.6) > 1d (∼0.5) > 1c (∼0.4), which rationalises the weak emissive nature of the complex 1d. High Φ in case of 1e can also be rationalised by other reasons (vide infra). Overall from these discussions, it can be inferred that PL properties of Pt(C^N^N) complexes bearing same arylacetylide ligands can be successfully tuned by varying the proximal C^ and N^-donors (1g–1j, Chart 1).
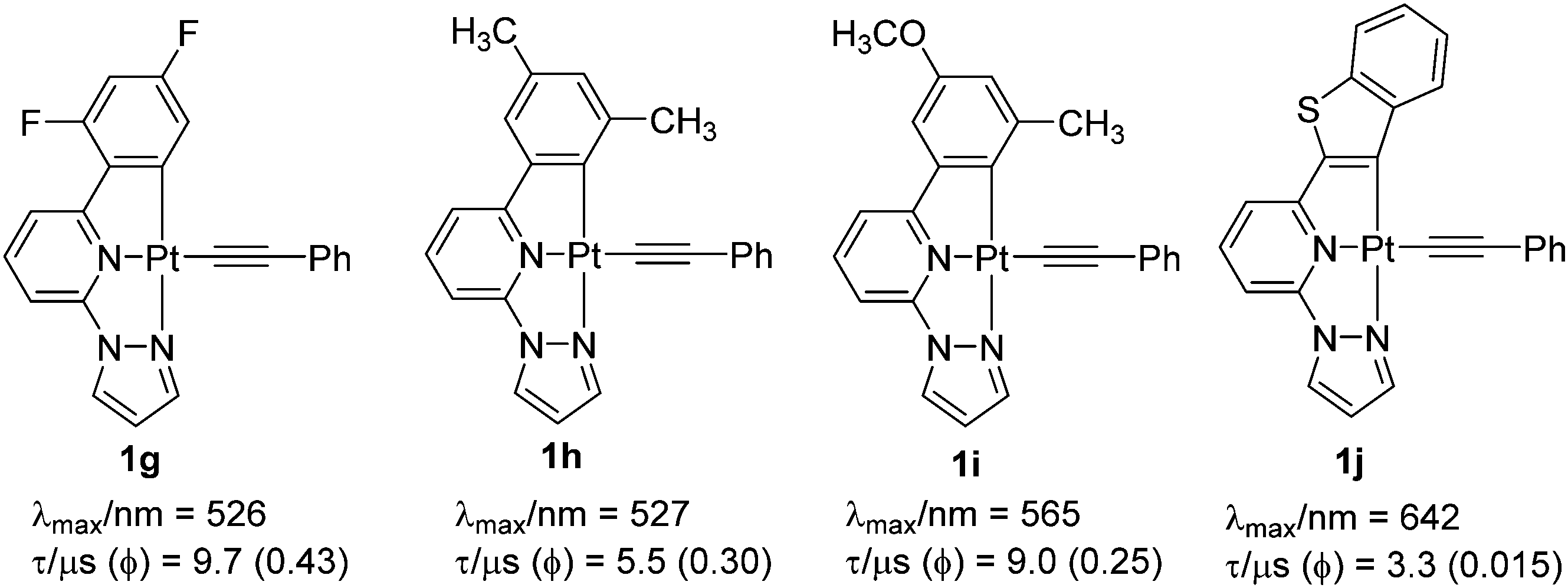 | ||
| Chart 1 Some recent examples of cyclometallated Pt(II) complexes bearing modified C^ and N^-donors.18 | ||
Another important aspect of modulating the optical properties is to expand or contract the size of a metallacycle. Both theoretical and experimental studies have demonstrated that the change in the size of metallacycle (5-5-membered → 5-6-membered) via insertion of an electronegative bridging atom could tune the emission colour and modulate the phosphorescence quantum yield (Φ).19 For example, a dramatic change in the emission properties is observed upon replacement of lateral phenyl unit from 1b by a diphenylamine unit (2a), a modification which led to change in the size of metallacycle (Fig. 3).20 This modification significantly lowered the quantum yield (Φ = 0.004), attributed to the formation of 5-6-membered metallacycles. However, another 5-6-membered complex 2b (Φ = 0.43), bearing a pyrazole as one of the N-donors emerged as being more efficient (albeit less than 5-5-counterparts). However, all these complexes exhibited values much higher than that of their chlorinated analogues. Since the combination of 5,5-membered metallacycle along with a pyrazole unit as one of the N-coordinating sites can improve the luminescence efficiency, this might be a reason why 1e showed superb performance (Φ = 0.62 at room temperature, in CH2Cl2). Based on this idea, attempts have been made to further improve the performance of the complexes by structural modification.21
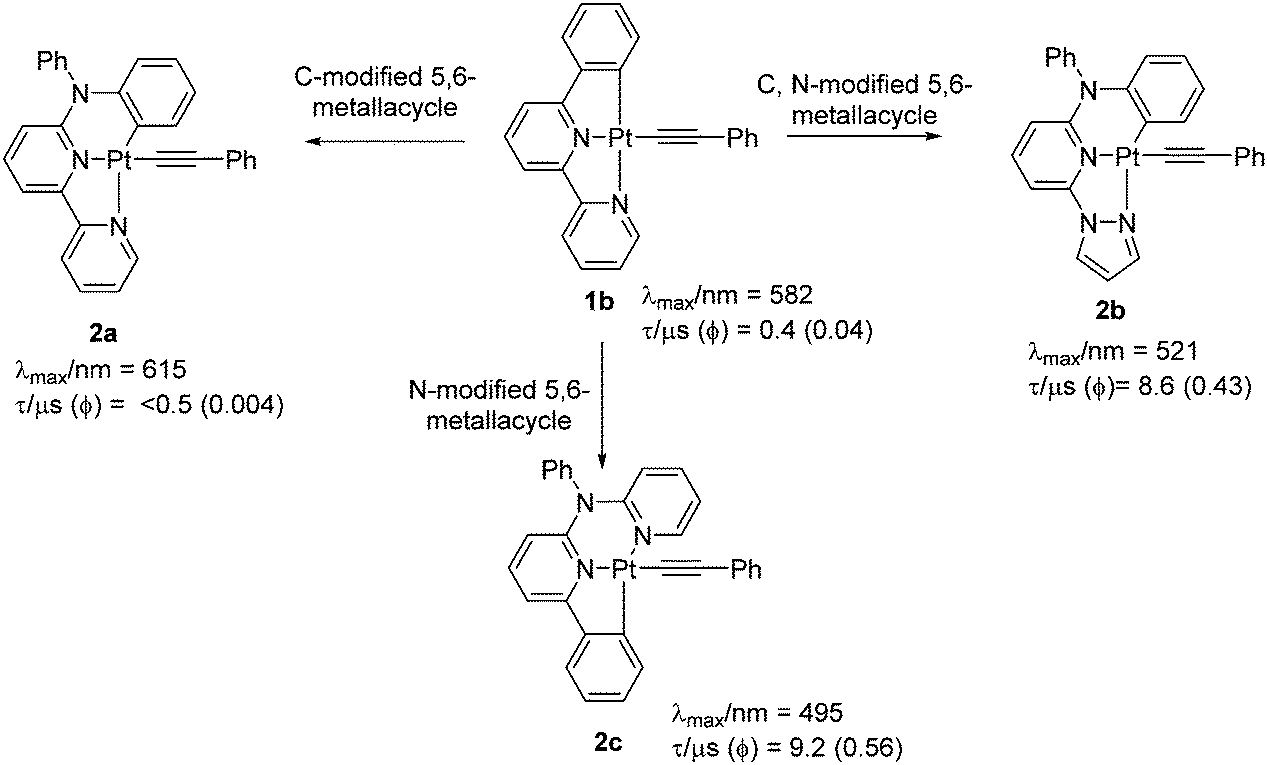 | ||
| Fig. 3 Complexes exhibiting metallacycle-dependent PL properties. | ||
These observations are considered to be due to the effect of the different extent of the structural distortions in the ground and excited states.22 Despite the latest research, complexes with Φ ∼ 1 are quite rare. It was suggested that a combination of the rigid scaffold and the presence of strong donor atoms could significantly minimize the structural distortion in the excited-state and so produce highly emissive complexes.22 To test this hypothesis, Chow et al.22 reported a series of complexes with extended π-conjugation (through both coordinating and auxiliary ligands). It was noted that complexes bearing extended conjugation through lateral aryl ring show improved PL properties. For example, complexes 3a–c (Chart 2) bearing a pentafluorophenylacetylide ligand showed a quantum yield (Φ = 0.08–0.99) which is substantially better than the chloro (Φ = 0.03–0.25), DMSO and N,N-dimethylimidazolium-bearing counterparts.23 It was proposed that exchanging a C^-donor (phenyl group by a naphthyl unit) assists in stabilizing the geometrical distortion of the excited state leading to enhanced kr values. Interestingly, auxiliary group dependent emitting excited states were suggested for these complexes. For example, excited states with mixed 3MLCT (dπ→π*(R–C^N^N–R′)Cl)/ILCT/XLCT (pπCl− → π*(R–C^N^N–R′)) parentages (XLCT = halogen-to-ligand charge transfer) were attributed to a chlorinated precursor, while mixed 3MLCT/ILCT/LLCT (LLCT = πCCAr → π*(R–C^N^N–R′)) was proposed for the alkynylated complexes. Among all the reported complexes, 3c exhibited relatively high kr (1.8 × 105) and negligibly small knr value leading to quantum yield close to unity (Φ = 0.99).
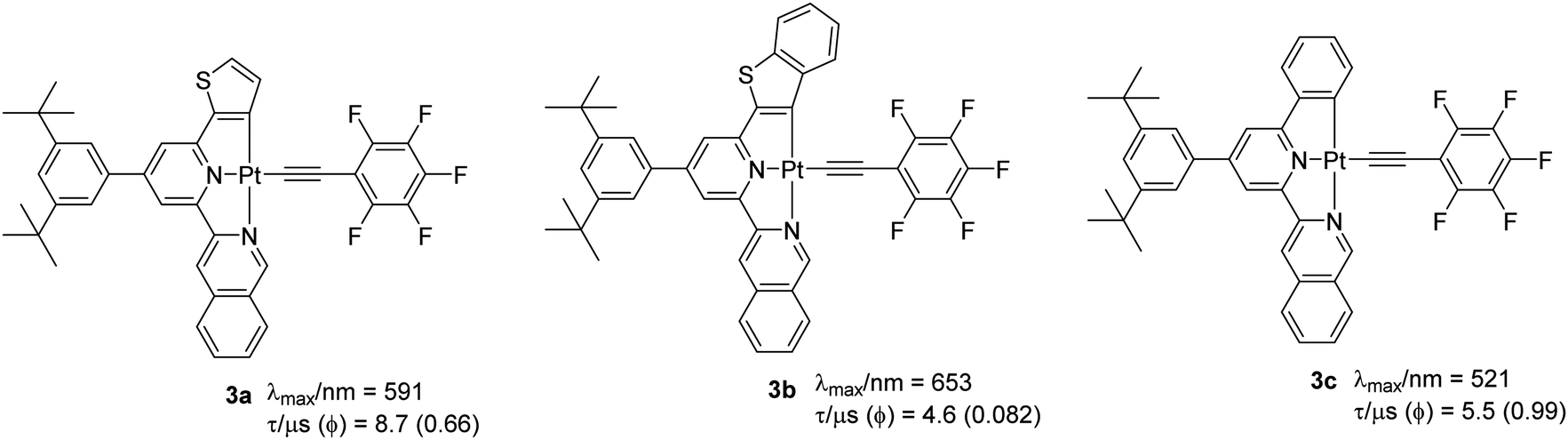 | ||
| Chart 2 Some examples of Pt(II) complexes bearing a more conjugated ligand through modification of the lateral aryl rings. | ||
One additional strategy to improve PL properties is to attach an electron withdrawing unit (such as terpyridine, TPY) to a C^N^N ligand via a phenylacetylide linkage (Chart 3). Bi et al.24 recently reported complexes 4a–b (Φ = 0.026–0.045, λmax = 596–600 nm) with modulated kr and knr factors compared to their chlorinated analogue (Φ = 0.019 at λmax = 566 nm). Strong phosphorescence (Φ = 0.045 at λmax = 596 nm) originated from the dπ(Pt) → π*(C^N^N) MLCT mixing with the π(C^C-Ar) → π*(C^N^N) LLCT transition. Interestingly, a switch in the excited state in which the acceptor ligand in the CT process switches from the N^N^C ligand to the pyridyl acetylide (from mixed L′LCT/MLCT → ML′CT) has also been noted when other acceptor ligands are attached to the phenyl unit.25
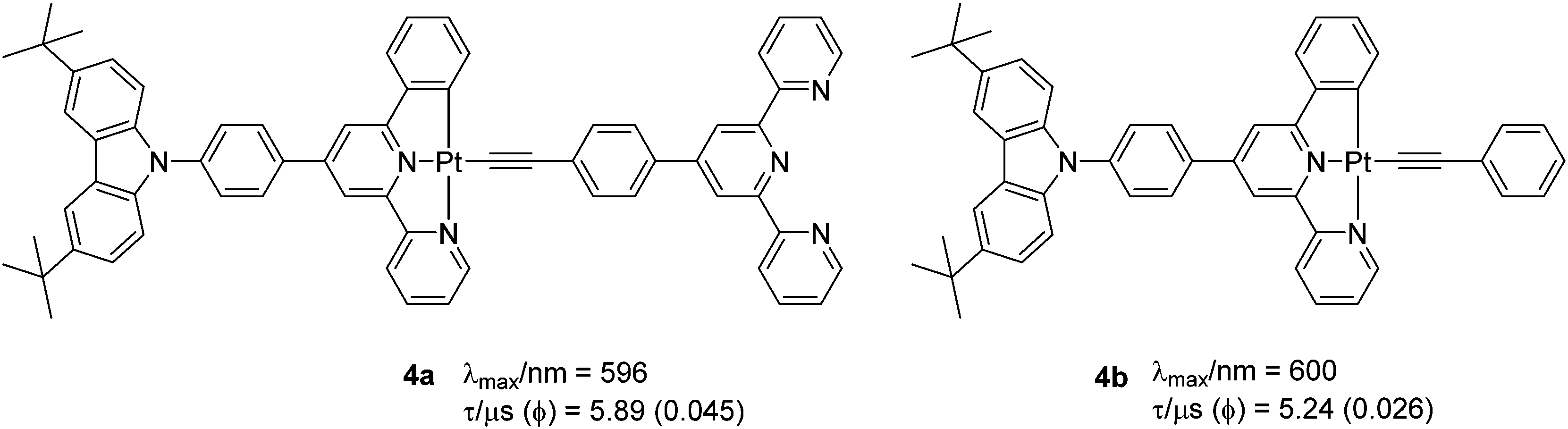 | ||
| Chart 3 Cyclometallated Pt(II) complexes with switchable excited states. | ||
2.3. Platinum(II) alkynyl complexes with N^C^N pincer ligands
Although complexes supported by N^C^N-type ligands have been known since the early 1990s,9 focussed research on cyclometallated Pt(II) complexes started in earnest only recently.26 As for the isomeric C^N^N counterpart, excited state properties are governed mainly by the geometrical rigidity, inter- and intramolecular interactions, the level of conjugation, the presence of heteroatoms, and the topology of the ligands. In general, cyclometallated Pt(II) complexes bearing N^C^N ligands produce stronger luminescence (with higher Φ and τ) than the isoelectronic tridentate (C^N^N and C^N^C) and related bidentate ligands.27 As discussed previously, the relatively high performance is due to the shortening of Pt–C (central anionic phenyl ring of the coordinating ligand) bond. To counterbalance this the Pt–C (alkynyl ligand) bond is elongated compared to its counterparts.28 The shortening of the Pt–C bond raises the energy of the d–d state and decreases the knr values.29 Yersin et al.30 proved that the Huang–Rhys S-parameter, which quantifies the level of molecular distortion in the excited state compared to the ground state, for a tridentate Pt(II) complex is half (S ≈ 0.1) that of a bidentate complex (S ≈ 0.2) (Fig. 4). This difference suggests that in a tridentate N^C^N-based Pt(II) complex, the emissive triplet states have less distortion than in the singlet ground state. It has also been demonstrated that the electronic nature (withdrawing/donating) of the substituent attached to the aryl ring as well as the alkynyl ligand produce changes in the excited states, thereby modifying the emission colour, purity and quantum yield.31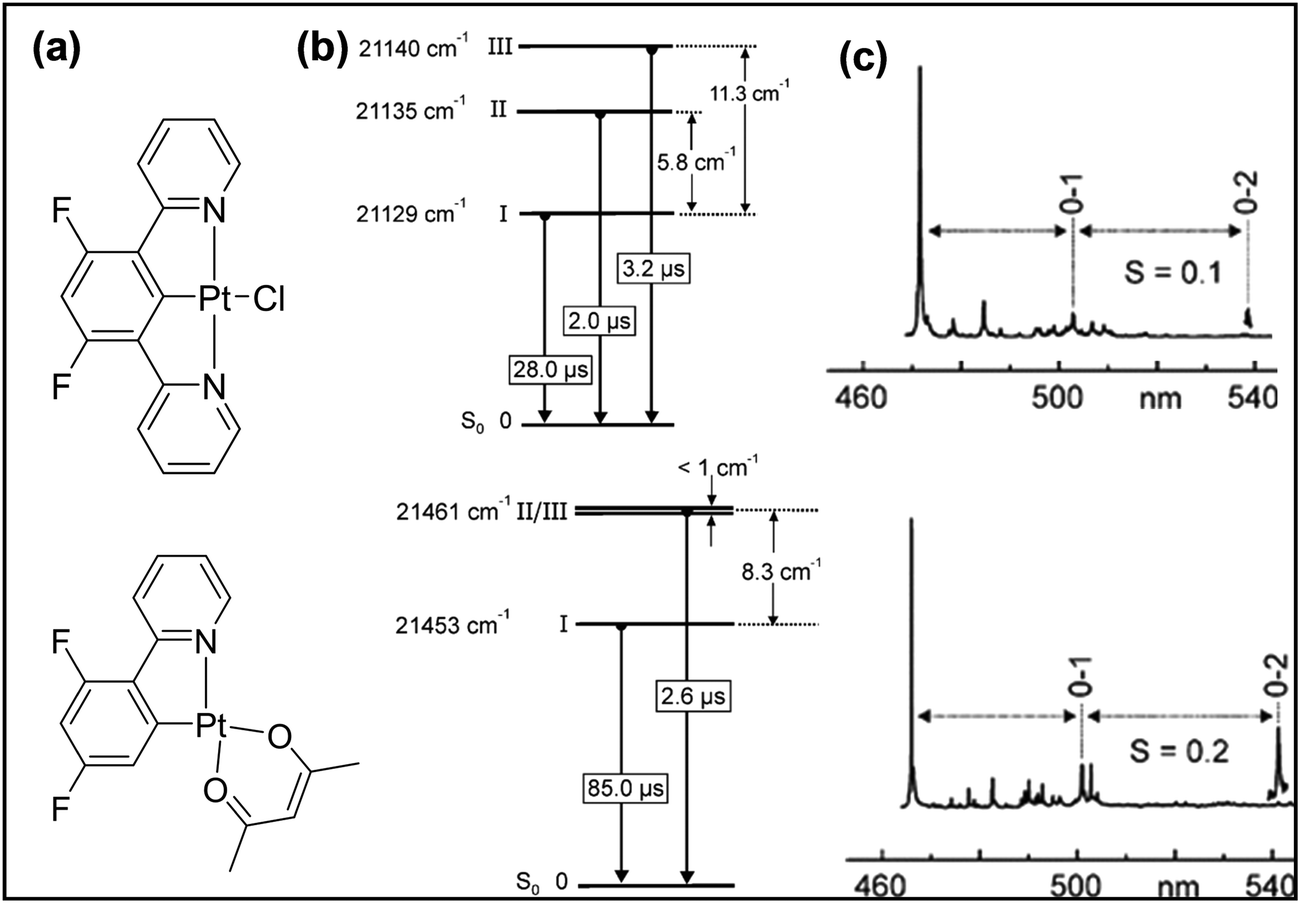 | ||
| Fig. 4 (a) Chemical structure, (b) energy level diagram and (c) S-parameters for bi- and tridentate cyclometallating Pt(II) complexes. This figure has been reproduced with permission from ref. 30 with permission from the American Chemical Society. | ||
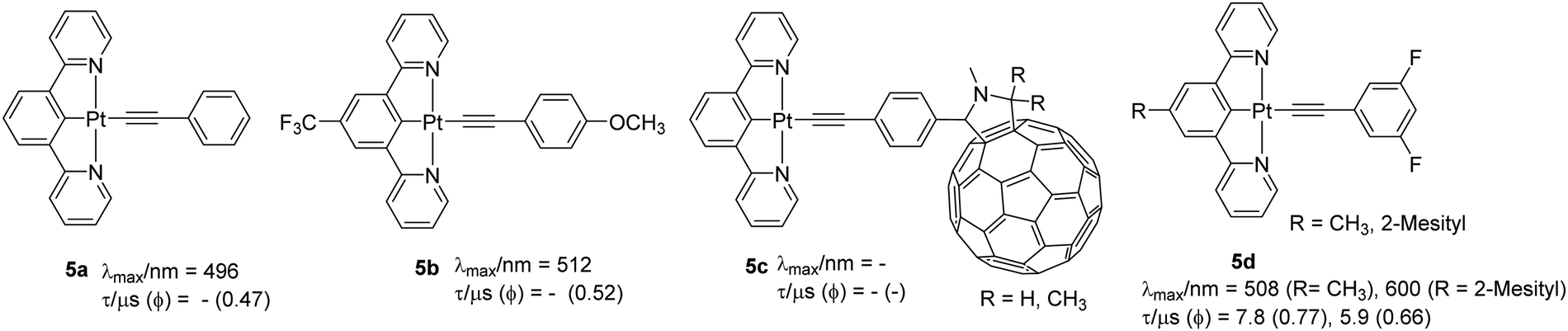
In addition to the geometric distortion-dependent PL properties in solution, cyclometallated Pt(II) complexes display solid-state-dependent emission due to the different extent of intermolecular interaction such as metallophilic and other non-covalent interactions. Unarguably, the introduction of alkynyl units enhances the propensity of such interactions and increases the crystallinity of the material, leading to synergistic modulation of PL properties. Because of these solid-state interactions, the majority of complexes show different luminescent properties in the solid-state compared to those observed in solution. The solid-state luminescence can be attributed to various phenomena including monomeric/excimeric luminescence, aggregation caused quenching (ACQ) or aggregation induced emission (AIE). This inherent luminescence in the solid-state leads to various properties including vapochromism, mechanochromism and photochromism which have resulted in new solid-state sensors and switches.2 For instance, complexes 5a (Φ = 0.47) and 5b (Φ = 0.52) (Chart 4) with varying level of intermolecular interactions showed different emission profiles.32 Compared to 5a, which exhibited only π⋯π interaction, 5b exhibited more structural features (such as the presence of Pt(II)⋯Pt(II), π⋯π and CH⋯π interactions). Such interactions assist in forming photoresponsive p-type semiconductor sheets, in which the complex stacked each other to make 1D columns containing an infinite linear chain of interacting Pt(II) centers. Motivated by this, Tashiro et al.33 designed a fullerene-based dyad 5c that forms alternating multilayers of electron-rich and -deficient molecular components. Replacing a CF3 group by a mesityl group in the coordinating ligand produces greater steric hindrance that serves to reduce ACQ. For instance, complexes 5d have superior Φ values than the corresponding chloride complexes, were the first examples of N^C^N-based OLED materials.34 As with other complexes of this type, these complexes too exhibited intense IL 1π–π* transitions (cyclometalated core and acetylide) at higher energy and mixed CT/LC character at lower energy. Substitution of chloride by the phenylacetylide introduces a πCC/π*N^C^N ligand-to-ligand charge-transfer (LLCT) transition suggesting that acetylide unit raises the energy of HOMO level effectively. Compared to [Pt(dpyb)Cl] (dpyb = 1,3-di(2-pyridyl)benzene, Φ = 0.6) and [Pt(dpyb)CC–Ph] (Φ = 0.21), these two complexes displayed green LC 3π–π* emission with Φ = 0.77 and 0.66, respectively. Owing to the AIE effect, the materials were found to be useful for OLED application.
 | ||
| Chart 4 Cyclometallated Pt(II) complexes with differing levels of intermolecular interactions. | ||
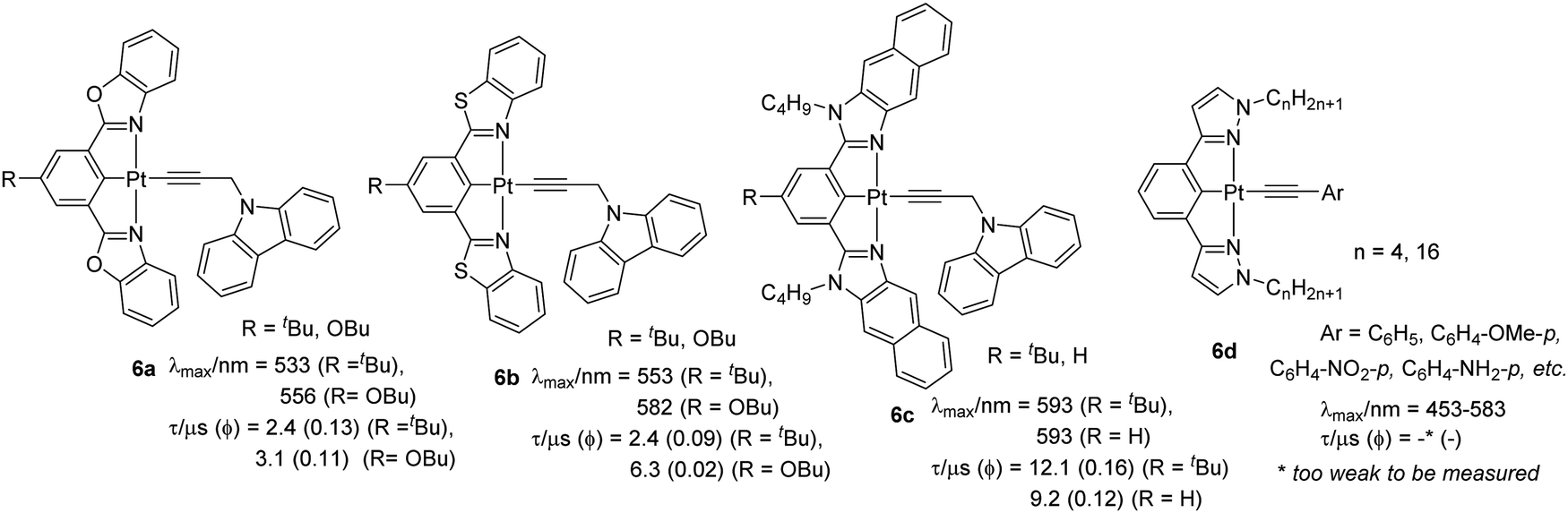
Carbazole and its derivatives exhibit interesting emission properties and act as triplet emitters due to their rigidity and electron donor properties. Yam et al.31 found that the absorption and emission properties of complexes bearing 9-(prop-2-ynyl)-9H-carbazole as the auxiliary ligand is governed by substituents attached to the central arene ring (p-butoxy, butyl or H) as well as the heteroatoms (N, O, S) present in the cyclometallating core (Chart 5). Electrochemical results suggested that the inductive effect of σ-donating substituents at the 5-position of the aryl ring of the N^C^N ligand destabilizes the lowest unoccupied molecular orbital (LUMO). This observation was supported by absorption and emission data. The electronic absorption spectra of the Pt(II) complexes at room temperature displayed a high energy intense absorption at around λ = 295–342 nm (ε ≈ 104 dm3 mol−1 cm−1) attributed to IL [π → π*] transitions of the cyclometallated N^C^N ligand. In addition, moderately intense low energy absorption bands at about 410–470 nm (ε ≈ 103–104 dm3 mol−1 cm−1) were assigned to an admix of π–π transitions of the cyclometallated N^C^N ligand (mainly) with the [dπ(Pt) → π*(N^C^N)] MLCT transition. They found that the introduction of alkynyl linked/separated carbazoles reduced the [dπ/pπ(Pt)-π*(N^C^N)] energy gap leading to the formation of low-lying LLCT excited states. The chloride complexes exhibited well-resolved vibronic-structured emission bands at around λ = 529–601 nm (Φ = 0.03–0.17) originating from 3IL [π → π*(N^C^N)]/[dπ(Pt) → π*(N^C^N)] excited state. The corresponding alkynyl complexes 6a–c (Chart 5) (Φ = 0.1–0.2) showed similar emission energy profile with emission originating from an admix of metal perturbed 3IL [π → π*(N^C^N)] excited state and 3MLCT [dπ(Pt) → π*(N^C^N)]. A significant blue shift in optical properties is noted when the coordinating ligand is changed to 1,3-bis(1-n-alkylpyrazol-3-yl)benzene (bpzb) and the fourth coordination position is occupied by substituted phenylacetylides (6d) (Chart 5).28 However, the origin of the absorption remains the same. Whereas complexes with substituted phenylethynyl ligand possess [π(CC) → π*(bpzb)] LLCT transitions, complexes having no phenyl ring attached to the alkynyl lack this transition. This observation was attributed to the low-lying HOMO of the 2-propyn-1-yl ligand when compared to that of the various phenylethynyl and bpzb ligands. Unlike previous example, with the inclusion of the nitrophenyl acetylide ligand, the emission energy for the complex was found to be less sensitive to the substituent attached to the alkynyl unit but their origin also remains the same. Furthermore, complexes with longer alkyl chains were non-emissive in nature. Interestingly, complex with the nitro group on the alkynyl ligand showed a structureless emission band with a long lifetime (593 nm and 0.7 μs at 298 K and 561 nm and 547.3 μs at 77 K in glass). Based on the absorption, electrochemical and emission results, this was assigned as derived from predominantly metal-perturbed π → π* 3IL state of the p-nitrophenylacetylene ligand with some mixing of metal-to-p-nitrophenylethynyl MLCT state. However, no metallophilic interaction was present, and structural analysis indicated that these complexes possess rare short intermolecular X–H⋯Pt contacts (X = N or C) along with weak π–π interactions. Functionalization of 4-ethynylphenyl with cholesteryl unit resulted in CD-active metallogels.
 | ||
| Chart 5 Cyclometallated Pt(II) complexes bearing carbazole as auxiliary and pyrazole as coordinating ligands. | ||
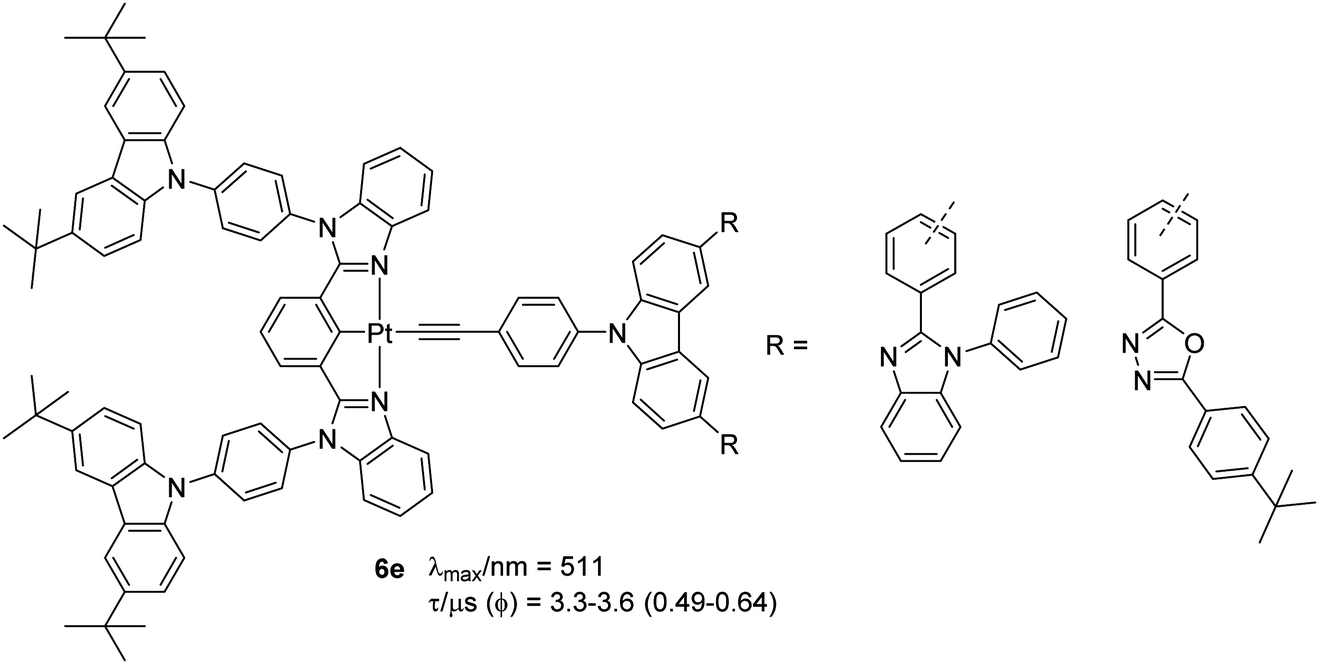
Complexes 6e (Chart 6) with carbazoles attached to both the coordinating and the auxiliary ligand have recently been reported as OLED materials.35 To achieve a varying level of donor–acceptor (D–A) interaction as well as charge transport, electron-accepting unit, phenylbenzimidazole (PBI) or oxadiazole (OXD) was linked to the carbazole ring of the auxiliary ligand. It was found that the topology of the ligands (m-or p-linkages of the acceptor) defined the properties and performance of the materials. Like previous examples of carbazole-based complexes, these complexes exhibited high energy IL [π → π*] transitions (λ < 300 nm) and low energy π → π* transitions (λ ∼ 305–390 nm) from the electron-donating carbazole moiety to the electron-accepting (PBI) or OXD moiety, with admix of IL, MLCT and LLCT character. Emission at 511 nm was assigned to be the 3IL [π → π*(N^C^N)]/[dπ(Pt) → π*(N^C^N)] excited states. Complexes bearing OXD exhibited better PL parameters in solution (Φ = 0.59 for meta while 0.64 for ortho) than PBI-based complexes (Φ = 0.59 for meta while 0.49 for ortho). The same group noted that by forming dendrons, the properties of the complexes can be further modulated. It has been noted that the incorporation of carbazole dendrons into the Pt(II) centre can significantly suppress intermolecular interactions in solid-state thin films, giving rise to emission spectra (λmax/nm = 512, lifetime = 2.4–2.7 and Φ = 0.42–0.71 μs) that are similar to those found in solution.36
 | ||
| Chart 6 Carbazole containing cyclometallated Pt(II) complex designed using a D–A strategy. | ||
Another example of changes in topology altering the emission properties has been demonstrated using the complex 7. Yang et al.37 found a correlation between the topology of the ligand and the T1 state energy. They noted that the presence of a pentiptycene substituent on the pincer ligands was more efficient at lowering the LC T1 state energy than the same substituent on the ancillary ligand in the fourth coordination site. Both 7a (0.66) and 7b (0.55) (Chart 7) have high Φ similar to the parent chlorinated Pt(II) complex (0.60). However, in the presence of the bromine substituent on the N^C^N pincer (7c), the Φ values decreased by 2–5-fold. This was attributed to increased knr, presumably due to the heavy bromine atom that facilitates the non-radiative T1 → S0 ISC process. The absorption spectra display an intense band (ε > 104 dm3 mol−1 cm−1) at 300–330 nm and a broad shoulder (ε ∼ 103–104 dm3 mol−1 cm−1) at 370–450 nm. These two bands have been assigned to a LC π → π* transition and mixed ILCT, LLCT, and MLCT transitions, respectively, on the basis of TDDFT calculations. The emission spectra are located in the 480–650 nm range with varied degrees of vibrational structure, revealing significant LC character for the T1 state.
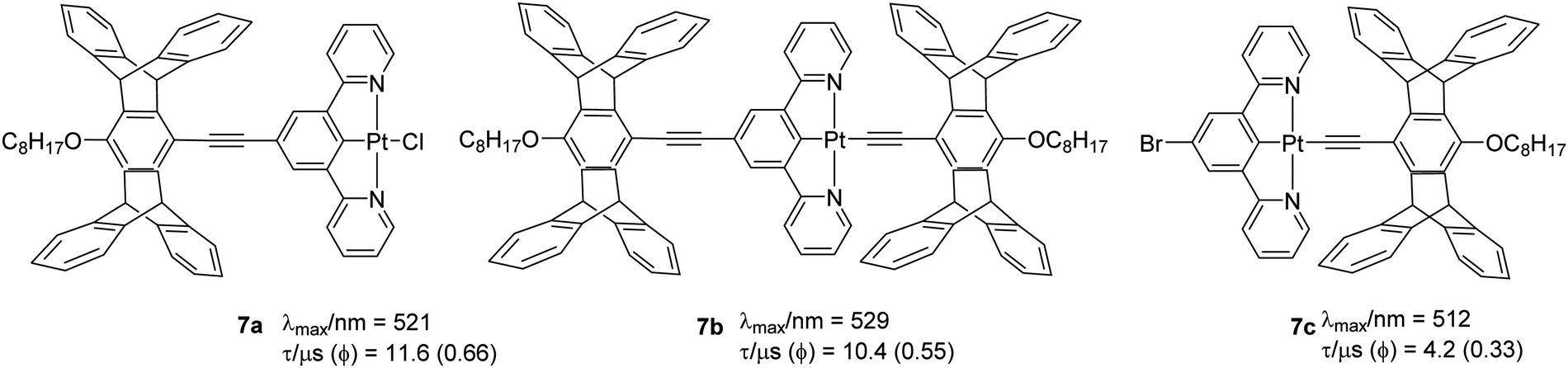 | ||
| Chart 7 Pentiptycene containing cyclometallated Pt(II) complexes that showed halide-dependent PL properties. | ||
2.4. Platinum(II) alkynyl complexes with C^N^C pincer ligands
Cyclometallated Pt(II) complexes based on the pincer C^N^C-type ligands (i.e. 2,6-diphenylpyridine based) bearing acetylide functionality as the fourth coordinating ligand are rather limited.38 Depending upon the type of auxiliary ligand, these complexes can be neutral or cationic or anionic (Chart 8). Compared to complexes with the cyclometallated N^N^N core, complexes based on this class of ligands exhibit long-lived 3MLCT. Lalinde and co-workers39 synthesized a series of anionic complexes 8a bearing acetylide and other anionic ligands. The reported complexes adopted 2D and 3D architectures through weak intermolecular interactions. All reported complexes were non-emissive in solution at room temperature but emitted intensely at 77 K (lifetime = 9.5–20.1 μs). All the alkynyl-bearing complexes showed a low energy and alkynyl unit insensitive band (dπ(Pt) → π* 1MLCT transition) and a high energy band attributed to ligand-centred transitions (1IL π → π*). Replacing the arene groups by N-heterocyclic carbene (NHC) ligands40 resulted in complex 8b that exhibited strong 3MLCT/3LLCT phosphorescence in degassed acetonitrile with the emission energy readily tuned by the variation of the alkynyl ligands. Unlike 8a, most of the reported NHC-based complexes exhibit emission at room temperature with moderate quantum yield (0.001–0.37) and lifetime (0.44–4.18 μs). The incorporation of the alkynyl ligand plays an important role in the luminescence enhancement. Recently, Che et al.41 designed NHC-based C^N^C-type ligands and their corresponding Pd(II) complexes. Absorption studies of 8c (R = Ph) showed profile similar to Pt(II) counterpart 8b (R = Ph), i.e.1IL transitions below 330 nm. However, 1MLCT band (>350 nm) was more intense for Pt(II) complex. With two lateral NHC binding motifs, the [Pd(C^N^C)]–acetylide complexes displayed enhanced emission and higher stability compared to its N^N^N analogues. The presence of metallophilic interaction led to the formation of 1D chain structure with alternating Pd⋯Pd contacts and π⋯π interactions.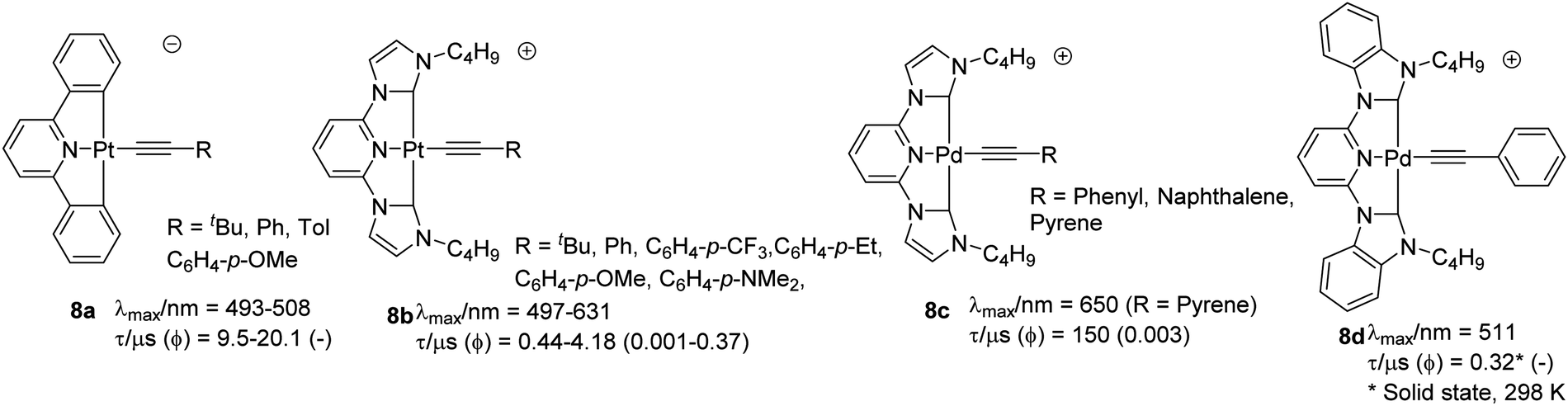 | ||
| Chart 8 Examples of cyclometallated Pt(II) and palladium(II) alkynyl complexes with C^N^C pincer ligands. | ||
3. Emerging applications
3.1. Organic electronics
The intense tunable phosphorescence of cyclometallated tridentate Pt(II) arylacetylide complexes of the types [Pt(C^N^N)(CCR)] and [Pt(N^C^N)(CCR)], together with their thermal stability and neutrality, render them good candidates as triplet emitters for both evaporated and solution-processed OLEDs. The Pt–CCR interaction would facilitate tuning of the 3MLCT energies by varying the R substituent. Complexes 1b and 1c (Fig. 2) are stable towards sublimation and thus are suitable for vacuum deposition in OLED fabrication. Orange (1b) and red (1c) electrophosphorescent doped OLEDs (4 wt% in 4,4′-bis(carbazol-9-yl)biphenyl, CBP) were obtained with low turn-on voltages in the 3.6–4.5 V range and current efficiency (CE) of 2.4 and 0.6 cd A−1 at 30 mA cm−2, respectively.42 Phosphors 3a and 3c (Chart 2) were also used as dopants for high-efficiency OLEDs, in which yellowish-green and saturated red devices were recorded with peak external quantum efficiencies (EQE) of 17.2 and 22.8%, respectively.22
Complexes of the type [Pt(N^C^N)(CCR)] are also promising dopants for OLED studies and 5a, 5b and 5d (Chart 4) are some good examples. Since the emission from aggregation states is significant at high doping concentrations for both 5a and 5b, solution-processed green OLEDs were fabricated at low doping levels (doped in poly(9-vinylcarbazole), PVK, at 2 and 4 wt%).32 Complexes 5a and 5b exhibit different kinds of intermolecular interactions. It was shown that complex 5a without intermolecular Pt⋯Pt interaction in the solid state furnished monochromatic OLEDs with higher efficiency than 5b due to its higher concentration threshold for aggregate formation. By utilizing {4-[{4-[5-(4-tert-butylphenyl)-4-phenyl-4H-1,2,4-triazol-3-yl]phenyl}(diphenyl)-silyl]phenyl}diphenylamine (p-TAZSiTPA) as the bipolar host, peak CE of 26.6 cd A−1, with low efficiency roll-off of 16.7% at 1000 cd m−2, has been obtained. This outstanding device performance has been ascribed to the high Φ of 5a as well as the bipolar transporting properties, high triplet energy and high-lying HOMO level of the host. Two similar Pt(II) complexes in 5d were also studied as metallophosphors in OLEDs. By using the sterically more bulky mesityl group on the central ring of the N^C^N ligand, a drastic enhancement in the device efficiency was observed owing to the inhibition of aggregate-induced quenching, with the maximum EQE reaching about 12% at 5 wt% level in {4,4′,4′-tris(N-carbazolyl)triphenylamine} (TCTA) host.34
A series of dendritic carbazole-containing [Pt(N^C^N)(CCR)] complexes of up to fourth generation 9a–9d (Chart 9) has been reported by Yam et al. with high Φ of up to 80% in thin films and applied as phosphorescent dyes for fabricating high-performance solution-processed OLEDs with peak EQE and CE of up to 10.4% and 37.6 cd A−1, which is comparable to that of the evaporated devices based on the small-molecule counterparts.36 The high efficiency is attributed to the presence of carbazole dendrimers that can significantly suppress intermolecular interactions of the complexes in the solid film.
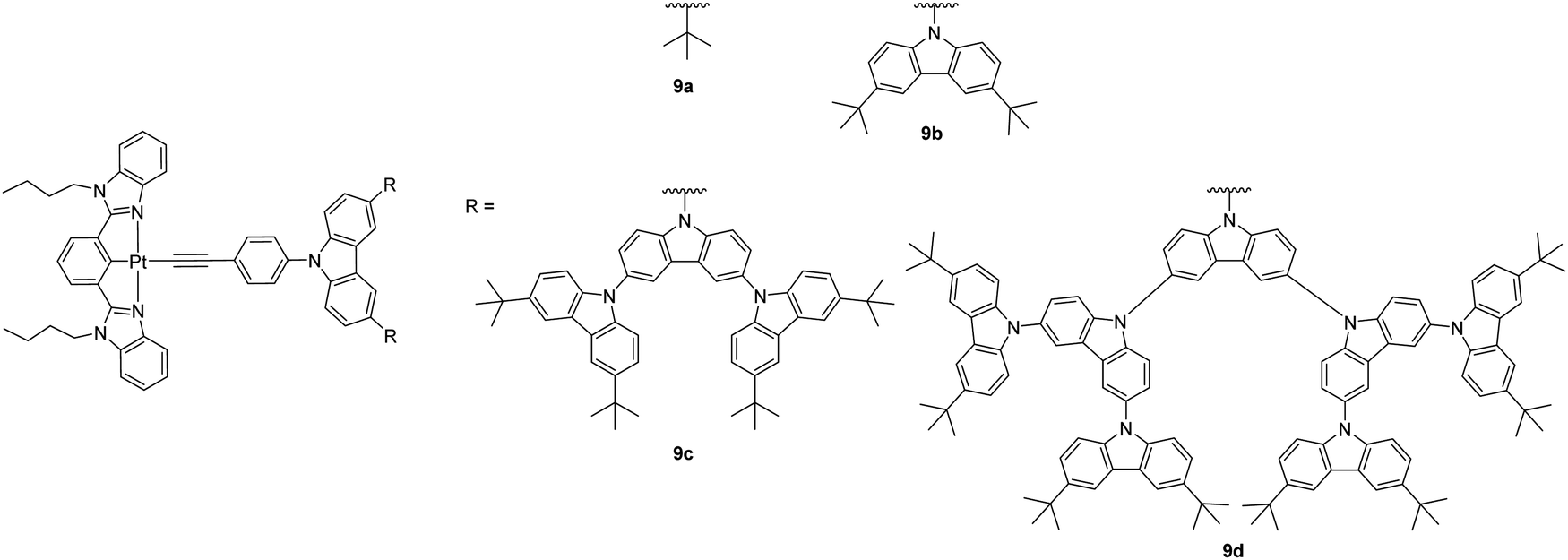 | ||
| Chart 9 Dendritic carbazole-containing [Pt(N^C^N)(CCR)] complexes. | ||
C) interactions. But, complexes 5b and 10c form quasi-1D nanowires or nanofibers whereas 5a gives mixtures of nanoparticles and nanobelts. In view of this, an organic field-effect transistor (OFET) in the bottom-contact mode was fabricated based on 5a which showed the p-type hole-transporting characteristic for such semiconductor.
 | ||
| Chart 10 A family of charge-neutral organoplatinum(II) complexes. | ||
Fabrication of bottom-contact OFETs was also made from the self-assembled 5c (R = CH3) and its co-assembly with 5a in order to study their charge-transporting properties.33 It was found that the co-assembled sample revealed an ambipolar charge-transporting character when charge carriers were photogenerated. However, the self-assembled 5c transports only electrons but not holes even under photoirradiation while 5a alone did not show any detectable FET responses under the same conditions.
The donor–acceptor properties of the cyclometallated pincer-type Pt(II) complexes can also be used to fabricate organic memory devices. For example, the memory device in the configuration of Al/11a/ITO displays binary memory performance with low operating voltages of about 3.5 V, high ON/OFF ratios of over 105 and long retention times of over 104 s.44
3.2. Nonlinear optics (NLO)
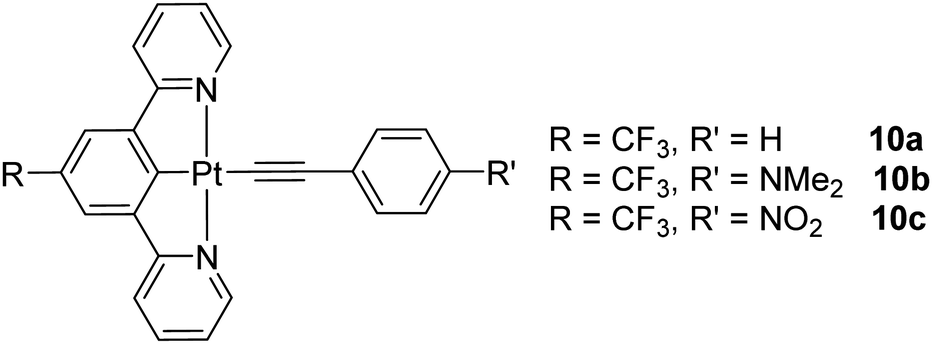
Currently, it is desirable to develop active molecular materials for various photonic device applications (e.g. light modulation, optical power limiting) based on nonlinear optics (NLO).1–3 In addition to the improved emission properties of the [Pt(C^N^N)X]-type complexes, [Pt(C^N^N)(CCR)] have been demonstrated to display broader triplet excited-state absorption, higher triplet excited-state quantum yield and thus enhanced reverse saturable absorption (RSA) for nanosecond (ns) laser pulses as compared to their Pt(II)-N^N^N analogues. So, 6-phenyl-2,2′-bipyridyl ligand is potentially a better tridentate ligand for nonlinear absorption applications. Another good reason for using a C^N^N ligand over the N^N^N ligand for Pt(II) complex formation is that neutral [Pt(C^N^N)(CCR)] complexes will be formed, which are more soluble in common organic solvents and thus facilitate the product purification. Along this line, the nonlinear absorption properties of a large library of cyclometallated [Pt(C^N^N)(CCR)] complexes containing 4,6-diphenyl-2,2′-bipyridine or 6-phenyl-4-(9,9-dihexylfluoren-2-yl)-2,2′-bipyridine 11–16 were extensively studied by Sun et al (Chart 11). They are generally good reverse saturable absorbers for ns laser pulses at 532 nm and exhibit the broadband triplet excited-state absorption in the visible to the NIR region. For 11a–11c, the molar extinction coefficients for dinuclear 11c are much larger than those for the mononuclear counterparts 11a–11b, suggesting the significant electronic coupling through the bridging ligand. The degree of RSA follows the trend 11a > 11b > 11c.45 The influence of alkoxy substituent on the C^N^N ligand on the photophysics and NLO properties of 12a–12d was also examined by the same team and the results were compared to those for the chloro derivative. The excited-state lifetime of 12a is much longer than that of its corresponding alkoxy-free analogue 13 in CH3CN.45 Complexes 12a–12d show broad triplet transient difference absorption in the near-UV to the NIR region, where strong RSA could occur but compound 13 only has weak RSA because of its larger ground-state absorption cross-section and its low triplet excited-state quantum yield. This work provides good evidence for the advantages of the alkynyl attachment to Pt(II) and alkoxy substitution in improving the nonlinear absorption features.
The potential application of these Pt(II) complexes on photonic devices that require broadband spectral response is limited because of the absence of ground-state absorption of these complexes above 600 nm, which prevents the population of the triplet excited state via one-photon absorption. To tackle this problem, it is important to choose an appropriate alkynyl ligand that would red-shift the low-energy absorption band while maintaining the triplet excited-state absorption. Sun and coworkers have introduced the electron-donating phenothiazinyl acetylide co-ligand in 14a and 14b.46 Both complexes show broadband triplet excited-state absorption. Complex 14b displays a stronger RSA than 14a does due to the smaller ground-state absorption cross-section and larger triplet excited-state absorption at 532 nm for 14b relative to 14a. Another complex 15 was reported as a potentially good broadband, nonlinear absorbing material that shows large ratios of excited-state absorption to ground-state absorption from 430 to 680 nm and strong two-photon absorption in the NIR region (from 740 to 910 nm).46 The two photon absorption (TPA) cross-section value of 1200 GM at 850 nm is among the largest reported for related Pt(II) complexes.
In another context, the second-order NLO properties of a family of [Pt(N^C^N)(CCR)] complexes 16a–16f were probed by electric-field-induced second harmonic (EFISH) generation and harmonic light scattering (HLS) measurements.47 These emitters are characterized by a good optical transparency in the visible window. A rational substitution of the cyclometallated or phenylacetylide ligands allows the fine-tuning of the second-order NLO response. The presence of CF3 substituents on the 5-position of the pyridine ring of the tridentate ligand can help in increasing the dipole moment and affording large μβEFISH values. This study offers a class of organometallic second-order NLO chromophores that possess additional flexibility relative to the organic congeners.
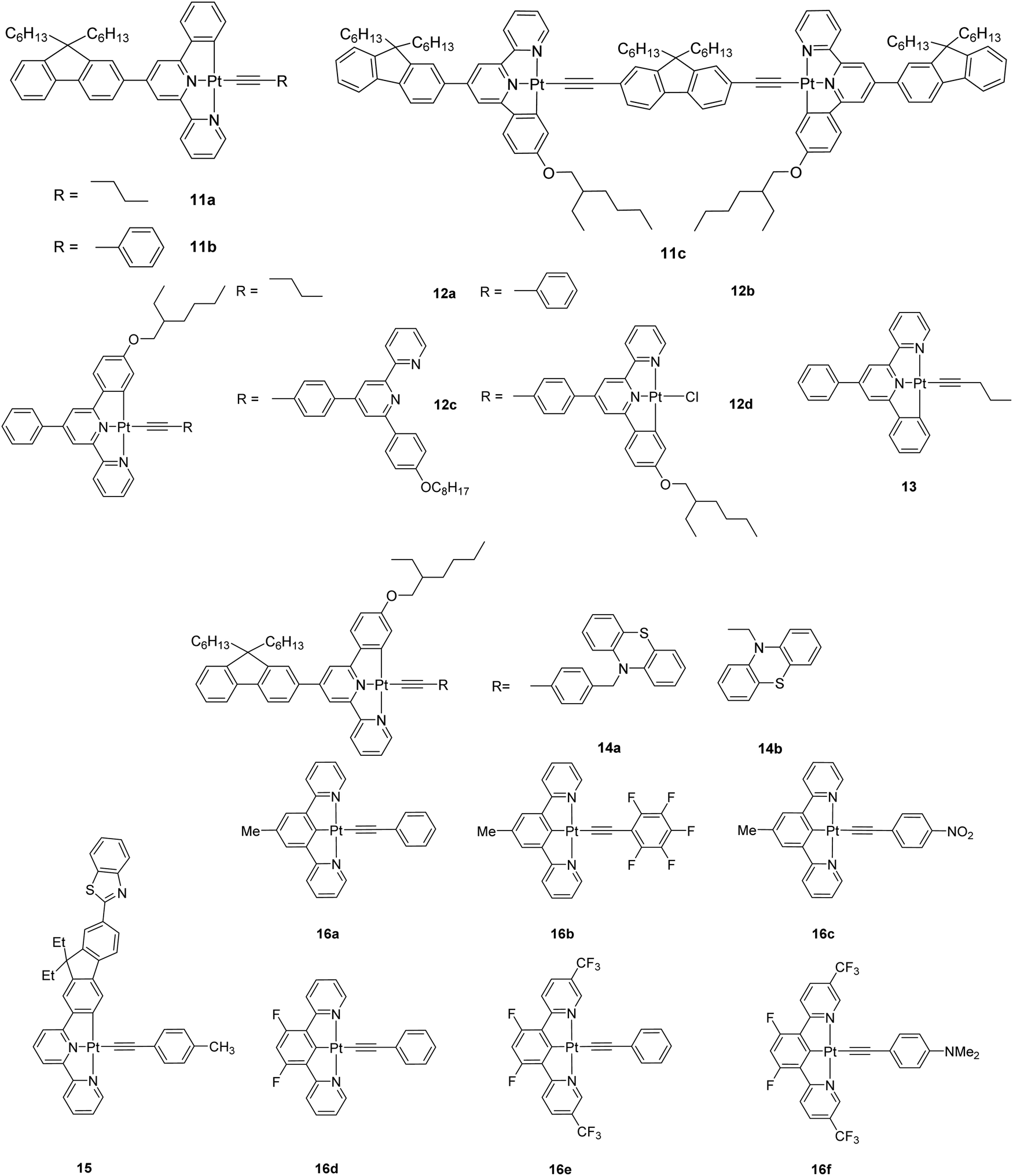 | ||
| Chart 11 A library of cyclometallated [Pt(C^N^N)(CCR)] complexes. | ||
3.3. Triplet photosensitizers
Generally, the intense visible-light absorption and the long-lived T1 excited state of phosphorescent metal complexes are important for applications in sensitizing photophysical processes that need triplet excited states, such as photovoltaics, photocatalysis and upconversion based on triplet–triplet annihilation (TTA), etc. Unlike other upconversion approaches involving two-photon absorption dyes or rare earth compounds, TTA upconversion is particularly useful due to the low excitation power, the tunable excitation/emission wavelengths and the high upconversion quantum yields. A good way to prepare such complexes is to attach a light-harvesting organic chromophore to the transition metal centre in a dyad. However, conventional Pt(N^N)–bis(acetylide) complexes are not suitable for this purpose since they usually display weak visible-light harvesting ability and short T1 lifetimes (<5 μs). A dinuclear Pt complex 17 (Chart 12), in which two Pt(N^C^N)(CC) units are connected to the π-chromophore of boron-dipyrromethene (BODIPY), was reported by Zhao et al. and shown to possess strong absorption at 574 nm (ε = 53![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800 M−1 cm−1) and a long-lived T1 excited state (128.4 μs), plus a room-temperature BODIPY NIR phosphorescence at 770 nm.48 The TTA upconversion with complex 17 as the triplet sensitizer was visible to the naked eye and the upconversion quantum yield was measured to be 5.2%. But, no upconversion was detected using the corresponding model compound of 17, namely, [Pt(N^C^N)Cl]. The same author also described another series of visible-light harvesting transition metal compounds 18a–18c (Chart 12) that show long-lived T1 excited states (9.2, 83.7 and 135.7 μs, respectively). The visible-light absorption of the pyrene- and naphthalimide-bearing complexes 18b and 18c was enhanced relative to that for the phenyl-substituted analogue 18a. These complexes can be used as triplet photosensitizers for TTA-based upconversion with upconversion quantum yields up to 19.5% for 18c.49
800 M−1 cm−1) and a long-lived T1 excited state (128.4 μs), plus a room-temperature BODIPY NIR phosphorescence at 770 nm.48 The TTA upconversion with complex 17 as the triplet sensitizer was visible to the naked eye and the upconversion quantum yield was measured to be 5.2%. But, no upconversion was detected using the corresponding model compound of 17, namely, [Pt(N^C^N)Cl]. The same author also described another series of visible-light harvesting transition metal compounds 18a–18c (Chart 12) that show long-lived T1 excited states (9.2, 83.7 and 135.7 μs, respectively). The visible-light absorption of the pyrene- and naphthalimide-bearing complexes 18b and 18c was enhanced relative to that for the phenyl-substituted analogue 18a. These complexes can be used as triplet photosensitizers for TTA-based upconversion with upconversion quantum yields up to 19.5% for 18c.49
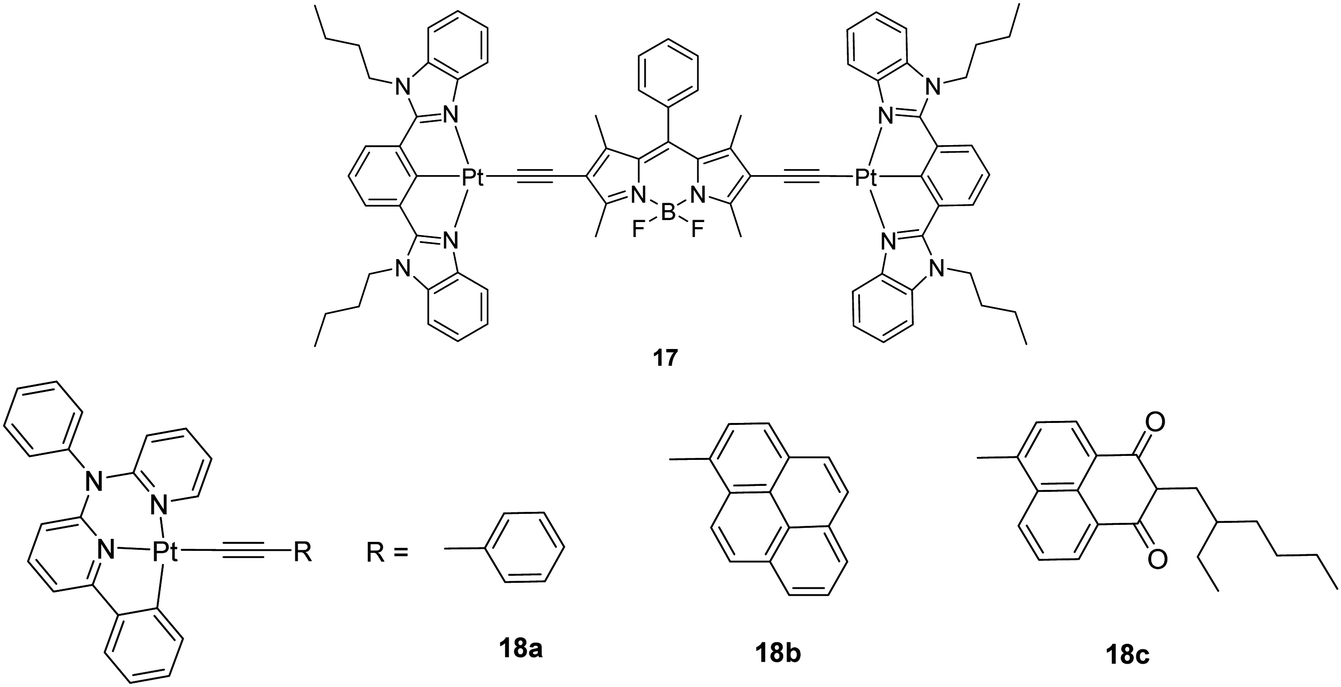 | ||
| Chart 12 A series of visible-light harvesting Pt(II) complexes with pyrene and naphthalimide ligands. | ||
3.4. Sensors for metal ions
A chemosensor is usually composed of two parts, one is the receptor unit and the other is the signaling unit. The former one is used to bind to the analyte selectively whereas the latter one reveals the changes in the optical or other properties induced by the interaction between the receptor and the analyte. The two parts can be rationally incorporated into the metal complex platforms. Receptor groups containing nitrogen or oxygen atoms are commonly used in the molecular design owing to their strong binding ability to metal ions. Phosphorescent metal complexes are generally good signaling systems because phosphorescent signals show many advantages such as sensitive emission properties in response to the local environment, strong single-photon excitation in the visible region, relatively long emission lifetimes and large Stokes shifts for easy separation of excitation and emission wavelengths. In this context, the design of neutral [Pt(C^N^N)(CCR)] complexes proved to be useful over the cationic [Pt(N^N^N)(CCR)]+ congener in terms of the affinity for metal cations. One example is the unprecedented Pt(II)–ethynylflavone system featuring various polyether arms 19a–19c (Fig. 5) that can switch from triplet to singlet emission in response to Pb2+ binding.50 The tBu groups sterically inhibit the undesirable intermolecular interactions that usually cause concentration-dependent self-quenching of the emission. Complex 19b shows 3IL phosphorescence associated with the anchored flavone perturbed by the Pt centre (τ ∼ 20 μs), but switches largely to flavone-localized 1IL fluorescence (τ ∼ 2 ns) after selective binding of Pb2+. The complexation of the metal ion to 19b is believed to occur via the formation of a metal chelate involving the O atom of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group of the benzopyrone unit and the terminal O atom of the polyether arm. This is supported by the observation that there is no such affinity for divalent metal ions in 19a. Hence, fine-tuning of the cation affinities of the system can be achieved by the facile chemical modification of the polyether arm.
O group of the benzopyrone unit and the terminal O atom of the polyether arm. This is supported by the observation that there is no such affinity for divalent metal ions in 19a. Hence, fine-tuning of the cation affinities of the system can be achieved by the facile chemical modification of the polyether arm.
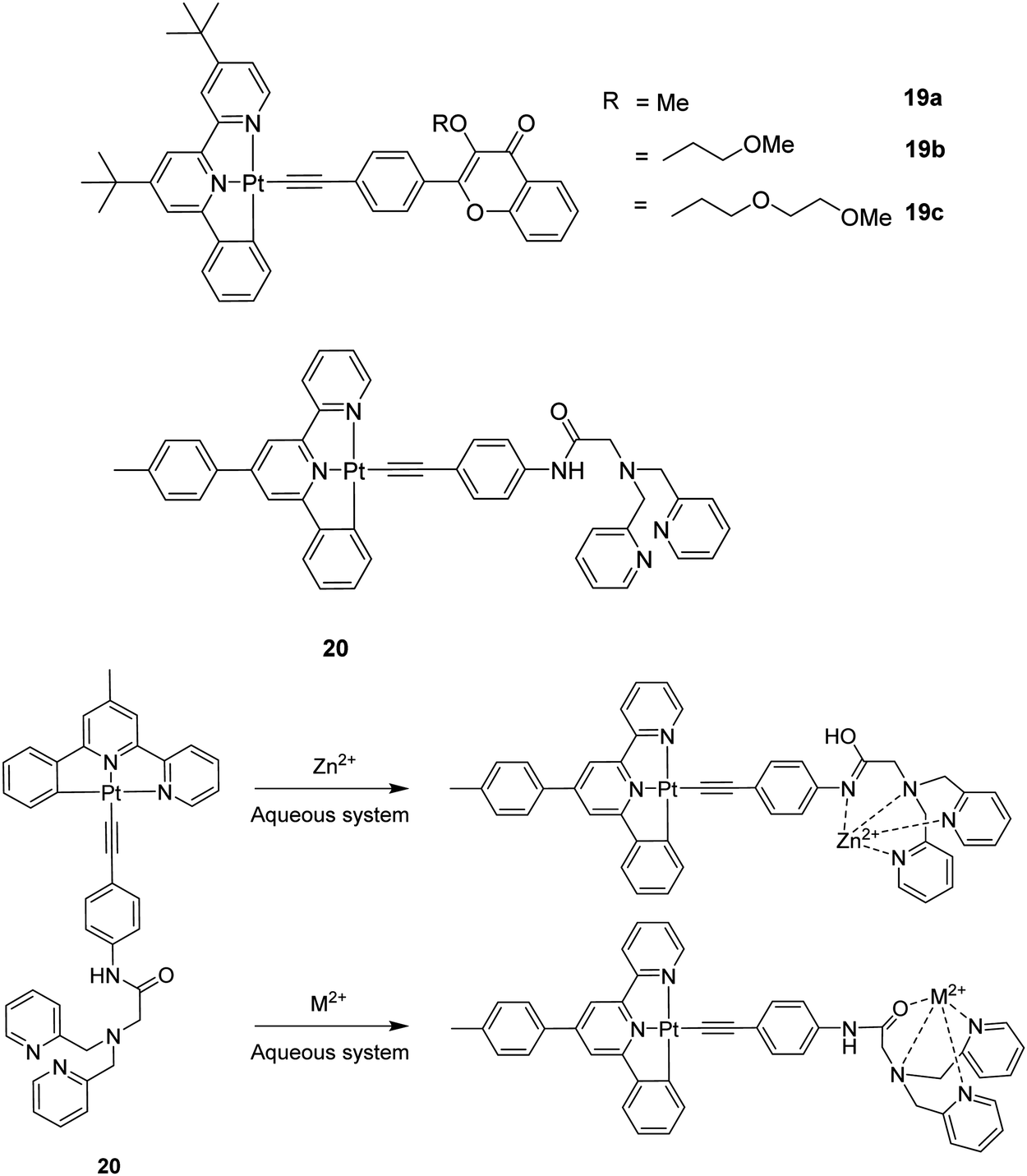 | ||
| Fig. 5 Possible binding modes of 20 with different heavy and transition metal ions. | ||
Another highly selective and sensitive luminescent Zn2+ ions chemosensor was reported based on complex 20 featuring an amide-DPA (DPA = 2,2-dipicolylamine) receptor.51 In the proposed mechanism, the tautomerization of amide favours detection of Zn2+ ions among other heavy and transition metal ions in aqueous systems (Fig. 5). The difference in the excited state lifetimes for amide and imidic acid provides a good avenue to probe the binding sites for Zn2+ ions recognition. Sensor 20 possesses a high affinity towards Zn2+ ions with an equilibrium dissociation constant (Kd) of 16.3 nM and a high selectivity for Zn2+ ions with a big enhancement of the emission in CH3CN.
3.5. Miscellaneous
Some luminescent pincer-type complexes of [Pt(C^N^N)(CCR)] have been shown to be efficient photocatalysts for organic transformations. For example, compound 3c (Chart 2) is both a powerful photoreductant and a strong photooxidant which can be excited by visible light. It serves as an effective photocatalyst for visible-light induced reductive C–C bond formation of benzyl chloride/unactivated alkyl bromides.22 Also, 3c shows two-photon-induced emission properties (two-photon absorption cross section ∼17 GM at 756 nm) and encapsulation of 3c in mesoporous silica nanoparticles (MSNs) for two-photon-excited cellular imaging has been demonstrated.22
4. Conclusions and outlook
This tutorial review highlights recent advances in the design strategy of cyclometalled platinum(II) pincer complexes with alkynyl groups in the fourth coordination site on the square planar metal centre. Using this strategy highly luminescent materials have been produced that perform more effectively in a range of opto-electronic devices than terpyridyl pincer analogues.Over the last decade, following on from the pioneering work that established the benefits of having a planar ligand arrangement around the platinum(II) centre, and strong field ligands that result in an increased energy gap between the lowest-lying excited state and the d–d state, all of which enhance the luminescence of the materials,9 it has been possible to manipulate the pincer molecules in solution and in the solid state to optimise specific desirable properties. The benefits of having cyclometallated pincer ligands of the general type N^C^N with the cyclometallated Pt–C bond trans to the fourth ligand position have been shown to maximize the ligand field strength. Using this pincer ligand, to produce neutral molecules (with a strong field anionic ligand such as cyanide or alkynyl in the fourth coordination site) has facilitated the development of solid state luminescent materials where intermolecular Pt⋯Pt and π⋯π stacking adds a new dimension to the luminescent properties of the materials.11 Fine tuning can be achieved by adding electron donating or accepting groups as substituents to the rings of the pincer ligands themselves, and altering the substituents on the alkynyl ligand, in the fourth coordination site, has led to the use of the pincer materials in a number of applications.
As we have seen the cyclometallated platinum pincer materials have already shown promise in the areas of OLEDs and OFETs, as NLO materials, as triplet photosensitizers and as sensors for metal ions. All these new materials have been produced through the manipulation of the design principles outlined in this review and there is considerable potential, using the knowledge gained over the last two decades, to produce new classes of materials with properties tailored for specific applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
MSK acknowledges the British Petroleum, Oman (Grant No. EG/SQU-BP/SCI/CHEM/19/01) and His Majesty's Trust Fund for Strategic Research (Grant No. SR/SQU/SCI/CHEM/16/02) for financial support. WYW thanks the financial support from the Science, Technology and Innovation Committee of Shenzhen Municipality (JCYJ20180507183413211), Hong Kong Research Grants Council (PolyU 153062/18P and C6009-17G), the National Natural Science Foundation of China (51873176), the Hong Kong Polytechnic University (1-ZE1C) and Ms Clarea Au for the Endowed Professorship in Energy (847S). PRR is grateful to The Engineering and Physical Sciences Research Council (UK) for continued funding (EP/K004956/1).References
- G. R. Whittell, M. D. Hager, U. S. Schubert and I. Manners, Nat. Mater., 2011, 10, 176–188 CrossRef CAS PubMed and references therein.
- A. Haque, R. A. Al-Balushi, I. J. Al-Busaidi, M. S. Khan and P. R. Raithby, Chem. Rev., 2018, 118, 8474–8597 CrossRef CAS PubMed and references therein.
- C.-L. Ho, Z.-Q. Yu and W.-Y. Wong, Chem. Soc. Rev., 2016, 45, 5264–5295 RSC and references therein.
- V. W.-W. Yam, V. K.-M. Au and S. Y.-L. Leung, Chem. Rev., 2015, 115, 7589–7728 CrossRef CAS PubMed and references therein.
- F. N. Castellano, I. E. Pomestchenko, E. Shikhova, F. Hua, M. L. Muro and N. Rajapakse, Coord. Chem. Rev., 2006, 250, 1819–1828 CrossRef CAS and references therein.
- Z. Gao, Y. F. Han, Z. C. Gao and F. Wang, Acc. Chem. Res., 2018, 51, 2719–2729 CrossRef CAS PubMed and references therein.
- S. Huo, J. Carroll and D. A. Vezzu, Asian J. Org. Chem., 2015, 4, 1210–1245 CrossRef CAS and references therein.
- J. A. G. Williams, in Photochemistry and Photophysics of Coordination Compounds π, ed. V. Balzani and S. Campagna, 2007, vol. 281, pp. 205–268 Search PubMed.
- J. G. Williams, Chem. Soc. Rev., 2009, 38, 1783–1801 RSC and references therein.
- J. S. Wilson, N. Chawdhury, M. R. Al-Mandhary, M. Younus, M. S. Khan, P. R. Raithby, A. Kohler and R. H. Friend, J. Am. Chem. Soc., 2001, 123, 9412–9417 CrossRef CAS PubMed.
- M. Bryant, J. Skelton, L. Hatcher, C. Stubbs, E. Madrid, A. R. Pallipurath, L. Thomas, C. Woodall, J. Christensen and S. Fuertes, Nat. Commun., 2017, 8, 1800 CrossRef CAS PubMed and references therein.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS PubMed.
- J. G. Williams, S. Develay, D. L. Rochester and L. Murphy, Coord. Chem. Rev., 2008, 252, 2596–2611 CrossRef and references therein.
- K. Li, G. S. Ming Tong, Q. Wan, G. Cheng, W.-Y. Tong, W.-H. Ang, W.-L. Kwong and C.-M. Che, Chem. Sci., 2016, 7, 1653–1673 RSC.
- W. Lu, M. C. Chan, N. Zhu, C.-M. Che, C. Li and Z. Hui, J. Am. Chem. Soc., 2004, 126, 7639–7651 CrossRef CAS PubMed.
- W. Lu, B.-X. Mi, M. C. Chan, Z. Hui, C.-M. Che, N. Zhu and S.-T. Lee, J. Am. Chem. Soc., 2004, 126, 4958–4971 CrossRef CAS PubMed.
- S.-W. Lai, M. C.-W. Chan, T.-C. Cheung, S.-M. Peng and C.-M. Che, Inorg. Chem., 1999, 38, 4046–4055 CrossRef CAS.
- R. Mroz, D. A. Vezzu, B. Wallace, R. D. Pike and S. Huo, Chin. J. Inorg. Chem., 2018, 38, 171–182 CAS.
- W. Li, J. Wang, X. Yan, H. Zhang and W. Shen, Appl. Organomet. Chem., 2018, 32, e3929 CrossRef.
- C. F. Harris, D. A. Vezzu, L. Bartolotti, P. D. Boyle and S. Huo, Inorg. Chem., 2013, 52, 11711–11722 CrossRef CAS PubMed.
- W. Wu, X. Wu, J. Zhao and M. Wu, J. Mater. Chem. C, 2015, 3, 2291–2301 RSC.
- P. K. Chow, G. Cheng, G. S. M. Tong, W. P. To, W. L. Kwong, K. H. Low, C. C. Kwok, C. Ma and C. M. Che, Angew. Chem., Int. Ed., 2015, 54, 2084–2089 CrossRef CAS PubMed.
- M. Krause, D. Kourkoulos, D. González-Abradelo, K. Meerholz, C. A. Strassert and A. Klein, Eur. J. Inorg. Chem., 2017, 5215–5223 CrossRef CAS.
- D. Bi, Y. Feng, Q. Zhao, H. Wang, Y. Zhu, X. Bao, H. Fan, L. Yu, Q. Yang and D. Qiu, RSC Adv., 2017, 7, 46980–46988 RSC.
- C. Latouche, P.-H. Lanoe, J. G. Williams, V. Guerchais, A. Boucekkine and J.-L. Fillaut, New J. Chem., 2011, 35, 2196–2202 RSC.
- J. G. Williams, A. Beeby, E. S. Davies, J. A. Weinstein and C. Wilson, Inorg. Chem., 2003, 42, 8609–8611 CrossRef CAS PubMed.
- W. H. Lam, E. S.-H. Lam and V. W.-W. Yam, J. Am. Chem. Soc., 2013, 135, 15135–15143 CrossRef CAS PubMed.
- Y. Ai, Y. Li, H. Ma, C.-Y. Su and V. W.-W. Yam, Inorg. Chem., 2016, 55, 11920–11929 CrossRef CAS PubMed.
- E. S. H. Lam, D. P. K. Tsang, W. H. Lam, A. Y. Y. Tam, M. Y. Chan, W. T. Wong and V. W. W. Yam, Chem. – Eur. J., 2013, 19, 6385–6397 CrossRef CAS PubMed.
- A. F. Rausch, L. Murphy, J. G. Williams and H. Yersin, Inorg. Chem., 2011, 51, 312–319 CrossRef PubMed.
- A. K. W. Chan, E. S. H. Lam, A. Y. Y. Tam, D. P. K. Tsang, W. H. Lam, M. Y. Chan, W. T. Wong and V. W. W. Yam, Chem. – Eur. J., 2013, 19, 13910–13924 CrossRef CAS PubMed.
- G. Cheng, Y. Chen, C. Yang, W. Lu and C. M. Che, Chem. – Asian J., 2013, 8, 1754–1759 CrossRef CAS PubMed.
- S. Sato, T. Takei, Y. Matsushita, T. Yasuda, T. Kojima, M. Kawano, M. Ohnuma and K. Tashiro, Inorg. Chem., 2015, 54, 11581–11583 CrossRef CAS PubMed.
- E. Rossi, A. Colombo, C. Dragonetti, D. Roberto, R. Ugo, A. Valore, L. Falciola, P. Brulatti, M. Cocchi and J. G. Williams, J. Mater. Chem., 2012, 22, 10650–10655 RSC.
- F. K.-W. Kong, M.-C. Tang, Y.-C. Wong, M. Ng, M.-Y. Chan and V. W.-W. Yam, J. Am. Chem. Soc., 2017, 139, 6351–6362 CrossRef CAS PubMed.
- F. K.-W. Kong, M.-C. Tang, Y.-C. Wong, M.-Y. Chan and V. W.-W. Yam, J. Am. Chem. Soc., 2016, 138, 6281–6291 CrossRef CAS PubMed.
- C.-J. Lin, Y.-H. Liu, S.-M. Peng, T. Shinmyozu and J.-S. Yang, Inorg. Chem., 2017, 56, 4978–4989 CrossRef CAS PubMed.
- V. W. W. Yam, R. P. L. Tang, K. M. C. Wong, X. X. Lu, K. K. Cheung and N. Zhu, Chem. – Eur. J., 2002, 8, 4066–4076 CrossRef CAS.
- J. R. Berenguer, E. Lalinde and J. Torroba, Inorg. Chem., 2007, 46, 9919–9930 CrossRef CAS PubMed.
- S. Y. L. Leung, E. S. H. Lam, W. H. Lam, K. M. C. Wong, W. T. Wong and V. W. W. Yam, Chem. – Eur. J., 2013, 19, 10360–10369 CrossRef CAS PubMed.
- F. F. Hung, S. X. Wu, W. P. To, W. L. Kwong, X. Guan, W. Lu, K. H. Low and C. M. Che, Chem. – Asian J., 2017, 12, 145–158 CrossRef CAS PubMed.
- W. Lu, B.-X. Mi, M. C. Chan, Z. Hui, N. Zhu, S.-T. Lee and C.-M. Che, Chem. Commun., 2002, 206–207 RSC.
- Y. Chen, K. Li, W. Lu, S. S.-Y. Chui, C.-W. Ma and C.-M. Che, Angew. Chem., Int. Ed., 2009, 48, 9909–9913 CrossRef CAS PubMed.
- A. K.-W. Chan, M. Ng, Y.-C. Wong, M.-Y. Chan, W.-T. Wong and V. W.-W. Yam, J. Am. Chem. Soc., 2017, 139, 10750–10761 CrossRef CAS PubMed.
- P. Shao, Y. Li, J. Yi, T. M. Pritchett and W. Sun, Inorg. Chem., 2010, 49, 4507–4517 CrossRef CAS PubMed.
- B. Zhang, Y. Li, R. Liu, T. M. Pritchett, A. Azenkeng, A. Ugrinov, J. E. Haley, Z. Li, M. R. Hoffmann and W. Sun, Chem. – Eur. J., 2012, 18, 4593–4606 CrossRef CAS PubMed.
- E. Rossi, A. Colombo, C. Dragonetti, S. Righetto, D. Roberto, R. Ugo, A. Valore, J. G. Williams, M. G. Lobello and F. De Angelis, Chem. – Eur. J., 2013, 19, 9875–9883 CrossRef CAS PubMed.
- W. Wu, J. Zhao, H. Guo, J. Sun, S. Ji and Z. Wang, Chem. – Eur. J., 2012, 18, 1961–1968 CrossRef CAS PubMed.
- W. Wu, D. Huang, X. Yi and J. Zhao, Dyes Pigm., 2013, 96, 220–231 CrossRef CAS.
- P.-H. Lanoë, J.-L. Fillaut, L. Toupet, J. A. G. Williams, H. L. Bozec and V. Guerchais, Chem. Commun., 2008, 4333–4335 RSC.
- L.-K. Zhang, L.-B. Xing, B. Chen, Q.-Z. Yang, Q.-X. Tong, L.-Z. Wu and C.-H. Tung, Dalton Trans., 2013, 42, 4244–4247 RSC.
Footnote |
| † A. Haque and L. Xu contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |