
Organic and hybrid resistive switching materials and devices

Abstract
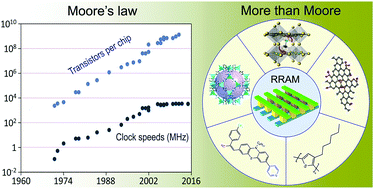
The explosive increase in digital communications in the Big Data and internet of Things era spurs the development of universal memory that can run at high speed with high-density and nonvolatile storage capabilities, as well as demonstrating superior mechanical flexibility for wearable applications. Among various candidates for the next-generation information storage technology, resistive switching memories distinguish themselves with low power consumption, excellent downscaling potential, easy 3D stacking, and high CMOS compatibility, fulfilling key requirements for high-performance data storage. Employing organic and hybrid switching media in addition allows light weight and flexible integration of molecules with tunable device performance via molecular design-cum-synthesis strategy. In this review, we present a timely and comprehensive review of the recent advances in organic and hybrid resistive switching materials and devices, with particular attention on their design principles for electronic property tuning and flexible device performance. The current challenges posed with development of organic and hybrid resistive switching materials and flexible memory devices, together with their future perspectives, are also discussed.
- This article is part of the themed collection: Materials chemistry in flexible electronics
 Please wait while we load your content...
Please wait while we load your content...

