
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydrogel bioelectronics
Hyunwoo
Yuk†
 a,
Baoyang
Lu†
ab and
Xuanhe
Zhao
*ac
a,
Baoyang
Lu†
ab and
Xuanhe
Zhao
*ac
aDepartment of Mechanical Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139, USA. E-mail: zhaox@mit.edu
bSchool of Pharmacy, Jiangxi Science and Technology Normal University, Nanchang, 330013, China
cDepartment of Civil and Environmental Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139, USA
First published on 26th November 2018
Abstract
Bioelectronic interfacing with the human body including electrical stimulation and recording of neural activities is the basis of the rapidly growing field of neural science and engineering, diagnostics, therapy, and wearable and implantable devices. Owing to intrinsic dissimilarities between soft, wet, and living biological tissues and rigid, dry, and synthetic electronic systems, the development of more compatible, effective, and stable interfaces between these two different realms has been one of the most daunting challenges in science and technology. Recently, hydrogels have emerged as a promising material candidate for the next-generation bioelectronic interfaces, due to their similarities to biological tissues and versatility in electrical, mechanical, and biofunctional engineering. In this review, we discuss (i) the fundamental mechanisms of tissue–electrode interactions, (ii) hydrogels’ unique advantages in bioelectrical interfacing with the human body, (iii) the recent progress in hydrogel developments for bioelectronics, and (iv) rational guidelines for the design of future hydrogel bioelectronics. Advances in hydrogel bioelectronics will usher unprecedented opportunities toward ever-close integration of biology and electronics, potentially blurring the boundary between humans and machines.
 Hyunwoo Yuk | Hyunwoo Yuk is currently a PhD student in Mechanical Engineering at Massachusetts Institute of Technology. He received his MS from Massachusetts Institute of Technology in 2016 and BS from Korea Advanced Institute of Science and Technology in 2014, both in Mechanical Engineering. His research focuses on the broad topics of mechanics and materials science and engineering in human–machine interfaces including robust adhesion of soft materials, hydrogel bioelectronics, and 3D printing of advanced soft materials. He is a recipient of the Materials Research Society Graduate Student Award. |
 Baoyang Lu | Baoyang Lu is an associate professor at Jiangxi Science and Technology Normal University in China. He received his PhD in Materials Science and Engineering from Shandong University in 2015. He has been working as a postdoctoral fellow in Mechanical Engineering at Massachusetts Institute of Technology since 2016. Dr Lu's current research interests include the design and synthesis of novel conjugated polymer-based molecular systems, and fabrication and characterization of organic optoelectronic devices. |
 Xuanhe Zhao | Xuanhe Zhao is an associate professor in Mechanical Engineering at Massachusetts Institute of Technology and the founder of Soft Active Materials Laboratory (http://zhao.mit.edu/). His research group is advancing the science and technology to seamlessly interface and merge humans and machines, particularly through inventing soft materials and machines. Dr Zhao is the recipient of the early career award or young investigator award from National Science Foundation, Office of Naval Research, Society of Engineering Science, Adhesion Society, and American Vacuum Society. He held the Hunt Faculty Scholar at Duke and d'Arbeloff Career Development Chair and Robert N. Noyce Career Development Professor at Massachusetts Institute of Technology. |
1 Introduction
Electronic activities in nervous systems are the principal constituents of our daily lives, ranging from routine regulation of muscular movements to complex intelligence such as memory and reasoning. Since the discovery of bioelectricity by Luigi Galvani's landmark experiment, a better understanding of electronic communications between biology and electronics, so-called bioelectronic interfacing or bioelectronics, has been one of the grand but ongoing challenges in science and technology. The majority of existing and emerging bioelectronic interfaces involve various forms of electrodes interacting with biological tissues.1,2 Electrodes with ever-refined performances and control have been available in a wide range of form factors and sizes, greatly aided by innovations and advances in modern electronics.2–4 Based on these electrodes, numerous wearable and implantable electronic devices have recently emerged to collect and deliver various bioelectronic signals in different parts of the human body as diverse as skin,5,6 brain,7 spinal cord,8 and heart.9 Particularly, various wearable epidermal bioelectronic devices have been commercialized and routinely used for diverse clinical purposes.10 Miniaturized implantable devices have driven breakthroughs in treatments for neurological disorders and damage including deep brain stimulation probes for Parkinson's disease11–13 and essential tremors,14 neural interfaces for robotic prostheses,15–18 flexible electrode arrays for heart failures,9,19,20 and closed-loop electrode arrays for spinal cord injuries.21,22Despite remarkable advances in the recent few decades, the intrinsic differences between biological tissues and man-made electronics pose immense challenges in materials, design, and manufacturing of the next generation bioelectronics. For instance, the human body consists of a broad range of soft and high water-containing tissues and organs. In contrast, almost all of commercially available and the majority of lab-level bioelectronic devices rely on rigid and dry electronic components such as silicon and metals.23–26 These stark disparities between the two realms have introduced significant difficulties towards seamless interfacing between biology and electronics.27–30
The central and peripheral nervous systems ceaselessly generate and receive electrical and biochemical signals through complex networks of neuronal cells. Unlike conventional electronics in which electrons serve as carriers of information, bioelectronic activities in the biological systems are essentially ionic through electrolytic media.10,31–33 Ever-evolving microenvironments due to diffusive and convective exchange of mobile ionic and biochemical species in water-rich tissues further highlight the distinctive nature of the biological systems over electronic systems. Together with mechanical and compositional dissimilarities, these inherent mismatches between biology and electronics signify the high hurdles to bring the two realms closer.
Hydrogels, crosslinked polymer networks infiltrated with water, have been extensively studied in tissue engineering and biomedicine due to their resemblance to biological tissues (Fig. 1).34,35 The soft and flexible nature of hydrogels allows minimization of mechanical mismatch with biological tissues, and the high water contents of hydrogels provide wet and ion-rich physiological environments. Moreover, the remarkable flexibility in the design of their electrical, mechanical, and biological properties renders hydrogels a unique bridging material to biological world.36 Powered by these exceptional advantages, hydrogels have recently attracted growing attention in bioelectronics, as part of continued endeavor toward seamless interfacing between biology and electronics.
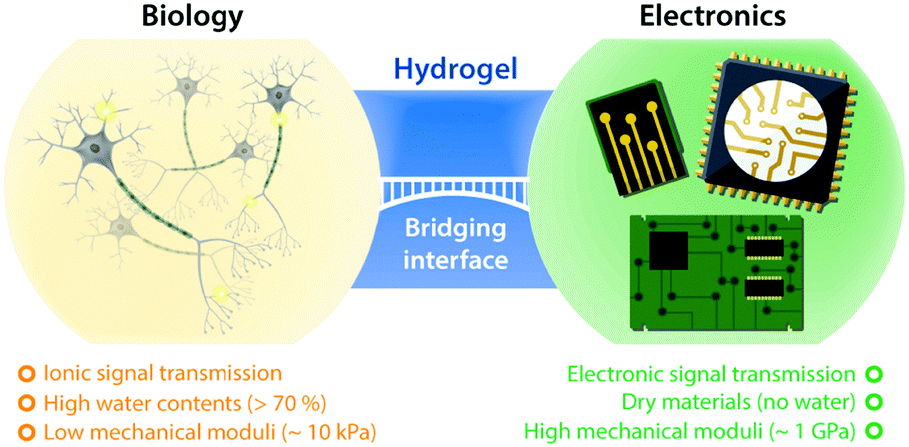 | ||
| Fig. 1 Hydrogels at the interface between biology and electronics. Hydrogels possess a unique set of properties to bridge the gap between biology and electronics, providing promising opportunities for bioelectronics applications. | ||
While a growing volume of bioelectronic devices and applications have adopted hydrogels to achieve improved interfacing with the human body, many of such approaches are still limited in an Edisonian manner. Simple additive combination of hydrogels in existing devices often results in non-optimal performance and a significant increase in the burden of development processes. Future innovations in hydrogel bioelectronics require us to harness hydrogels’ unique advantages based on the fundamental understanding of tissue–electrode interactions, in order to rationally guide the engineering of various properties and design parameters. Along with fundamental understanding, advances in hydrogel bioelectronics progress hand-in-hand with breakthroughs in material developments, synergistically benefiting each other.
This review is aimed to provide a set of rational guidelines for the design of future hydrogels in bioelectronic applications based on understanding the fundamental mechanisms of bioelectronic communications between biological and electronic systems. We start with a brief overview on the fundamental mechanisms of tissue–electrode interactions to provide a rational foundation that justifies hydrogels’ unique benefits to bioelectronics. Thereafter, the recent advances in hydrogel bioelectronics are categorized into four classes: (i) hydrogel coatings and encapsulations, (ii) ionically conductive hydrogels, (iii) conductive nanocomposite hydrogels, and (iv) conducting polymer hydrogels, followed by discussions on important issues in hydrogel-device interfaces. Last but not least, we propose a number of directions and guidelines for the design of next-generation hydrogel bioelectronic materials and devices, and conclude with a perspective discussion on the remaining opportunities and challenges. By highlighting hydrogels’ importance in bioelectronics spanning from tissue–electrode interactions to advances in materials, we hope to bring new insights into seamless merging between biology and electronics.
2 Tissue–electrode interfaces
Biological tissues are typically considered as volume conductors with a moderate level of conductivity (e.g., electrical conductivity σ = 0.1 to 1 S m−1) in electrical models, owing to mobile ionic species dissolved in the media.10,31,37,38 As electrons cannot serve as charge carriers in electrolytic tissue media, electrical communications between neuronal cells mostly rely on ionic fluxes.1,39,40 To establish electrical communications between neural tissues and external electronics, electrodes have been the most commonly used terminals, through which signals and information can transmit from cells to electronics and vice versa. However, unlike biological tissues, almost all conventional electronic systems depend on electronically conductive materials such as metals, in which free electrons act as mobile charge carriers.1,26,33,41,42 This characteristic difference introduces a unique interface between tissue and electrode, at which ionically and electronically carried signals are exchanged with each other.Bioelectronic activities in tissue–electrode interfaces involve various ionic and electronic interactions across a wide range of length scales (Fig. 2A). Starting from the tissue side of the picture, spikes of ionic currents or action potentials (APs) in excitable cell membranes provide the most basic element of bioelectronic activities at the nm length scale.33,39,43 The transmembrane potential of an individual neuronal cell maintains a negative value (∼−60 to −75 mV) in the resting state due to the imbalance of ions (typically an excess of intracellular K+ and extracellular Na+). The transmembrane potential fluctuates from the resting potential by excitatory (depolarization to more positive potential) or inhibitory (hyperpolarization to more negative potential) inputs from other neurons or electrodes.10,39 When the net excitatory inputs provide enough depolarization beyond the threshold (mostly −50 to −55 mV), voltage-gated Na+ channels open allowing a rapid influx of Na+ ions into the cells, which generates a spike of depolarization. Upon reaching a potential around +30 to +40 mV, the transmembrane potential repolarizes to the resting potential via slow efflux of K+ ions.39 These processes create impulsive local ionic currents and potentials (or action potentials) across the cellular membrane as well as the surrounding extracellular space (Table 1).
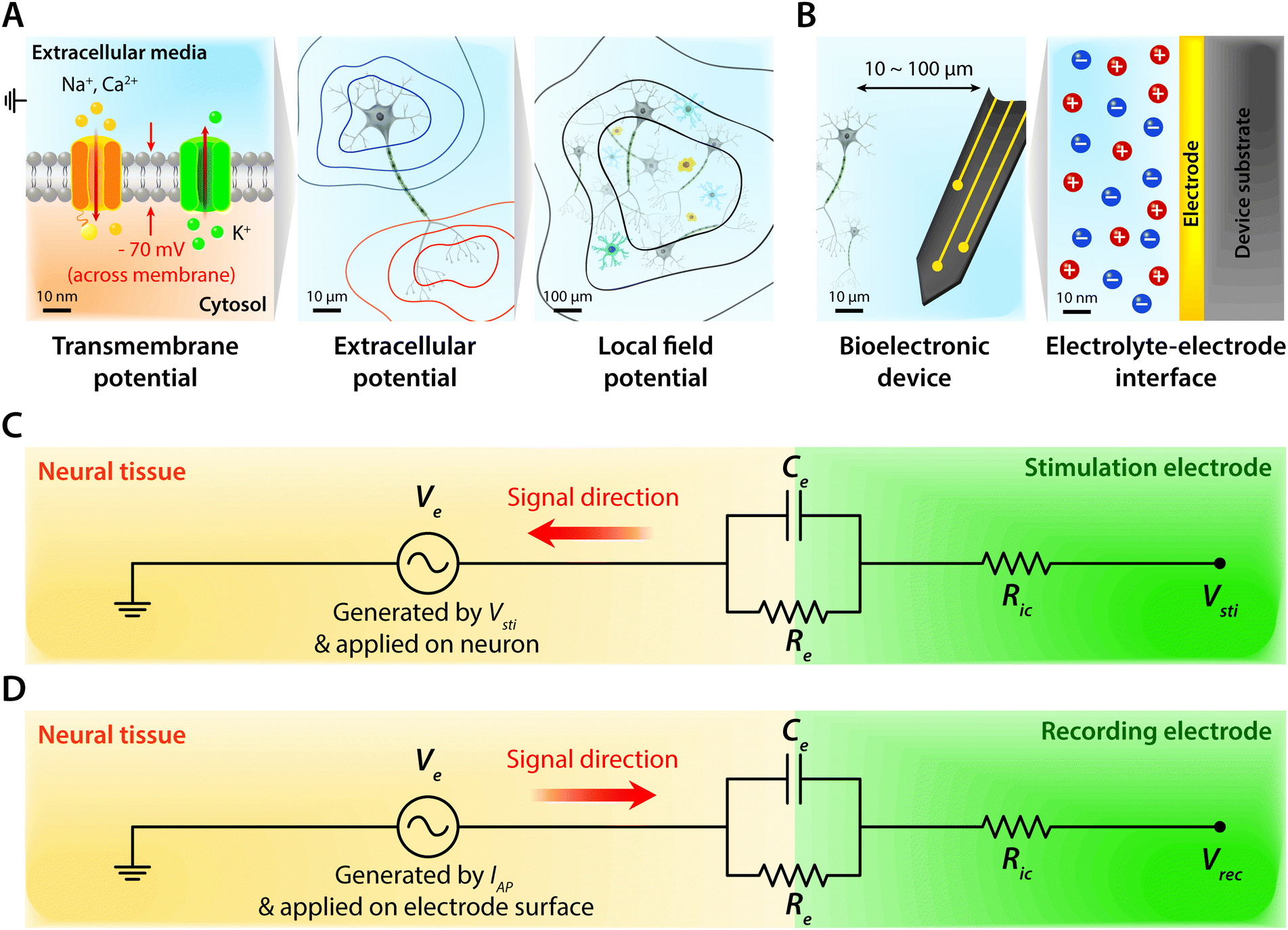 | ||
| Fig. 2 Schematic overview of tissue–electrode interfaces. (A) On the tissue side, the transmembrane potential comprises the most basic bioelectronic activity at the nm scale. Distribution of ionic species in the extracellular media and cytosol generates the transmembrane potential which is tightly regulated by ion channels. At the μm scale, electrically active neural cells generate an extracellular potential. The red and blue contours correspond to equipotential lines for positive and negative values for local field potential amplitude, respectively. At the larger scale, collective activities of electrically active neuronal cells generate a local field potential which is represented as black lines. (B) Toward the electrode side, bioelectronic devices are typically introduced within 100 μm of target cells, and the exchange between ionic signals in biological systems and electronic signals in electronic systems happens at the electrolyte–electrode interface at the nm scale. (C) Equivalent circuit model of tissue–electrode interfaces for bioelectronic stimulation. Vsti represents the input for bioelectronic stimulation with the interconnect resistance Ric. Ve represents the electric potential within electrolytic media generated by Vsti applied on the outer membrane of the targeted neurons. (D) Equivalent circuit model of tissue–electrode interfaces for bioelectronic recording. At the interface between biology and electronics, the electrolyte–electrode interface is considered as a parallel circuit of the EDL capacitance, Ce, and the leakage resistance, Re. Vrec represents the output for bioelectronic recording with the interconnect resistance Ric. Ve represents the electric potential within electrolytic media generated by bioelectronic activities of neurons (i.e., ionic currents by action potentials, IAP) applied on the electrode surface. For both bioelectronic stimulation and recording, the electrolyte–electrode interface is considered as a parallel circuit of the EDL capacitance, Ce, and the leakage resistance, Re. | ||
| Terminology | Definition |
|---|---|
| Stimulation | Induced firings of excitable neural cells by electrical inputs from the external electronic terminal |
| Recording | Electric potential readings by the external electronic terminal generated by neural activities |
| Volume conductor | Three-dimensional conductor with isotropic and homogeneous electrical property |
| Ohmic | Electrical current flow in a purely resistive manner. The extracellular medium is primarily Ohmic in the 1–10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 kHz frequency range 000 kHz frequency range |
| Transmembrane potential | Electric potential between the extracellular space and the intracellular space in neural cells |
| Local field potential | Electric potential generated by the summed ionic currents from multiple nearby neurons within a small volume of neural tissue |
| Action potential (AP) | Impulsive change of electric potential along the membrane of excitable neuronal cells |
| Electrochemical reaction | Chemical reactions at the electrolyte–electrode interface by the applied electric potential |
| Electrical double layer (EDL) | Accumulation of charged ions around the surface of the electrode within the electrolytic medium under the applied potential |
| Current source density | Rate of current flow in a given direction through the unit surface area |
| Charge injection capacity | Amount of charge that the electrode can inject per unit area without causing irreversible electrochemical reactions or tissue damage |
| Signal-to-noise ratio (SNR) | Ratio between the amplitude of recorded signal and the noise from the recording |
Extracellular potentials decay rapidly away from the source (e.g., soma or axon of neurons) at cellular length scales (e.g., 10 to 100 μm).10,33 Superposition of all concurrent activities of neurons generates tissue-scale (e.g., mm to cm) oscillations and rhythms of electrical potentials, which are called local field potentials10,31,38,44 (Fig. 2A). Overall, the tissue side of bioelectronic activities relies on spatial and temporal changes of ionic fluxes across different length scales.
Moving toward the electrode side, interactions between neural tissues and electrodes are less specific in a larger distance (e.g., mm to cm). Local field potentials convey information from collective activities of numerous cells and the contribution from an individual neuron is mostly indistinguishable.10,41,45 Closer to a cellular distance (e.g., <100 μm), extracellular potentials from each neuron present without significant decay, and the electrodes begin to be able to sense or actuate individual neuron.33,43 At an even smaller length scale (e.g., 1 to 100 nm), ionic tissue media and electrodes form an electrolyte–electrode interface at which exchange between ionic and electronic signals takes place (Fig. 2B).
Electrodes communicate with biological tissues in a bidirectional manner. In one way, electrodes stimulate excitable cells (e.g., neurons) by delivering electrical inputs toward the tissues. In another way, electrical signals from electrically active cells propagate toward and are recorded by the electrodes. During this bidirectional communication, electrical signals are transmitted via ionic currents and electric potentials in the electrolytic tissue media and electronic currents and electric potentials in the conducting electrodes.1,32,45,46 In addition, the potential difference at the electrolyte–electrode interface is equilibrated by the electron flow in the electrodes, ion flow in the tissue media, and possible reactions of the electrolyte and electrodes.42 Therefore, in stimulation applications, the applied potential on the electrode drives ionic current injected toward the tissue media and generates the corresponding electric potential on the outer membrane of the targeted neurons, by which the neurons are stimulated31,39,47,48 (Fig. 3). Conversely, in recording applications, the ionic flux by action potentials of neurons drives electron flows in the electrode by building up the electrolyte–electrode potential difference (Fig. 4).
 | ||
| Fig. 3 Schematic overview of bioelectronic stimulation. (A) Stimulation of excitable cells is conducted via charge injection from the electrode toward the neural tissues to depolarize the cellular membrane of the targeted neurons, which results in firing of action potentials. (B) Faradaic charge injection mechanism relies on surface-confined electrochemical reactions of the electrode. (C) Capacitive charge injection mechanism relies on charging/discharging of the electric double layer (EDL) by accumulation of ions on the electrode without electrochemical reactions. | ||
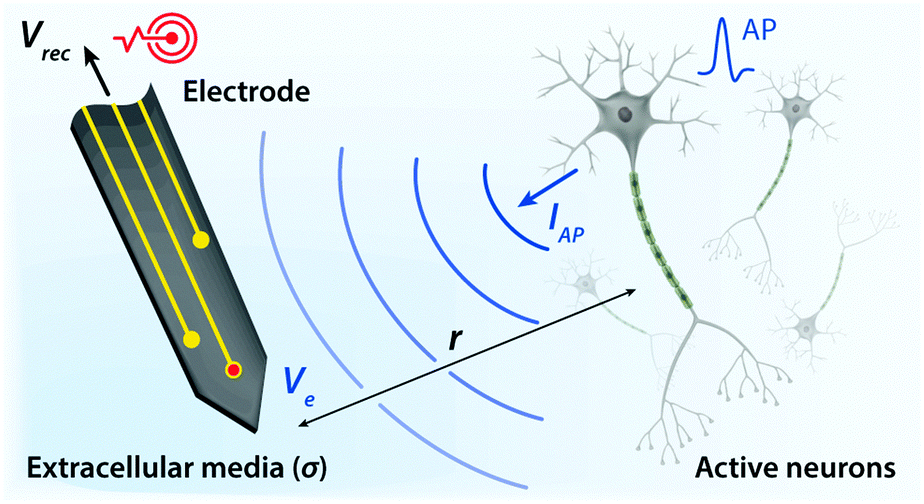 | ||
| Fig. 4 Schematic overview of bioelectronic recording. Upon firing of APs, electrically active neurons inject charges into extracellular media with the corresponding extracellular potential and local field potential. The resultant potential Ve within electrolytic tissue media applied on the electrolyte–electrode interface is transmitted via electronic interconnects and recorded as the output signal, Vrec. | ||
The tissue–electrode interfaces are often depicted by using the equivalent circuit models in order to more quantitatively discuss complex bioelectronic phenomena. Fig. 2C and D show the simplest equivalent circuit models for bioelectronic stimulation and recording, respectively. The Vsti and Vrec are the electric potentials of the external electronics, which serve as an input for stimulation applications (e.g., stimulation waveforms) and an output for recording applications (e.g., neural activity recordings), respectively.41 The electrolyte–electrode interface is depicted as a parallel circuit of the leakage resistance Re and the electrical double layer (EDL) capacitance Ce in the simplest form.1,41,43,49 The interconnect resistance Ric captures the resistance of the higher order electronic circuitry connected to the electrode. In stimulation applications, electrical signals flow from the electrode toward the tissues and generate the electric potential Ve in electrolytic media and applied on the outer membrane of the targeted neurons (Fig. 2C). On the other hand, in recording applications, electrical signals are originated by the bioelectronic activities of neural cells (i.e., ionic currents by action potentials, IAP) and generate the electric potential Ve in electrolytic media and applied on the electrode surface (Fig. 2D). In addition to the electrical interactions (e.g., stimulations and recordings), there also exist biomechanical interactions between the tissues and electrodes. In the following sections, we discuss various tissue–electrode interactions in hydrogel bioelectronics (Table 2).
| Parameter | Definition |
|---|---|
| V sti | Electric potential of the external electronics as an input for bioelectronic stimulation |
| V rec | Electric potential of the external electronics as an output for bioelectronic recording |
| R ic | Interconnect resistance for higher order electronic circuitry connected to the electrode |
| C e | Capacitance of the electrode that charges/discharges by capacitive charge injection |
| R e | Leakage resistance of the electrode |
| V e (stimulation) | Electric potential within electrolytic media generated by Vsti on the outer membrane of the targeted neurons |
| V e (recording) | Electric potential within electrolytic media generated by IAP at the electrolyte–electrode interface |
| σ | Conductivity of extracellular media |
| r | Distance between the target cells and the electrode surface |
| I in | Ionic current injected by the stimulation electrode toward the tissues |
| I AP | Ionic current injected by action potentials (APs) toward the recording electrode |
| I e | Electronic current flow induced by Ve toward the recording electrode in bioelectronic recording |
| τ EDL | Characteristic time for the EDL capacitor charging at the electrolyte–electrode interface |
2.1 Bioelectronic stimulation
Bioelectronic stimulation finds critical importance as a versatile tool for a wide range of neuroscientific studies and clinical treatments.11,12,16,22,50 Electrical stimulation of the biological systems can largely be divided into two major categories based on the invasiveness of tissue–electrode interactions: invasive and non-invasive stimulations. Implanted stimulation electrodes provide more invasive but direct interfacing with the tissues. Implanted electrodes in the proximity of target neurons have been adopted in sensory stimulations (e.g., cochlear implants and retinal implants),51–53 neuromuscular stimulations (e.g., control of limbs for rehabilitation), and treatments for neurodegenerative disorders (e.g., deep brain stimulations for Parkinson's disease).54 Epidermal stimulation electrodes offer non-invasive but less specific interfacing with the tissues. One classical example of epidermal bioelectronic stimulation is transcutaneous electrical nerve stimulation (TENS), which has been widely adopted in clinical settings for pain relief.55–58 Despite the broadness in forms and applications, all electrode-based bioelectronic stimulations share common physical principles.Electrodes can stimulate neurons by injecting ionic currents in electrolytic tissue media and generating electric potential on the outer membrane of the targeted neurons. While bioelectronic stimulation has a broad range of applications from targeted stimulation of neurons around the implanted electrodes to less specific stimulation via large area epidermal electrodes, all electrical stimulations rely on triggered firing of APs by the membrane depolarization of excitable cells.39,59 Depolarization of the transmembrane potential is induced by the local field potential Ve applied on the outer membrane of the targeted neuron (Fig. 3A). In principle, Ve at a certain spatial point within the neural tissue is determined by electric fields in the surrounding electrolytic media.10,48,60 Since the physical origin of the volume-conducting electric field is ionic potentials, Ve correlates with the ionic current source density J (unit in A m−2) at a point of stimulating electrode by Maxwell's equations of electromagnetism.10,31,32 Under standard electro-quasi-static conditions (i.e., the magnetic contributions are neglected), Maxwell's equations provide the simplified relationship between Ve and J as J = −∇(σVe), where σ is the conductivity of the tissue media (units in S m−1).38 For the simplest case of homogeneous and isotropic Ohmic volume conducting media (i.e., ionic current flows in a purely resistive manner),41,43Ve is dictated by Laplace's equation, ∇2Ve = 0. Solving this equation for a single point current source yields the governing equation for Ve as
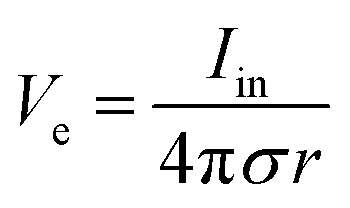 | (1) |
As Ve is proportional to Iin in eqn (1), the key criterion in bioelectronic stimulation is the ability to inject more ionic current at the electrolyte–electrode interface without inducing adverse outcomes such as electrode degradation, water hydrolysis, and tissue damage.1,43 Two major mechanisms are widely adopted to introduce the ionic current injected at the electrolyte–electrode interfaces: (i) faradaic charge injection based on surface-confined electrochemical reactions (i.e., oxidation and reduction) and (ii) capacitive charge injection based on charging/discharging of the electrolyte–electrode interface1,33,42,61 (Fig. 3B and C). Since the faradaic mechanism inevitably involves oxidation and reduction of the electrode to allow direct electron transfer across the electrolyte–electrode interface, the capacitive mechanism is generally more desired in bioelectronic stimulations.1 For more detailed discussion on various charge injection mechanisms and their characteristics, the readers are guided to several comprehensive reviews in the literature.1,33,42
Capacitive charge injection can rely on either areal capacitance (i.e., EDL formation on the surface of the electrode) or volumetric capacitance (i.e., EDL formation within the micro/nano structure of the electrode) depending on the electrode material.62–64 When Vsti builds up a potential difference at the electrolyte–electrode interface, surface aggregation of oppositely charged ions rapidly forms a compact layer (or Stern layer) with the corresponding potential drop. The residual potential difference further creates a diffusive layer of ions (or Gouy–Chapman layer) over a short distance (typically a few nm), beyond which the potential difference is equilibrated to the bulk electrolyte media65 (Fig. 3C). Essentially, the formation of EDL is analogous to the charging of a capacitor with the same capacitance Ce as in the equivalent circuit model (Fig. 2C). The electrodes can inject ionic currents Iin until the EDL is fully charged with the characteristic time as
| τEDL = ReCe | (2) |
Following the simplest equivalent circuit model in Fig. 2C, we can further express the governing equation for Iin as
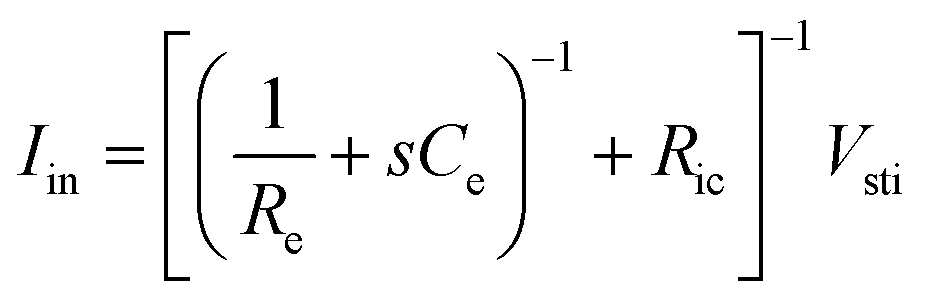 | (3) |
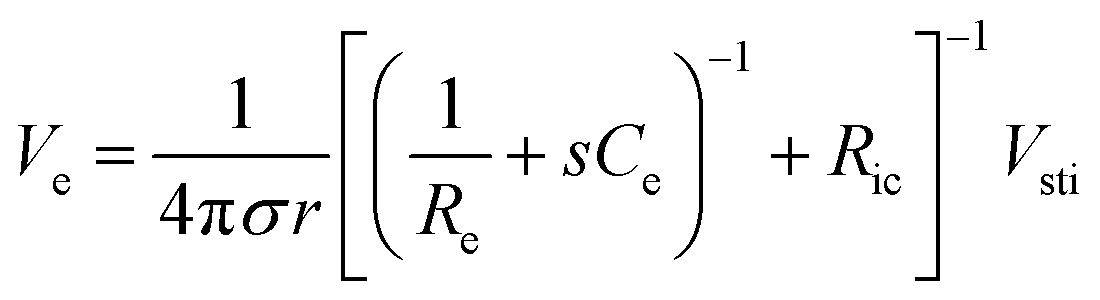 | (4) |
2.2 Bioelectronic recording
In addition to bioelectronic stimulation, the ability to record neural activities constitutes an essential component for bidirectional communication between biology and electronics.1,41,43 Similar to stimulation, recording of bioelectronic signals is generally realized by both invasive and non-invasive tissue–electrode interfaces. Epidermal recording electrodes are the most widespread form of bioelectronic recording terminals in clinical applications, largely because of their non-invasive nature.10 For example, electroencephalography (EEG) via large area epidermal electrodes is one of the oldest and most popular methods for bioelectronic recording of brain activities.67 Moreover, similar epidermal recording electrodes are also adopted for the recording of cardio-muscular activities such as electrocardiography (ECG) and electromyography (EMG), which are routinely conducted in various clinical settings for diagnostic purposes. With the rapidly growing demand for more specific interrogations of single neuron activities, a wide range of recording electrodes implanted in the vicinity of targeted neural tissues (typically <100 μm distance) have been developed.2,24,43 Such a short distance between recording electrodes and neurons allows acquisition of neural signals in greater details, though with a significantly increased invasiveness in tissue–electrode interactions.28,30,68The physical mechanisms of bioelectronic stimulation and recording of biological tissues are alike but proceed in a reverse manner.1,43 In bioelectronic stimulation, the stimulation electrode induces ionic currents toward electrolytic tissue media to elicit membrane depolarization of nearby neurons by building a potential Ve on the outer membrane of the targeted neurons41 (Fig. 2C and 3A). In contrast, in bioelectronic recording, Ve represents the potential generated by electrically active neurons (i.e., extracellular potential and local field potential by APs of an individual neuron) applied on the electrolyte side of the electrolyte–electrode interface45 (Fig. 2D and 4). Hence, the governing equation for Ve in bioelectronic recording can be expressed as
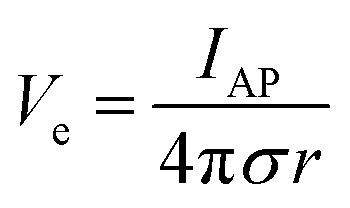 | (5) |
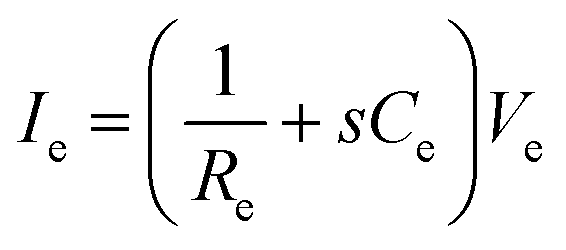 | (6) |
| Vrec = IeRic | (7) |
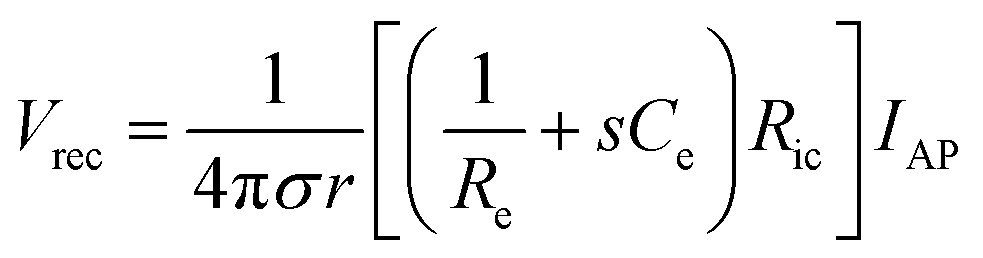 | (8) |
From eqn (8), the amplitude of Vrec is proportional to Ce and Ric and inversely proportional to Re and r. One crucial metric to evaluate the quality of bioelectronic recording is the signal-to-noise ratio (SNR), defined as Vrec/Vnoise, whose amplitude determines the sensitivity of the electronic signals.10,41 To guarantee high quality recording, SNR should be kept above 1 (i.e., the amplitude of the signal should be larger than noise). Noise in bioelectronic recording is generally unavoidable while its degree and source vary widely including biological noise (e.g., APs from distant cells, ionic activities in tissues) and device noise (e.g., thermal noise from electrodes, amplifier noise).41 Particularly, the device noise from the interconnect resistance, Ric, can be described by the Johnson noise, Vnoise2 = 4kBTRicΔf, where kB is the Boltzmann constant, T is the temperature, and Δf is the bandwidth.10,33 We can define the threshold interconnect resistance, RSNR, as
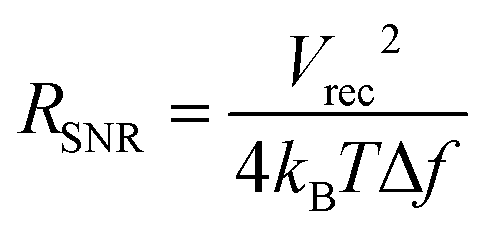 | (9) |
2.3 Biomechanical interactions
On top of electrical interactions, tissue–electrode interfaces also involve a variety of biomechanical interactions. Electrodes can biomechanically interact with tissues in a less invasive manner such as epidermal attachments as well as in invasive ways such as implantation within tissues. Less invasive tissue–electrode interfaces invite relatively simple biomechanical interactions. For example, epidermal electrodes experience dynamic mechanical deformations (e.g., bending, stretching, compression) in conformal contact with soft and curvilinear skin tissues.23 Invasive electrodes typically bring more complicated biomechanical interactions due to their direct disruption of tissue microenvironments.68,69 Implantation of the electrodes into the target tissues introduces acute damage (e.g., stab wounds and microvasculature disruptions), which immediately elicits activation of nearby cells (e.g., microglia and macrophage) and their attachment on the electrode surface.2,30,43 These acute injuries and activated microglia shortly initiate the foreign body responses including the microglial secretion of proinflammatory cytokines and the influx of blood-serum proteins by blood–brain-barrier breach.30 In a longer implantation period, a dense fibrous encapsulation (or a scar tissue) forms around the implanted electrode as a result of progressive chronic biomechanical interactions.28–30 For more detailed discussions on acute and chronic inflammatory responses, the readers are guided to several comprehensive reviews in the literature.26,28–30Adverse biomechanical interactions can significantly compromise the reliability and performance of bioelectronic applications. One critical origin of such adverse biomechanical interactions is the mismatch of physical and mechanical properties between man-made electrodes and human tissues that interface with the electrodes (i.e., skin, muscle, heart, spinal cord and brain).23,24,43,70 Biological tissues and cells are soft (e.g., E < 100 kPa) and with high water contents (e.g., >70%).70 Moreover, most tissues continuously experience highly dynamic mechanical stimuli. For example, skin, muscle, spinal cord, and peripheral nerves can experience as much as 30% tensile strain and displacement during routine postural movements.24 Heart and vascular tissues experience continuous cyclic mechanical deformations during cardiovascular activities. Brain tissues and neurons undergo μm-scale micromotions due to respiratory and cardiovascular cycles in the brain.2,24 In contrast, most conventional electrode materials such as metals and silicon are rigid (e.g., E > 1 GPa) and static (e.g., elastic strain limit <5%).23
Dissimilar physical and mechanical properties between implanted electrodes and human tissues introduce two major consequences in bioelectronics: (i) the increase in interfacial impedance due to the scar tissue formation and (ii) the reduction in stimulation/recording efficacy due to the increasing tissue–electrode distance43 (Fig. 5). Electrodes with much higher elastic moduli and lower flexibility than biological tissues are prone to induce more injuries during the initial implantation.24,69 Furthermore, significant mismatch in mechanical properties can provoke a higher degree of tissue scarring in long-term via elevated foreign body responses.28,30 A dense scar tissue around the electrode can substantially increase the interfacial impedance, which is given by the equivalent circuit model as Zinterface = (1/Re + sCe)−1, by increasing the resistance (higher Re) and/or degrading the charge injection capacity (lower Ce) of the electrode.2,41 Hence, a higher interfacial impedance is detrimental to both bioelectronic stimulation and recording based on eqn (4) and (8). Formation of the dense scar encapsulation further harms bioelectronic performance by increasing the distance between the target neural tissue and the electrode (i.e., higher r in eqn (4) and (8)) (Fig. 5).
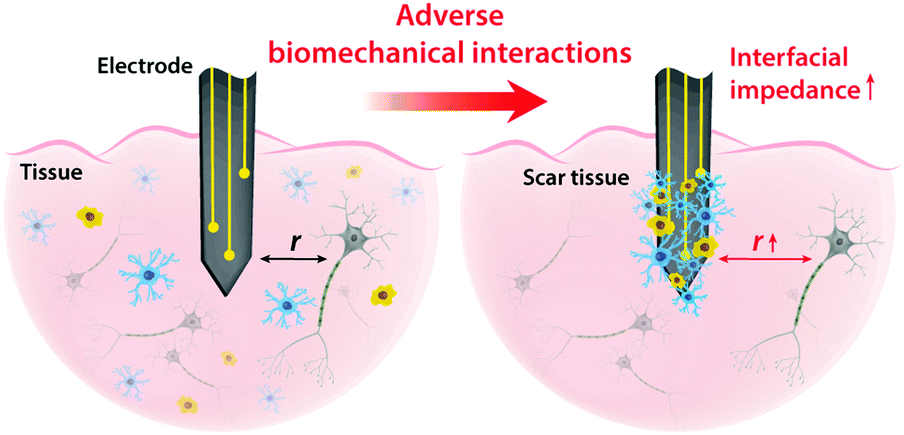 | ||
| Fig. 5 Mismatch between biological tissues and electrode can elicit various adverse biomechanical interactions including scar tissue formation. Such adverse biomechanical interactions can increase interfacial impedance and distance between the electrode and the target cells, which significantly hampers the performance and efficacy of bioelectronic stimulation and recording. | ||
Mechanical mismatch adversely affects less invasive epidermal bioelectronic applications as well. The stretchable, non-planar, and highly dynamic nature of human skin provides a substantially challenging environment for mechanically dissimilar electrodes to keep conformal contact over time.23,24 Differences in stretchability and/or bending stiffness at the tissue–electrode interface can generate mechanical failures such as delamination, which impede bioelectronic performance.
2.4 Design guideline for tissue–electrode interfaces
Considering the complexity and multitude of tissue–electrode interfaces, a rationally guided approach is crucial for successful development of next-generation bioelectronic materials and devices. As summarized in Table 3, fundamental tissue–electrode interactions discussed in Sections 2.1–2.3 provide a set of requirements and corresponding desired electrode features for bioelectronic stimulation and recording. High Ce values as well as low Re and E are desirable for both bioelectronic stimulation and recording, which provides the first-order design guideline for future developments. In the following section, we will use these design guidelines to discuss hydrogels’ unique advantages in bioelectronic interfaces and their recent advances.| Tissue–electrode interaction | Requirements | Desired electrode features |
|---|---|---|
| Bioelectronic stimulation | High Ve | Low Re |
| High Ce | ||
| Low Ric | ||
| High Vsti (Vsti < Vreac) | ||
| Low r | ||
| Low mismatch | Low E | |
| Bioelectronic recording | High SNR | Low Re |
| High Ce | ||
| High Ric (Ric < RSNR) | ||
| Low r | ||
| Low mismatch | Low E | |
3 Hydrogels as next-generation bioelectronic interfaces
Early bioelectronic interfaces mostly relied on relatively simple electrodes such as insulated metallic microwires or needles.24,43 The explosive technological developments in the electronic industry have subsequently introduced much advanced neural probes based on microfabricated silicon and metals. These commercially available microelectrodes such as the Michigan-type probes,71 Utah arrays,72 and deep brain stimulation probes12 are still considered as state-of-the-art tools for neuroscientific studies and clinical treatments. In spite of their great success, these commercially available bioelectronic interfaces have called the need for further advances due to their sub-optimal performance (e.g., insufficient charge injection capacity and high interfacial impedance) and numerous deleterious outcomes from substantial biomechanical mismatch to neural tissues (e.g., device encapsulation by scar tissue and chronic degradation of performance).24,43,68 The majority of conventional electrode materials (e.g., silicon, gold, platinum, titanium nitride, iridium, tungsten, and tin) exhibit very high Young's moduli over 1 GPa, which are several orders of magnitude higher than that of neural tissues.2,23,24,43 In the last decade, a number of novel materials and laboratory-level devices have been developed to minimize the biomechanical dissimilarities between electronics and biology.3,69–71 For example, polymeric materials such as plastics (e.g., polycarbonate, polyimide, and parylene C) and elastomers (e.g., epoxy, PDMS, and polyurethane) have been adopted to reduce the modulus gap.23 However, their Young's moduli (typically 1 MPa to 1 GPa) are still much higher than those of neural tissues (E ∼ 10 kPa) and not sufficient to provide truly mechanically matching interfaces.With close similarity to biological tissues, hydrogels have attracted growing interest in the field of bioelectronics.23,26 Hydrogels’ unique tissue-like mechanical property and biocompatible nature endow a promising route to minimize biomechanical mismatch at tissue–electrode interfaces34–36 (Fig. 6). Unlike other dry electrode materials, the water- and ion-rich hydrogels have the potential to offer unconventional but improved stimulation/recording performance via integrative use of both electronic and ionic activities.62 While hydrogels consist of a broad range of material designs and chemistries, hydrogels in bioelectronics find four major embodiments: (i) hydrogel coatings and encapsulations, (ii) ionically conductive hydrogels, (iii) conductive nanocomposite hydrogels, and (iv) conducting polymer hydrogels (Table 4).
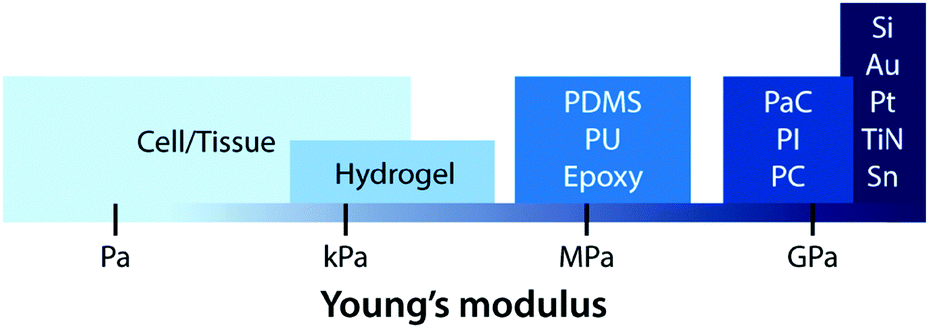 | ||
| Fig. 6 Young's moduli of cell/tissue and common electrode materials. Conventional materials for electrodes are much stiffer than cell/tissue. Hydrogels exhibit a similar level of Young's moduli, potentially minimizing the mechanical mismatch with biological tissues. | ||
| Type of interface | Electrical property | Mechanical property | Ref. | |||
|---|---|---|---|---|---|---|
| Impedance (Ω) | Charge injection capacity (mC cm−2) | Conductivity (S m−1) | Young's modulus (kPa) | |||
| Hydrogel coatings and encapsulations | 103–107 | 48–55 | 10−3–10−2 | 10−1–102 | 43 and 80 | |
| Ionically conductive hydrogels | 102–105 | N/A | 10−1–101 | 100–102 | 95 and 96 | |
| Conductive nanocomposite hydrogels | Metal | 101–103 | N/A | 10−1–101 | 100–101 | 102 and 104 |
| CNT | 103–107 | 1–10 | 10−3–101 | 101–102 | 100 and 109 | |
| Graphene | 103–105 | 2–50 | 10−3–100 | 101–102 | 113 and 120 | |
| Conducting polymer hydrogels | IPN | 102–106 | 30–560 | 10−3–102 | 102–106 | 1, 138 and 154 |
| Pure | 101–104 | 2–230 | 100–103 | 102–103 | 178 and 181 | |
3.1 Hydrogel coatings and encapsulations
The simplest way of introducing hydrogels in bioelectronics is through coating and encapsulation of existing bioelectronic devices with hydrogels. Despite the simplicity of the embodiment, recent advances in hydrogel coatings and encapsulations demonstrate a promising route to alleviate adverse biomechanical interactions between existing electrodes and biological tissues.Hydrogel coatings have found a ubiquitous usage in commercially available epidermal stimulation and recording electrodes (Fig. 7). Skin–electrode interfacial impedance is significantly affected by various factors including the conformal contact with the skin and the degree of epidermis hydration.73 Poor interfacial contact between skin and electrodes increases r in eqn (1), which results in substantial reduction in both stimulation and recording performances. Dehydrated epidermis acts as a barrier to ionic currents (i.e., lower Ce and higher Re in the equivalent circuit) and increases the interfacial impedance.73 Soft, adhesive, and wet hydrogel coatings possess particular suitability to ensure conformal contact and hydrated epidermis, and therefore, have been widely adopted in various forms of EEG, EMG, ECG, and TENS electrodes. More recent advances in epidermal bioelectronic devices also benefit from hydrogel coatings as a unique bridging interface to skin, including long-term conformal EMG sensors,74 electronic skin,75,76 and highly stretchable wearable devices.77
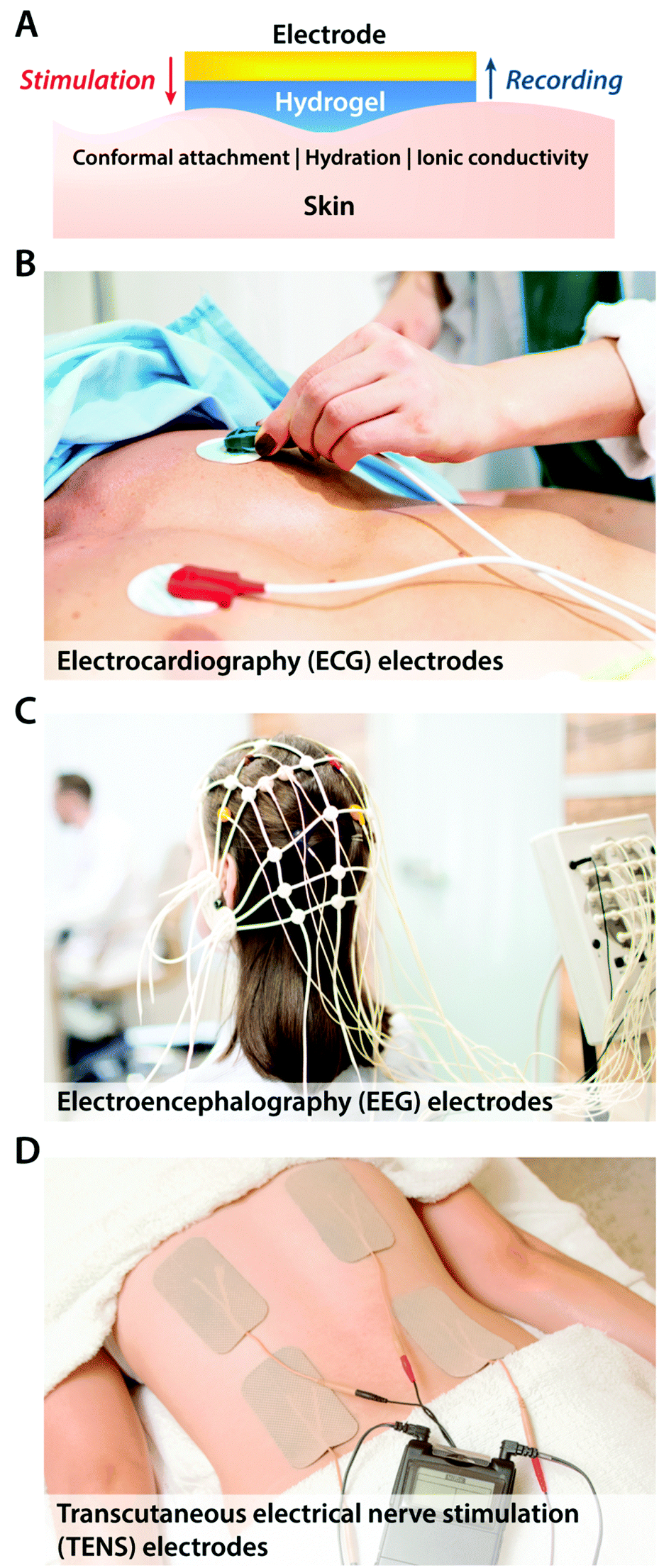 | ||
| Fig. 7 Hydrogel-coated electrodes in epidermal bioelectronic applications. (A) In order to provide soft, wet, adhesive, and conformal interfaces between the electrode and the skin, epidermal electrodes with a hydrogel layer are widespread in various clinical bioelectronic applications for both stimulation and recording. (B) Hydrogel electrodes for electrocardiography (ECG) recording. (C) Hydrogel electrodes for electroencephalography (EEG) recording. (D) Hydrogel electrodes for transcutaneous electrical nerve stimulation (TENS). | ||
In more invasive applications, hydrogel coatings with matching mechanical property can improve the biocompatibility of neural implants by attenuating neuroinflammatory responses. Soft poly(ethylene glycol)78,79 and poly(vinyl alcohol)80 hydrogel coatings around stiffer implants (e.g., glass, silicon, and PDMS) effectively reduce the degree of glial scarring and neural cell loss by minimizing the induced strain field during brain micromotions (Fig. 8). Together with tissue-like mechanical property, hydrogels’ ability to provide biofunctional interfaces further benefits the use of hydrogel coatings and encapsulations. Neural adhesion molecules incorporated in hydrogel coatings can significantly lower the degree of astrogliosis and the loss of neuronal bodies around neural implants.81 The water-rich nature of hydrogels also allows controlled delivery of biofunctional substances. Hydrogel coatings and encapsulations are engineered to deliver nerve growth factors82 and anti-inflammatory drugs83 into the surrounding neural tissues, which substantially alleviate neuroinflammatory responses in the case of chronic in vivo implantations.
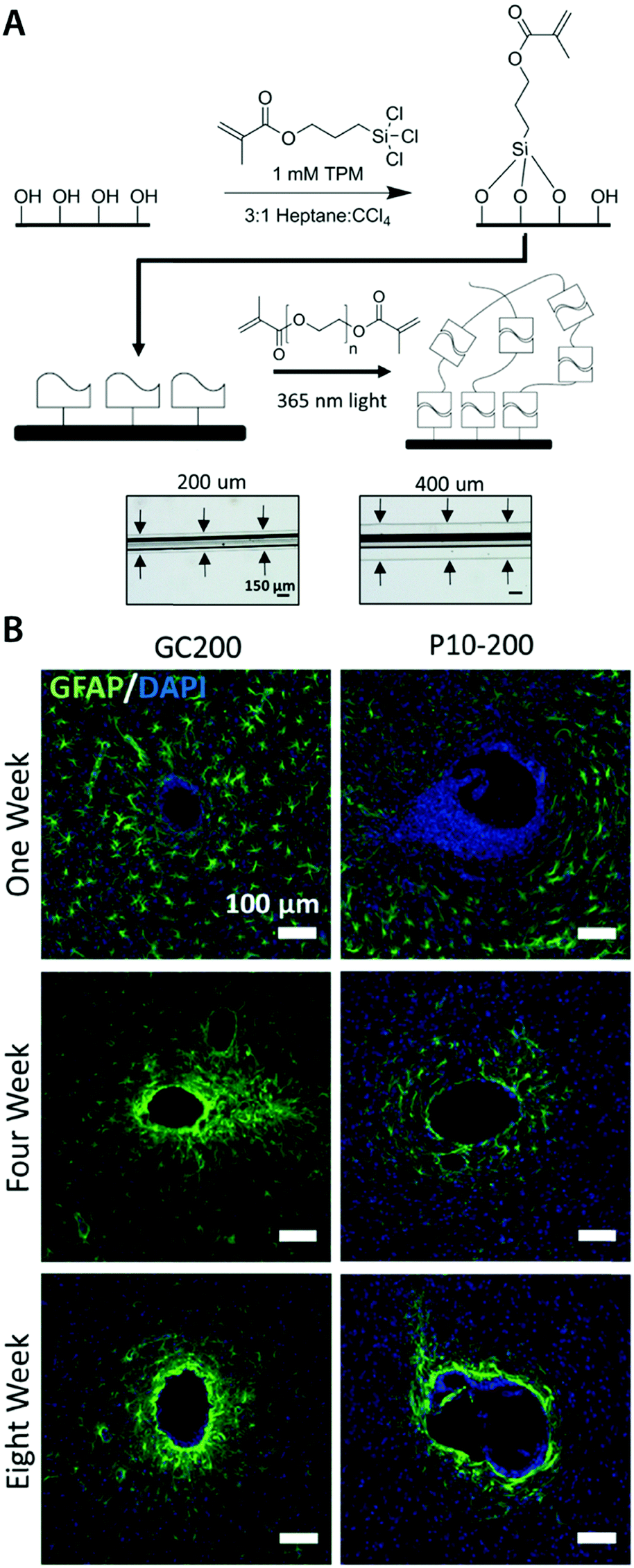 | ||
| Fig. 8 Mechanically matched hydrogel coatings with improved biocompatibility of neural implants. (A) Soft PEG hydrogel coatings are formed on glass capillary via surface grafting of the silane coupling agent followed by UV crosslinking of the hydrogel precursor. The thickness of the hydrogel coatings can be controlled by adjusting mold geometry. (B) Representative immunofluorescence images depicting the GFAP reactivity at the implant location at 1, 4, and 8 weeks post implantation. Glial scarring is significantly reduced for the hydrogel coated implant (P10-200) compared to the uncoated glass capillary (GC200). Reproduced with permission from ref. 78. Copyright 2017 Macmillan Publishers Limited. | ||
In addition to improvements in biocompatibility, hydrogel coatings and encapsulations have been utilized to provide better attachment and integration between the implanted electrodes and the surrounding tissues.84,85 Hydrogels’ superior biocompatibility and biofunctionality can provide bridging interfaces that closely mimic the mechanical and chemical properties of the extracellular matrix.34,86,87 Extracellular matrix-like hydrogel coatings and encapsulations can serve as tissue scaffolds that promote cell adhesion, proliferation, and tissue in-growth, providing bio-integrated interfaces between the implanted devices and tissues.88,89 Together with enhanced biocompatibility, such improved biomechanical interfaces by hydrogel coatings and encapsulations can offer significantly enhanced performances for bioelectronic applications by imparting long-term stability in interfacial impedance.90,91
3.2 Ionically conductive hydrogels
Unlike conventional dry engineering materials, the high water contents and nano- or micro-porous architecture of hydrogels permit the ionic conductivity by dissolved mobile ionic species (e.g., salt ions) similar to electrolytic tissue media.92,93 However, hydrogels are elastic solids whose mechanical properties are originated from the crosslinked polymer networks, distinguishing them from liquid-phase electrolytes.94 This unique combination of ionic conductivity and soft elastic nature, together with other advantageous properties such as optical transparency, renders ionically conductive hydrogels as a promising material in various bioelectronic applications.95,96Ionically conductive hydrogels are most commonly prepared by dissolving ionic salts (e.g., NaCl, LiCl) into the hydrogels.94,95 The resultant ionically conductive hydrogels can exhibit high stretchability (>10 times the original length), tissue-like softness (E < 100 kPa), and high ionic conductivity (>10 S m−1).94,95 While these ionically conductive hydrogels possess good electrical properties and favourable mechanical properties, they have been mostly utilized in non-invasive bioelectronic applications such as wearable devices97 and epidermal electrodes74 due to the need of relatively high salt concentrations (>1 M) compared to the physiological ionic concentrations (<300 mM).94,95 Hydrogels are capable of exchanging dissolved substances such as ions with the surrounding tissue media via diffusion over time. Hence, the high ionic concentrations in ionically conductive hydrogels can potentially cause biocompatibility issues as well as unstable bioelectronic performances in more invasive bioelectronic applications (e.g., implantable devices).98
Recently, another type of ionically conductive hydrogels with improved stability in physiological environments is developed based on the phase-separation between polyethylene glycol hydrogels and aqueous salt solutions98 (Fig. 9A). The phase-separated aqueous ionic solutions can provide soft, stretchable, and highly ionically conductive circuits within the biocompatible polyethylene glycol hydrogels without undesirable diffusion of ions into the surrounding tissue media. The improved stability in physiological environments enables in vivo bioelectronic stimulation of muscles by the ionically conductive hydrogel circuits in direct contact with tissues over extended periods of time (Fig. 9B and C).
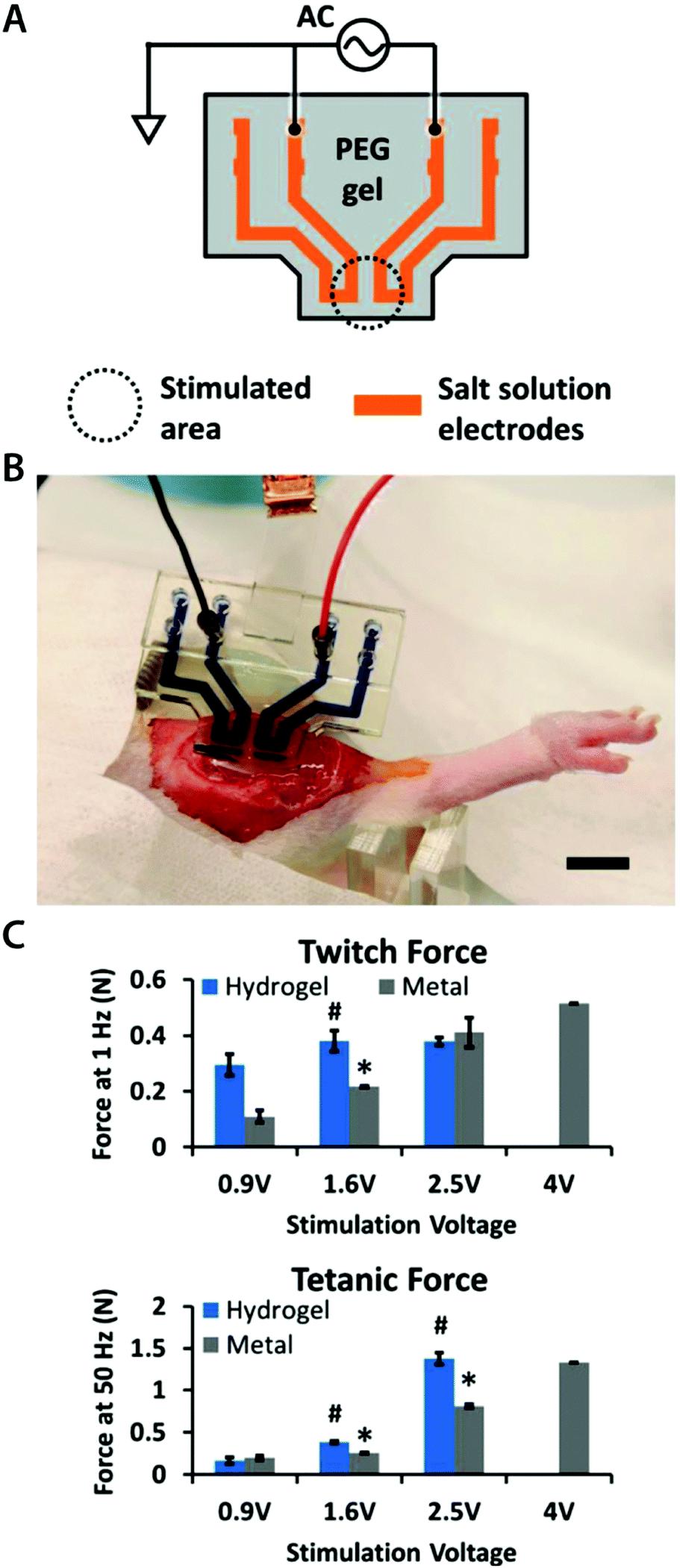 | ||
| Fig. 9 Hydrogel ionic circuits for in vivo electrical stimulation of muscles. (A) Ionic salt solution encapsulated within the PEG hydrogel serves as an electrode. (B) The hydrogel ionic stimulating electrode is placed on the rat muscle with external electronic connections. (C) The hydrogel ionic stimulation electrode exhibits better or comparable muscular activation under stimulation inputs compared to standard gold electrodes. Reproduced with permission from ref. 98. Copyright 2018 Wiley-VCH. | ||
3.3 Conductive nanocomposite hydrogels
Hydrogels can allow ionic currents via dissolved ions in water but typically act as electronic insulators (i.e., no electron flow). Consequently, the electrical properties of hydrogels in the physiologically relevant conditions are mostly similar to those of the tissue media, and much inferior to common electronic conductors (e.g., metals).92,93 Hence, the presence of hydrogels at tissue–electrode interfaces can potentially compromise electrical performance in spite of enhanced biomechanical interactions. Owing to this limitation, hydrogel coatings and encapsulations commonly serve as biomechanical enhancers, while bioelectronic stimulation and recording still rely on conventional electrodes.73,79,80 Introducing enhanced electrical property (e.g., lower interfacial impedance and higher charge injection capacity) without compromising their desirable biomechanical features (e.g., low mechanical modulus and biocompatibility) can provide opportunities to further improve tissue–electrode interfaces.One possible route to enhance the electrical property of hydrogels is by incorporating electronic conductivity on top of their ionic conductivity. Hydrogels are nano- or micro-porous 3D polymeric networks infiltrated with water, and such inherent porous architecture offers ample room for composite formation with a wide range of materials. In recent years, several types of electronically conductive nanomaterials including metallic nanoparticles and nanowires, carbon nanotubes, and graphene have been composited with the hydrogel matrix to endow electronic conductivity.99–102 By integrating these conductive nanomaterials into the hydrogel matrix, the resultant hydrogel composites can possess both ionic and electronic conductivity while retaining hydrogels’ unique biomechanical benefits. In this section, we introduce several recent advances in conductive nanocomposite hydrogels based on metallic and carbon nanomaterials, and their properties and applications in bioelectronics.
To avoid the undesirable trade-off between mechanical and electrical properties in metal–hydrogel composites, metallic fillers are typically introduced in the form of nanoscale particles or fibers.104,105 For example, silver nanowires (AgNWs) have been successfully incorporated into the poly(acrylamide) hydrogel to form highly flexible micropatterned electrode arrays104 (Fig. 10A). The conductive silver provides superior electrical conductivity, and nanoscale interactions between highly flexible AgNWs and hydrogel polymer networks allow great flexibility and low mechanical modulus comparable to the original poly(acrylamide) hydrogel.104 As another example, gold nanowires (AuNWs) have been composited with the alginate hydrogel to develop a bioelectronic cardiac patch102 (Fig. 10B). The alginate hydrogel provides a biocompatible three-dimensional extracellular matrix-like tissue scaffold while the nano-composited AuNWs endow the hydrogel high electrical conductivity, which can be used to synchronously stimulate imbedded cardiomyocytes.102 The resultant AuNW–alginate nanocomposite hydrogel shows improved cardiac tissue growth and maturation compared to the pristine alginate hydrogel.102
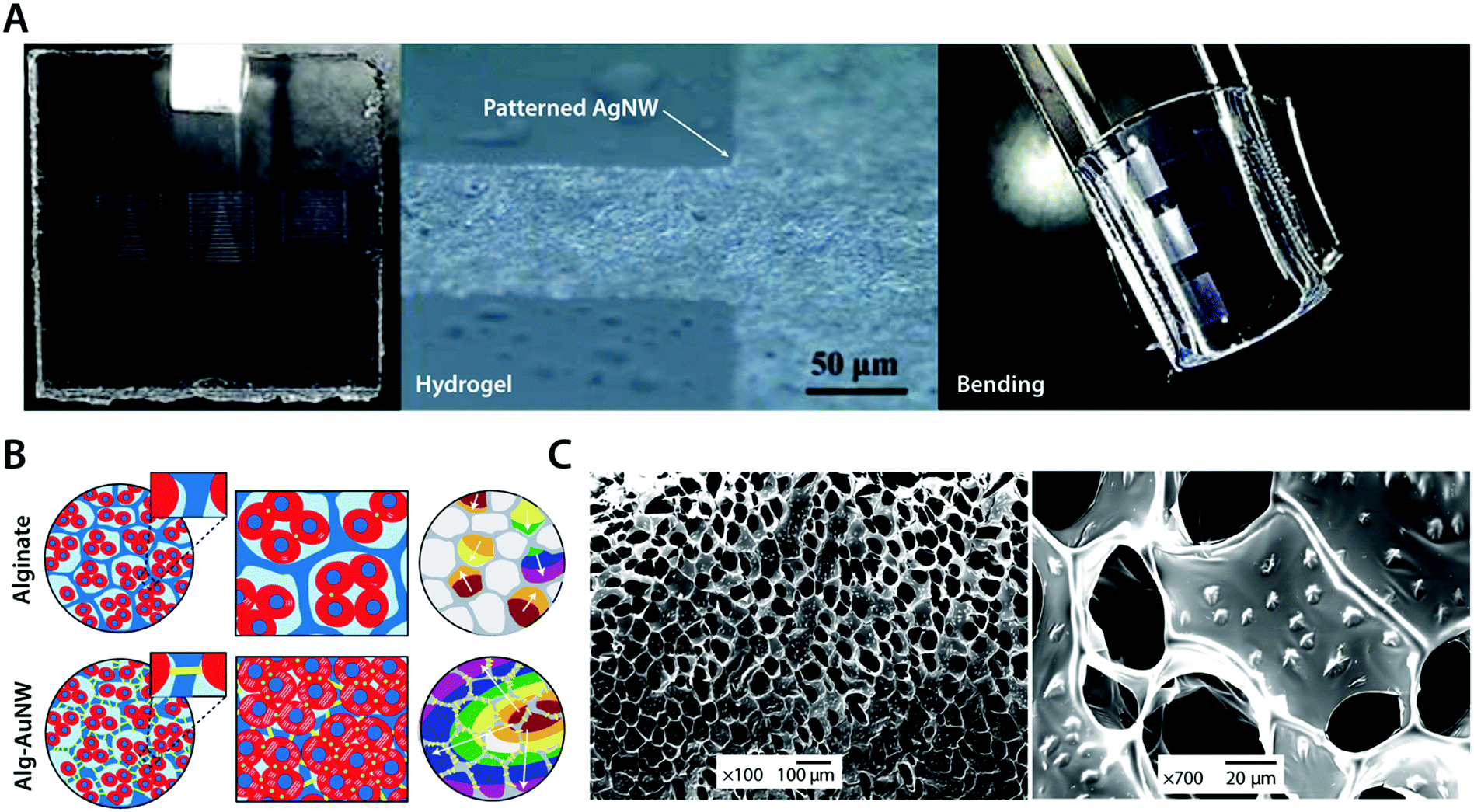 | ||
| Fig. 10 Conductive metal nanocomposite hydrogels. (A) Highly conductive and flexible microelectrodes on hydrogels are achieved via lithography-patterned AgNWs. The resultant metal nanocomposite hydrogel exhibits high flexibility and electrical conductivity. Reproduced with permission from ref. 104. Copyright 2014 American Chemical Society. (B) Isolated cardiomyocytes in pristine alginate hydrogel scaffolds typically form small clusters with asynchronous beating and random polarization due to the absence of sufficient electrical communication capability between cardiomyocyte clusters. The alginate–AuNW nanocomposite hydrogel provides sufficient electrical communication between isolated cardiomyocyte clusters that can enable the formation of organized cardiac-like tissue with synchronous beating. (C) SEM images of AuNWs assembled within the pore walls of the alginate hydrogel scaffold. Reproduced with permission from ref. 102. Copyright 2011 Macmillan Publishers Limited. | ||
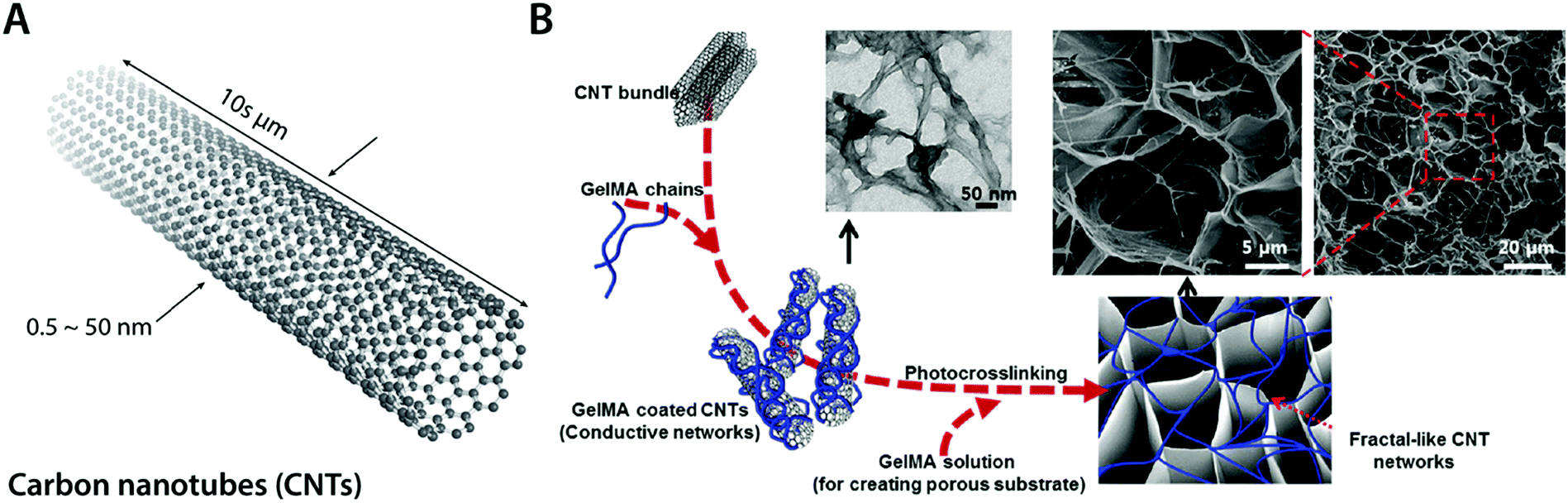 | ||
| Fig. 11 Conductive carbon nanocomposite hydrogels based on carbon nanotubes. (A) Carbon nanotubes (CNTs) are cylindrical carbon-based nanostructures with exceptionally high aspect ratio, which makes them commonly be called as 1D materials. CNTs typically have a diameter of 0.5–50 nm with 10 s of μm in length. CNTs show extraordinary mechanical properties and high thermal and electronic conductivity. (B) Conductive hydrogel composite can be achieved by embedding the fractal-like CNT network within the GelMA hydrogel. The embedded CNT network provides significantly enhanced electrical property to the hydrogel without affecting GelMA hydrogels’ favorable mechanical properties and biocompatibility as a tissue scaffold. Reproduced with permission from ref. 100. Copyright 2013 American Chemical Society. | ||
Recent advances in CNT-based conductive hydrogel nanocomposites have provided a versatile engineering platform for bioelectronic applications in nervous and cardio-muscular systems. For example, Shin et al. developed a functional cardiac patch by seeding cardiomyocytes onto a CNT–gelatin methacrylate (GelMA) conductive nanocomposite hydrogel100 (Fig. 11B). The CNT–GelMA nanocomposite hydrogel shows greatly improved excitatory responses and synchronous beating of the myocardial tissue cultured onto it.100 The CNT–GelMA nanocomposite hydrogel was also adopted by Ahadian et al. for skeletal muscle cells, but with vertically aligned CNTs to provide strong anisotropy in electrical properties.110 Vertically aligned CNTs in the conductive hydrogel nanocomposite contribute to enhanced myogenic maturation and responses compared to random or horizontally aligned CNTs in hydrogel composites.110 Significantly improved electrical property in CNT nanocomposite hydrogels further enables cardiovascular defect repairs by engineering the electrical property of CNT–gelatin–chitosan hydrogels similar to the native cardiac tissues.111
Graphene, a single- or multi-layered 2D nanocarbon structure, is one of the most important forms of carbon-based nanomaterials (Fig. 12A). Similar to CNTs, graphene boasts high electronic conductivity, extraordinary mechanical property, and good stability in a wet environment. Moreover, the π–π interactions between graphene nanosheets further provide a rich room for self-assembly assisted formation of hydrogel nanocomposites.112,113 Graphene sheets can form a self-assembled network by themselves or interact with other polymers via electrostatic and/or chemical bonds. Taking advantage of this unique self-assembly capability, graphene has been made into nanocomposite hydrogels using hydrothermal113,114 and chemical approaches115,116 as well as interpenetration with non-conductive hydrogel templates similar to other nanofillers.117–119
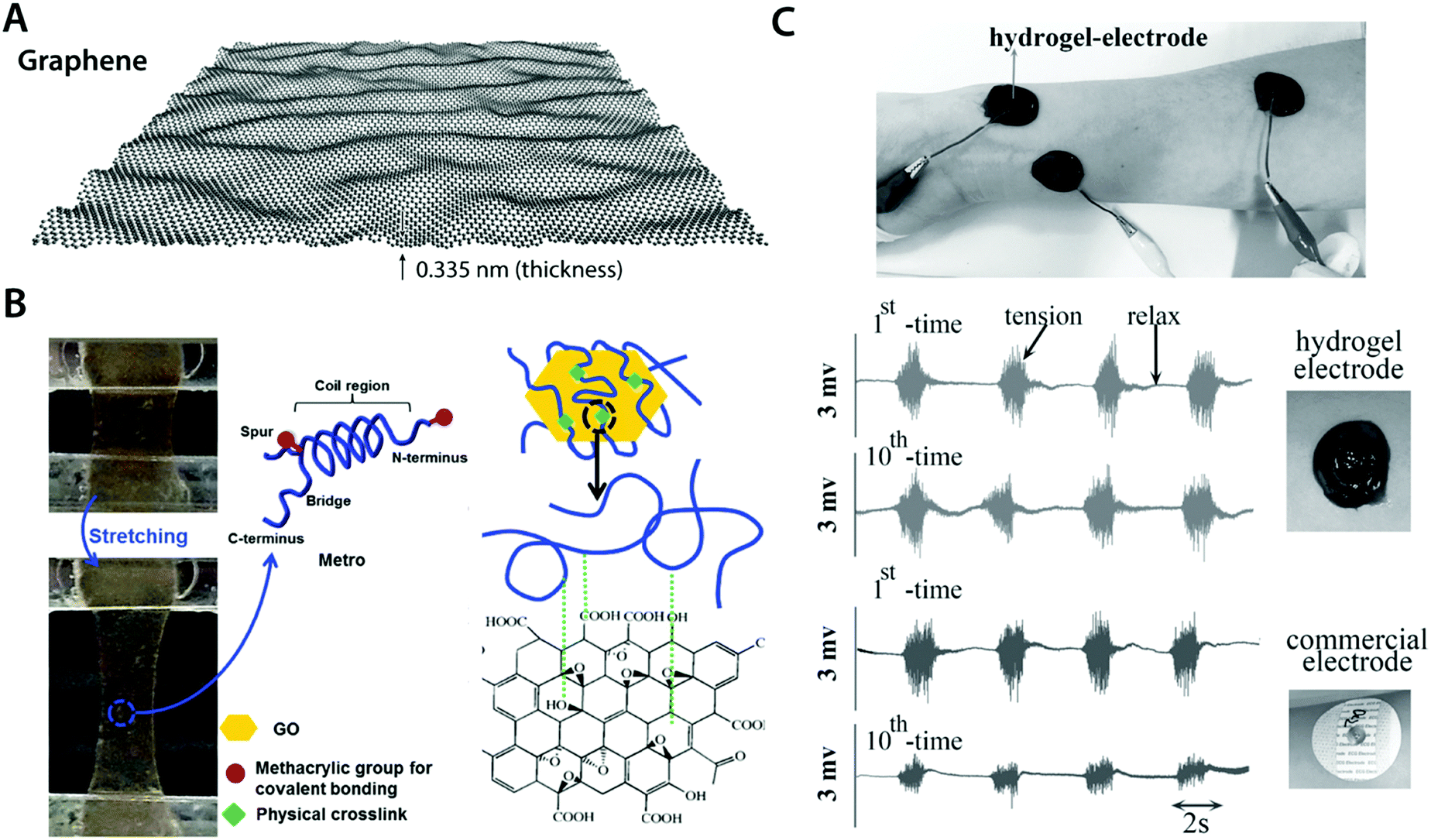 | ||
| Fig. 12 Conductive carbon nanocomposite hydrogels based on graphene. (A) Graphene is a carbon nanomaterial consisting of a single layer of carbon atoms arranged in a hexagonal lattice with a thickness of 0.335 nm. Due to its atomically thin nature, graphene is commonly referred as a 2D material. Graphene has extraordinary mechanical and electrical properties with a remarkable ability to form self-assembled structures owing to strong π–π interactions between graphene sheets. (B) Highly elastic and conductive hydrogels are achieved based on the hybrid nanocomposite between graphene and human-based proteins. Protein molecules interact with graphene via hydrophobic bonding, which provides physically crosslinked conductive nanocomposite hydrogels. Reproduced with permission from ref. 118. Copyright 2015 Wiley-VCH. (C) Conductive, self-adhesive, and self-healable hydrogel is prepared by introducing graphene into polydopamine hydrogel. The resultant conductive nanocomposite hydrogel can serve as an EMG electrode whose performance is comparable to that of commercially available electrodes. Reproduced with permission from ref. 121. Copyright 2016 Wiley-VCH. | ||
Works by Annabi et al.118 and Jo et al.120 successfully demonstrate that graphene-based nanocomposite hydrogels can synergistically combine high electrical conductivity and favorable mechanical property (e.g., stretchability and low modulus) with good in vitro as well as in vivo biocompatibility (Fig. 12B). This success has encouraged the development of more functional bioelectronic devices based on graphene nanocomposite hydrogels. For instance, Han et al. developed self-healable and self-adhesive graphene nanocomposite hydrogels for a range of bioelectronic applications including epidermal EMG recording and implantable intramuscular stimulation121 (Fig. 12C). More recently, Xiao et al. showed that the graphene–poly(vinyl alcohol)–poly(ethylene glycol) nanocomposite hydrogel can match the signal recording quality of the commercially available ECG recording electrodes with improved biocompatibility.122
3.4 Conducting polymer hydrogels
Composite formation between hydrogels and conductive nanomaterials provides a simple and effective route to introduce electronic conductivity, but it has several limitations as well. Nano-scale filler size is still much larger than individual polymer chains in hydrogel networks (typically sub-nm scale), and such disparity in length scales can be the source of inhomogeneity in mechanical and electrical properties.123,124 Furthermore, several nanomaterials such as CNTs have been found to be potentially cytotoxic via their unfavourable mechanical interactions with the cellular membrane.125,126 Interestingly, intrinsically conducting polymers (or simply conducting polymers) such as polypyrrole (PPy), polyaniline (PAni), and poly(3,4-ethylene-dioxythiophene) (PEDOT) have recently gained increasing popularity in hydrogel bioelectronics as an attractive alternative43,127–130 (Fig. 13A).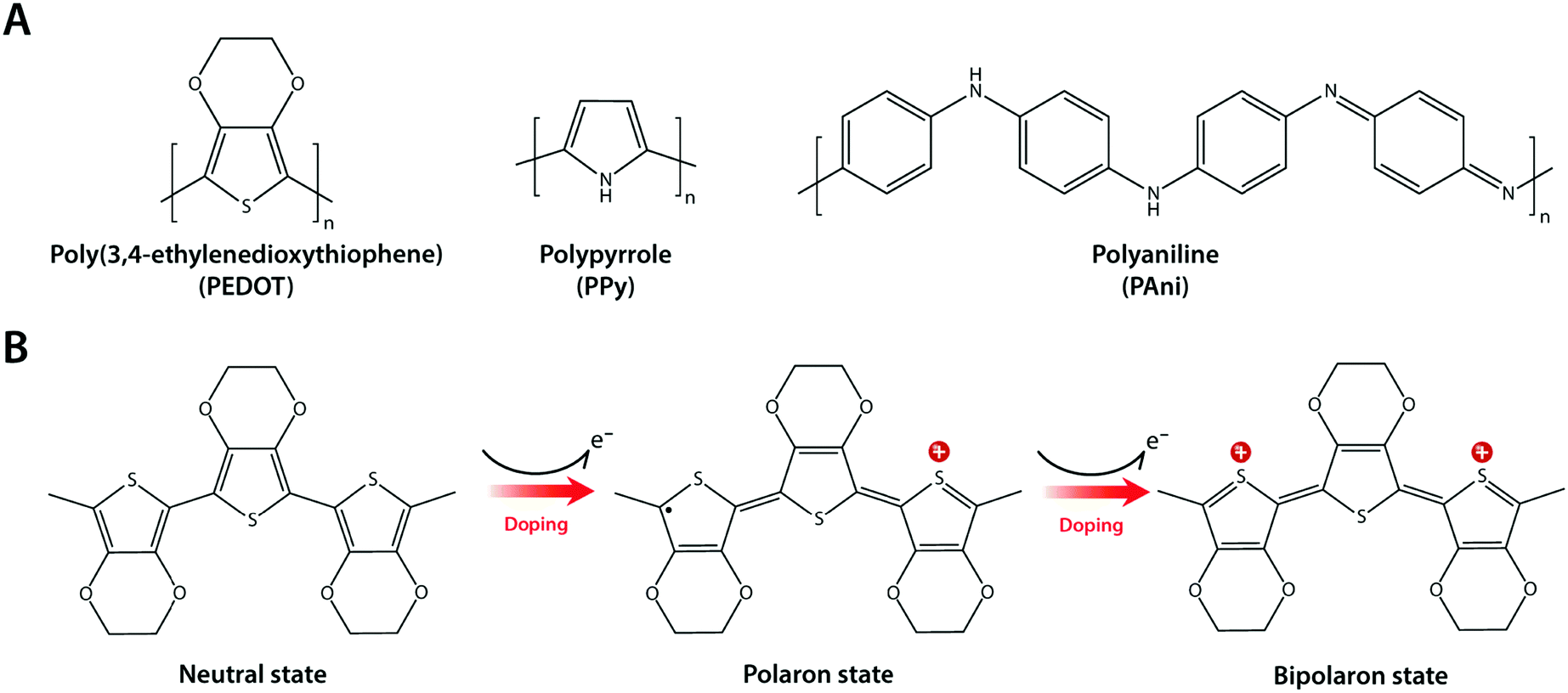 | ||
| Fig. 13 Conducting polymers and their mechanism of electronic conductivity. (A) Chemical structures of commonly used conducting polymers including poly(3,4-ethylenedioxythiophene) (PEDOT), polypyrrole (PPy), and polyaniline (PAni). (B) Conduction mechanism of conducting polymers exemplified by PEDOT. Electronic conductivity in conducting polymers originates from the delocalization of π-bonded electrons over the conjugated backbone structures with alternating single and double bonds. Doping of conducting polymers by ionic species gives rise to polarons and bipolarons, which significantly enhance conductivity via more charge carriers in the form of extra electrons or holes. | ||
Conducting polymers are π-conjugated polymers with alternating single and double covalent bonds, which can conduct electrons. The physical origin of the electronic conductivity in conducting polymers is the delocalization of π-bonded electrons over the conjugated backbone structures (Fig. 13B). To enhance the electronic conductivity, conducting polymers are commonly used together with various ionic dopants. The incorporation of ionic dopants further oxidizes (p-doping) or reduces (n-doping) the conducting polymer at the π-bond sites by removing or donating electrons. Such additional ionic doping creates free radicals that can pair with the dopant to form polarons, which can further be oxidized to bipolarons. These polaron/bipolaron sites facilitate electronic conduction by providing additional paths for charge carriers along with the conducting polymer backbone under the applied electric potential.131–133
Conducting polymers offer several unique advantages for hydrogel bioelectronics. Unlike metallic or carbon nanomaterials, the polymer chain-level intrinsic electronic conductivity allows extraordinary flexibility and compatibility in their use with other polymer systems like hydrogels. The organic and polymeric nature of conducting polymers enables ready access to rich and well-established chemical modification toolboxes. Some conducting polymers are hygroscopic or water-swellable, and oftentimes take a hydrogel form in a wet environment. High water contents within the bulk conducting polymers can provide soft mechanical property as well as high ionic conductivity together with the inherent electronic conductivity.63,134,135
Notably, the co-presence of polymer chain-level ionic and electronic conductivity makes conducting polymers a particularly charming candidate for bioelectronic applications. The water-rich and polymeric nature allows prompt penetration of ions into the bulk conducting polymers, and the electronically conductive polymer backbones create nm or sub-nm scale molecular-level EDL throughout the bulk material. This unique mechanism yields high volumetric capacitance (i.e., Ce proportional to the volume of the electrode) unlike the areal capacitance of metals (i.e., Ce proportional to the surface area of the electrode)62,64,136,137 (Fig. 14). Following the same governing equation and the equivalent circuit models discussed in Section 2, the high volumetric capacitance can substantially decrease the interfacial impedance and enhance the performance of both bioelectronic stimulation and recording, especially for small-scale electrodes with limited surface areas.
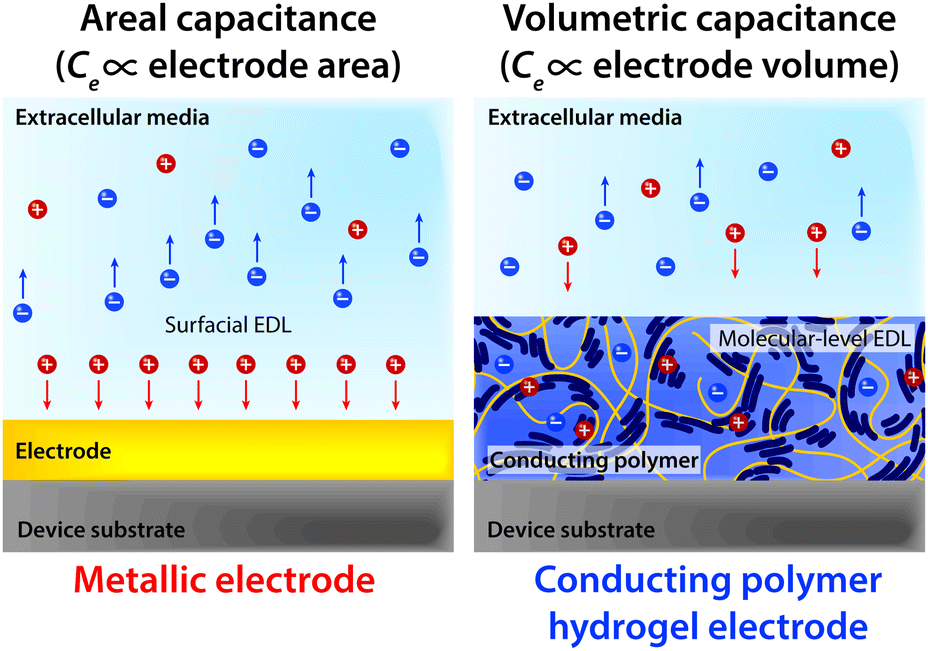 | ||
| Fig. 14 Capacitive charge injection by metallic electrode materials relies on the formation of surficial EDL whose capacitance is proportional to the electrode area (areal capacitance). In contrast, capacitive charge injection by a conducting polymer hydrogel electrode relies on the formation of molecular-level EDL throughout the conducting polymer hydrogel whose capacitance is proportional to the electrode volume (volumetric capacitance). | ||
Driven by the unique combination of favourable chemical, mechanical, and electrical properties, conducting polymer hydrogels have already found widespread usages in a number of applications such as neural implants, implantable sensors, prosthetic interfaces, and controlled drug delivery.128,138–151 This broad spectrum in applications is largely aided by their marked flexibility in processing and preparation. Conducting polymers can take a wide range of forms including polymerizable aromatic monomers, long-chain polymers, and crosslinked networks, and the choice of appropriate forms greatly affects the development of conducting polymer hydrogels. Conducting polymer hydrogels are most commonly prepared by forming molecular-level composites or interpenetrating networks (IPNs) between conducting polymers and non-conducting hydrogel templates. More recently, pure conducting polymer hydrogels (i.e., hydrogels prepared only with conducting polymers without non-conducting hydrogel templates) are developed to further improve the electrical property. In this section, we discuss the recent progress in conducting polymer hydrogels and their applications in bioelectronics based on two major preparation strategies.
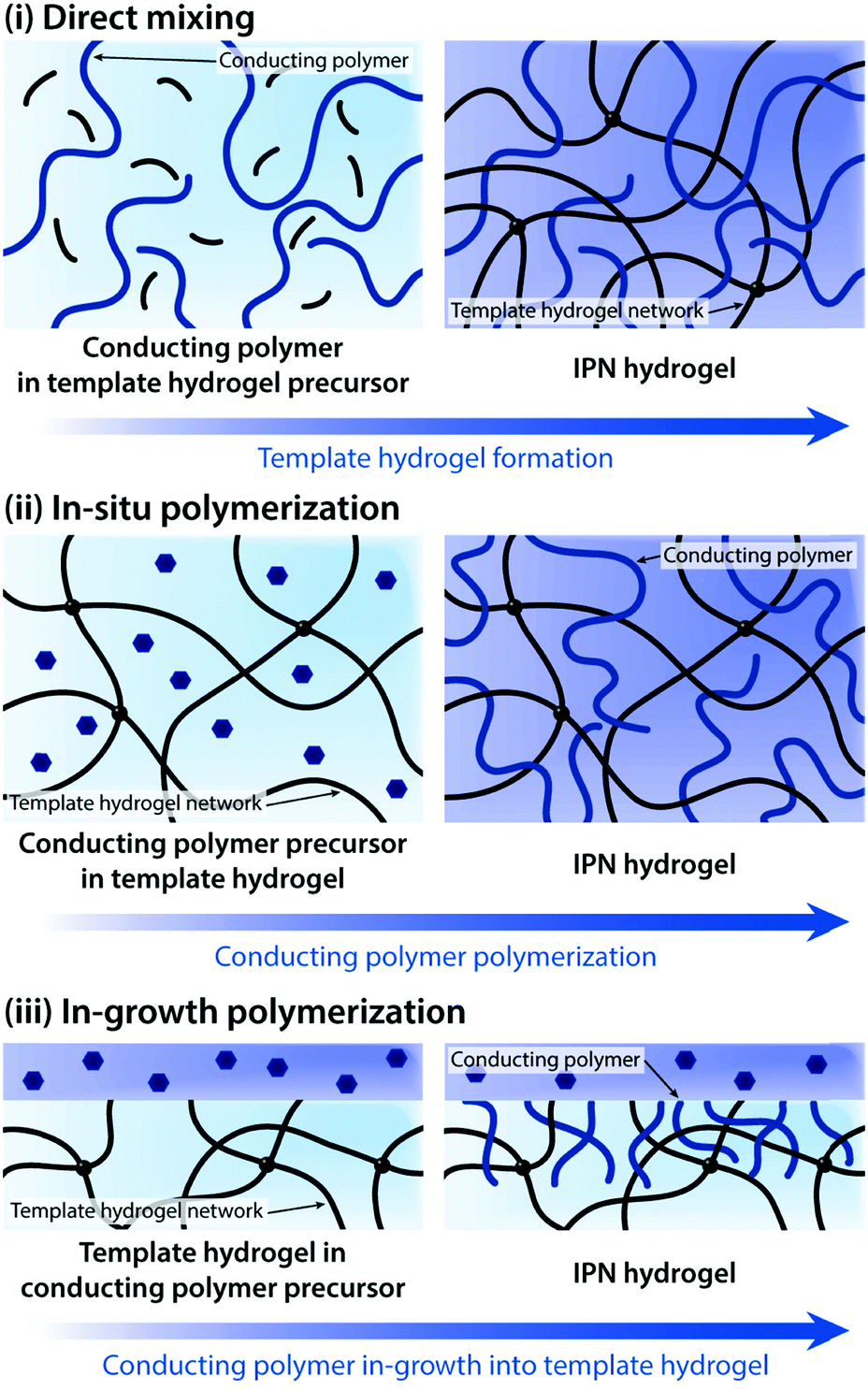 | ||
| Fig. 15 Various preparation routes toward conducting polymer IPN hydrogels. IPN hydrogels are typically prepared through three major approaches: (i) direct mixing between conducting polymers and the template hydrogel precursor, (ii) in situ polymerization of conducting polymers within the hydrogel template, and (iii) in-growth polymerization of conducting polymers into the hydrogel template. | ||
Direct mixing of conducting polymers provides a straightforward and chemically simple route to form IPN hydrogels. A wide range of hydrogels (e.g., poly(acrylamide), poly(acrylic acid), and gellan gum) and conducting polymers (e.g., PEDOT:PSS and PAni) have been used to form IPN hydrogels via the direct mixing strategy.161–163 The electrical performance of IPN hydrogels (e.g., electrical conductivity and volumetric capacitance) is typically proportional to the amount of conducting polymers in the hydrogel template. However, the direct mixing approach oftentimes faces limitations in terms of increasing conducting polymer contents. To address this challenge, in situ polymerization to form conducting polymers within the hydrogel template has been proposed.155,156,158,160,164,165In situ polymerization is typically achieved by introducing oxidative reagents (e.g., ferric ions and persulfate salts) into the hydrogel template containing aromatic monomers of conducting polymers.
In some bioelectronic applications, localized formation of IPN hydrogels is strongly preferred over the bulk IPN hydrogel. For example, complex patterning of conducting polymer hydrogels or selective introduction of electronic conductivity to existing hydrogel coatings and encapsulations represents a more spatially controlled strategy other than direct mixing or in situ polymerization. Notably, polymerization of conducting polymers can also be realized by electropolymerization, in which electrochemical oxidation by the applied electric potential serves the same role as oxidative reagents in in situ polymerization. Utilizing electropolymerization, selective in-growth of conducting polymers into the hydrogel template has been applied to develop conductive hydrogel patterning166,167 and neural implant coatings138,168 (Fig. 16).
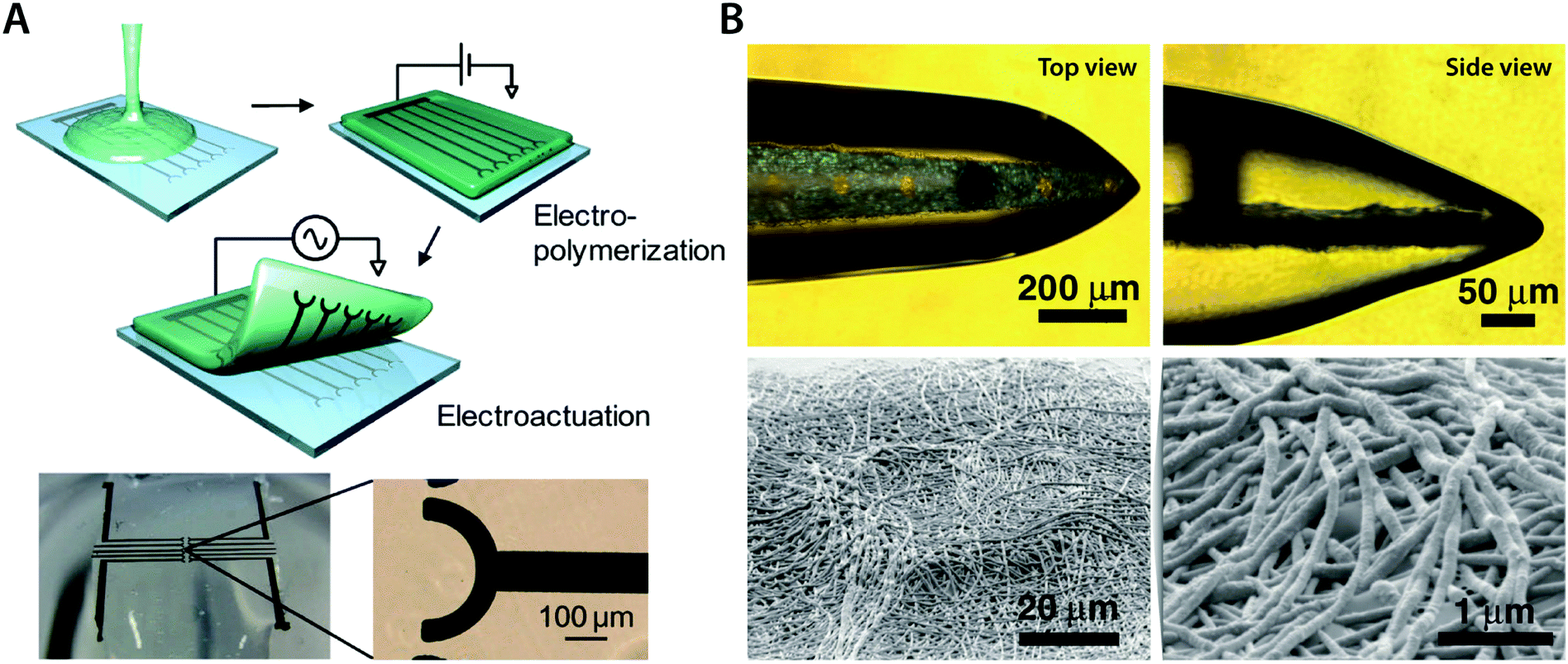 | ||
| Fig. 16 Conducting polymer IPN hydrogels via the in-growth polymerization approach. (A) Microelectrode pattern of PEDOT can be formed onto a biocompatible agarose hydrogel by electrosynthesizing PEDOT onto the hydrogel substrate. Applying a potential between the top agarose hydrogel and the bottom Pt microelectrode with EDOT monomers allows the in-growth polymerization of PEDOT into the agarose template hydrogel. Reproduced with permission from ref. 166. Copyright 2010 American Chemical Society. (B) Conductive IPN hydrogel between alginate and PEDOT (black) can be selectively formed by electropolymerizing EDOT monomers into the alginate encapsulation around the neural probe. Reproduced with permission from ref. 138. Copyright 2009 Wiley-VCH. | ||
Together with enhanced electrical performance, mechanical reliability of the electrode also plays a critical role in bioelectronic applications. Several recent efforts have aimed to develop mechanically robust IPN hydrogels. Early attempts simply adopted mechanically robust hydrogels as the template for conducting polymer IPN formation157,169,170 (Fig. 17A). Interestingly, IPN formation between two networks is a common strategy to achieve mechanically robust hydrogels via double-network formation.171,172 Inspired by this similarity, several recent works demonstrated that conducting polymers can serve as the second network for tough double-network hydrogels to synergistically impart both electronic conductivity and mechanical robustness173–176 (Fig. 17B).
 | ||
| Fig. 17 Mechanically robust conducting polymer IPN hydrogels. (A) Electrically conductive and mechanically tough hydrogel is prepared by in situ polymerization of PEDOT:PSS within a poly(ethylene glycol)methyl ether methacrylate–poly(acrylic acid) (PPEGMA–PAA) DN hydrogel template. Adapted with permission from ref. 157. Copyright 2012 American Chemical Society. (B) Mechanically strong conducting polymer IPN hydrogel is prepared by using the in situ polymerized PEDOT:PSS network as the second network within the first PAAm network to form a tough DN hydrogel. The PAAm single network (SN) hydrogel is brittle while the PEDOT:PSS reinforced DN hydrogel exhibits greatly improved mechanical robustness. Reproduced with permission from ref. 173. Copyright 2010 Elsevier. | ||
Among conducting polymers, PEDOT:PSS is one of the most widespread materials for pure conducting polymer hydrogels. PEDOT is generally not water-soluble, but a stable water dispersion of PEDOT can be achieved by introducing polyanionic dopants such as poly(styrene sulfonate) (PSS). Benefited by its ability to form stable and benign aqueous dispersion with superior biocompatibility and processability, PEDOT:PSS has been widely utilized in bioelectronics.127,142,182 Very recently, several preparation approaches are proposed to achieve pure conducting polymer hydrogels from PEDOT:PSS aqueous dispersions. Yao et al. showed that pure PEDOT:PSS hydrogels can be prepared by adding concentrated sulfuric acid into PEDOT:PSS aqueous solution followed by thermal treatment181 (Fig. 18A). Remarkably, the resultant pure PEDOT:PSS hydrogels exhibit an extraordinary electronic conductivity of 8.8 S cm−1 with over 95% water contents. As another example, Xu et al. demonstrated pure PEDOT:PSS hydrogels by using poly(ethylene glycol)–peptide as a physical crosslinker for aqueous PEDOT:PSS dispersion183 (Fig. 18B). Notably, peptide-based noncovalent crosslinking of the hydrogel provides self-healing and injectable property as well as superior biocompatibility.
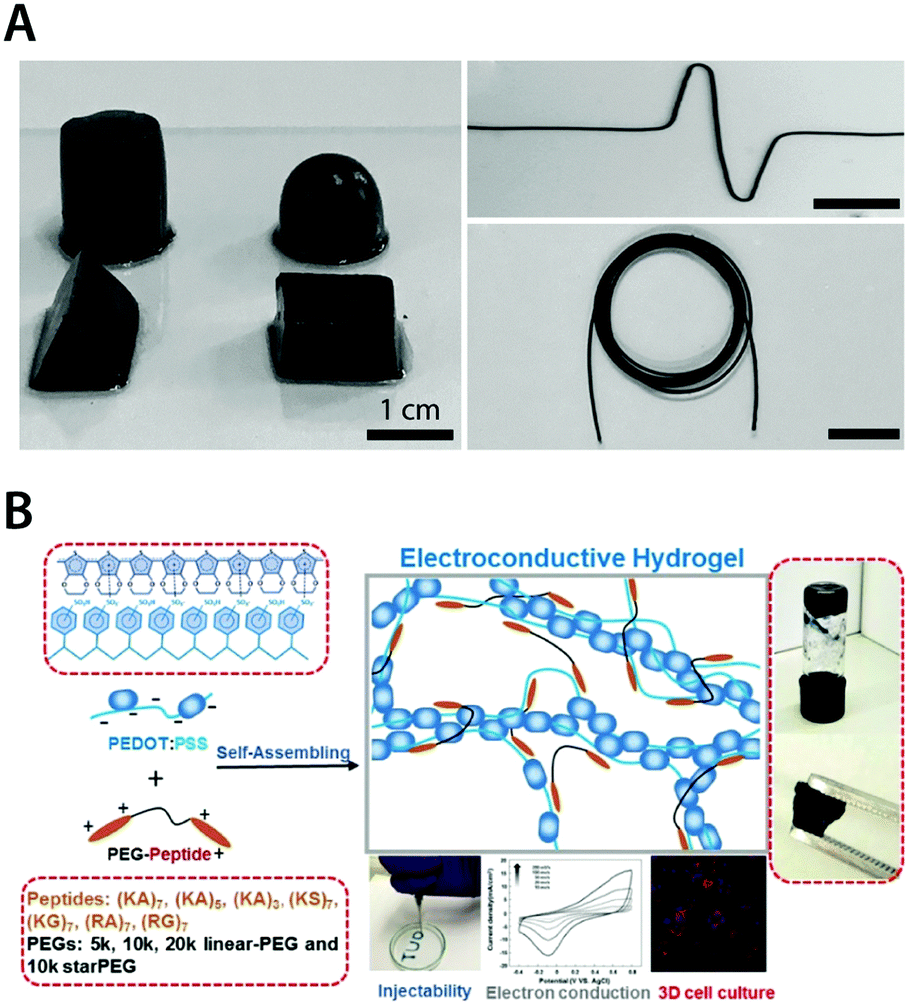 | ||
| Fig. 18 Pure conducting polymer hydrogels based on PEDOT:PSS. (A) Pure conducting polymer hydrogel with a high electric conductivity of 8.8 S cm−1 is prepared by adding concentrated sulfuric acid into aqueous PEDOT:PSS dispersion. Reproduced with permission from ref. 181. Copyright 2017 Wiley-VCH. (B) Non-covalent interactions between PEDOT:PSS and PEG–peptide form a physically crosslinked pure conducting polymer hydrogel with good electrical properties. The resultant hydrogel is self-healable and injectable via a needle due to the physically crosslinked nature and exhibits excellent biocompatibility as a conductive 3D cell culture scaffold. Adapted with permission from ref. 183. Copyright 2018 American Chemical Society. | ||
Other conducting polymers such as PAni and PPy have also been adopted to prepare pure conducting polymer hydrogels. Pan et al. demonstrated pure PAni hydrogels with high electronic conductivity (0.11 S cm−1) by using phytic acid as both dopant and crosslinker for PAni178 (Fig. 19A). The resultant pure PAni hydrogels also exhibit superior specific capacitance (∼480 F g−1) and glucose sensing capability. Lu et al. showed that elastic pure conducting polymer hydrogels can be realized by aging of the weakly gelled PPy suspension184 (Fig. 19B). Interestingly, the mechanical stability and elasticity of the pure PPy hydrogels stem from the gradual maturation of PPy networks during the aging process of polymerized pyrrole solution with deficient oxidant (ferric nitrite).
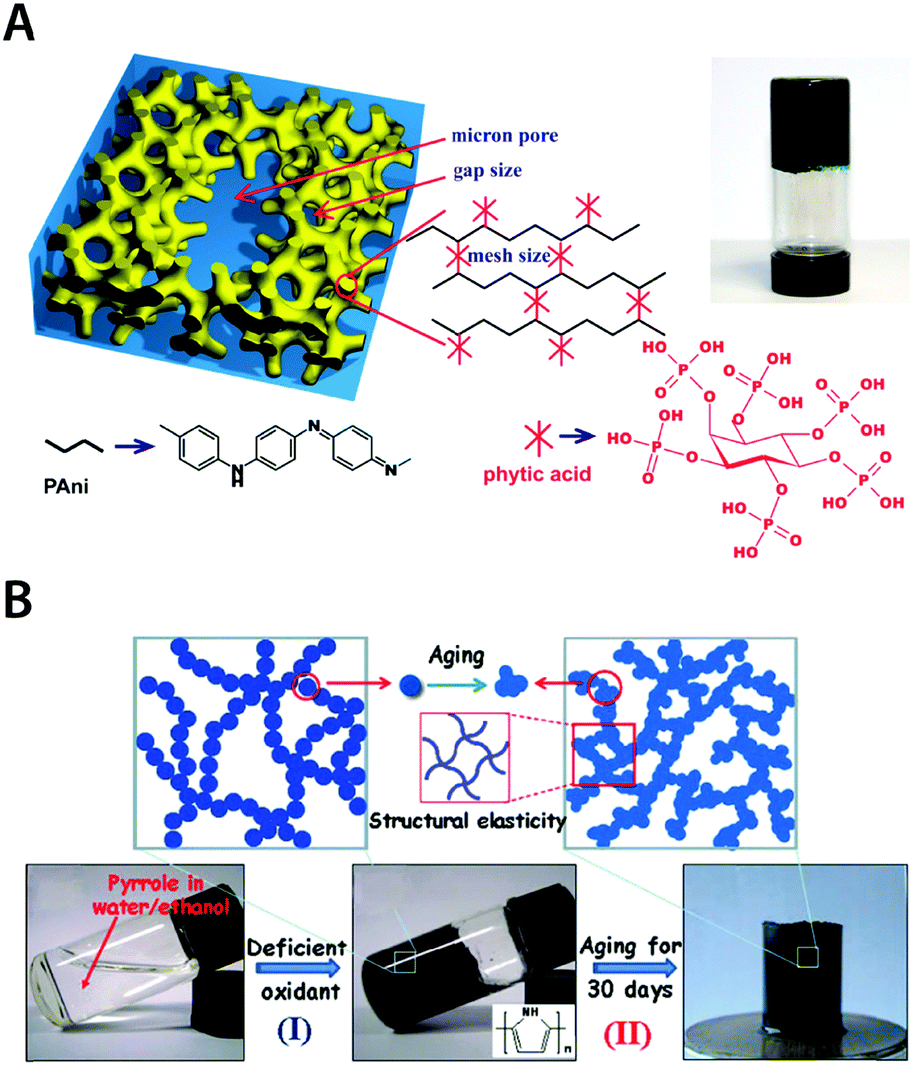 | ||
| Fig. 19 Pure conducting polymer hydrogels based on PAni and PPy. (A) Hierarchical nanostructured pure conducting polymer hydrogel with high electrochemical activity is prepared based on PAni doped with phytic acid. Phytic acid serves as both dopant and crosslinker for PAni which provides a unique hierarchical nanostructure within the hydrogel. Adapted with permission from ref. 178. Copyright 2012 National Academy of Sciences. (B) Elastic and conductive pure PPy hydrogel can be achieved by deficient oxidative polymerization of pyrrole followed by long-term aging. The aging process of the as-prepared PPY hydrogel provides improved mechanical and electrical properties via further aggregation formation during aging. Adapted with permission from ref. 184. Copyright 2014 Macmillan Publishers Limited. | ||
4 Hydrogel-device interfaces
In hydrogel bioelectronics, the hydrogels form interfaces not only with the biological tissues but also with the electronic systems, typically in the form of metallic electrodes, interconnects, and external terminals. Together with hydrogel–tissue interfaces, hydrogel-device interfaces also play a critical role in order to achieve desirable bioelectronic performances and functionalities. While hydrogel–tissue interfaces mostly involve bioelectronic and biomechanical interactions as discussed in Section 2, hydrogel-device interfaces invite a distinctive set of challenges. For example, the soft and high water-containing nature of hydrogels poses a number of difficulties in the long-term stability of hydrogel bioelectronic applications including weak adhesion and dehydration. Furthermore, incorporation of hydrogel interfaces in the fabrication of bioelectronic devices with high spatial resolution and complexity is oftentimes a non-trivial task. In this section, we highlight several key issues in hydrogel-device interfaces and recent advances including adhesion, dehydration, and fabrication of hydrogel interfaces.4.1 Adhesion of hydrogel interfaces
Engineering devices typically consist of numerous materials based on their corresponding merits in functionality. Not surprisingly, existing bioelectronic devices are also highly integrated systems made out of a wide range of materials, such as metallic interconnects, silicon substrates, and polymeric passivation layers. Likewise, hydrogel bioelectronic devices also involve a broad spectrum of materials, many of which are not hydrogels. In such integrated systems, robust assembly (or interfacial adhesion) between distinctive components is one of the most critical factors that determines the overall reliability and robustness of the devices. Even for existing hydrogel coatings and encapsulations for bioelectronic devices, insufficient interfacial adhesion poses significant challenges such as delamination failure of hydrogel coatings.Due to the physical and mechanical uniqueness of hydrogels such as very low elastic moduli and high water contents, robust interfacial adhesion between hydrogels and other engineering or biological materials has been a formidable challenge.36 Recently, tough bonding of hydrogels on diverse solids has been proposed by covalently anchoring stretchy polymer networks of hydrogels on solid surfaces185–187 (Fig. 20A). Furthermore, robust hydrogel hybrids have been achieved with a broad range of materials including metals76,185 (Fig. 20B), elastomers75,187–189 (Fig. 20C) and biological tissues.190,191 Such robust interfacial adhesion of hydrogels on various devices can offer more reliable bioelectronic performances and functionalities, particularly for dynamic and highly deformable tissues such as skin.75,77,192
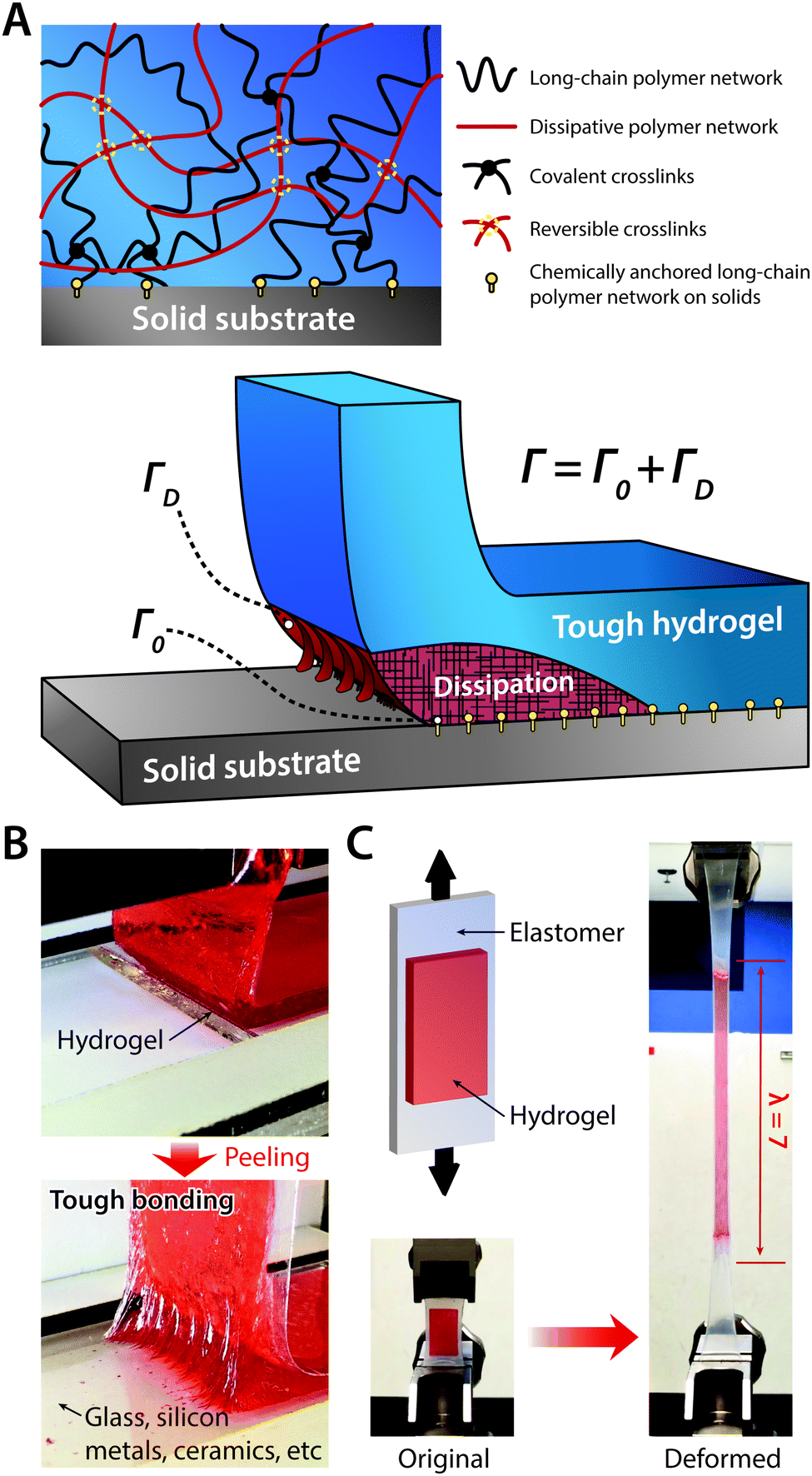 | ||
| Fig. 20 Tough bonding of hydrogels on diverse solids. (A) Tough bonding of hydrogels can be achieved by covalently anchoring stretchy polymer networks of hydrogels on solid surfaces. (B) The resultant tough bonding of hydrogels can provide the interfacial toughness over 1000 J m−2 on various solids including glass, silicon, metals, and ceramics. Reproduced with permission from ref. 185. Copyright 2016 Macmillan Publishers Limited. (C) Tough bonding of hydrogels can also be applied on highly stretchable materials such as elastomers. Reproduced with permission from ref. 187. Copyright 2016 Macmillan Publishers Limited. | ||
4.2 Dehydration of hydrogel interfaces
High volume fraction of water is one of the most essential requirements for hydrogels to retain their advantageous properties such as mechanical compliance and ionic conductivity. While implanted hydrogel bioelectronic interfaces can keep the hydrated state by continuously exchanging water from the surrounding wet environments, epidermal hydrogel interfaces are susceptible to dehydration due to the evaporation of water molecules into the atmosphere.96 Dehydration of hydrogel interfaces in dry conditions and subsequent changes in mechanical and electrical properties can adversely affect bioelectronic performances. For example, dehydrated hydrogels become stiff and less stretchable with significantly decreased ionic conductivity, prohibiting their use as bioelectronic stimulation and recording electrodes.73To avoid dehydration of hydrogel interfaces, several methods have been proposed at both material-level and device-level. At the material-level, the addition of hydroscopic elements such as glycerol,193 cellulose,194 and highly concentrated salts195,196 into hydrogels can provide enhanced retention of water in dry conditions. At the device-level, the encapsulation of hydrogel interfaces with stretchable evaporation barriers such as thin elastomeric coatings187,189,197 has been explored.
4.3 Fabrication of hydrogel interfaces
Transforming bulk materials into functional devices with complex designs and architectures is a highly non-trivial process. Among numerous challenges, appropriate fabrication strategies to process bulk materials into desired patterns and shapes in a highly integrated manner are one of the most crucial technological requirements. With remarkable advances and efforts during the last few decades, several well-established fabrication techniques such as micromachining, lithography, and thermal drawing have served as workhorses to drive innovation and establishment of state-of-the-art bioelectronic devices. These advanced fabrication techniques have enabled the development of existing commercially available bioelectronic devices (e.g., Michigan-type probes,71 Utah arrays,72 and DBS probes12) as well as laboratory-level devices (e.g., flexible electronics3,5,198–200 and multifunctional polymer probes201,202). However, many of these established fabrication approaches are not compatible with hydrogels or require significant modifications, mostly due to hydrogels’ distinctive properties compared to conventional engineering materials. For example, the soft and compliant nature of hydrogels virtually prevents the use of micromachining of high-precision small-scale features.203 Moreover, high water contents in hydrogels generally obviate the co-processing with other engineering materials in a high temperature environment (e.g., thermal drawing).In order to incorporate hydrogel interfaces into bioelectronic devices with sufficient spatial resolution and complexity, several fabrication approaches have been adopted in hydrogel bioelectronics. As one example, ink-jet printing has been utilized to deposit hydrogels on the device surface with micron precision.204,205 More recently, several 3D printing techniques have been introduced to hydrogel bioelectronics as a new route to construct complex 3D structures in small scales. Stereolithography (SLA) printing can generate highly complex designs of hydrogel constructs with high resolution.206,207 The direct ink writing (DIW) 3D printing technique provides a particularly attractive approach owing to its superior flexibility in terms of material choice and processing (Fig. 21).208–213
 | ||
| Fig. 21 Direct ink writing 3D printing of hydrogels. (A) 3D printed hydrogels with micro-scale vascular networks. Adopted with permission from ref. 208. Copyright 2014 Wiley-VCH. (B) Multi-material high resolution 3D printing of hydrogels into various geometries. Reproduced with permission from ref. 211. Copyright 2018 Wiley-VCH. (C) 3D printed ionically conductive hydrogel circuits on silicone elastomer substrate. Adopted with permission from ref. 209. Copyright 2017 Wiley-VCH. | ||
5 Future development directions for hydrogel bioelectronics
During the last decade, the tremendous amount of efforts highlighted above has driven rapid progress in this nascent field. Hydrogels have found their unique role in numerous stimulation and recording applications as bioelectronic and biomechanical bridging interfaces. A myriad of novel hydrogel interfaces have also shown unprecedented integration of outstanding electrical and mechanical properties. Despite these recent advances, hydrogel bioelectronics still faces numerous challenges ahead. For example, the majority of works on hydrogel bioelectronics have mostly focused on material developments accompanied only with proof-of-concept level demonstrations. However, the successful implementation ultimately depends on the device-level translation of technologies specific to the application of interest. Not surprisingly, such upcoming device-level implementation of hydrogel bioelectronics may likely invite a number of new issues and tasks, including a higher requirement of electrical and mechanical properties as well as device assembly and fabrication.On the other hand, these remaining challenges provide an ample room for future developments, particularly as the field is growing beyond its early stage. Continuous efforts to further improve the electrical and biomechanical performances of hydrogels may be one obvious direction, while the addition of new functional properties such as biodegradability can also open untapped opportunities. Regarding the road toward device-level translation of hydrogel bioelectronics, technological innovations for the successful device realization will require intense future developments too. For instance, robust interfacial assembly between hydrogels and various devices and tissues is still an open question in the field. Furthermore, hydrogel bioelectronics yet resides outside of many well-established advanced manufacturing techniques, which are essential for fabrication of complex functional devices. Here, we classify future development directions for hydrogel bioelectronics into three major categories: (i) development of hydrogels with improved property, (ii) robust integration between hydrogels and other device components, and (iii) advanced fabrication methodologies for hydrogel bioelectronic devices.
5.1 Improved property
Aided by recent advances, the improvements in the electrical and biomechanical properties of hydrogels have been remarkable. Electronic conductivity is now routinely introduced into a wide range of hydrogels based on a variety of novel chemistries and materials. The mechanical property of engineering hydrogels has started to closely match that of native biological tissues while retaining extraordinary biocompatibility. However, there is still plenty of space for further enhancement. As one example, conductive hydrogels are still limited in relatively low electrical conductivity (typically less than 1 S cm−1, which is several orders of magnitude lower than that of dry conducting polymers or metals), and oftentimes require the employment of acutely toxic substances for their preparation.181 While hydrogels with extraordinary stretchability and toughness have been recently developed,172,214 synergistically incorporating such mechanical performance and favourable electrical property is yet an unresolved challenge in the field. For example, most of pure conducting polymer hydrogels with good electrical conductivity typically suffer from poor mechanical properties, which greatly hinders their utilization in bioelectronic devices. Moreover, fine-tuning of their mechanical properties to match specific target tissues is also a remaining challenge to explore.Adding a new property into bioelectronic hydrogels also provides a promising direction for future development. On top of biocompatibility of hydrogels, biodegradability can be an appealing feature in bioelectronic applications. Biodegradable or transient devices substantially benefit bioelectronic applications by avoiding undesirable surgical removal of implanted devices from neural tissues.215,216 Biodegradability has been introduced in a few types of bulk conductive hydrogels,217–219 but incorporation of biodegradability in high-performance hydrogel bioelectronic devices may still require extensive efforts to be realized. Introduction of self-healing property can also be a promising direction to explore. Numerous self-healing hydrogels have been developed to mimic the self-repairing function of living tissues,220,221 but self-healing property is yet rarely studied in hydrogel bioelectronics.222,223
5.2 Robust integration
Despite the recent progress in the adhesion of hydrogel interfaces, robust integration of hydrogels into various bioelectronic devices and biological tissues still has unaddressed challenges and new opportunities for future developments and investigations. While a few early studies including covalent coupling of hydrogel coatings for neural implants78 and mussel-inspired adhesion of conductive hydrogels for cardiac patches224 have shown initial efforts toward this direction, there are more to be done. The unique chemistry of many hydrogels with promising electrical and mechanical properties (e.g., pure conducting polymer hydrogels) necessitates further technological innovations to realize robust interfacial integration. Together with device-level integration of hydrogels, reliable interfacial assembly of hydrogels with target tissues can also be a possible direction for future developments. For example, attachments of implantable hydrogel bioelectronic devices on highly dynamic and deforming wet tissues (e.g., heart, muscle, and spinal cord) typically rely on sub-optimal tissue adhesives or suturing due to the lack of capability to form robust and instant interfacial adhesion. Addressing these challenges in robust integration of hydrogel bioelectronics will greatly benefit the development of next-generation devices and applications by alleviating potential mechanical and electrical failures at the interface.5.3 Advanced fabrication
Development of advanced fabrication methodologies for hydrogel bioelectronics is still in its infancy, and future advances toward this direction will significantly support the success and establishment of the field. While several advanced fabrication approaches such as ink-jet printing and 3D printing have been introduced to hydrogel bioelectronics, further developments are necessary in various aspects. For example, ink-jet printing and 3D printing have been applicable for a few limited types of hydrogel bioelectronic interfaces such as ionically conductive hydrogels. Hence, expanding the coverage of such advanced fabrication methods can be one promising direction for future development. Interestingly, a few conducting polymers such as PEDOT:PSS and PAni have been used in ink-jet printing for fabrication of electronic devices in dry environments,178,225,226 which provides an encouraging starting point for future developments. Moreover, incorporating hydrogel bioelectronic interfaces into well-established conventional fabrication techniques can also be a potential direction to explore. For instance, conducting polymers such as PEDOT:PSS have been adopted in lithography-based fabrication processes to achieve high-resolution electronic devices operating in dry conditions.226 Similar approaches may provide opportunity to take advantage of well-established fabrication methods in hydrogel bioelectronics.6 Concluding remarks
During the last few decades, the remarkable similarities of hydrogels to biological tissues made them one of the most heavily studied engineering materials in a variety of fields including tissue engineering, biomedicine, biomechanics and physics. However, hydrogels have found their growing importance in bioelectronics relatively recently. Interestingly, the advances in hydrogel bioelectronics progress hand-in-hand with our understanding of interactions between biology and electronics. With our previously limited knowledge of biological systems and their interactions in contact with electronics, commonly used engineering materials and devices (e.g., metal- and silicon-based probes) were deemed sufficient. Consequently, developments in bioelectronic devices have mostly focused on the advancements of functionalities and form factors within the boundary of conventional electronic materials. As we understand more about the complex nature of tissue–electrode interactions, our comprehension of the source of myriad of adverse outcomes and failures of bioelectronic devices grows together.One critical lesson from the recent progress in neuroscience and engineering is that physical and mechanical mismatch at the tissue–electrode interface acts as one of the major sources of neuroinflammatory responses and corresponding failures in bioelectronic applications. This new perspective in designing enhanced bioelectronic interfaces has motivated the development of various unconventional bioelectronic devices with lower elastic moduli and higher flexibility. Not surprisingly, the continued efforts in the search of better solutions to minimize the mismatch between biology and electronics have led toward the rapid emergence of hydrogel-based bioelectronics, owing to their unique properties discussed in previous sections.
In recognition of this recent but significant development, we provide a comprehensive overview of hydrogel bioelectronics, starting from the fundamental tissue–electrode interactions to the latest advances in the field. Historically, advances and discoveries in burgeoning fields typically rely on either trial-and-error approaches or serendipity. However, it should be emphasized that the future progress in hydrogel bioelectronics will mostly benefit from rationally guided design principles based on understanding the fundamental mechanisms of tissue–electrode interactions. Despite a remarkable growth of hydrogel bioelectronics in recent years, many opportunities remain unexplored. Translation of bulk hydrogels with extraordinary electrical and biomechanical properties into realistic functional bioelectronic devices is still an open challenge in the field. The ample room for further technological innovations for improved property, robust integration, and advanced fabrication necessitates continuous efforts from researchers in multiple disciplines. Throughout the future developments, hydrogel bioelectronics will face daunting challenges and tasks to overcome, but in parallel, it will also provide the exciting promise of seamless merging between biology and electronics.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Shaoting Lin for the insightful discussion on the mechanisms of tissue–electrode interfaces. This work is supported by the National Science Foundation (CMMI-1661627), Office of Naval Research (N00014-17-1-2920), and the U.S. Army Research Office through the Institute for Soldier Nanotechnologies at MIT (W911NF-13-D-0001). H. Y. acknowledges the financial support from Samsung Scholarship. B. L. acknowledges the financial support from the National Natural Science Foundation of China (51763010), Science Foundation for Excellent Youth Talents in Jiangxi Province (20162BCB23053), Key Research and Development Program of Jiangxi Province (20171BBH80007), Natural Science Foundation of Jiangxi Province (20171BAB216018), and China Scholarship Council (201608360062).Notes and references
- S. F. Cogan, Annu. Rev. Biomed. Eng., 2008, 10, 275–309 CrossRef CAS PubMed.
- R. Feiner and T. Dvir, Nat. Rev. Mater., 2017, 3, 17076 CrossRef.
- D.-H. Kim, R. Ghaffari, N. Lu and J. A. Rogers, Annu. Rev. Biomed. Eng., 2012, 14, 113–128 CrossRef CAS PubMed.
- J. J. Jun, N. A. Steinmetz, J. H. Siegle, D. J. Denman, M. Bauza, B. Barbarits, A. K. Lee, C. A. Anastassiou, A. Andrei and Ç. Aydın, Nature, 2017, 551, 232 CrossRef CAS PubMed.
- D.-H. Kim, N. Lu, R. Ma, Y.-S. Kim, R.-H. Kim, S. Wang, J. Wu, S. M. Won, H. Tao and A. Islam, Science, 2011, 333, 838–843 CrossRef CAS PubMed.
- R. C. Webb, A. P. Bonifas, A. Behnaz, Y. Zhang, K. J. Yu, H. Cheng, M. Shi, Z. Bian, Z. Liu and Y.-S. Kim, Nat. Mater., 2013, 12, 938 CrossRef CAS PubMed.
- D.-H. Kim, J. Viventi, J. J. Amsden, J. Xiao, L. Vigeland, Y.-S. Kim, J. A. Blanco, B. Panilaitis, E. S. Frechette and D. Contreras, Nat. Mater., 2010, 9, 511 CrossRef CAS PubMed.
- I. R. Minev, P. Musienko, A. Hirsch, Q. Barraud, N. Wenger, E. M. Moraud, J. Gandar, M. Capogrosso, T. Milekovic and L. Asboth, Science, 2015, 347, 159–163 CrossRef CAS PubMed.
- L. Xu, S. R. Gutbrod, A. P. Bonifas, Y. Su, M. S. Sulkin, N. Lu, H.-J. Chung, K.-I. Jang, Z. Liu, M. Ying, C. Lu, R. C. Webb, J.-S. Kim, J. I. Laughner, H. Chen, Y. Liu, A. Ameen, J.-W. Jeong, G.-T. Kim, Y. Huang, I. R. Efimov and J. A. Rogers, Nat. Commun., 2014, 5, 3329 CrossRef PubMed.
- G. Buzsáki, C. A. Anastassiou and C. Koch, Nat. Rev. Neurosci., 2012, 13, 407 CrossRef PubMed.
- A. L. Benabid, Curr. Opin. Neurobiol., 2003, 13, 696–706 CrossRef CAS PubMed.
- M. S. Okun, N. Engl. J. Med., 2012, 367, 1529–1538 CrossRef CAS PubMed.
- C. De Hemptinne, N. C. Swann, J. L. Ostrem, E. S. Ryapolova-Webb, M. San Luciano, N. B. Galifianakis and P. A. Starr, Nat. Neurosci., 2015, 18, 779 CrossRef CAS PubMed.
- W. Ondo, J. Jankovic, K. Schwartz, M. Almaguer and R. Simpson, Neurology, 1998, 51, 1063–1069 CrossRef CAS PubMed.
- V. Gilja, C. Pandarinath, C. H. Blabe, P. Nuyujukian, J. D. Simeral, A. A. Sarma, B. L. Sorice, J. A. Perge, B. Jarosiewicz and L. R. Hochberg, Nat. Med., 2015, 21, 1142 CrossRef CAS PubMed.
- D. Farina, I. Vujaklija, M. Sartori, T. Kapelner, F. Negro, N. Jiang, K. Bergmeister, A. Andalib, J. Principe and O. C. Aszmann, Nat. Biomed. Eng., 2017, 1, 0025 CrossRef.
- E. L. Graczyk, M. A. Schiefer, H. P. Saal, B. P. Delhaye, S. J. Bensmaia and D. J. Tyler, Sci. Transl. Med., 2016, 8, 362ra142 CrossRef PubMed.
- S. Srinivasan, M. Carty, P. Calvaresi, T. Clites, B. Maimon, C. Taylor, A. Zorzos and H. Herr, Science Robotics, 2017, 2, eaan2971 CrossRef.
- L. Xu, S. R. Gutbrod, Y. Ma, A. Petrossians, Y. Liu, R. C. Webb, J. A. Fan, Z. Yang, R. Xu and J. J. Whalen, Adv. Mater., 2015, 27, 1731–1737 CrossRef CAS PubMed.
- H. Fang, K. J. Yu, C. Gloschat, Z. Yang, E. Song, C.-H. Chiang, J. Zhao, S. M. Won, S. Xu and M. Trumpis, Nat. Biomed. Eng., 2017, 1, 0038 CrossRef PubMed.
- C. E. Bouton, A. Shaikhouni, N. V. Annetta, M. A. Bockbrader, D. A. Friedenberg, D. M. Nielson, G. Sharma, P. B. Sederberg, B. C. Glenn and W. J. Mysiw, Nature, 2016, 533, 247–250 CrossRef CAS PubMed.
- M. Capogrosso, T. Milekovic, D. Borton, F. Wagner, E. M. Moraud, J.-B. Mignardot, N. Buse, J. Gandar, Q. Barraud and D. Xing, Nature, 2016, 539, 284 CrossRef PubMed.
- J.-W. Jeong, G. Shin, S. I. Park, K. J. Yu, L. Xu and J. A. Rogers, Neuron, 2015, 86, 175–186 CrossRef CAS PubMed.
- S. P. Lacour, G. Courtine and J. Guck, Nat. Rev. Mater., 2016, 1, 16063 CrossRef CAS.
- T. Someya, Z. Bao and G. G. Malliaras, Nature, 2016, 540, 379 CrossRef CAS PubMed.
- S. M. Wellman, J. R. Eles, K. A. Ludwig, J. P. Seymour, N. J. Michelson, W. E. McFadden, A. L. Vazquez and T. D. Y. Kozai, Adv. Funct. Mater., 2018, 28, 1701269 CrossRef PubMed.
- W. M. Grill, S. E. Norman and R. V. Bellamkonda, Annu. Rev. Biomed. Eng., 2009, 11, 1–24 CrossRef CAS PubMed.
- A. Sridharan, S. D. Rajan and J. Muthuswamy, J. Neural Eng., 2013, 10, 066001 CrossRef PubMed.
- Y. Xie, N. Martini, C. Hassler, R. D. Kirch, T. Stieglitz, A. Seifert and U. G. Hofmann, Front. Neuroeng., 2014, 7, 34 Search PubMed.
- J. W. Salatino, K. A. Ludwig, T. D. Kozai and E. K. Purcell, Nat. Biomed. Eng., 2017, 1, 862 CrossRef.
- J. Malmivuo and R. Plonsey, Bioelectromagnetism: principles and applications of bioelectric and biomagnetic fields, Oxford University Press, USA, 1995 Search PubMed.
- A. Grodzinsky, Field, Forces and Flows in Biological Systems, Garland Science, 2011 Search PubMed.
- M. D. Ferro and N. A. Melosh, Adv. Funct. Mater., 2018, 28, 1704335 CrossRef.
- K. Y. Lee and D. J. Mooney, Chem. Rev., 2001, 101, 1869–1880 CrossRef CAS PubMed.
- D. Seliktar, Science, 2012, 336, 1124–1128 CrossRef CAS PubMed.
- Y. S. Zhang and A. Khademhosseini, Science, 2017, 356, eaaf3627 CrossRef PubMed.
- P. F. Grant and M. M. Lowery, IEEE Trans. Biomed. Eng., 2010, 57, 2386–2393 Search PubMed.
- E. C. Greco and J. W. Clark, IEEE Trans. Biomed. Eng., 1977, 18–23 Search PubMed.
- A. L. Hodgkin and A. F. Huxley, J. Physiol., 1952, 117, 500–544 CrossRef CAS.
- H. Ye and A. Steiger, J. Neuroeng. Rehabil., 2015, 12, 65 CrossRef PubMed.
- M. E. J. Obien, K. Deligkaris, T. Bullmann, D. J. Bakkum and U. Frey, Front. Neurosci., 2015, 8, 423 Search PubMed.
- D. C. Martin and G. G. Malliaras, ChemElectroChem, 2016, 3, 686–688 CrossRef CAS.
- J. Rivnay, H. Wang, L. Fenno, K. Deisseroth and G. G. Malliaras, Sci. Adv., 2017, 3, e1601649 CrossRef PubMed.
- P. C. Miranda, A. Mekonnen, R. Salvador and G. Ruffini, NeuroImage, 2013, 70, 48–58 CrossRef PubMed.
- N. Joye, A. Schmid and Y. Leblebici, Neurocomputing, 2009, 73, 250–259 CrossRef.
- H. Meffin, B. Tahayori, D. B. Grayden and A. N. Burkitt, J. Neural Eng., 2012, 9, 065005 CrossRef PubMed.
- M. H. Histed, V. Bonin and R. C. Reid, Neuron, 2009, 63, 508–522 CrossRef CAS PubMed.
- H. Meffin, B. Tahayori, E. N. Sergeev, I. M. Mareels, D. B. Grayden and A. N. Burkitt, J. Neural Eng., 2014, 11, 065004 CrossRef PubMed.
- S. Moulton, J. Barisci, A. Bath, R. Stella and G. Wallace, Electrochim. Acta, 2004, 49, 4223–4230 CrossRef CAS.
- K. Ashkan, P. Rogers, H. Bergman and I. Ughratdar, Nat. Rev. Neurol., 2017, 13, 548 CrossRef PubMed.
- B. S. Wilson and M. F. Dorman, IEEE Sens. J., 2008, 8, 131–147 Search PubMed.
- A. K. Ahuja, J. Dorn, A. Caspi, M. McMahon, G. Dagnelie, P. Stanga, M. Humayun, R. Greenberg and A. I. S. Group, Br. J. Ophthalmol., 2011, 95, 539–543 CrossRef CAS PubMed.
- E. Zrenner, K. U. Bartz-Schmidt, H. Benav, D. Besch, A. Bruckmann, V.-P. Gabel, F. Gekeler, U. Greppmaier, A. Harscher and S. Kibbel, Proc. R. Soc. London, Ser. B, 2011, 278, 1489–1497 CrossRef PubMed.
- E. B. Montgomery Jr and J. T. Gale, Neurosci. Biobehav. Rev., 2008, 32, 388–407 CrossRef PubMed.
- M. I. Johnson, eLS, 2012 Search PubMed , http://www.els.net/WileyCDA/ElsArticle/refId-a0024044.html.
- A. Akhtar, J. Sombeck, B. Boyce and T. Bretl, Science Robotics, 2018, 3, eaap9770 CrossRef.
- M. Johnson, Rev. Pain, 2007, 1, 7–11 CrossRef PubMed.
- B. M. Doucet, A. Lam and L. Griffin, Yale J. Biol. Med., 2012, 85, 201 Search PubMed.
- T. M. Pinto, R. S. Wedemann and C. M. Cortez, PLoS One, 2014, 9, e96194 CrossRef PubMed.
- D. R. Cantrell, S. Inayat, A. Taflove, R. S. Ruoff and J. B. Troy, J. Neural Eng., 2007, 5, 54 CrossRef PubMed.
- D. R. Merrill, Implantable Neural Prostheses 2, Springer, 2010, pp. 85–138 Search PubMed.
- A. V. Volkov, K. Wijeratne, E. Mitraka, U. Ail, D. Zhao, K. Tybrandt, J. W. Andreasen, M. Berggren, X. Crispin and I. V. Zozoulenko, Adv. Funct. Mater., 2017, 27, 1700329 CrossRef.
- S. Inal, G. G. Malliaras and J. Rivnay, Nat. Commun., 2017, 8, 1767 CrossRef PubMed.
- K. Tybrandt, I. V. Zozoulenko and M. Berggren, Sci. Adv., 2017, 3, eaao3659 CrossRef PubMed.
- R. Pradhan, A. Mitra and S. Das, J. Appl. Solution Chem. Model., 2012, 1, 74–78 CAS.
- P. F. Grant and M. M. Lowery, Conf. Proc. IEEE Eng. Med. Biol. Soc., 2009, 6497–6500 Search PubMed.
- E. Niedermeyer and F. L. da Silva, Electroencephalography: basic principles, clinical applications, and related fields, Lippincott Williams & Wilkins, 2005 Search PubMed.
- T. D. Kozai, A. S. Jaquins-Gerstl, A. L. Vazquez, A. C. Michael and X. T. Cui, ACS Chem. Neurosci., 2015, 6, 48–67 CrossRef CAS PubMed.
- H. Lee, R. V. Bellamkonda, W. Sun and M. E. Levenston, J. Neural Eng., 2005, 2, 81 CrossRef PubMed.
- T. P. Prevost, A. Balakrishnan, S. Suresh and S. Socrate, Acta Biomater., 2011, 7, 83–95 CrossRef PubMed.
- K. D. Wise, IEEE Eng. Med. Biol. Mag., 2005, 24, 22–29 Search PubMed.
- P. K. Campbell, K. E. Jones, R. J. Huber, K. W. Horch and R. A. Normann, IEEE Trans. Biomed. Eng., 1991, 38, 758–768 CrossRef CAS PubMed.
- N. A. Alba, R. J. Sclabassi, M. Sun and X. T. Cui, IEEE Trans. Neural Syst. Rehabil. Eng., 2010, 18, 415–423 Search PubMed.
- K. Nagamine, S. Chihara, H. Kai, H. Kaji and M. Nishizawa, Sens. Actuators, B, 2016, 237, 49–53 CrossRef CAS.
- D. Wirthl, R. Pichler, M. Drack, G. Kettlguber, R. Moser, R. Gerstmayr, F. Hartmann, E. Bradt, R. Kaltseis and C. M. Siket, Sci. Adv., 2017, 3, e1700053 CrossRef PubMed.
- S. Lin, H. Yuk, T. Zhang, G. A. Parada, H. Koo, C. Yu and X. Zhao, Adv. Mater., 2016, 28, 4497–4505 CrossRef CAS PubMed.
- S. H. Kim, S. Jung, I. S. Yoon, C. Lee, Y. Oh and J. M. Hong, Adv. Mater., 2018, 30, 1800109 CrossRef PubMed.
- K. C. Spencer, J. C. Sy, K. B. Ramadi, A. M. Graybiel, R. Langer and M. J. Cima, Sci. Rep., 2017, 7, 1952 CrossRef PubMed.
- L. Rao, H. Zhou, T. Li, C. Li and Y. Y. Duan, Acta Biomater., 2012, 8, 2233–2242 CrossRef CAS PubMed.
- Y. Lu, D. Wang, T. Li, X. Zhao, Y. Cao, H. Yang and Y. Y. Duan, Biomaterials, 2009, 30, 4143–4151 CrossRef CAS PubMed.
- E. Azemi, C. F. Lagenaur and X. T. Cui, Biomaterials, 2011, 32, 681–692 CrossRef CAS PubMed.
- J. O. Winter, S. F. Cogan and J. F. Rizzo, J. Biomed. Mater. Res., Part B, 2007, 81, 551–563 CrossRef PubMed.
- Y. Zhong and R. V. Bellamkonda, Brain Res., 2007, 1148, 15–27 CrossRef CAS PubMed.
- U. A. Aregueta-Robles, A. J. Woolley, L. A. Poole-Warren, N. H. Lovell and R. A. Green, Front. Neuroeng., 2014, 7, 15 Search PubMed.
- J. Leach, A. K. H. Achyuta and S. K. Murthy, Front. Neuroeng., 2010, 2, 18 Search PubMed.
- S. S. Rao and J. Winter, Front. Neuroeng., 2009, 2, 6 CAS.
- A. Subramanian, U. M. Krishnan and S. Sethuraman, J. Biomed. Sci., 2009, 16, 108 CrossRef PubMed.
- M. Laura, N. D. Leipzig and M. S. Shoichet, Mater. Today, 2008, 11, 36–43 Search PubMed.
- E. Purcell, J. Seymour, S. Yandamuri and D. Kipke, J. Neural Eng., 2009, 6, 026005 CrossRef CAS PubMed.
- H. Zhou, T. Li and Y. Y. Duan, Sens. Actuators, B, 2012, 161, 198–202 CrossRef CAS.
- S. Sommakia, J. Gaire, J. L. Rickus and K. J. Otto, Front. Neuroeng., 2014, 7, 33 Search PubMed.
- J. Gong, N. Komatsu, T. Nitta and Y. Osada, J. Phys. Chem. B, 1997, 101, 740–745 CrossRef CAS.
- H. Li, A. Erbaş, J. Zwanikken and M. Olvera de la Cruz, Macromolecules, 2016, 49, 9239–9246 CrossRef CAS.
- C. Keplinger, J.-Y. Sun, C. C. Foo, P. Rothemund, G. M. Whitesides and Z. Suo, Science, 2013, 341, 984–987 CrossRef CAS PubMed.
- C. Yang and Z. Suo, Nat. Rev. Mater., 2018, 3, 125–142 CrossRef CAS.
- H. R. Lee, C. C. Kim and J. Y. Sun, Adv. Mater., 2018, 1704403 CrossRef PubMed.
- J. Y. Sun, C. Keplinger, G. M. Whitesides and Z. Suo, Adv. Mater., 2014, 26, 7608–7614 CrossRef CAS PubMed.
- S. Zhao, P. Tseng, J. Grasman, Y. Wang, W. Li, B. Napier, B. Yavuz, Y. Chen, L. Howell and J. Rincon, Adv. Mater., 2018, 30, 1800598 CrossRef PubMed.
- J. Lee, H. Kwon, J. Seo, S. Shin, J. H. Koo, C. Pang, S. Son, J. H. Kim, Y. H. Jang and D. E. Kim, Adv. Mater., 2015, 27, 2433–2439 CrossRef CAS PubMed.
- S. R. Shin, S. M. Jung, M. Zalabany, K. Kim, P. Zorlutuna, S. b. Kim, M. Nikkhah, M. Khabiry, M. Azize and J. Kong, ACS Nano, 2013, 7, 2369–2380 CrossRef CAS PubMed.
- L. Wang, J. Jiang, W. Hua, A. Darabi, X. Song, C. Song, W. Zhong, M. M. Xing and X. Qiu, Adv. Funct. Mater., 2016, 26, 4293–4305 CrossRef CAS.
- T. Dvir, B. P. Timko, M. D. Brigham, S. R. Naik, S. S. Karajanagi, O. Levy, H. Jin, K. K. Parker, R. Langer and D. S. Kohane, Nat. Nanotechnol., 2011, 6, 720 CrossRef CAS PubMed.
- A. Skardal, J. Zhang, L. McCoard, S. Oottamasathien and G. D. Prestwich, Adv. Mater., 2010, 22, 4736–4740 CrossRef CAS PubMed.
- Y. Ahn, H. Lee, D. Lee and Y. Lee, ACS Appl. Mater. Interfaces, 2014, 6, 18401–18407 CrossRef CAS PubMed.
- A. C. Balazs, T. Emrick and T. P. Russell, Science, 2006, 314, 1107–1110 CrossRef CAS PubMed.
- C. Cha, S. R. Shin, N. Annabi, M. R. Dokmeci and A. Khademhosseini, ACS Nano, 2013, 7, 2891–2897 CrossRef CAS PubMed.
- S. Goenka, V. Sant and S. Sant, J. Controlled Release, 2014, 173, 75–88 CrossRef CAS PubMed.
- A. Vashist, A. Kaushik, A. Vashist, V. Sagar, A. Ghosal, Y. Gupta, S. Ahmad and M. Nair, Adv. Healthcare Mater., 2018, 7, 1701213 CrossRef PubMed.
- A. A. Adewunmi, S. Ismail and A. S. Sultan, J. Inorg. Organomet. Polym. Mater., 2016, 26, 717–737 CrossRef CAS.
- S. Ahadian, J. Ramón-Azcón, M. Estili, X. Liang, S. Ostrovidov, H. Shiku, M. Ramalingam, K. Nakajima, Y. Sakka and H. Bae, Sci. Rep., 2014, 4, 4271 CrossRef PubMed.
- S. Pok, F. Vitale, S. L. Eichmann, O. M. Benavides, M. Pasquali and J. G. Jacot, ACS Nano, 2014, 8, 9822–9832 CrossRef CAS PubMed.
- S. H. Lee, H. W. Kim, J. O. Hwang, W. J. Lee, J. Kwon, C. W. Bielawski, R. S. Ruoff and S. O. Kim, Angew. Chem., 2010, 122, 10282–10286 CrossRef.
- Y. Xu, K. Sheng, C. Li and G. Shi, ACS Nano, 2010, 4, 4324–4330 CrossRef CAS PubMed.
- P. Chen, J.-J. Yang, S.-S. Li, Z. Wang, T.-Y. Xiao, Y.-H. Qian and S.-H. Yu, Nano Energy, 2013, 2, 249–256 CrossRef CAS.
- H.-P. Cong, X.-C. Ren, P. Wang and S.-H. Yu, ACS Nano, 2012, 6, 2693–2703 CrossRef CAS PubMed.
- W. Chen and L. Yan, Nanoscale, 2011, 3, 3132–3137 RSC.
- Y. Xu, Q. Wu, Y. Sun, H. Bai and G. Shi, ACS Nano, 2010, 4, 7358–7362 CrossRef CAS PubMed.
- N. Annabi, S. R. Shin, A. Tamayol, M. Miscuglio, M. A. Bakooshli, A. Assmann, P. Mostafalu, J. Y. Sun, S. Mithieux and L. Cheung, Adv. Mater., 2016, 28, 40–49 CrossRef CAS PubMed.
- H. S. Song, O. S. Kwon, J.-H. Kim, J. Conde and N. Artzi, Biosens. Bioelectron., 2017, 89, 187–200 CrossRef CAS PubMed.
- H. Jo, M. Sim, S. Kim, S. Yang, Y. Yoo, J.-H. Park, T. H. Yoon, M.-G. Kim and J. Y. Lee, Acta Biomater., 2017, 48, 100–109 CrossRef CAS PubMed.
- L. Han, X. Lu, M. Wang, D. Gan, W. Deng, K. Wang, L. Fang, K. Liu, C. Chan, Y. Tang, L. T. Weng and H. Yuan, Small, 2017, 13, 1601916 CrossRef PubMed.
- X. Xiao, G. Wu, H. Zhou, K. Qian and J. Hu, Polymers, 2017, 9, 259 CrossRef.
- X. Niu, S. Peng, L. Liu, W. Wen and P. Sheng, Adv. Mater., 2007, 19, 2682–2686 CrossRef CAS.
- N. Matsuhisa, D. Inoue, P. Zalar, H. Jin, Y. Matsuba, A. Itoh, T. Yokota, D. Hashizume and T. Someya, Nat. Mater., 2017, 16, 834 CrossRef CAS PubMed.
- S. Smart, A. Cassady, G. Lu and D. Martin, Carbon, 2006, 44, 1034–1047 CrossRef CAS.
- M. Hussain, M. Kabir and A. Sood, Curr. Sci., 2009, 96, 00113891 Search PubMed.
- M. Berggren and A. Richter-Dahlfors, Adv. Mater., 2007, 19, 3201–3213 CrossRef CAS.
- M. R. Abidian, K. A. Ludwig, T. C. Marzullo, D. C. Martin and D. R. Kipke, Adv. Mater., 2009, 21, 3764–3770 CrossRef CAS PubMed.
- R. Green and M. R. Abidian, Adv. Mater., 2015, 27, 7620–7637 CrossRef CAS PubMed.
- J. Rivnay, S. Inal, A. Salleo, R. M. Owens, M. Berggren and G. G. Malliaras, Nat. Rev. Mater., 2018, 3, 17086 CrossRef CAS.
- H. Shirakawa, E. J. Louis, A. G. MacDiarmid, C. K. Chiang and A. J. Heeger, J. Chem. Soc., Chem. Commun., 1977, 578–580 RSC.
- A. Dyer, J. Reynolds, T. Skotheim and J. Reynolds, Conjugated Polymers: Theory, Synthesis, Properties, and Characterization, CRC Press Taylor & Francic Group, Boca Raton, FL, USA, 3rd edn, 2007 Search PubMed.
- A. Elschner, S. Kirchmeyer, W. Lovenich, U. Merker and K. Reuter, PEDOT: principles and applications of an intrinsically conductive polymer, CRC Press, 2010 Search PubMed.
- E. Stavrinidou, P. Leleux, H. Rajaona, D. Khodagholy, J. Rivnay, M. Lindau, S. Sanaur and G. G. Malliaras, Adv. Mater., 2013, 25, 4488–4493 CrossRef CAS PubMed.
- J. Rivnay, S. Inal, B. A. Collins, M. Sessolo, E. Stavrinidou, X. Strakosas, C. Tassone, D. M. Delongchamp and G. G. Malliaras, Nat. Commun., 2016, 7, 11287 CrossRef PubMed.
- C. M. Proctor, J. Rivnay and G. G. Malliaras, J. Polym. Sci., Part B: Polym. Phys., 2016, 54, 1433–1436 CrossRef CAS.
- J. Rivnay, P. Leleux, M. Ferro, M. Sessolo, A. Williamson, D. A. Koutsouras, D. Khodagholy, M. Ramuz, X. Strakosas and R. M. Owens, Sci. Adv., 2015, 1, e1400251 CrossRef PubMed.
- M. R. Abidian and D. C. Martin, Adv. Funct. Mater., 2009, 19, 573–585 CrossRef CAS.
- M. R. Abidian, J. M. Corey, D. R. Kipke and D. C. Martin, Small, 2010, 6, 421–429 CrossRef CAS PubMed.
- M. R. Abidian, D. H. Kim and D. C. Martin, Adv. Mater., 2006, 18, 405–409 CrossRef CAS PubMed.
- M. R. Abidian and D. C. Martin, Biomaterials, 2008, 29, 1273–1283 CrossRef CAS PubMed.
- S. M. Richardson-Burns, J. L. Hendricks, B. Foster, L. K. Povlich, D.-H. Kim and D. C. Martin, Biomaterials, 2007, 28, 1539–1552 CrossRef CAS PubMed.
- G. M. Cheong, K. S. Lim, A. Jakubowicz, P. J. Martens, L. A. Poole-Warren and R. A. Green, Acta Biomater., 2014, 10, 1216–1226 CrossRef PubMed.
- X. Liu, Z. Yue, M. J. Higgins and G. G. Wallace, Biomaterials, 2011, 32, 7309–7317 CrossRef CAS PubMed.
- R. A. Green, N. H. Lovell and L. A. Poole-Warren, Acta Biomater., 2010, 6, 63–71 CrossRef CAS PubMed.
- R. T. Richardson, A. K. Wise, B. C. Thompson, B. O. Flynn, P. J. Atkinson, N. J. Fretwell, J. B. Fallon, G. G. Wallace, R. K. Shepherd and G. M. Clark, Biomaterials, 2009, 30, 2614–2624 CrossRef CAS PubMed.
- J. Isaksson, P. Kjäll, D. Nilsson, N. Robinson, M. Berggren and A. Richter-Dahlfors, Nat. Mater., 2007, 6, 673 CrossRef CAS PubMed.
- D. T. Simon, S. Kurup, K. C. Larsson, R. Hori, K. Tybrandt, M. Goiny, E. W. Jager, M. Berggren, B. Canlon and A. Richter-Dahlfors, Nat. Mater., 2009, 8, 742 CrossRef CAS PubMed.
- P. M. George, D. A. LaVan, J. A. Burdick, C. Y. Chen, E. Liang and R. Langer, Adv. Mater., 2006, 18, 577–581 CrossRef CAS.
- J. Goding, A. Gilmour, P. Martens, L. Poole-Warren and R. Green, Adv. Healthcare Mater., 2017, 6, 1601177 CrossRef PubMed.
- J. Stejskal, Chem. Pap., 2017, 71, 269–291 CrossRef CAS.
- K. Gilmore, A. Hodgson, B. Luan, C. Small and G. Wallace, Polym. Gels Networks, 1994, 2, 135–143 CrossRef CAS.
- A. Guiseppi-Elie, Biomaterials, 2010, 31, 2701–2716 CrossRef CAS PubMed.
- J. Stejskal, P. Bober, M. Trchová, A. Kovalcik, J. i. Hodan, J. i. Hromádková and J. Prokeš, Macromolecules, 2017, 50, 972–978 CrossRef CAS.
- G.-P. Hao, F. Hippauf, M. Oschatz, F. M. Wisser, A. Leifert, W. Nickel, N. Mohamed-Noriega, Z. Zheng and S. Kaskel, ACS Nano, 2014, 8, 7138–7146 CrossRef CAS PubMed.
- J. Duan, X. Liang, J. Guo, K. Zhu and L. Zhang, Adv. Mater., 2016, 28, 8037–8044 CrossRef CAS PubMed.
- S. Naficy, J. M. Razal, G. M. Spinks, G. G. Wallace and P. G. Whitten, Chem. Mater., 2012, 24, 3425–3433 CrossRef CAS.
- Y. Shi, C. Ma, L. Peng and G. Yu, Adv. Funct. Mater., 2015, 25, 1219–1225 CrossRef CAS.
- S. Zeng, H. Chen, F. Cai, Y. Kang, M. Chen and Q. Li, J. Mater. Chem. A, 2015, 3, 23864–23870 RSC.
- Y. Wu, Y. X. Chen, J. Yan, D. Quinn, P. Dong, S. W. Sawyer and P. Soman, Acta Biomater., 2016, 33, 122–130 CrossRef CAS PubMed.
- H. Hu, J. L. Cadenas, J. M. Saniger and P. Nair, Polym. Int., 1998, 45, 262–270 CrossRef CAS.
- Y. Y. Lee, H. Y. Kang, S. H. Gwon, G. M. Choi, S. M. Lim, J. Y. Sun and Y. C. Joo, Adv. Mater., 2016, 28, 1636–1643 CrossRef CAS PubMed.
- H. Warren, MRS Online Proceedings Library Archive, 2013, 1569, 219–223 Search PubMed.
- J. Hur, K. Im, S. W. Kim, J. Kim, D.-Y. Chung, T.-H. Kim, K. H. Jo, J. H. Hahn, Z. Bao and S. Hwang, ACS Nano, 2014, 8, 10066–10076 CrossRef CAS PubMed.
- S. Yang, L. Jang, S. Kim, J. Yang, K. Yang, S. W. Cho and J. Y. Lee, Macromol. Biosci., 2016, 16, 1653–1661 CrossRef CAS PubMed.
- S. Sekine, Y. Ido, T. Miyake, K. Nagamine and M. Nishizawa, J. Am. Chem. Soc., 2010, 132, 13174–13175 CrossRef CAS PubMed.
- Y. Ido, D. Takahashi, M. Sasaki, K. Nagamine, T. Miyake, P. Jasinski and M. Nishizawa, ACS Macro Lett., 2012, 1, 400–403 CrossRef CAS.
- D. H. Kim, M. Abidian and D. C. Martin, J. Biomed. Mater. Res., Part A, 2004, 71, 577–585 CrossRef PubMed.
- R. Kishi, K. Hiroki, T. Tominaga, K. I. Sano, H. Okuzaki, J. G. Martinez, T. F. Otero and Y. Osada, J. Polym. Sci., Part B: Polym. Phys., 2012, 50, 790–796 CrossRef CAS.
- R. Kishi, K. Kubota, T. Miura, T. Yamaguchi, H. Okuzaki and Y. Osada, J. Mater. Chem. C, 2014, 2, 736–743 RSC.
- J. P. Gong, Y. Katsuyama, T. Kurokawa and Y. Osada, Adv. Mater., 2003, 15, 1155–1158 CrossRef CAS.
- X. Zhao, Soft Matter, 2014, 10, 672–687 RSC.
- T. Dai, X. Qing, H. Zhou, C. Shen, J. Wang and Y. Lu, Synth. Met., 2010, 160, 791–796 CrossRef CAS.
- W. Li, F. Gao, X. Wang, N. Zhang and M. Ma, Angew. Chem., 2016, 128, 9342–9347 CrossRef.
- W. Zhao, Z. Han, L. Ma, S. Sun and C. Zhao, J. Mater. Chem. B, 2016, 4, 8016–8024 RSC.
- F. Zhu, J. Lin, Z. L. Wu, S. Qu, J. Yin, J. Qian and Q. Zheng, ACS Appl. Mater. Interfaces, 2018, 10, 13685–13692 CrossRef CAS PubMed.
- S. Ghosh and O. Inganäs, Adv. Mater., 1999, 11, 1214–1218 CrossRef CAS.
- L. Pan, G. Yu, D. Zhai, H. R. Lee, W. Zhao, N. Liu, H. Wang, B. C.-K. Tee, Y. Shi, Y. Cui and Z. Bao, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 9287–9292 CrossRef CAS PubMed.
- D. Mawad, E. Stewart, D. L. Officer, T. Romeo, P. Wagner, K. Wagner and G. G. Wallace, Adv. Funct. Mater., 2012, 22, 2692–2699 CrossRef CAS.
- Y. Wang, Y. Shi, L. Pan, Y. Ding, Y. Zhao, Y. Li, Y. Shi and G. Yu, Nano Lett., 2015, 15, 7736–7741 CrossRef CAS PubMed.
- B. Yao, H. Wang, Q. Zhou, M. Wu, M. Zhang, C. Li and G. Shi, Adv. Mater., 2017, 29, 1700974 CrossRef PubMed.
- C. E. Schmidt, V. R. Shastri, J. P. Vacanti and R. Langer, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 8948–8953 CrossRef CAS.
- Y. Xu, X. Yang, A. K. Thomas, P. A. Patsis, T. Kurth, M. Kräter, K. Eckert, M. Bornhäuser and Y. Zhang, ACS Appl. Mater. Interfaces, 2018, 10, 14418–14425 CrossRef CAS PubMed.
- Y. Lu, W. He, T. Cao, H. Guo, Y. Zhang, Q. Li, Z. Shao, Y. Cui and X. Zhang, Sci. Rep., 2014, 4, 5792 CrossRef PubMed.
- H. Yuk, T. Zhang, S. Lin, G. A. Parada and X. Zhao, Nat. Mater., 2016, 15, 190 CrossRef CAS PubMed.
- T. Zhang, H. Yuk, S. Lin, G. A. Parada and X. Zhao, Acta Mech. Sin., 2017, 33, 543–554 CrossRef CAS.
- H. Yuk, T. Zhang, G. A. Parada, X. Liu and X. Zhao, Nat. Commun., 2016, 7, 12028 CrossRef PubMed.
- X. Liu, T.-C. Tang, E. Tham, H. Yuk, S. Lin, T. K. Lu and X. Zhao, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 2200–2205 CrossRef CAS PubMed.
- Q. Liu, G. Nian, C. Yang, S. Qu and Z. Suo, Nat. Commun., 2018, 9, 846 CrossRef PubMed.
- J. Li, A. Celiz, J. Yang, Q. Yang, I. Wamala, W. Whyte, B. Seo, N. Vasilyev, J. Vlassak and Z. Suo, Science, 2017, 357, 378–381 CrossRef CAS PubMed.
- J. Yang, R. Bai and Z. Suo, Adv. Mater., 2018, 1800671 CrossRef PubMed.
- Y. Lee, S. H. Cha, Y.-W. Kim, D. Choi and J.-Y. Sun, Nat. Commun., 2018, 9, 1804 CrossRef PubMed.
- M. Y. Choi and C. K. Chan, Environ. Sci. Technol., 2002, 36, 2422–2428 CrossRef CAS PubMed.
- A. Gregorová, N. Saha, T. Kitano and P. Sáha, Carbohydr. Polym., 2015, 117, 559–568 CrossRef PubMed.
- L. Gong, R. Wang, Z. Xia and C. Chen, J. Chem. Eng. Data, 2010, 55, 2920–2923 CrossRef CAS.
- Y. Bai, B. Chen, F. Xiang, J. Zhou, H. Wang and Z. Suo, Appl. Phys. Lett., 2014, 105, 151903 CrossRef.
- P. Le Floch, X. Yao, Q. Liu, Z. Wang, G. Nian, Y. Sun, L. Jia and Z. Suo, ACS Appl. Mater. Interfaces, 2017, 9, 25542–25552 CrossRef CAS PubMed.
- J. A. Rogers, T. Someya and Y. Huang, Science, 2010, 327, 1603–1607 CrossRef CAS PubMed.
- C. Xie, J. Liu, T.-M. Fu, X. Dai, W. Zhou and C. M. Lieber, Nat. Mater., 2015, 14, 1286 CrossRef CAS PubMed.
- M. Kaltenbrunner, T. Sekitani, J. Reeder, T. Yokota, K. Kuribara, T. Tokuhara, M. Drack, R. Schwödiauer, I. Graz and S. Bauer-Gogonea, Nature, 2013, 499, 458 CrossRef CAS PubMed.
- A. Canales, X. Jia, U. P. Froriep, R. A. Koppes, C. M. Tringides, J. Selvidge, C. Lu, C. Hou, L. Wei and Y. Fink, Nat. Biotechnol., 2015, 33, 277 CrossRef CAS PubMed.
- S. Park, Y. Guo, X. Jia, H. K. Choe, B. Grena, J. Kang, J. Park, C. Lu, A. Canales and R. Chen, Nat. Neurosci., 2017, 20, 612 CrossRef CAS PubMed.
- Z. Zhu, S. Ling, J. Yeo, S. Zhao, L. Tozzi, M. J. Buehler, F. Omenetto, C. Li and D. L. Kaplan, Adv. Funct. Mater., 2018, 28, 1704757 CrossRef.
- T. Boland, T. Xu, B. Damon and X. Cui, Biotechnol. J., 2006, 1, 910–917 CrossRef CAS PubMed.
- K. Pataky, T. Braschler, A. Negro, P. Renaud, M. P. Lutolf and J. Brugger, Adv. Mater., 2012, 24, 391–396 CrossRef CAS PubMed.
- B. Dhariwala, E. Hunt and T. Boland, Tissue Eng., 2004, 10, 1316–1322 CrossRef CAS PubMed.
- V. Chan, P. Zorlutuna, J. H. Jeong, H. Kong and R. Bashir, Lab Chip, 2010, 10, 2062–2070 RSC.
- D. B. Kolesky, R. L. Truby, A. S. Gladman, T. A. Busbee, K. A. Homan and J. A. Lewis, Adv. Mater., 2014, 26, 3124–3130 CrossRef CAS PubMed.
- K. Tian, J. Bae, S. E. Bakarich, C. Yang, R. D. Gately, G. M. Spinks, M. in het Panhuis, Z. Suo and J. J. Vlassak, Adv. Mater., 2017, 29, 1604827 CrossRef PubMed.
- H. Yuk and X. Zhao, Adv. Mater., 2018, 30, 1704028 CrossRef PubMed.
- X. Liu, H. Yuk, S. Lin, G. A. Parada, T. C. Tang, E. Tham, C. de la Fuente-Nunez, T. K. Lu and X. Zhao, Adv. Mater., 2018, 30, 1704821 CrossRef PubMed.
- S. Agarwala, J. M. Lee, W. L. Ng, M. Layani, W. Y. Yeong and S. Magdassi, Biosens. Bioelectron., 2018, 102, 365–371 CrossRef CAS PubMed.
- S. Agarwala, J. M. Lee, W. Y. Yeong, M. Layani and S. Magdassi, MRS Adv., 2018, 1–7 Search PubMed.
- J.-Y. Sun, X. Zhao, W. R. Illeperuma, O. Chaudhuri, K. H. Oh, D. J. Mooney, J. J. Vlassak and Z. Suo, Nature, 2012, 489, 133 CrossRef CAS PubMed.
- S.-W. Hwang, H. Tao, D.-H. Kim, H. Cheng, J.-K. Song, E. Rill, M. A. Brenckle, B. Panilaitis, S. M. Won and Y.-S. Kim, Science, 2012, 337, 1640–1644 CrossRef CAS PubMed.
- S.-K. Kang, R. K. Murphy, S.-W. Hwang, S. M. Lee, D. V. Harburg, N. A. Krueger, J. Shin, P. Gamble, H. Cheng and S. Yu, Nature, 2016, 530, 71–76 CrossRef CAS PubMed.
- B. Guo, A. Finne-Wistrand and A.-C. Albertsson, Biomacromolecules, 2011, 12, 2601–2609 CrossRef CAS PubMed.
- B. Guo, A. Finne-Wistrand and A.-C. Albertsson, Chem. Mater., 2011, 23, 1254–1262 CrossRef CAS.
- B. Guo, L. Glavas and A.-C. Albertsson, Prog. Polym. Sci., 2013, 38, 1263–1286 CrossRef CAS.
- T. C. Tseng, L. Tao, F. Y. Hsieh, Y. Wei, I. M. Chiu and S. h. Hsu, Adv. Mater., 2015, 27, 3518–3524 CrossRef CAS PubMed.
- D. L. Taylor and M. in het Panhuis, Adv. Mater., 2016, 28, 9060–9093 CrossRef CAS PubMed.
- R. Dong, X. Zhao, B. Guo and P. X. Ma, ACS Appl. Mater. Interfaces, 2016, 8, 17138–17150 CrossRef CAS PubMed.
- T. Wang, Y. Zhang, Q. Liu, W. Cheng, X. Wang, L. Pan, B. Xu and H. Xu, Adv. Funct. Mater., 2018, 28, 1705551 CrossRef.
- S. Liang, Y. Zhang, H. Wang, Z. Xu, J. Chen, R. Bao, B. Tan, Y. Cui, G. Fan, W. Wang, W. Wang and W. Liu, Adv. Mater., 2018, 0, 1704235 CrossRef PubMed.
- H. Sirringhaus, T. Kawase, R. Friend, T. Shimoda, M. Inbasekaran, W. Wu and E. Woo, Science, 2000, 290, 2123–2126 CrossRef CAS PubMed.
- Y. Wang, C. Zhu, R. Pfattner, H. Yan, L. Jin, S. Chen, F. Molina-Lopez, F. Lissel, J. Liu and N. I. Rabiah, Sci. Adv., 2017, 3, e1602076 CrossRef PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |