
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAcceptor reactivity in glycosylation reactions
Stefan
van der Vorm
 ,
Thomas
Hansen
,
Jacob M. A.
van Hengst
,
Herman S.
Overkleeft
,
Gijsbert A.
van der Marel
and
Jeroen D. C.
Codée
*
,
Thomas
Hansen
,
Jacob M. A.
van Hengst
,
Herman S.
Overkleeft
,
Gijsbert A.
van der Marel
and
Jeroen D. C.
Codée
*
Leiden Institute of Chemistry, Leiden University, Einsteinweg 55, 2333 CC Leiden, The Netherlands. E-mail: jcodee@chem.leidenuniv.nl
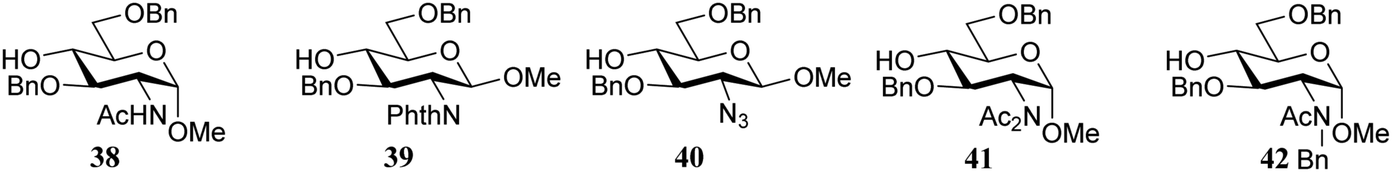
First published on 9th July 2019
Abstract
The outcome of a glycosylation reaction critically depends on the reactivity of all reaction partners involved: the donor glycoside (the electrophile), the activator (that generally provides the leaving group on the activated donor species) and the glycosyl acceptor (the nucleophile). The influence of the donor on the outcome of a glycosylation reaction is well appreciated and documented. Differences in donor reactivity have led to the development of chemoselective glycosylation reactions and the reactivity of donor glycosides has been tuned to affect stereoselective glycosylation reactions. The quantification of donor reactivity has enabled the conception of streamlined one-pot glycosylation sequences. In contrast, although it has long been known that the nature and the reactivity of the nucleophile influence the outcome of a glycosylation, the knowledge of acceptor reactivity and insight into the consequences thereof are often circumstantial or anecdotal. This review documents how the reactivity impacts the glycosylation reaction outcome both in terms of chemical yield and stereoselectivity. The effect of acceptor nucleophilicity on the reaction mechanism is described and steric, conformational and electronic influences are outlined. Quantitative and computational approaches to comprehend acceptor nucleophilicity are assessed. The increasing insight into the stereoelectronic effects governing glycoside reactivity will eventually enable the conception of effective stereoselective glycosylation methodology that can be tuned to the reaction partners at hand.
 From left to right: Jacob van Hengst, Jeroen Codée, Hermen Overkleeft, Thomas Hansen, Gijs van der Marel and Stefan van der Vorm | Jacob van Hengst obtained his BS degree in Molecular Science and Technology (2014) and a MS degree in Chemistry (2017) from Leiden University. In 2017 he started his PhD research under the guidance of Jeroen Codée and Gijs van der Marel, investigating how the stereochemistry and protecting group pattern of both the donor and acceptor glycoside building blocks affect the course of glycosylation reactions. Jeroen Codée obtained his PhD degree from Leiden University (2004) under the guidance of Jacques van Boom and Stan van Boeckel investigating thioglycosides in the assembly of heparin and heparan sulfates. After a post-doctoral stay at the ETH Zürich with Peter Seeberger, working on automated and flow synthesis, he returned to Leiden University, where he currently is Associate Professor. He was awarded the Carbohydrate Research Award for Creativity in Carbohydrate Chemistry in 2017 and his research interests include synthetic carbohydrate chemistry, reaction mechanisms, glycobiology and glycoimmunology. Hermen Overkleeft received his PhD degree from the University of Amsterdam (1997) and, after post-doctoral stays at Leiden University (1997–1999, with Van Boom and Van der Marel) and Harvard Medical School (1999–2001, with Hidde Ploegh) he became Full Professor at Leiden University, where he currently is the director of the Leiden Institute of Chemistry. Trained as an organic chemist, his work centers around the design and development of covalent and competitive glycosidase and peptidase inhibitors for chemical biology research. He is the recipient of the Jeremy Knowles Award of the Royal Society of Chemistry to promote interdisciplinary research between chemistry and the life sciences. He is a Fellow of the Royal Society of Chemistry (UK) and an elected member of the Royal Netherlands Academy of Arts and Sciences (Koninklijke Nederlandse Akademie van Wetenschappen – KNAW). Thomas Hansen obtained his bachelor degree from the Leiden University of Applied Sciences (2013) and his master degree summa cum laude from Leiden University in 2015. Directly after he started his PhD research under guidance of Jeroen Codée and Gijs van der Marel developing computational tools to study the glycosylation reaction mechanism. He has recently introduced a computational strategy to study the conformational behavior and reactivity of glycosyl oxocarbenium ions. His research interests include computational chemistry, synthetic carbohydrate chemistry and unraveling reaction mechanisms. Gijs van der Marel is Full Professor in Organic Chemistry at Leiden University. He trained with Prof. Jacques van Boom on the development of synthetic methods for oligonucleotides and obtained his PhD degree in 1981. His research has been directed at the development of synthetic chemistry to assemble all types of biopolymers: nucleic acids, peptides and carbohydrates as well as hybrids and analogues thereof to study their role in biology. He has supervised > 100 PhD students and his research interests include synthetic organic chemistry methodology, biopolymer synthesis, glycobiology and chemical biology and immunology. Stefan van der Vorm obtained his BSc Molecular Science and Technology degree from Leiden University in 2010. In 2013 he completed his Master degree in Chemistry and in the same year he started his PhD research with Jeroen Codée and Gijs van der Marel. In 2018 he completed his PhD thesis “Reactivity and Selectivity in Glycosylation Reactions” which describes the development of tools to study glycosylation reactions mechanisms, and the effect of donor and acceptor reactivity thereon. Reactivity scales have been introduced to map the effect of acceptor nucleophilicity on the stereoselectivity of glycosylation reactions. Currently Stefan is lecturer in organic chemistry at Leiden University. |
Introduction
Synthetic oligosaccharides and glycoconjugates are extremely valuable research tools for biomedical and biotechnological purposes and synthetic oligosaccharides have made it into the clinic to replace naturally sourced oligosaccharides that are structurally less well-defined and more heterogeneous. Notwithstanding these successes, the assembly of complex oligosaccharides continues to be a time and labor consuming process, as a result of the lack of general glycosylation procedures and the many variables that play a role in a chemical glycosylation reaction.1–3 In a traditional (Lewis) acid catalyzed reaction, the donor is activated to produce a reactive electrophilic species which then reacts with the incoming nucleophile, the “acceptor”. Over the years significant progress has been made in understanding and harnessing the reactivity of the donor glycoside and insight into the effect of the ring substituents and protecting group patterns on the reactivity of the donor building block has allowed the generation of effective chemoselective and orthogonal glycosylation strategies as well as enabled the development of stereoselective glycosylation methodology.4 The reactivity of the acceptor, on the other hand, is less well studied and often taken for granted.5 At the same time, it is well appreciated that the nature of the acceptor can have a major influence on the outcome of a glycosylation reaction, both in terms of isolated yield and stereoselectivity. Numerous examples of glycosylation reactions have shown that the reactivity of the acceptor, like that of glycosyl donors, can be manipulated by changing protecting groups.6 Unfortunately, most studies that report on new glycosylation methods, strategies or mechanisms, employ a rather variable set of acceptors, often chosen because of ease of availability, or used because a target oriented approach is taken. As a result, the acceptors used in these studies differ greatly in steric and electronic properties, making it difficult to establish clear structure–reactivity relationships.7 Unexpected stereoselectivities and/or poor yields, as a result of ill-understood acceptor reactivity, are continuously being reported,8–12 and indicate the need for deeper insight into carbohydrate acceptor reactivity and its effect of the outcome of glycosylation reactions. At a time when the mechanism of the glycosylation reaction is understood better than ever before13 and insight in and control over donor reactivity has taken shape it is clear that understanding and harnessing the reactivity of the glycosyl acceptor is crucial for the development of more general glycosylation methodology and to remedy the need for ill-defined and time consuming reaction optimization procedures, that have thwarted the field for so long. This review aims to provide an overview of our current understanding of the structural features influencing acceptor reactivity and the effect thereof on the outcome of glycosylation reactions. It will survey the systematic approaches that have been undertaken to probe, analyze and quantify acceptor reactivity.Observations on acceptor reactivity
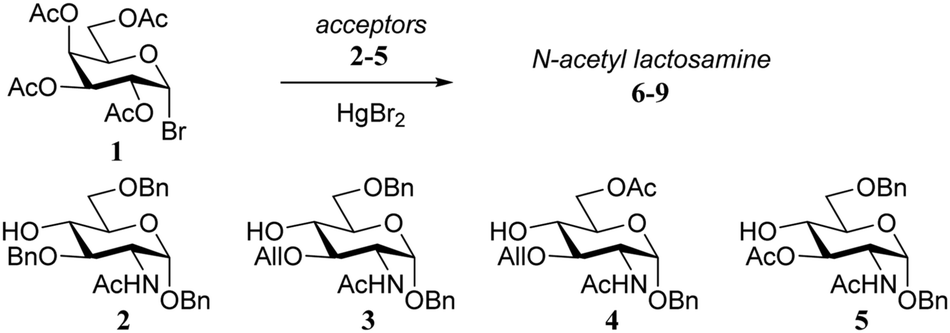
In one early example, Sinaÿ and co-workers14 described the clear influence of the protecting groups of the acceptor on the outcome of glycosylations of galactosyl bromide 1 (Table 1). N-Acetyl-glucosamine acceptors 2–5, with an O-benzyl (2) or O-allyl (3, 4) group at C-3 gave good yields, regardless of the nature of the protecting group at C-6 (O-benzyl or O-acetyl), but the yield of the condensation dropped to a mere 5% when the acceptor 5, bearing an O-acetyl at C-3 was used.|
|
||
|---|---|---|
| Acceptor | Product | Yield (%) |
| All glycosylations proceeded with exclusive β-selectivity. | ||
| 2 | 6 | 87 |
| 3 | 7 | 77 |
| 4 | 8 | 78 |
| 5 | 9 | 5 |
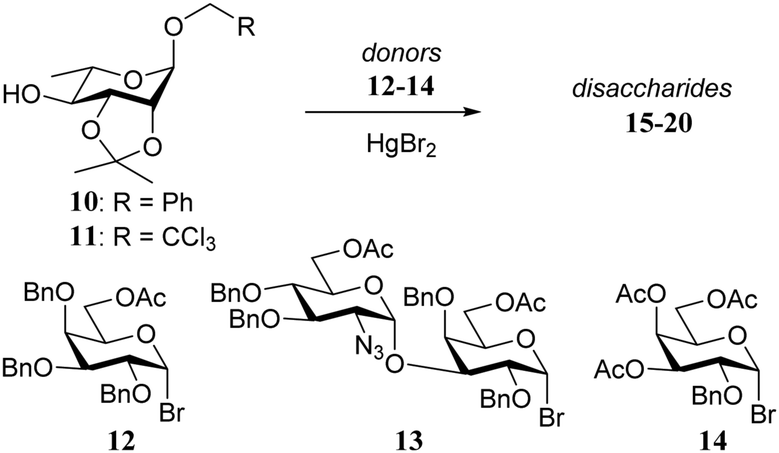
In 1981 Paulsen and Lockhoff examined a set of donors (12–14, Table 2) with two very similar rhamnosyl acceptors, differing only in the anomeric protection (O-benzyl in 10vs. O-trichloroethyl in 11).15 In this set of experiments both the influence of the reactivity of the donor (12 > 13 > 14) and acceptor (10 > 11) became evident. Formation of the β-linked products was explained by assuming a direct displacement of the anomeric α-bromides, while the α-galactosyl linkages were thought to arise from the corresponding β-bromides, formed by in situ anomerization of the α-bromides with HgBr2. Reactive acceptors can take the direct substitution pathway displacing the α-bromide, while less reactive nucleophiles require the more reactive β-bromides for an effective reaction. Following this line of reasoning, the trichloroethyl protected rhamnosyl acceptor 11, providing more of the α-linked products than its benzyl protected analogue 10, was found to be significantly less reactive than its benzyl counterpart.
|
|
||||
|---|---|---|---|---|
| Donor | Acceptor 10 | Acceptor 11 | ||
| Product | α![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :β (yield) :β (yield) |
Product | α:β (yield) |
|
| Yields of combined isolated anomers. Reagents and conditions: donor (1 eq.), acceptor (1 eq.), powdered 4 Å M.S., HgBr2 (0.1 eq.), DCM, room temperature (20), 0 °C (17), or −20 °C (15, 16, 18, 19). | ||||
| 12 | 15 | 19:81 (75%) |
18 | 81:19 (82%) |
| 13 | 16 | 34:66 (66%) |
19 | 100:0 (54%) |
| 14 | 17 | 100:0 (81%) |
20 | 100:0 (87%) |
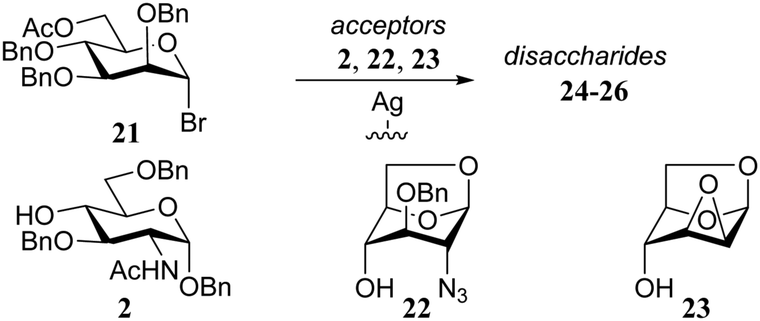
In another example, Paulsen and Lebuhn probed the silver-silicate promoted glycosylation of mannosyl bromide 21 with different glucose and glucosamine acceptors (Table 3). While the conformationally locked glucosamine acceptor 22 and mannose acceptor 23 proved to be capable of direct SN2-type displacement of activated α-bromide, leading to the synthesis of 1,2-cis-linked disaccharides 25 and 26, the use of N-acetyl glucosamine 2 only delivered the undesired α-product, possibly through the intermediacy of an oxocarbenium-like intermediate that is attacked from the α-face.16
|
|
|||
|---|---|---|---|
| Acceptor | Product | Yield (%) | α:β |
| Yields of the β-anomer. Reagents and conditions: donor (1.1 eq.), acceptor (1 eq.), powdered 4 Å M.S., silver-silicate, DCM, room temperature (or 35 °C for 24). | |||
| 2 | 24 | 75 | α only |
| 22 | 25 | 65 | 1:6 |
| 23 | 26 | 63 | 1:5.5 |
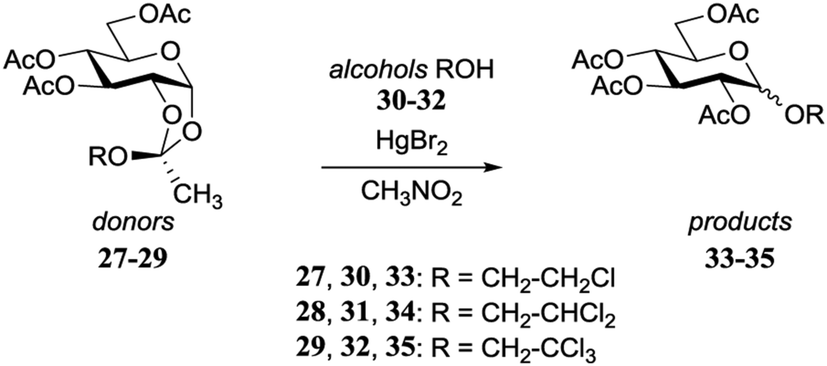
Garegg and Kvarnström provided an early example how the stereochemical outcome of a glycosylation reaction can be influenced by the reactivity of the acceptor nucleophile.17,18 Through a Kochetkov orthoester glycosylation reaction, different orthoesters (27–29) were converted in the presence of the corresponding alcohol (30–32) under the aegis of 0.33 eq. HgBr2 in refluxing CH3NO2, into the α/β-glycosides 33–35 (Table 4). A gradual change in stereoselectivity is observed depending on the orthoester/alcohol functionality. The dichloroethanol system provided an unselective glycosylation, while the more electron rich monochloroethanol showed moderate β-selectivity and the more electron poor trichloroethanol led to a slightly α-selective reaction.
|
|
||||
|---|---|---|---|---|
| Donor | Alcohol | Product | Yield (%) | α:β |
| Reagents and conditions: donor (1 eq.), alcohol (2 eq.), HgBr2 (0.33 eq.), CH3NO2 reflux, 15 min. | ||||
| 27 | 30 | 33 | 87 | 16:84 |
| 28 | 31 | 34 | 83 | 50:50 |
| 29 | 32 | 35 | 78 | 67:33 |
Over the years it has become clear that N-acetylglucosamine C-4-OH acceptors are generally very poor nucleophiles.5 In a detailed study by Crich and co-workers, several glucosamine acceptors, bearing different N-protecting groups (38–42, Table 5) were used to unearth the underlying reasons why these acceptors behave so poorly in glycosylation reactions.19 Glycosylations of these acceptors with mannosyl sulfoxide 36 are reported in Table 5 and the results showed glucosazide 40 to be superior to the other acceptors, based on the yield of the reactions. Diamides 41 and 39 were found to be more effective nucleophiles than acetamide 38. In a competition experiments in which 38, 39, and 40 competed for the same activated donor, products 46, 47 and 48 were formed in a 1:3:10 ratio, corroborating the results of the individual glycosylations.
|
|
||||
|---|---|---|---|---|
| Acceptor | Product | Yield (%) | α:β |
|
| Reagents and conditions: for 36: donor (0.2 mmol), DTBMP (0.4 mmol), Tf2O (0.22 mmol), DCM (8 mL), then acceptor (0.4 mmol, 2 mL DCM), −78 °C to 0 °C; for 37: donor (0.1 mmol), Ph2SO (0.28 mmol) Tf2O (0.15 mmol), toluene/DCM (3/1, 1 mL), −78 °C to −40 °C then TTBP (0.5 mmol, 0.5 mL DCM), acceptor (0.1 mmol, 1 mL DCM), −78 °C to room temperature. | ||||
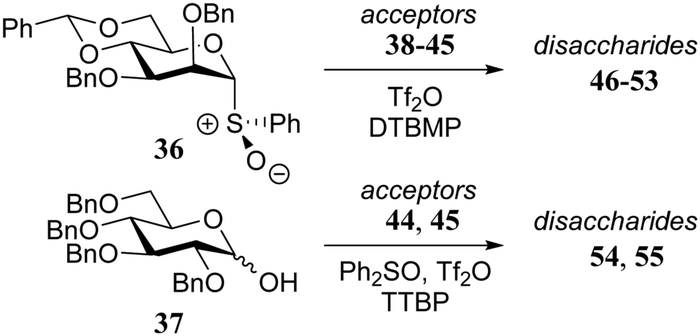
|
38 | 46 | 9 | β only |
| 39 | 47 | 53 | β only | |
| 40 | 48 | 70 | β only | |
| 41 | 49 | 47 | β only | |
| 42 | 50 | 39 | β only | |
| 43 | 51 | 8 | β only | |
| 44 | 52 | 63 | β only | |
| 45 | 53 | 39 | β only | |
| 44 | 54 | 87 | 1:1.2 |
|
| 45 | 55 | 18 | 1:2.4 |
|

|
||||
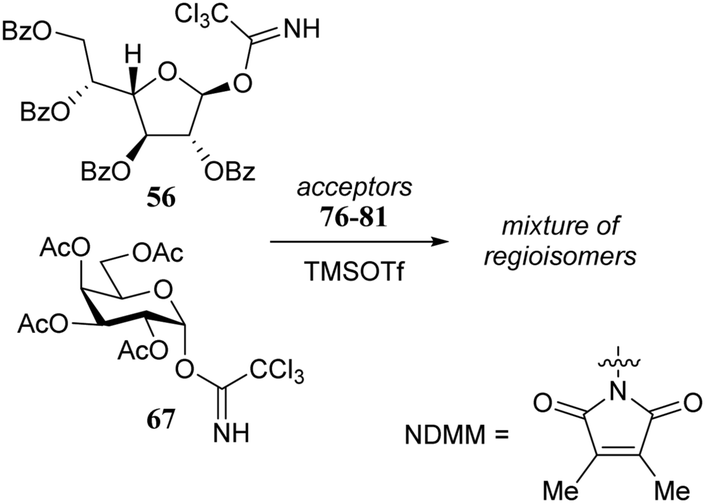
It was reasoned that the poor reactivity of acceptor 38 originated from an intermolecular hydrogen-bonding network involving the amide functionality. To substantiate this assumption, picolyl protected 43 and 44 were prepared to disrupt the intermolecular network by introducing an intramolecular hydrogen-bond between the picolyl nitrogen and the amide hydrogen. Experiments using acceptor 44, bearing the C-3-O-picolyl ether and its C-3-O-benzyl counterpart 45, showed that for the primary alcohol in 44 disruption of the intermolecular hydrogen-bond network is effective, leading to higher glycosylation yields for picolyl acceptor 44 to product 52. It sorted no effect in increasing the reactivity of the C-4-OH in 43 with respect to acceptor 38 as both glycosylations proceeded with a similarly poor yield. This result was explained by the possibility of the picolyl nitrogen in 43 to form either a hydrogen-bond with the C-4-OH or with the C-2-amide NH. Acceptors 44 and 45 were made to compete in a glycosylation with sulfoxide donor 36 and this experiment resulted in a 2:1 mixture of disaccharides 52:53, corroborating the findings of the individual glycosylations. Acceptors 44 and 45 were also used in dehydrative glycosylations with donor 37 to show how the reactivity difference between the two acceptors translates into a large difference in yield between products 55 and 56.
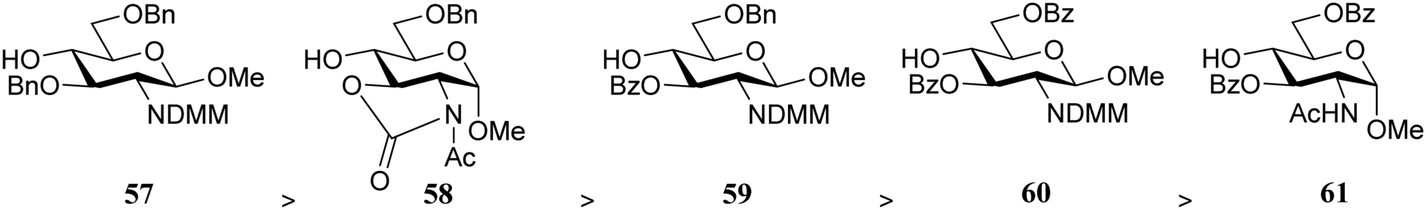
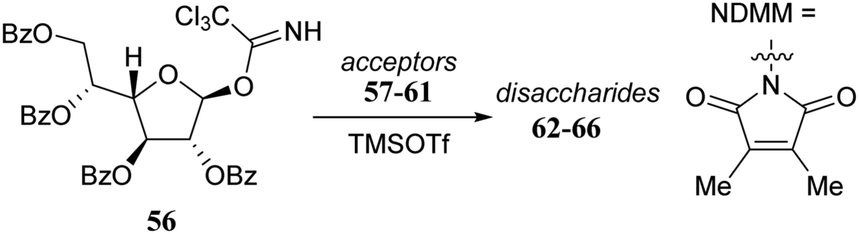
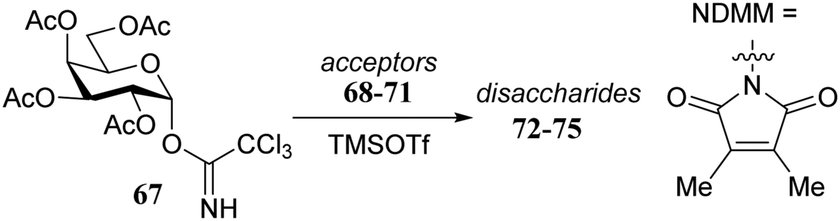
Rúveda and co-workers investigated the relative reactivities of a series of dimethylmaleimide (DMM) protected glucosamine acceptors (57, 59, and 60, Table 6) by competition experiments using galactofuranose donor 56.20 The reactivity of these nucleophiles was compared to that of N-acetyl glucosamine acceptor 61 and cyclic carbamate 58. The cyclic nature of the 2-N-3-O-carbamate in the latter glucosamine ties back the group at C-3, rendering the C-4-OH more accessible and thus a better nucleophile.21–23
|
|
||
|---|---|---|
| Acceptors | Products | Ratio |
| Reagents and conditions: two acceptors (1 eq. each), donor (1.2 eq.), TMSOTf (1.25 eq.), 4 Å M.S., DCM/CH3CN (29/1, 0.34 M), −30 °C. | ||
|
58:57 |
62:63 |
1:4 |
|
58:59 |
62:64 |
1.5:1 |
|
58:60 |
62:65 |
7:1 |
|
58:61 |
62:66 |
11:1 |
|
||
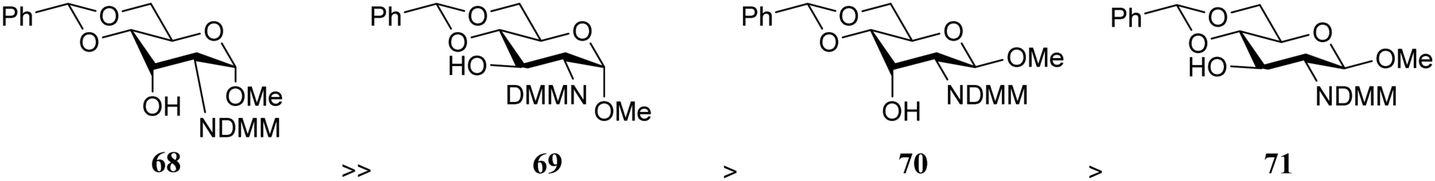
From the results in Table 6 it becomes clear that benzoyl groups in the acceptor have a retarding effect on the glycosylation rate. In this study the poor reactivity of N-actyl glucosamine 61 again becomes apparent. In a second set of competition experiments, the reactivity of allosamine and glucosamine acceptors bearing the DMM-protecting group, were assessed in glycosylations with galactopyranosyl donor 67 (Table 7).24 Allosamine 68 outcompeted the epimeric acceptors 69 and 70. This relatively high reactivity was related to an activating H-bond that can be formed between the DMM carbonyl and the axial C-3-OH in 68, which was supported by NMR and computational studies. Notably this reactivity series reveals, that axial-orientated hydroxyl groups are not always poorer nucleophiles; a commonly regarded notion that is primarily based on steric arguments.25
|
|
||
|---|---|---|
| Acceptors | Products | Ratio |
| Reagents and conditions: two acceptors (1 eq. each), donor (1.1 eq.), TMSOTf (0.28 eq.), 4 Å M.S., DCM, −25 °C. | ||
|
68:69 |
72:73 |
10:1 |
|
68:70 |
72:74 |
13:1 |
|
69:70 |
73:74 |
2:1 |
|
69:71 |
73:75 |
5:1 |
|
70:71 |
74:75 |
3:1 |
|
||
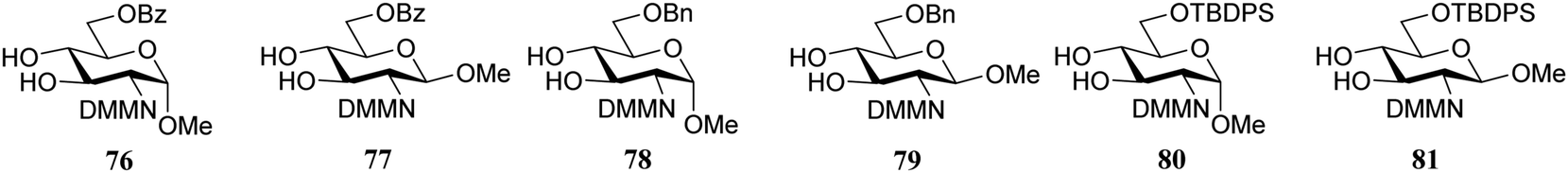
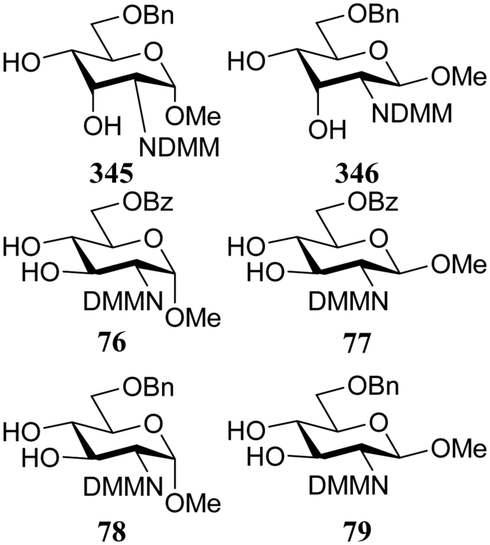
Rúveda and co-workers further explored the DMM-glucosamine series in a set of glycosylation reactions in which the relative reactivity of C-3-OH and C-4-OH nucleophiles were tested.26 The regioselectivity for glycosylation at the C-3-OH over the C-4-OH increased in the order of C-6-OBz > C-6-OTBDPS > C-6-OBn showing that the electron withdrawing benzoyl at C-6 diminishes the reactivity of the proximal C-4-OH with respect to the C-3-OH (C-3/C-4, 1:0 for 56, and 2:1 for 67, Table 8). The bulky TBDPS in acceptor 80 sterically hinders the nucleophilic attack of the C-4-OH, leading to increased C-3/C-4-regioselectivity with respect to the glycosylation of the C-6-OBn acceptor 78 (compare 5:1 for 80 and 3.2:1 for 78, with donor 56). Notably, the relative reactivity of the acceptors was more similar in glycosylations using donor 67 and the glycosylations of the β-anomeric acceptors (77, 79, 81) also showed different regioselectivities, favouring the C-4-OH nucleophile, which was attributed to the difference in hydrogen-bonding capacity of the DMM group with the C-3-OH in the different anomers.27,28
|
|
||||
|---|---|---|---|---|
| Donor | Acceptor | Yield (%) | (1 → 3):(1 → 4) |
|
| Reagents and conditions: donor (0.11 mmol, 1.1 eq.), acceptor (0.1 mmol, 1 eq.), 4 Å M.S., TMSOTf (0.21 mmol, 2.1 eq.), DCM/CH3CN (37/1), −25 °C. | ||||
|
56 | 76 | 68 | 1:0 |
| 56 | 77 | 75 | 1:1 |
|
| 56 | 78 | 71 | 3.2:1 |
|
| 56 | 79 | 71 | 1:2.9 |
|
| 56 | 80 | 73 | 5:1 |
|
| 56 | 81 | 87 | 1:2.2 |
|
| 67 | 76 | 91 | 2:1 |
|
| 67 | 77 | 74 | 1:13 |
|
| 67 | 78 | 56 | 1:1 |
|
| 67 | 79 | 88 | 0:1 |
|
| 67 | 80 | 50 | 1.6:1 |
|
| 67 | 81 | 88 | 0:1 |
|
Steric and conformational effects
It is difficult to separate individual steric or electronic contributions of the different functional groups on the overall reactivity of a glycosyl acceptor alcohol as these effects are heavily intertwined. In the following section, selected examples of glycosylation reactions are provided, of which the relative stereochemical outcome can be understood to result from changes in steric and conformational effects.As a first example the model thiodonors 82 and 83 are compared in glycosylation reactions with a set of acceptors (86–89) of increasing steric demands.29 Because donors 82 and 83 only carry a single electronwithdrawing substituent, they are rather reactive and substitution reactions on these donors likely proceed through a dissociative mechanism. Two observations merit attention. First, with increasing steric demand of the acceptor nucleophile more α-product is formed for both donors. Second, the uronic acid donor provides products with a larger degree of β-selectivity than the benzyloxymethyl donor. To account for the observed stereoselectivity of the reactions, the half-chairs 84 and 85 were proposed to be product forming intermediates. Structure 85 with its equatorial substituent is the predominant conformer when R is large, whereas in structure 84, the smaller carboxylic acid ester can provide better electronic stabilization of the positive charge at the anomeric center when taking up an axial orientation. Favourable top-face attack then delivers the β-glycoside. With larger acceptors a Curtin–Hammett-type scenario takes place, in which the energetically less favorable conformer contributes more to the final product distribution, because there are less steric interactions in the transition state leading from this oxocarbenium ion. The two half-chairs of the benzyloxymethyl ion are more similar in energy, explaining the higher α-selectivity of donor 83 (Table 9).
|
|
||||
|---|---|---|---|---|
| Donor | Acceptor | Product | Yield (%) | α:β |
| Reagents and conditions: donor (1 eq.), Ph2SO (1.2 eq.), TTBP (3 eq.), Tf2O (1.1 eq.), 3 Å M.S., DCM (0.05 M), −78 °C, then acceptor (4 eq.) in DCM (5 M) −78 °C, 15 min. | ||||
| 82 | 86 | 90 | 67 | 1:7.7 |
| 82 | 87 | 91 | 48 | 1:3.8 |
| 82 | 88 | 92 | 52 | 1:2.9 |
| 82 | 89 | 93 | 52 | 1:1.2 |
| 83 | 86 | 94 | 82 | 1:1.4 |
| 83 | 87 | 95 | 61 | 1.7:1 |
| 83 | 88 | 96 | 60 | 2.6:1 |
| 83 | 89 | 97 | 74 | 3:1 |
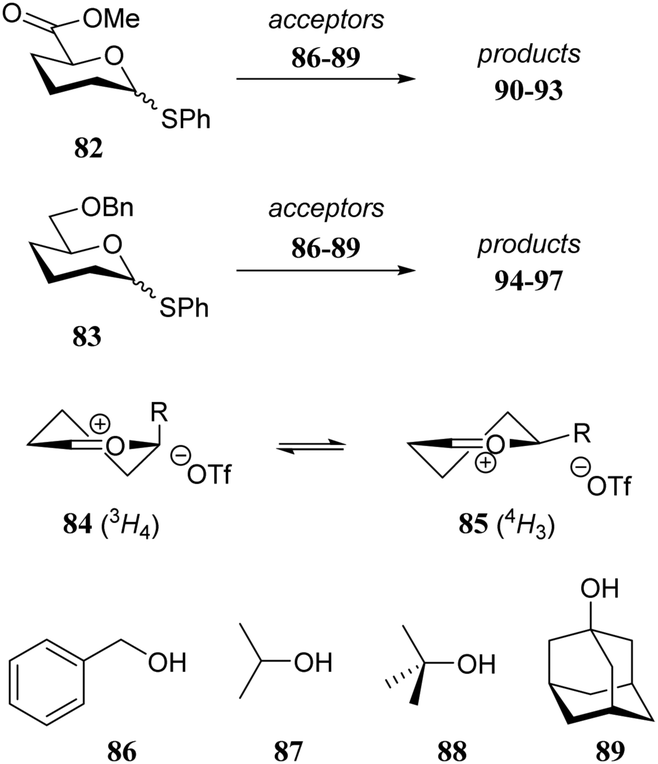
In the early 90's Spijker and van Boeckel were the first to report on the concept of double stereodifferentiation30 in synthetic carbohydrate chemistry.31 They unambiguously showed how the absolute chirality of the coupling partners can impact the outcome of a glycosylation reaction (Scheme 1). The condensations of the two enantiomeric donors, D-fucosyl bromide 98 and L-fucosyl bromide 99 with D-glucosamine acceptor 100 proceeded with a rather different outcome. The glycosylation of donor 99 and acceptor 100 provided the disaccharide product with the expected trans-selectivity (α:β, 1:8.4), while the use of the enantiomeric donor 98 led to an anomeric mixture (α:β, 2:1). Although neighboring group participation is generally a powerful stereocontrolling tool, here it falls short because of an apparent steric mismatch in the transition state. In the second example, the enantiomeric pairs of D- and L-diacetoneglucose 102/103 were condensed with D-glucosyl bromide 101 to show that the 1,2-cis-selectivity in these glycosylations also depend, albeit to a lesser extent, on the absolute chirality of the reaction partners.
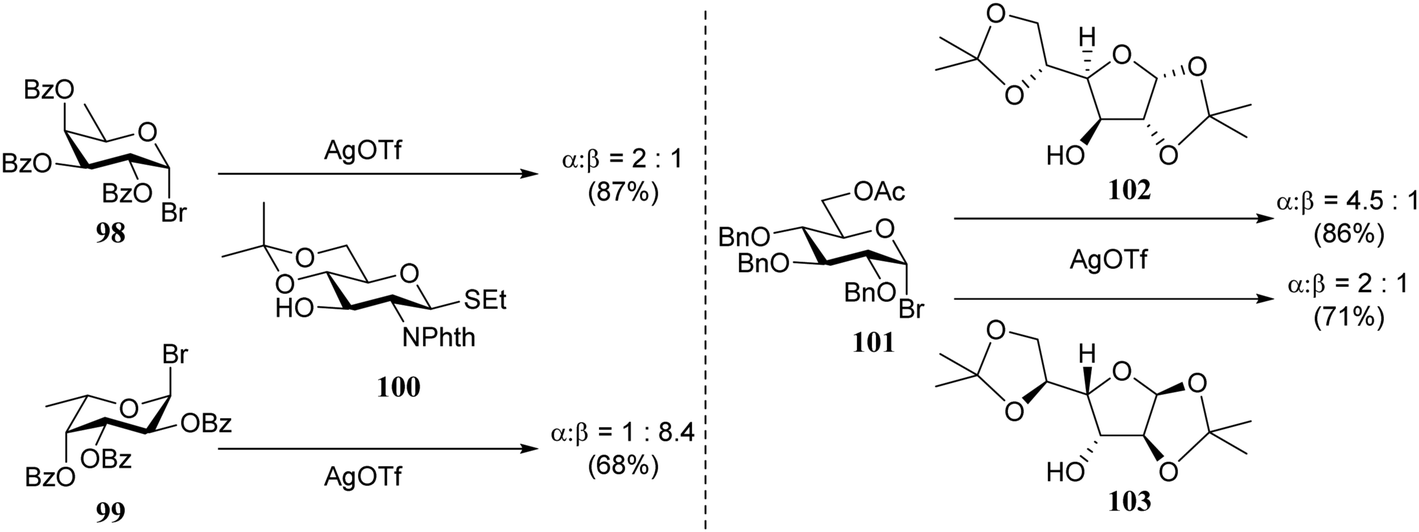 | ||
| Scheme 1 Double stereodifferentiation in glycosylation reactions. (Spijker and van Boeckel, 1991).31Reagents and conditions: AgOTf, 2,6-di-tert-butylpyridine (0.8 eq.), 4 Å M.S., DCM, −50 °C. | ||
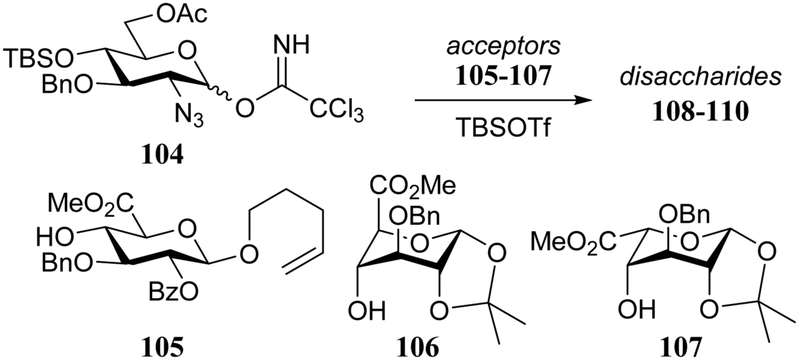
Another clear manifestation of the effect of the shape of the acceptor on the outcome of a glycosylation reaction can be observed when carbohydrate acceptors are locked in ‘inverted’ chair conformations. As was shown above, conformationally locking a glucose/glucosamine acceptor in a 1C4 chair places the C-4-OH in a position that is well accessible, thereby increasing its nucleophilicity.16,32 A well-established phenomenon in heparin synthesis is the excellent α-selectivity, generally observed in glycosylations of glucosazide donors with L-idose/L-iduronic acid acceptors taking up an 1C4 chair conformation. This important manifestation of double stereodifferentiation has been of great assistance in the assembly of synthetic heparin and heparan sulfate fragments.33,34 To transpose this stereodifferentiation to glycosylations involving D-glucuronic acid ester acceptors, Seeberger and co-workers locked these acceptors in a similar 1C4 chair conformation (Table 10).35 While the condensation of glucosazide donor 104 with D-glucuronic acid acceptor 105 provided an anomeric mixture (108; α:β, 3:1) in a relatively low yield, the glycosylation of the glucuronate acceptor in the 1C4 conformation (109), proceeded in high yield with excellent α-selectivity, in analogy to reaction of the L-iduronic acid acceptor 110.
|
|
|||
|---|---|---|---|
| Acceptor | Product | Yield (%) | α:β |
| Reagents and conditions: donor (1.25 eq.), acceptor (1 eq.), TBSOTf (0.125 eq.), 4 Å M.S., DCM, −78 °C to room temperature, 2.5 h. | |||
| 105 | 108 | 57 | 3:1 |
| 106 | 109 | 86 | α only |
| 107 | 110 | 91 | α only |
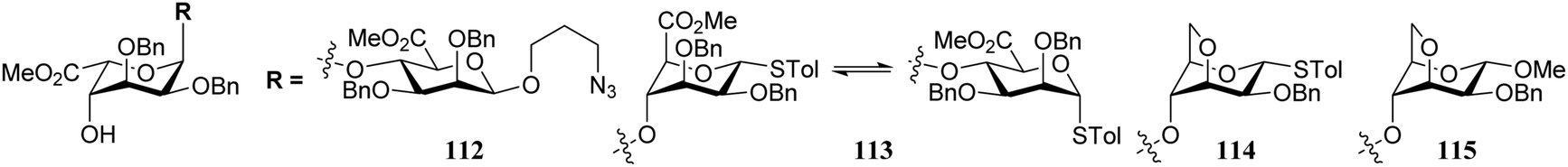
Conformational changes further away from the reacting alcohol may also impact the reactivity of the nucleophile.36–38 In the assembly of L-guluronic acid–D-mannuronic acid alginates Zhang et al. observed that the condensation of disaccharide acceptor 112 with mannuronic acid donor 111 proceeded in moderate yield (Table 11).39 When this condensation was performed with acceptor 113, having an α-S-tolyl group instead of the β-O-(azidopropyl) functionality at the ‘reducing’ end of the acceptor, a 91% yield was obtained. It was reasoned that the conformational flexibility of acceptor 113 was responsible for this large difference in reactivity.40,41 The use of model disaccharide acceptors having a conformationally locked 1C4 reducing end saccharide (as in 114 and 115) confirmed that the ‘ring inverted’ acceptors were apt nucleophiles. This study has shown that conformational flexibility of the reaction partners can be key to accommodate the stringent steric requirements in the crowded glycosylation reaction transition states.
|
|
|||
|---|---|---|---|
| Acceptor | Product | Yield (%) | |
| All glycosylations proceeded with exclusive β-selectivity. Reagents and conditions: donor (3 eq.), acceptor (1 eq.), TBSOTf (0.6 eq.), 4 Å M.S., DCM, −78 °C to −45 °C. | |||
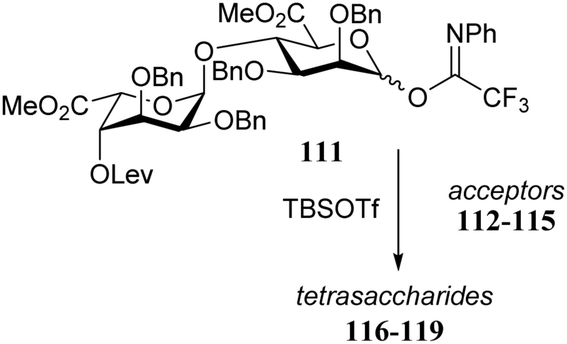
|
112 | 116 | 45 |
| 113 | 117 | 91 | |
| 114 | 118 | 71 | |
| 115 | 119 | 95 | |
It has been observed by Fraser-Reid and co-workers that the different hydroxyl groups in diol acceptors react with different specificity for a given donor system. Diol 120 can be regioselectively glycosylated at the equatorial hydroxyl with pentenyl orthoester donor 121, while the axial alcohol in 120 reacts selectively with pentenyl mannoside 123 (Scheme 2).42–45 Building on Paulsen notion that the reactivity of both reaction partners should be “matched” for an optimal glycosylation reaction,46–49 Fraser-Reid coined the concept of reciprocal donor acceptor selectivity (RDAS), to account for these observations.50–53 Although the concept still awaits a proper mechanistic explanation, the counter-intuitive outcome of several recent reactions have been related to this phenomenon.54–58 To provide a satisfactory explanation for these observations, more insight is required into the intrinsic reactivity of (carbohydrate) acceptors and better (computational) methodology should be developed to assess the transition states of glycosylation reactions.
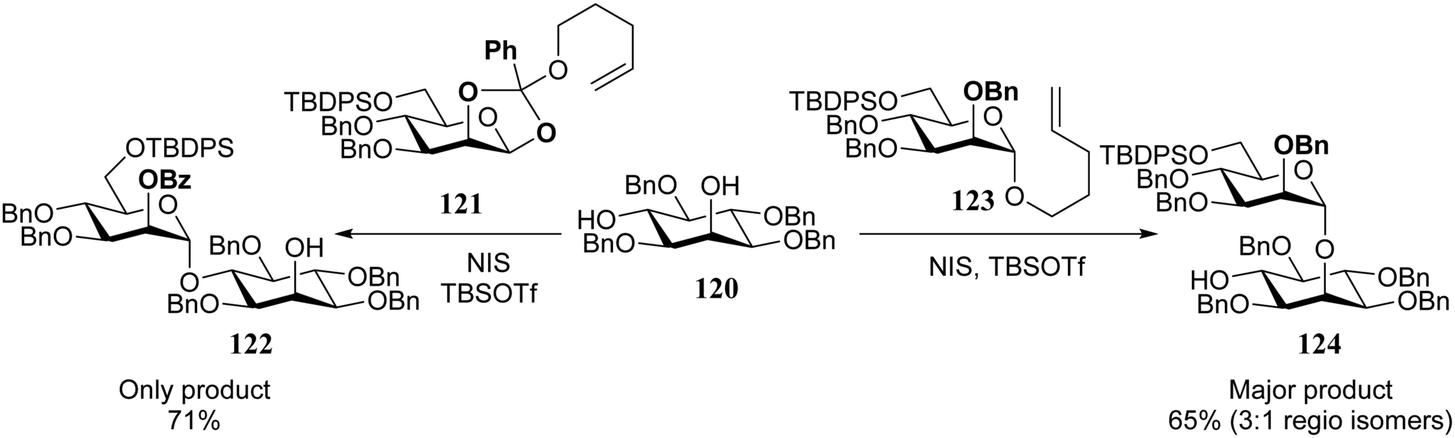 | ||
| Scheme 2 Donor–acceptor match and mismatch, led to the formulation of reciprocal donor–acceptor selectivity. (Fraser-Reid, 2000).42,43Reagents and conditions: donor (1.3 eq.), acceptor (1 eq.), NIS (1.3 eq.), TBSOTf (cat), DCM, room temperature. | ||
Systematic studies on acceptor reactivity
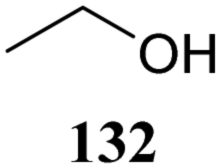
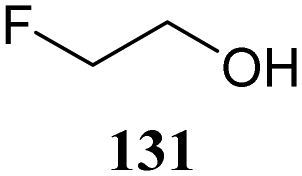
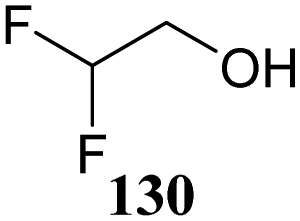
Although it is clear that the nature of the protecting groups on the acceptor glycosides has an influence on the glycosylation outcome it is often difficult to dissect electronic, steric and conformational effects.59 Woerpel and co-workers have reported a systematic study relating the effect of the nucleophilicity of the acceptor on the outcome of a glycosylation reaction, using both C- and O-nucleophiles.60–64Table 12 lists the results of glycosylation of both sets of nucleophiles, using 2-deoxyglucosyl acetate or ethanethiol donors 133 and 134. The nucleophilicity of the acceptor is assessed from the nucleophilicity parameter N,65–70 introduced by Mayr to quantitatively compare different nucleophiles. Following a logarithmic scale, stronger nucleophiles are characterized by a higher number N, obtained using a large set of kinetic experiments employing benzhydrilium ion electrophiles. Table 12 also reports the field inductive parameter F,71,72 a measure for the inductive electron-withdrawing power of the substituent (higher numbers indicating a stronger inductive effect). The trend that becomes apparent from the results in Table 12 is that weaker nucleophiles provide more α-product. To account for these results, it was reasoned that the weakest C- and O-nucleophiles 125 and 129, react in a stereoselective manner with the glucosyl oxocarbenium ion, taking up a 4H3 conformation (144). Increasing acceptor nucleophilicity leads to a decrease in α-selectivity. This erosion of stereoselectivity (from 135 to 138, and from 139 to 142) is caused by alternative reaction pathways becoming accessible for the stronger nucleophiles: either non-selective SN1 reactions in which both sides of oxocarbenium ion 144 are attacked, or SN2-type substitutions.|
|
|||||
|---|---|---|---|---|---|
| Acceptor | N | Product | Yield (%) | α:β |
|
| a Mayr's nucleophilicity parameter. b Field inductive parameter.71 Reagents and conditions for acetyl donors: donor (1 eq.), acceptor (4 eq.), BF3OEt2 (1.5 eq.), DCM, −42 °C to 0 °C. Reagents and conditions for thiodonors: donor (1 eq.), acceptor (4 eq.), NIS (2 eq.), CH3CN, 0 °C. | |||||
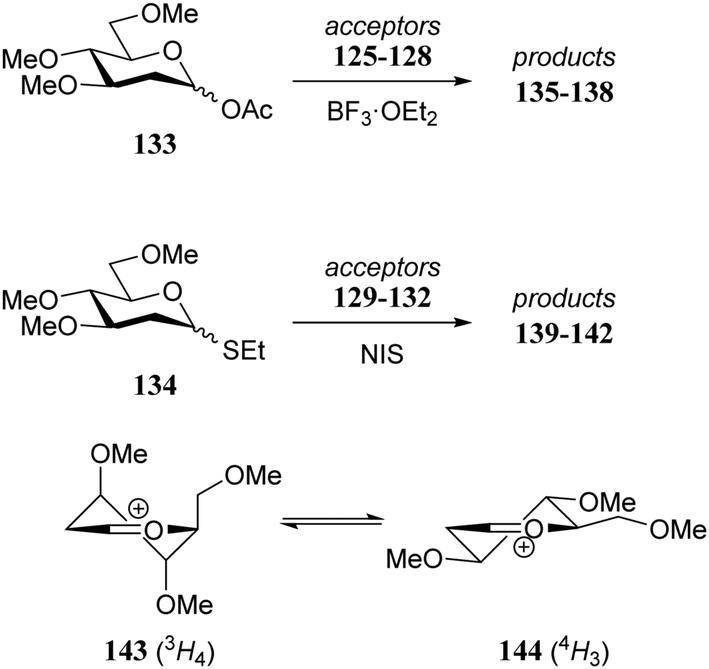
|
125 | 1.7 | 135 | 80 | 89:11 |
| 126 | 4.4 | 136 | 79 | 43:57 |
|
| 127 | 6.2 | 137 | 83 | 61:39 |
|
| 128 | 8.2 | 138 | 83 | 45:55 |
|

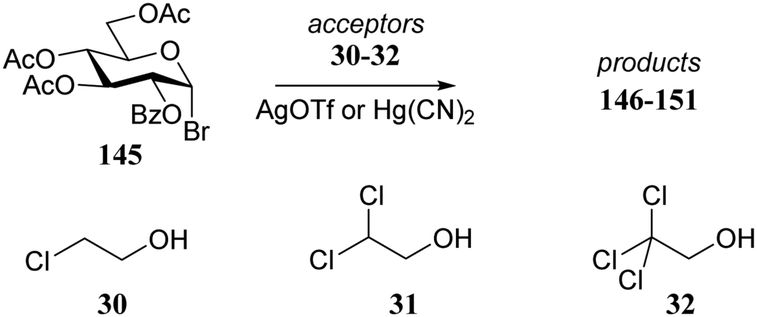
In an earlier study, Garegg et al.73 studied the stereoselectivity of Könings-Knorr reactions of bromide donor 145 with a series of chlorine containing alcohols 30–32 (see also Table 3). Table 13 shows a similar reactivity–stereoselectivity trend, as reported by Woerpel and co-workers, when a polar solvent (CH3CN) is used in combination with Hg(CN)2 as activator. Despite the fact there was a participating group present on the C-2 of donor 145, a substantial amount of the product α-anomer 148 was formed in the reaction with acceptor 32. In a more apolar solvent, DCM, employing AgOTf as activator, the pathway proceeding through the dioxolenium ion prevailed and the β-products were mainly formed for all three acceptors with only a slight shift in stereoselectivity.
|
|
|||||
|---|---|---|---|---|---|
| Acceptor | Product | Activator | Solvent | Yield (%) | α:β |
| Reagents and conditions: donor (1 eq.), alcohol (1 eq.), activator AgOTf or Hg(CN)2 (1 eq.), solvent CH3CN or DCM. Glycosylations with Hg(CN)2 were conducted at room temperature, glycosylations with AgOTf at −25 °C with 4 Å M.S. | |||||
| 30 | 146 | Hg(CN)2 | CH3CN | 88 | 5:95 |
| 31 | 147 | Hg(CN)2 | CH3CN | 83 | 17:83 |
| 32 | 148 | Hg(CN)2 | CH3CN | 74 | 67:33 |
| 30 | 149 | AgOTf | DCM | 89 | 0:100 |
| 31 | 150 | AgOTf | DCM | 89 | 1:99 |
| 32 | 151 | AgOTf | DCM | 81 | 4:96 |
In a subsequent study by the same group,74 the per-methylated glucosyl bromide 152 (Scheme 3) was used. In a series of competition reactions monochloroethanol 30 was shown to react faster than trichloroethanol 32. The weaker nucleophile provided slightly more α-product than the stronger nucleophile. Based on kinetic studies the authors proposed an ion pair mechanism to account for the observed reactivity and stereoselectivity.
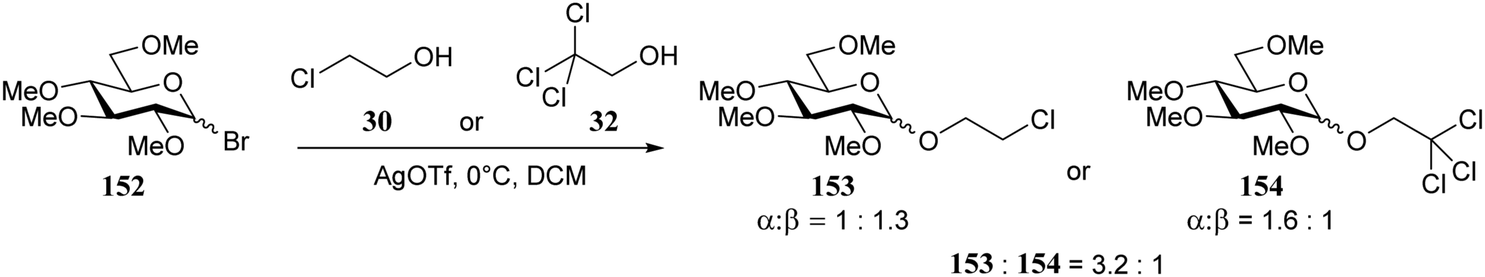 | ||
| Scheme 3 Competition reactions of different nucleophiles. (Konradsson, 2000).74 | ||
In line with the above described results, Seeberger and co-workers found that the stereoselectivity of condensations of donor 155 with linkers 156 and 157 strongly depended on the reactivity of the nucleophile. While the reactive primary alcohol 156 provided a β-selective reaction, the weaker nucleophile 157 mainly provided the α-product (Scheme 4).75 By tweaking the reaction temperature and solvent, nearly complete α- or β-stereoselectivity could be obtained.76 A variety of different donors provided a similar reactivity–stereoselectivity trend.
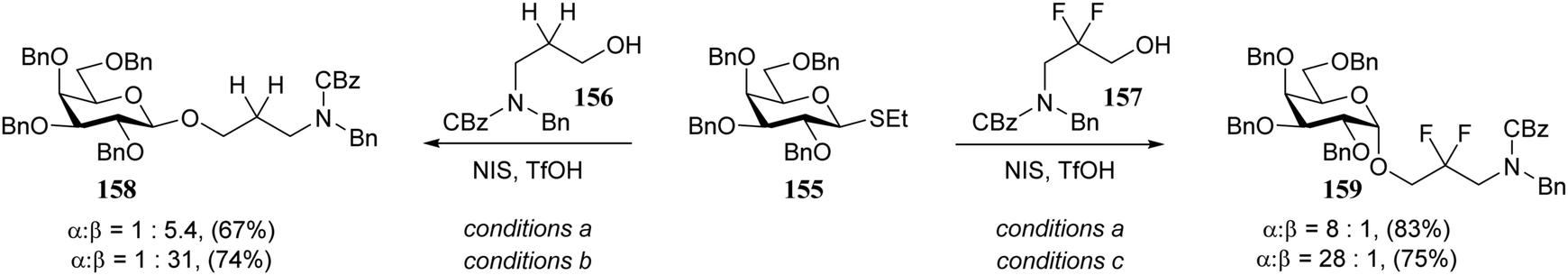 | ||
| Scheme 4 Linkers of varying nucleophilicity gave opposite glycosylation stereoselectivity. (Seeberger, 2016).75Reagents and conditions: donor (1.5 eq.), acceptor (1 eq.), NIS (1.5 eq.), TfOH (0.2 eq.); (a) DCM, −20 °C; (b) CH3CN −40 °C; (c) toluene/dioxane (3/1), room temperature. | ||
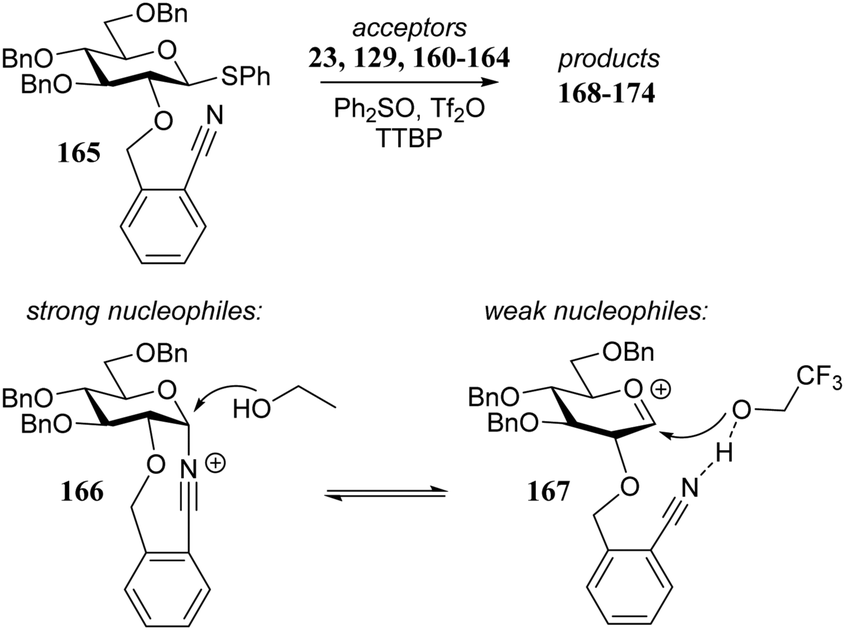
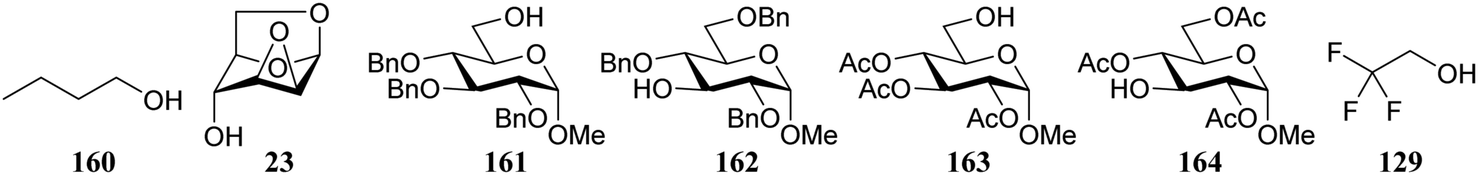
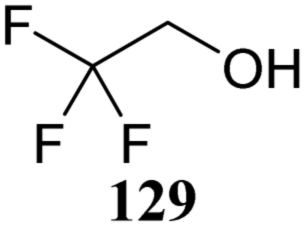
Le Mai Hoang and Liu introduced donors equipped with a 2-cyanobenzyl group at the C-2-OH and investigated these donors, in a pre-activation glycosylation scheme, with a panel of acceptors (Table 14).77 Next to the model acceptors n-butanol 160 and trifluoroethanol 129, this study also included carbohydrate acceptors bearing either benzyl ether of acetyl ester protection groups. It was observed that the stronger nucleophiles stereoselectively provided the β-linked product, while the use of the weaker nucleophiles led to the generation of the α-linked products in a fully stereoselective manner.78 The authors reasoned that the stronger nucleophiles (23, 160–162) can partake in an SN2-like substitution of the intermediate α-nitrilium ion 166, to selectively provide the β-products. SN2-like substitution of the intermediate α-triflate, or a closely related contact ion pair, will provide a similar outcome. The α-selectivity of the weaker, acetyl bearing acceptors 163 and 164 and trifluoroethanol 129 was accounted for by a assuming a hydrogen-bond with the cyano functionality on the C-2-O-protecting group, guiding the acceptor to the α-face of the donor (as in 167). An alternative explanation can be found in the diastereoselective attack of the weaker acceptors on the intermediate oxocarbenium ion.
|
|
||||
|---|---|---|---|---|
| Acceptor | Product | Yield (%) | α:β |
|
| Reagents and conditions: donor (1 eq.), acceptor (1.3 eq.), Ph2SO (1.4 eq.), TTBP (3 eq.), Tf2O (2.8 eq.), toluene −60 °C.a Et2O was used as solvent. | ||||
|
160 | 168 | 90 | β only |
| 23 | 169 | 86 | β only | |
| 161 | 170 | 89 | β only | |
| 162 | 171 | 86 | β only | |
| 163 | 172 | 87 | α only | |
| 164 | 173 | 81 | α only | |
| 129a | 174 | 71 | α only | |
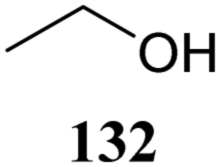
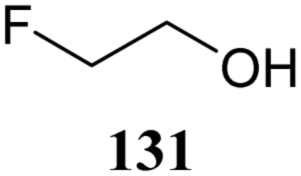
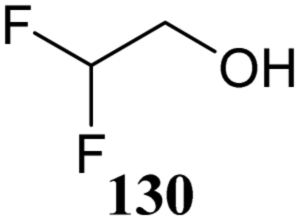
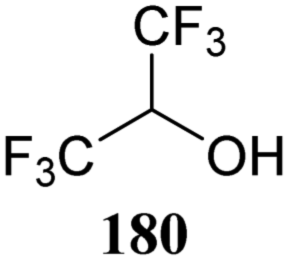
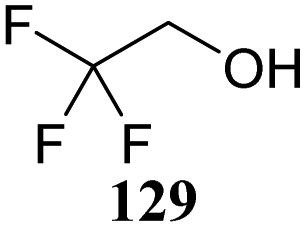
The systematic study described above lay bare the intrinsic dependence of the stereoselectivity of glycosylation reactions on the nature of the nucleophile. To relate the reactivity of carbohydrate acceptors to the set of partially fluorinated ethanol model acceptors, we have investigated a set of glycosyl donors in combination with both the model ethanol acceptors (129–132) as well as a set of carbohydrate alcohols.79 We investigated benzylidene mannose and benzylidene glucose donors, 175 and 177, because the reaction pathways of these donors have been well characterized.80 In addition, mannuronic acid donor 176 was probed, as previous results indicated this donor to provide highly selective 1,2-cis-glycosylations through reaction pathways, likely involving oxocarbenium ion intermediates.81Scheme 5 displays the general pre-activation glycosylation protocol used for glycosylations described in Tables 15–17. Table 15 summarizes the results of the condensation reactions and it shows that the stereoselectivity of the reactions of the benzylidene glucose donor strongly depend on the nucleophilicity of the acceptor alcohol. Glucosylations with the most reactive acceptor, ethanol 132, provides product 195 with high β-selectivity. Going down the table with decreasing nucleophilicity of acceptors 131, 130, 129 and 180, the glucosylation selectivity gradually changes to exclusively form the α-anomers of 198 and 199. In contrast, the reactions of the benzylidene mannose 175 and mannuronic acid 176 donors are less sensitive to the reactivity of the nucleophiles and a amaller change in selectivity is observed for donors 175 and 176, (185–189, from 1:5 to 3:1, α:β; and 190–194, from 1:8 to 1:1, α:β) when moving down the nucleophilicity scale in Table 15. It can be reasoned that the most important pathway for substitutions of the strong nucleophiles follows an SN2-like itinerary, displacing the anomic triflates of the donor glycosides. The weaker nucleophiles require a stronger electrophile bearing more oxocarbenium ion character. The benzylidene glucose oxocarbenium ion will preferentially take up a 4H3-like half-chair conformation that is preferentially attacked on the α-face.82 This accounts for the gradually shifting stereoselectivity from the β-side to the α-side when the nucleophilicity of the acceptor alcohols decreases. The benzylidene mannose oxocarbenium ion on the other hand may take up a B2,5 conformation,83,84 that can be attacked form the β-face. The mannuronic acid oxocarbenium ion will adopt a 3H4-like half-chair structure, that preferentially follows a reaction itinerary through attack on its β-face. The stereoselectivity of the reactions of the latter two oxocarbenium ions will therefore be similar to the stereoselectivity of the SN2-type displacement of the intermediate α-triflates and the reactions thus relatively insensitive to the nucleophilicity of the acceptors. The parallels that can be found in the stereoselectivity of the reactions of the carbohydrate acceptors and those of the model ethanol acceptors shows that the reactivity of the carbohydrate alcohols falls somewhere in between the reactivity of monofluoro- and trifluoro-ethanol.
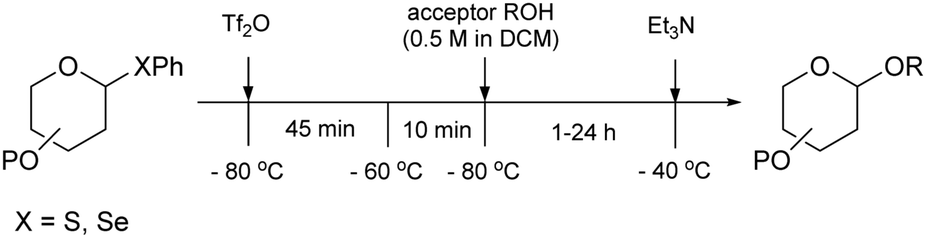 | ||
| Scheme 5 Glycosylation protocol for the reactions described in Tables 15–17. Reagents and conditions: donor (1 eq.), Ph2SO (1.3 eq.), TTBP (2.5 eq.), Tf2O (1.3 eq.), 3 Å M.S., DCM (0.05 M), −80 °C to −60 °C, then acceptor (2 eq.) in DCM (0.5 M) −80 °C to −40 °C. | ||
|
|
|
|
|
|
|
|---|---|---|---|---|---|
| 175 | 176 | 177 | 178 | 179 | |
| Acceptor | Product α:β (yield) |
Product α:β (yield) |
Product α:β (yield) |
Product α:β (yield) |
Product α:β (yield) |
|
185 | 190 | 195 | 200 | 205 |
| 1:5 |
1:8 |
1:10 |
<1:20 |
<1:20 |
|
| (70%) | (95%) | (68%) | (65%) | (83%) | |
|
186 | 191 | 196 | 201 | 206 |
| 1:5 |
1:6 |
1:3 |
1:5 |
1:6.7 |
|
| (86%) | (70%) | (70%) | (79%) | (90%) | |
|
187 | 192 | 197 | 202 | 207 |
| 1:5 |
1:5 |
5:1 |
2.7:1 |
2.9:1 |
|
| (90%) | (87%) | (70%) | (76%) | (64%) | |
|
188 | 193 | 198 | 203 | 208 |
| 1:4 |
1:2.5 |
>20:1 |
>20:1 |
>20:1 |
|
| (78%) | (85%) | (64%) | (82%) | (94%) | |
|
189 | 194 | 199 | 204 | 209 |
| 3:1 |
1:1 |
>20:1 |
>20:1 |
>20:1 |
|
| (56%) | (52%) | (65%) | (34%) | (53%) | |
|
210 | 215 | 220 | 225 | 230 |
| 1:10 |
<1:20 |
1:3 |
1:14 |
<1:20 |
|
| (97%) | (71%) | (81%) | (92%) | (89%) | |
|
211 | 216 | 221 | 226 | 231 |
| 1:9 |
<1:20 |
1:1 |
1:3 |
1:7 |
|
| (75%) | (61%) | (79%) | (81%) | (88%) | |
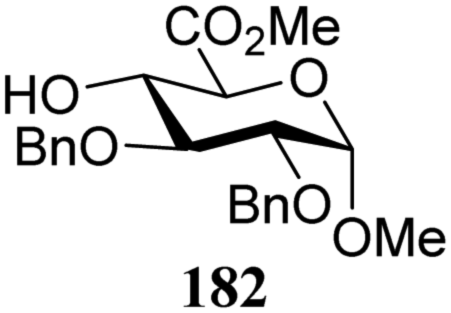
|
212 | 217 | 222 | 227 | 232 |
| 1:10 |
1:10 |
5:1 |
3.3:1 |
1.1:1 |
|
| (87%) | (71%) | (90%) | (84%) | (93%) | |
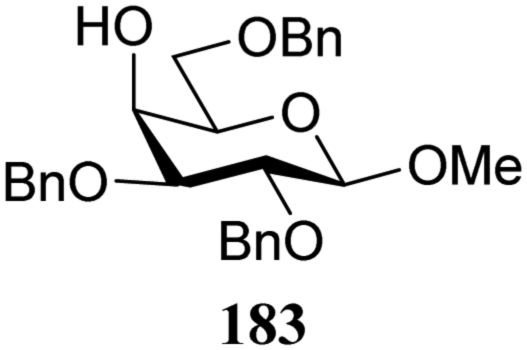
|
213 | 218 | 223 | 228 | 233 |
| <1:20 |
<1:20 |
>20:1 |
7:1 |
9:1 |
|
| (70%) | (76%) | (83%) | (52%) | (75%) | |
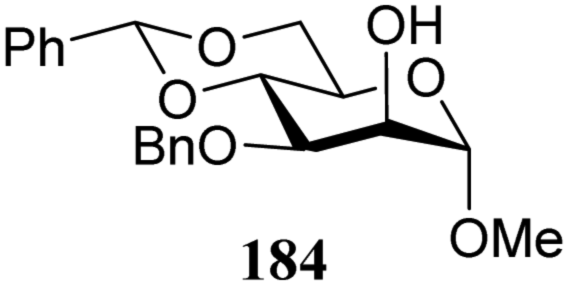
|
214 | 219 | 224 | 229 | 234 |
| <1:20 |
1:7 |
>20:1 |
>20:1 |
9:1 |
|
| (87%) | (80%) | (80%) | (85%) | (74%) | |
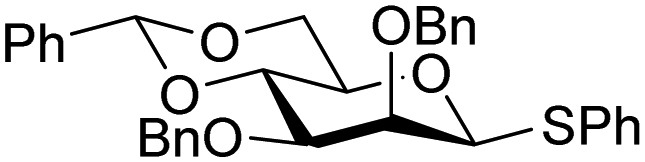
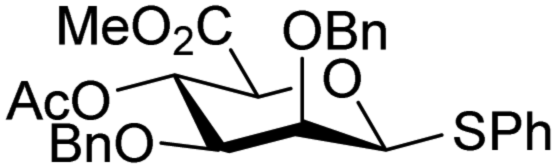
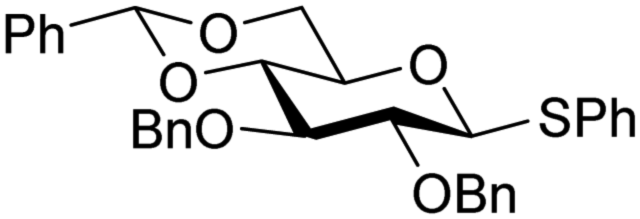
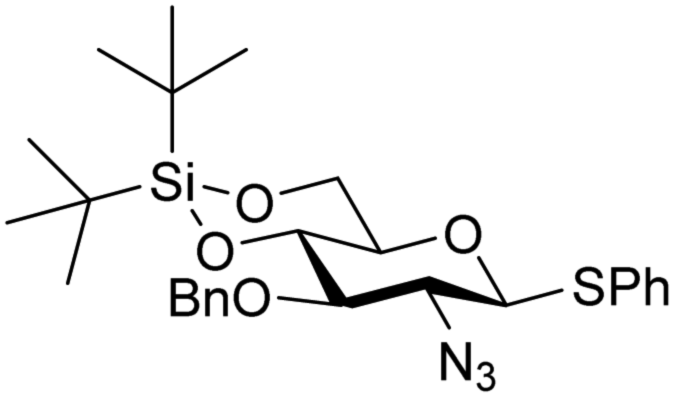
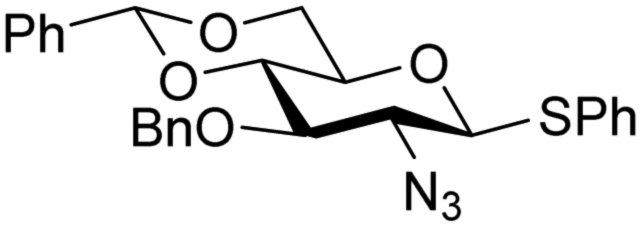
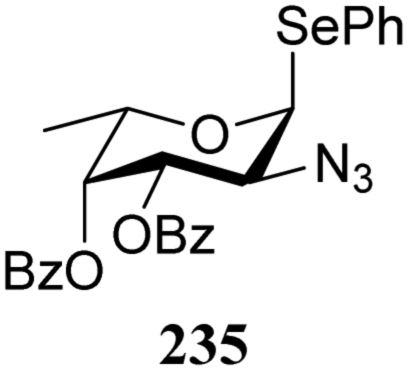
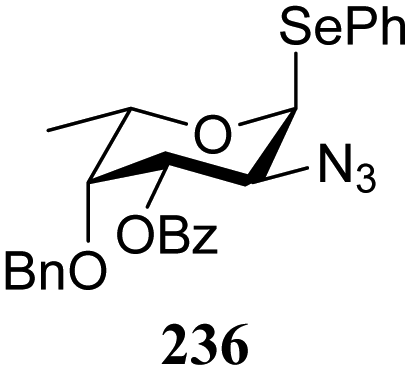
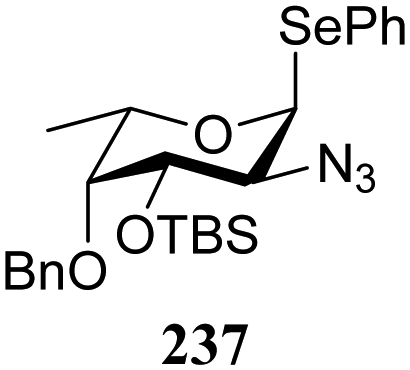
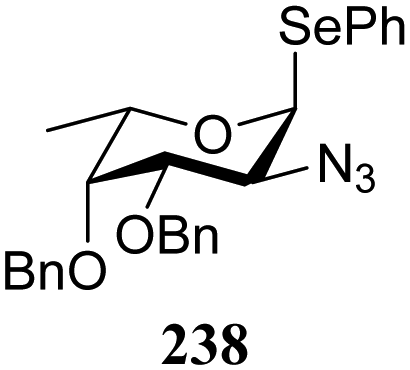
The reactivity–stereoselectivity trends observed for the glycosylation reactions of the benzylidene glucose donor also became apparent in the condensations of the analogous benzylidene glucosamine donors (178 and 179, Table 16). The presence of the azide at C-2 shifted the reaction mechanism balance towards the SN2-side as the electron-withdrawing azide stabilizes the covalent triflate with respect to the intermediate oxocarbenium ion. Glucosazide donors 178 and 179 provide relatively more β-product (disaccharides 200–204 and 205–209) than their glucose counterpart. The relatively weak nucleophiles 129 and 180 still only provided the α-products 203, 204, 208 and 209. The increased reactivity of silylidene donor 178 in comparison to that of benzylidene donor 179 translates to the formation of more of the SN1-product. A similar reactivity–stereoselectivity relationship was revealed for a set of fucosazide donors, that were studied in the context of the assembly of complex bacterial glycans.85,86 As Table 17 reveals, the 1,2-cis:1,2-trans-product ratio increases with increasing reactivity of the donor (237, 238 > 235, 236) and decreasing acceptor reactivity. This can be accounted for with a shift in product forming reaction pathways form a 1,2-trans-selective SN2-like reaction of the reactive nucleophiles and the anomeric α-fucosazide triflates to 1,2-cis-selective SN1-type reactions of the weaker nucleophiles, involving the 3H4-like half-chair L-fucosazide oxocarbenium ions as product forming intermediates.
|
|
|
|
|
|
|---|---|---|---|---|
| Acceptor | Product α:β (yield) |
Product α:β (yield) |
Product α:β (yield) |
Product α:β (yield) |
|
240 | 244 | 248 | 252 |
| 1:3 |
1:3 |
1:1 |
1:1 |
|
| (59%) | (58%) | (81%) | (88%) | |
|
241 | 245 | 249 | 253 |
| 1:2 |
1:1.5 |
1:1 |
1:1 |
|
| (34%) | (60%) | (80%) | (72%) | |
|
242 | 246 | 250 | 254 |
| 1.5:1 |
1:1 |
2:1 |
2:1 |
|
| (74%) | (80%) | (87%) | (81%) | |
|
243 | 247 | 251 | 255 |
| 10:1 |
>20:1 |
>20:1 |
>20:1 |
|
| (50%) | (45%) | (90%) | (80%) | |
|
256 | 258 | 260 | 262 |
| 4:1 |
4:1 |
>20:1 |
>20:1 |
|
| (38%) | (68%) | (74%) | (68%) | |
|
257 | 259 | 261 | 263 |
| >20:1 |
10:1 |
9:1 |
>20:1 |
|
| (64%) | (64%) | (64%) | (72%) | |
|
|
|
|
|
|
|
|||
|---|---|---|---|---|---|---|---|---|
| 177 | 179 | 177 | 179 | 177 | 179 | |||
| Acceptor | Product α:β (yield) |
Product α:β (yield) |
Acceptor | Product α:β (yield) |
Product α:β (yield) |
Acceptor | Product α:β (yield) |
Product α:β (yield) |
|
221 | 231 |
|
278 | 279 |
|
284 | 285 |
| 1:1 |
1:7 |
1:1.1 |
1:6 |
>20:1 |
6.7:1 |
|||
| (82%) | (88%) | (81%) | (88%) | (95%) | (77%) | |||
|
276 | 277 |
|
280 | 281 |
|
286 | 287 |
| 4:1 |
1:1.1 |
3.5:1 |
1.3:1 |
>20:1 |
>20:1 |
|||
| (92%) | (67%) | (88%) | (87%) | (95%) | (85%) | |||
|
222 | 232 |
|
282 | 283 |
|
288 | 289 |
| 5:1 |
1.1:1 |
4.8:1 |
1.2:1 |
>20:1 |
>20:1 |
|||
| (90%) | (93%) | (96%) | (82%) | (86%) | (93%) | |||
|
290 | 291 |
|
294 | 295 |
|
298 | 299 |
| 1:2.7 |
<1:20 |
6:1 |
1:1.3 |
8:1 |
1.1:1 |
|||
| (78%) | (70%) | (85%) | (88%) | (82%) | (70%) | |||
|
292 | 293 |
|
296 | 297 |
|
300 | 301 |
| >20:1 |
11:1 |
>20:1 |
11:1 |
20:1 |
>20:1 |
|||
| (100%) | (83%) | (83%) | (90%) | (100%) | (92%) | |||
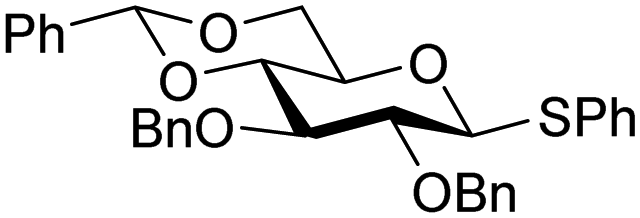
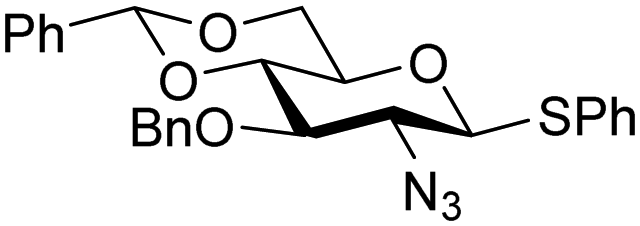
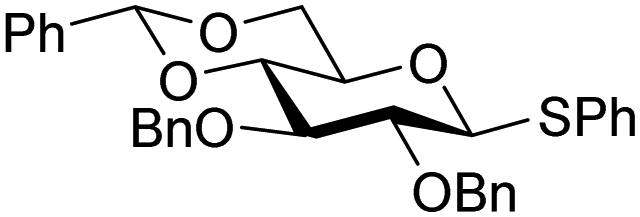
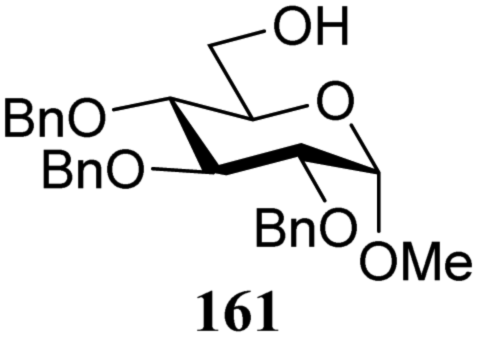
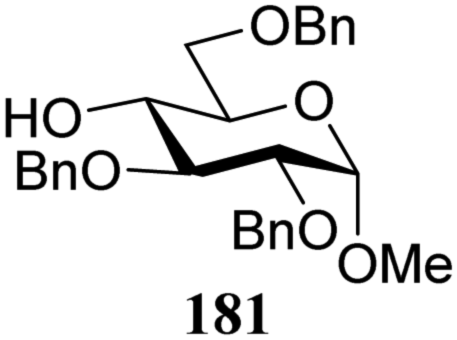
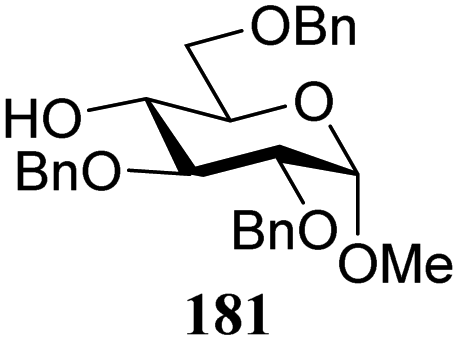
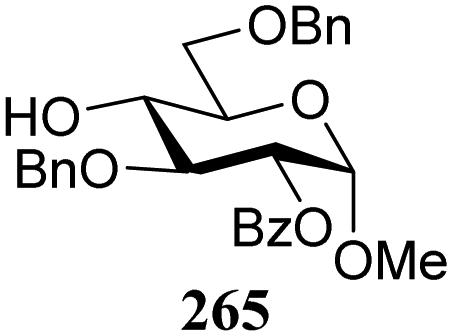
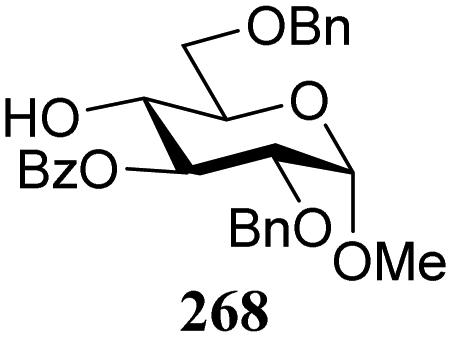
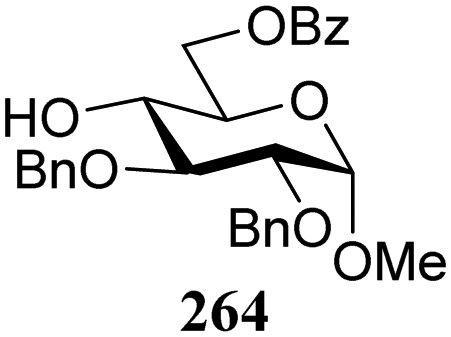
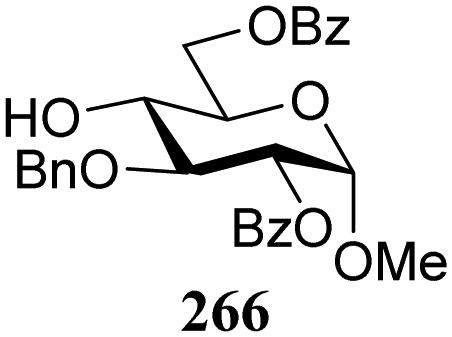
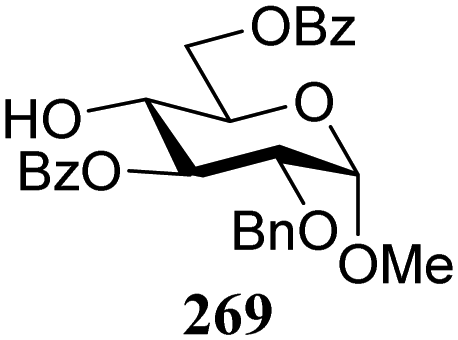
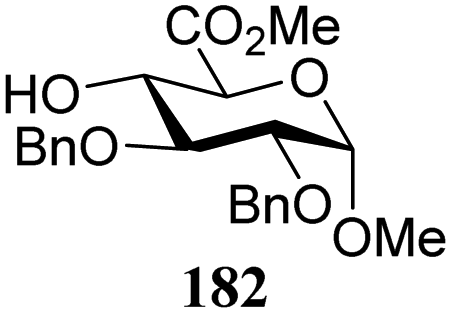
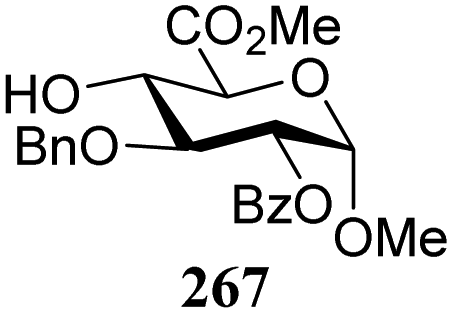
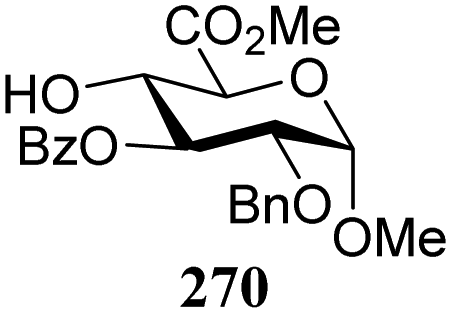
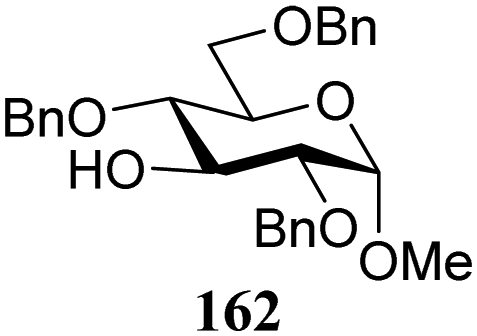
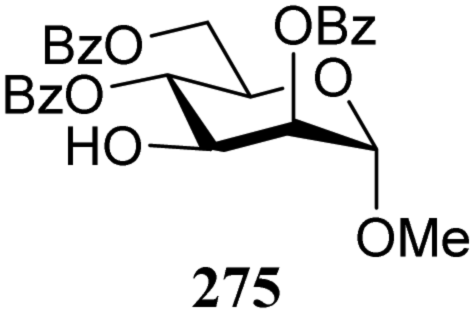
The gradually changing stereoselectivity of glycosylations of the benzylidene glucose/glucosazide donors as a function of acceptor nucleophilicity, opened up the possibility to use this system as a measure for the reactivity of carbohydrate alcohol acceptors.87 We have used this set-up to establish structure-stereoselectivity relationships for a large set of glycosyl acceptors, of which the structure in terms of functional and protecting group pattern was systematically changed. We initially investigated C-4-OH glucose acceptors with all possible permutations of benzyl and benzoyl protecting groups of which a selection of the results is given in Table 17. These groups differ significantly in their electronic properties while being sterically very similar. A clear dependence of the reactivity/stereoselectivity on the functional/protecting group pattern was uncovered, with the less-reactive, benzoyl protected acceptors generally providing more 1,2-cis linked products. Notably, replacing a single benzyl ether for a benzoyl group on the position closest to the nucleophilic oxygen (cf. acceptors 181 and 268) led to a drastic change in the stereoselectivity of the glycosylations, showing that non-selective reactions can be turned into highly selective reactive reactions by the judicious choice of protecting groups. Probing other regioisomeric glucosyl, mannosyl and galactosyl acceptors (162, 271–275) revealed the same recurring trend. Care should be taken to compare the results obtained for different regioisomeric or diastereomeric acceptors as the different steric requirements for the acceptors will also play an important role in shaping the overall glycosylation outcome. It is expected that the extension of this study will provide further detailed insight into structure–reactivity–stereoselectivity relationships of diversely functionalized carbohydrate acceptor alcohols which will pave the way to develop more predictable glycosylation methodology.
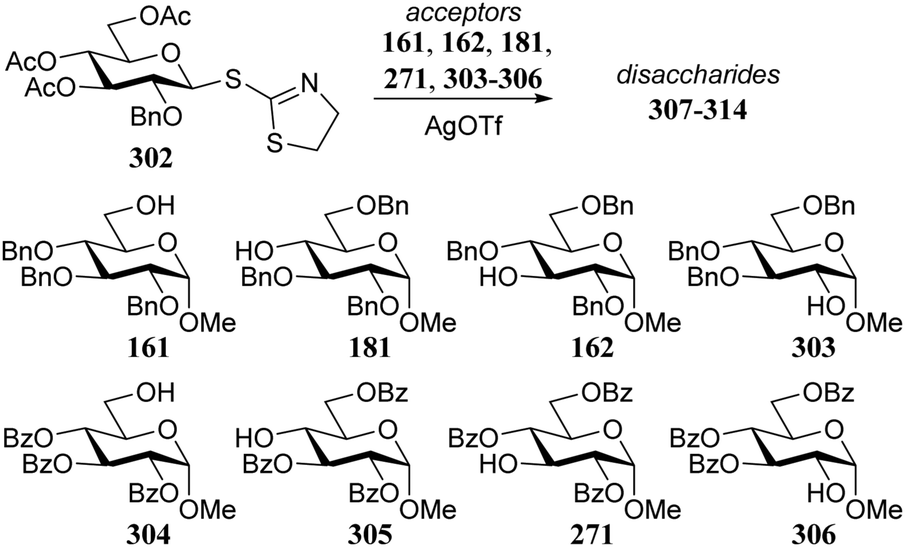
Demchenko and co-workers established similar protecting group effects on a smaller set of regioisomeric glucosyl acceptors in glycosylations with STaz donor 302 (Table 18).88 While the yields of the silver triflate mediated reactions proved independent of acceptor reactivity, the α/β-selectivity of the glycosylation reactions involving the benzyl protected acceptors is generally lower than the selectivity for the same acceptors bearing O-benzoyl groups. It was observed that the benzyl protected acceptors were converted faster to their respective products than their benzoyl protected counterparts.
|
|
||||
|---|---|---|---|---|
| Acceptor | Product | Time (h) | Yield (%) | α:β |
| Reagents and conditions: donor (0.11 mmol, 1.1 eq.), acceptor (0.10 mmol, 1 eq.), 3 Å M.S., AgOTf (0.22 mmol, 2 eq.), 1,2-dichloroethane (2 mL), room temperature. | ||||
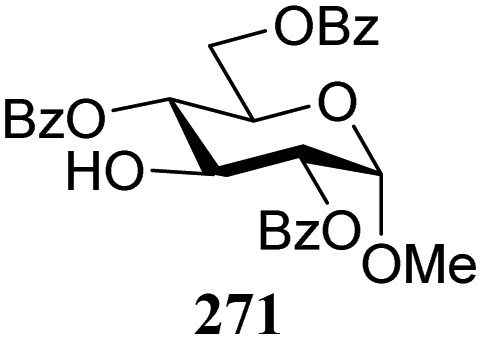
| 161 | 307 | 1.5 | 81 | 2.7:1 |
| 304 | 308 | 2 | 89 | 7.4:1 |
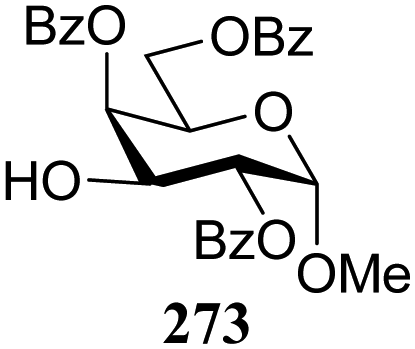
| 181 | 309 | 14 | 90 | 6.8:1 |
| 305 | 310 | 16 | 89 | 11.7:1 |
| 162 | 311 | 8 | 85 | 6.5:1 |
| 271 | 312 | 12 | 87 | 12.1:1 |
| 303 | 313 | 6 | 87 | 9.3:1 |
| 306 | 314 | 12 | 72 | 12.0:1 |
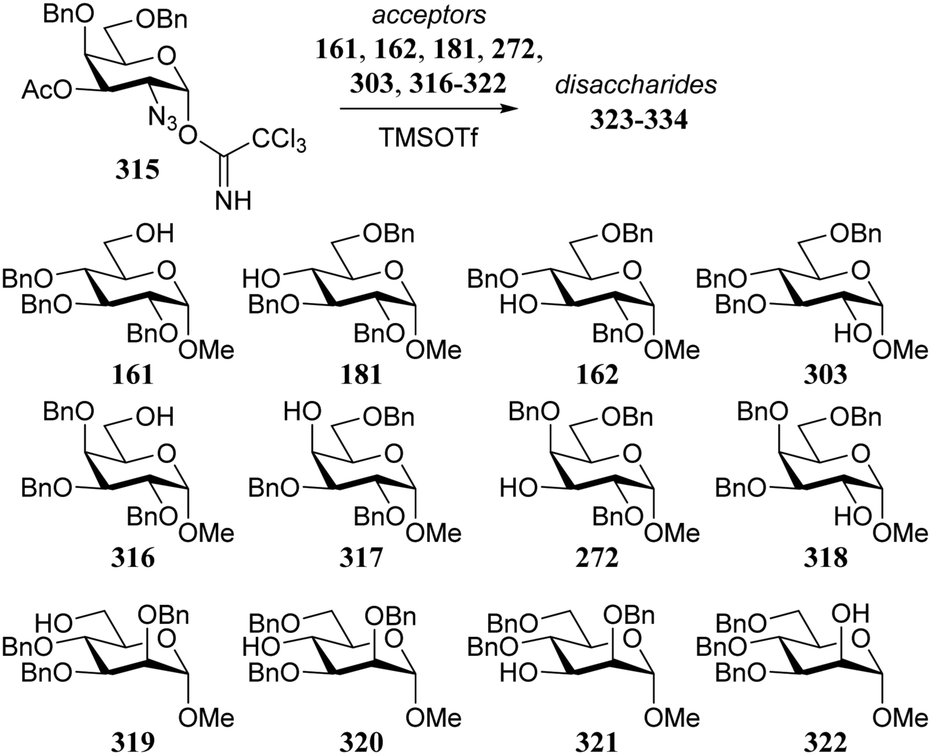
In similar vein, Kalikanda and Li investigated the effect of different regioisomeric and configurational glycosyl acceptors. They studied twelve tri-O-benzylated acceptors, having either a gluco-, galacto-, or manno-configuration in glycosylations with galactosazide donor 315 (Table 19).89 Again, it becomes clear that the most reactive alcohols react in a β-selective manner, while the least reactive nucleophiles provide α-linked products. Although the exact mechanism of these glycosylations are not clear, the results indicate the primary alcohols to be the most reactive and the secondary, axially orientated hydroxyls to be least reactive. The reactivity order, as assessed from the α/β-product ratio, in the glucose series matches that established in Demchenko's study described above.68
|
|
|||
|---|---|---|---|
| Acceptor | Product | Yield (%) | α:β |
| Reagents and conditions: donor (1.2 eq.), acceptor (1 eq.), M.S., TMSOTf (0.15 eq.), DCM (0.2 M), −78 °C. | |||
| 161 | 323 | 98 | β only |
| 181 | 324 | 56 | 1.8:1 |
| 162 | 325 | 53 | 1:3.4 |
| 303 | 326 | 68 | α only |
| 316 | 327 | 75 | 1:4 |
| 317 | 328 | 63 | α only |
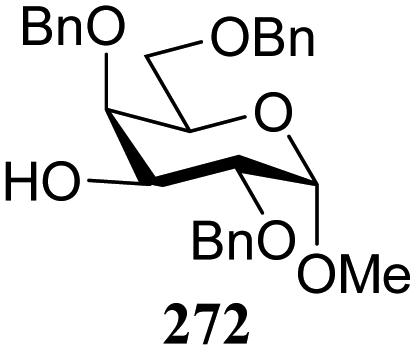
| 272 | 329 | 65 | 3:1 |
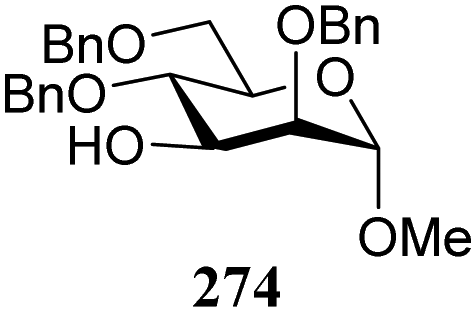
| 318 | 330 | 90 | 1.3:1 |
| 319 | 331 | 90 | 1:10 |
| 320 | 332 | 81 | 1.2:1 |
| 321 | 333 | 82 | 1:4.7 |
| 322 | 334 | 93 | α only |
Quantifying acceptor reactivity
Notwithstanding the progress that has been made in computational chemistry, only few attempts have been reported to date to investigate the nucleophilicity of glycosyl alcohol acceptors in a computational manner. The Fukui function provides a measure for the change in electron density at an atom of interest when an electron is subtracted (or added), and Fukui indices have been reported to account for the regioselectivity of electrophilic (f−) or nucleophilic (f+) reactions. Kalikanda and Li have computed Fukui f−-indices for a series of mannosyl diol nucleophiles to account for the regioselectivity observed in an acetylation and a glycosylation reaction (Table 20).90 The higher the f− value is for a particular atom, the higher the nucleophilicity of this atom is. As shown in Table 20, the calculated Fukui indices show that the relative nucleophilicity of the C-2 and C-3-alcohol functions depends on the protecting group pattern on the ring. A relatively large difference in Fukui values (such as for 338) indicates a more regioselective reaction as is borne out in the experiments, although it should be noted that only a very small set of nucleophiles and reactions has been probed.91|
|
|
|
||
|---|---|---|---|---|
| Entry | Electrophile | Ratio O-3/O-2 | Ratio O-3/O-2 | Ratio O-3/O-2 |
| Thiophenyl and trichloroimidate donors also gave trisaccharide byproducts, the disaccharides were formed with the same selectivity regardless of the donor. Atom-condensed Fukui values fm− were based on Mulliken charges and were obtained by DFT (B3LYP/6-31+G*). Reagents and conditions: donor (1 eq.), acceptor (1 eq.), 3 Å M.S., AgOTf (1 eq.), DCM, −30 °C. | ||||
| 1 | Ac2O (+pyridine) | 6:1 |
3:2 |
1:0 |
| 2 |
|
1:0 |
3:1 |
1:0 |
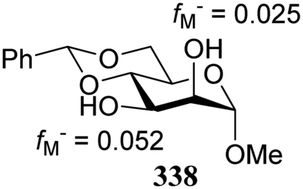
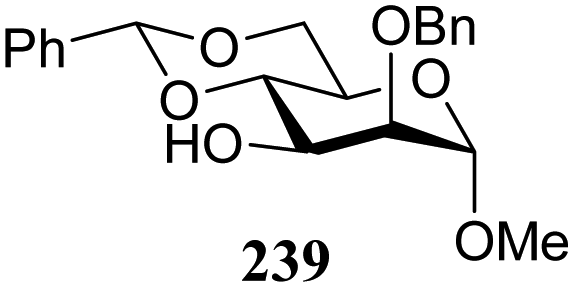
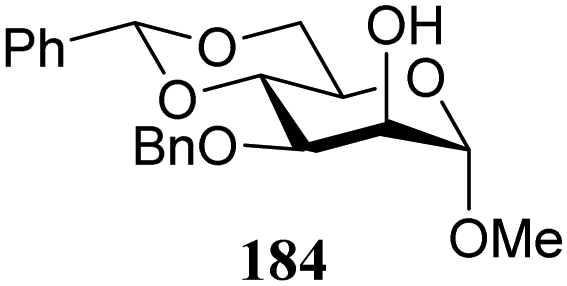
The group of Rúveda and Stortz also determined Fukui functions for a set of glucosamine acceptors (also see Table 6). They used the chemical hardness/softness (local chemical softness, s) of a reaction center and the atomic charge (q) as indicators for the relative reactivity of a series of acceptors (339–341, Fig. 1). In the examples studied, the atomic charge differed slightly between the alcohols in 339–341, and the chemical softness (s) seemed to correlate best with the relative reactivity (a lower sO-4 value indicates a more reactive acceptor), as determined in a glycosylation reaction using a per-benzoylated galactofuranose imidate donor 56 (see Table 6). The authors concluded that the interaction of their glycosyl acceptors with a glycosyl donor are better described by hard-hard (atomic charges) interactions than by frontier molecular orbital (soft–soft) interactions, and that all three descriptors have to be taken into account.20,27
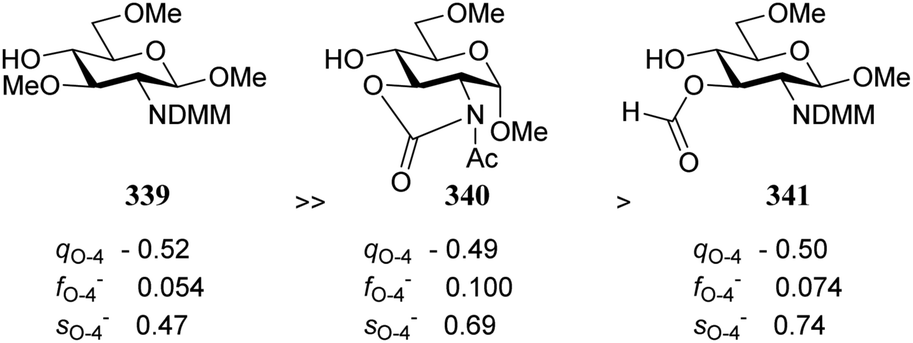 | ||
| Fig. 1 Computation evaluation of relative acceptor reactivities. (Rúveda, 2006).20 Atomic charge q, atom condensed Fukui value f and local chemical softness s are determined by multiple approaches, see the original publication for details. | ||
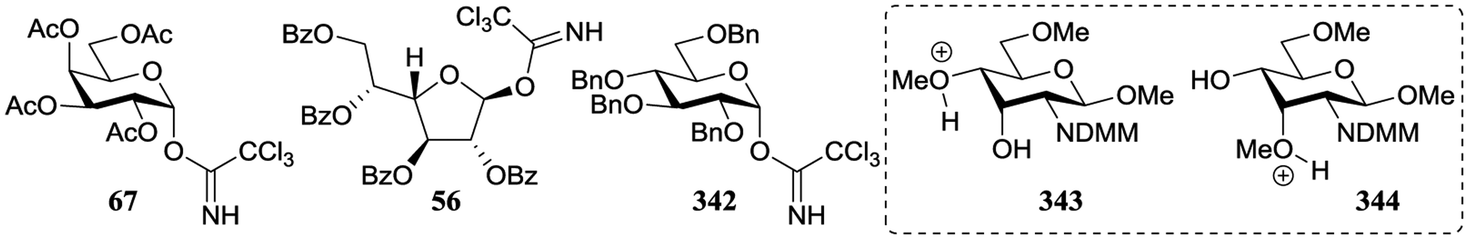
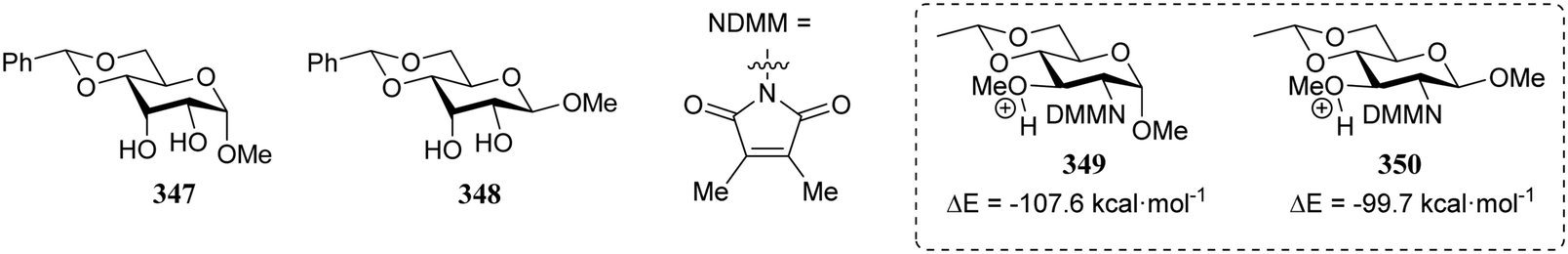
In a different approach the same group correlated the relative acceptor reactivity of a series of acceptors to the relative energy of the related methyloxonium ions (for example 343 and 344, energy difference between the C-3-OH(Me)(+) and C-4-OH(Me)(+) species is reported in the Table 21).92 The positively charged structures served to mimic the charge development in the glycosylation transition state and enabled the investigation of the influence of intramolecular hydrogen-bonding on the stability and geometry on the acceptor entity.93,94Table 21 reports computational results of a variety of diol acceptors and the experimental regioselectivity obtained in glycosylations with galactopyranose and furanose donors 67 and 56 (also see Table 8). Acceptors 345 and 346 were exclusive glycosylated at the axial C-3 in condensation reactions with donor 67, a result that correlates well with the calculated relative energy of the C-3-OH(Me)(+) and C-4-OH(Me)(+) species. The relative energy difference for glucosamine acceptors 76–79 proved to be smaller, and this correlated with a diminished regioselectivity in the reactions. Notably the regioselectivity proved dependent on the type of donor used, with the result obtained with the galactopyranose donor matching better to the computational results than the results obtained in the galactofuranose series. Benzylidene allose diols 347 and 348 were combined with glucose donor 342 revealing a slight preference for glycosylation at the C-3-OH both experimentally and computationally, although there clearly is no perfect agreement between both methods. The authors also calculated the energies of formation (from the neutral hydroxyl acceptor and a methyl cation) of structures 349 and 350 to compare the reactivity of individual acceptors with a single free hydroxyl group. The energy difference ΔΔE of 7.9 kcal mol−1 between the two systems is in agreement with the observed reactivity difference (Table 7; 69/71, 5:1). It appears that this relatively simple method is a promising way to estimate relative acceptor reactivities. With the advent of more accurate and powerful computational techniques, the extension to larger set of acceptors, and the use of a glycosylation system that follows well-defined and understood reaction paths, it may provide a more qualitative picture of acceptor reactivity.
|
|
||||
|---|---|---|---|---|
| Acceptor | O-3/O-4 donor 67 | O-3/O-4 donor 56 | E 3-O-Me − E4-O-Me (kcal mol−1) | |
| Energies obtained by DFT (B3LYP/6-31+G**). Reagents and conditions: donor (1.1 eq.), acceptor (1 eq.), TMSOTf (2.1 eq.), 4 Å M.S., DCM/CH3CN (29/1, 0.34 M), −25 °C. | ||||
|
345 | 1:0 |
−8.64 | |
| 346 | 1:0 |
−6.93 | ||
| 76 | 2:1 |
1:0 |
−4.60 | |
| 78 | 1:1 |
3.2:1 |
−1.85 | |
| 77 | 1:13 |
1:1 |
−0.03 | |
| 79 | 0:1 |
1:2.9 |
+2.15 | |
| Acceptor | O-3/O-2 donor 342 | E 3-O-Me − E2-O-Me (kcal mol−1) |
|---|---|---|
| 347 | 2.6:1 |
−4.39 |
| 348 | 1.2:1 |
−2.25 |
|
||
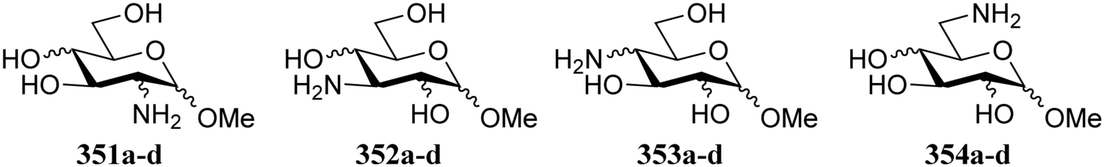
Bols and Inouye have taken a rather different approach to estimate the reactivity of different carbohydrate alcohols. They evaluated model systems in which specific hydroxyl groups were changed to amine functions.95,96 The pKas of the corresponding ammonium salts were determined by titration and these values are tabularized in Table 22. The pKaH values indicate the 6-NH2 group to be the most basic. The order of basicity in glucose found with aminoglycosides 351–354a/b, C-6-NH2 > C-3-NH2 > C-2-NH2 > C-4-NH2, roughly corresponds with the nucleophilicity on the parent hexoses (see Tables 17–19).97–99 To account for the pKaH trends recorded in Table 22, the authors identified that an anti-periplanar arrangement of the C-4-N and the C-5-O in 353a/b/d (Fig. 2), but also of C-2-N and C-1-O in 351a/b/d lead to a less basic NH2 group.100
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Position | α-Glc | pKaH | β-Glc | pKaH | α-Gal | pKaH | α-Man | pKaH |
| 2-NH2 | 351a | 7.5 | 351b | 7.2 | 355c | 7.9 | 359d | 7.2 |
| 3-NH2 | 352a | 7.8 | 352b | 7.6 | 356c | 8.0 | 360d | 8.1 |
| 4-NH2 | 353a | 6.8 | 353b | 6.7 | 357c | 7.3 | 361d | 7.2 |
| 6-NH2 | 354a | 8.9 | 354b | 8.6 | 358c | 8.9 | 362d | 9.0 |
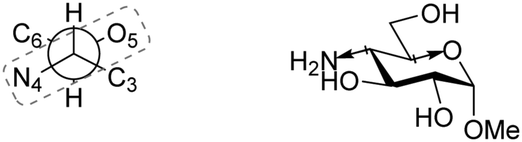 | ||
| Fig. 2 Anti-periplanar relationship between the ring oxygen and the C-4 substituent in methyl glucoside. | ||
Conclusions
The reactivity of a glycosyl acceptor is of fundamental importance to the outcome of a glycosylation reaction. The nucleophilicity of a carbohydrate alcohol is influenced by electronic aspects, through inductive effects and hydrogen-bonding, and by steric and conformational effects. The protecting groups on the acceptor play a pivotal role in shaping the acceptor reactivity. In contrast to the reactivity of glycosyl donors, for which relative reactivity values have been established4,101,102 to provide a numerical means to compare their reactivity, the relative reactivity of glycosyl acceptors remains relatively poorly understood and no numerical scales are available to assess acceptor reactivity. The insightful competition experiments performed by Rúveda did provide relative acceptor reactivities based on kinetics but to be more generally useful should be significantly expanded.26 It would also be of interest to see how relative acceptor values change with different donors. A systematic evaluation of different well established donor systems with the same set of acceptors may provide an accurate structure–reactivity–stereoselectivity map. Another approach would be to establish Kinetic Isotope Effects for donor–acceptor combinations or to perform cation-clock kinetics. Both methods have been used by the group of Crich, but only on the relatively nucleophilic and minimally intrusive iso-propanol.103–107 An extension of these methods spanning a wider range of acceptors, will provide the much needed insight how the reactivity of the acceptors determines the position of the operational reaction mechanisms along the SN2–SN1-continuum.108Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the European Research Council (ERC-CoG-726072-‘GLYCONTROL’, to J. D. C. C.)References
- A. V. Demchenko, Handbook of Chemical Glycosylation: Advances in Stereoselectivity and Therapeutic Relevance, Wiley-VCH Verlag GmbH & Co. KGaA, 2008 Search PubMed.
- L. K. Mydock and A. V. Demchenko, Org. Biomol. Chem., 2010, 8, 497–510 RSC.
- D. Crich, Acc. Chem. Res., 2010, 43, 1144–1153 CrossRef CAS.
- For selected examples, see: (a) B. Fraser-Reid and J. C. López, in Reactivity Tuning in Oligosaccharide Assembly, ed. B. Fraser-Reid and J. Cristóbal López, Springer, Berlin, Heidelberg, 2011, pp. 1–29 CrossRef; (b) P. Grice, S. V. Ley, J. Pietruszka, H. W. M. Priepke and E. P. E. Walther, Synlett, 1995, 781–784 CrossRef CAS; (c) N. L. Douglas, S. V. Ley, U. Lücking and S. L. Warriner, J. Chem. Soc., Perkin Trans. 1, 1998, 51–66 RSC; (d) Z. Zhang, I. R. Ollmann, X.-S. Ye, R. Wischnat, T. Baasov and C.-H. Wong, J. Am. Chem. Soc., 1999, 121, 734–753 CrossRef CAS; (e) K.-K. T. Mong and C.-H. Wong, Angew. Chem., 2002, 114, 4261–4264 CrossRef; (f) C. M. Pedersen, L. G. Marinescu and M. Bols, Chem. Commun., 2008, 2465–2467 RSC; (g) M. Heuckendorff, C. M. Pedersen and M. Bols, J. Org. Chem., 2013, 78, 7234–7248 CrossRef CAS.
- H. Paulsen, Angew. Chem., Int. Ed. Engl., 1982, 21, 155–173 CrossRef.
- For selected examples, see: (a) M. B. Cid, F. Alfonso and M. Martín-Lomas, Chem. – Eur. J., 2005, 11, 928–938 CrossRef CAS; (b) M. Islam, G. Gayatri and S. Hotha, J. Org. Chem., 2015, 80, 7937–7945 CrossRef CAS; (c) S. Buda, M. Nawój, P. Gołębiowska, K. Dyduch, A. Michalak and J. Mlynarski, J. Org. Chem., 2015, 80, 770–780 CrossRef CAS; (d) S. Buda, P. Gołębiowska and J. Mlynarski, Eur. J. Org. Chem., 2013, 3988–3991 CrossRef CAS; (e) B. S. Komarova, M. V. Orekhova, Y. E. Tsvetkov and N. E. Nifantiev, Carbohydr. Res., 2014, 384, 70–86 CrossRef CAS; (f) J. Y. Baek, B.-Y. Lee, M. G. Jo and K. S. Kim, J. Am. Chem. Soc., 2009, 131, 17705–17713 CrossRef CAS; (g) K. S. Kim, D. B. Fulse, J. Y. Baek, B.-Y. Lee and H. B. Jeon, J. Am. Chem. Soc., 2008, 130, 8537–8547 CrossRef CAS; (h) Y. J. Lee, K. Lee, E. H. Jung, H. B. Jeon and K. S. Kim, Org. Lett., 2005, 7, 3263–3266 CrossRef CAS; (i) I. M. Ryzhov, E. Y. Korchagina, I. S. Popova, T. V. Tyrtysh, A. S. Paramonov and N. V. Bovin, Carbohydr. Res., 2016, 430, 59–71 CrossRef CAS; (j) T. H. Schmidt and R. Madsen, Eur. J. Org. Chem., 2007, 3935–3941 CrossRef CAS; (k) T. Hashihayata, H. Mandai and T. Mukaiyama, Chem. Lett., 2003, 32, 442–443 CrossRef CAS; (l) P. I. Abronina, K. G. Fedina, N. M. Podvalnyy, A. I. Zinin, A. O. Chizhov, N. N. Kondakov, V. I. Torgov and L. O. Kononov, Carbohydr. Res., 2014, 396, 25–36 CrossRef CAS.
- For selected examples, see: (a) R. Dyapa, L. T. Dockery and M. A. Walczak, Org. Biomol. Chem., 2016, 15, 51–55 RSC; (b) M. Heuckendorff and H. H. Jensen, Carbohydr. Res., 2018, 455, 86–91 CrossRef CAS; (c) Y. Geng, A. Kumar, H. M. Faidallah, H. A. Albar, I. A. Mhkalid and R. R. Schmidt, Angew. Chem., Int. Ed., 2013, 52, 10089–10092 CrossRef CAS; (d) Y. Hu, K. Yu, L.-L. Shi, L. Liu, J.-J. Sui, D.-Y. Liu, B. Xiong and J.-S. Sun, J. Am. Chem. Soc., 2017, 139, 12736–12744 CrossRef CAS PubMed; (e) S. Medina, M. J. Harper, E. I. Balmond, S. Miranda, G. E. M. Crisenza, D. M. Coe, E. M. McGarrigle and M. C. Galan, Org. Lett., 2016, 18, 4222–4225 CrossRef CAS; (f) K. S. Kim, J. H. Kim, Y. J. Lee, Y. J. Lee and J. Park, J. Am. Chem. Soc., 2001, 123, 8477–8481 CrossRef CAS.
- R. Castelli, S. Schindler, S. M. Walter, F. Kniep, H. S. Overkleeft, G. A. Van der Marel, S. M. Huber and J. D. C. Codée, Chem. – Asian J., 2014, 9, 2095–2098 CrossRef CAS PubMed.
- A. Joosten, M. Boultadakis-Arapinis, V. Gandon, L. Micouin and T. Lecourt, J. Org. Chem., 2017, 82, 3291–3297 CrossRef CAS.
- N. Teumelsan and X. Huang, J. Org. Chem., 2007, 72, 8976–8979 CrossRef CAS.
- L. Guazzelli, O. McCabe and S. Oscarson, Carbohydr. Res., 2016, 433, 5–13 CrossRef CAS.
- B. Sylla, K. Descroix, C. Pain, C. Gervaise, F. Jamois, J.-C. Yvin, L. Legentil, C. Nugier-Chauvin, R. Daniellou and V. Ferrières, Carbohydr. Res., 2010, 345, 1366–1370 CrossRef CAS PubMed.
- (a) P. O. Adero, H. Amarasekara, P. Wen, L. Bohé and D. Crich, Chem. Rev., 2018, 118, 8242–8284 CrossRef CAS; (b) W.-L. Leng, H. Yao, J.-X. He and X.-W. Liu, Acc. Chem. Res., 2018, 51, 628–639 CrossRef CAS.
- P. Sinaÿ, Pure Appl. Chem., 1978, 50, 1437–1452 Search PubMed.
- H. Paulsen and O. Lockhoff, Chem. Ber., 1981, 114, 3079–3101 CrossRef CAS.
- H. Paulsen and R. Lebuhn, Liebigs Ann. Chem., 1983, 1047–1072 CrossRef CAS.
- P. J. Garegg and I. Kvarnström, Acta Chem. Scand., Ser. B, 1976, 30, 655–658 CrossRef.
- P. J. Garegg and I. Kvarnström, Acta Chem. Scand., Ser. B, 1977, 31, 509–513 CrossRef.
- D. Crich and V. Dudkin, J. Am. Chem. Soc., 2001, 123, 6819–6825 CrossRef CAS.
- M. L. Bohn, M. I. Colombo, C. A. Stortz and E. A. Rúveda, Carbohydr. Res., 2006, 341, 1096–1104 CrossRef CAS.
- R. Lucas, D. Hamza, A. Lubineau and D. Bonnaffé, Eur. J. Org. Chem., 2004, 2107–2117 CrossRef CAS.
- D. Crich and A. U. Vinod, Org. Lett., 2003, 5, 1297–1300 CrossRef CAS.
- S. S. Pertel, V. Y. Osel'skaya, V. Y. Chirva and E. S. Kakayan, Russ. Chem. Bull., 2015, 64, 1119–1124 CrossRef CAS.
- M. I. Colombo, C. A. Stortz and E. A. Rúveda, Carbohydr. Res., 2011, 346, 569–576 CrossRef CAS PubMed.
- U. Clara, M. Gómez Ana, L. J. Cristóbal and F.-R. Bert, Eur. J. Org. Chem., 2009, 403–411 Search PubMed.
- M. L. Bohn, M. I. Colombo, P. L. Pisano, C. A. Stortz and E. A. Rúveda, Carbohydr. Res., 2007, 342, 2522–2536 CrossRef CAS PubMed.
- M. L. Bohn, M. I. Colombo, E. A. Rúveda and C. A. Stortz, Org. Biomol. Chem., 2008, 6, 554–561 RSC.
- The β-anomers of DMM protected glucosamine 3,4-diols (see also Table 8) are selective towards O-4. Similar regioselectivity has been observed for the N-phthaloyl (O-3/O-4, 1:3.6) see S. Numomura, M. Iida, M. Numata, M. Sugimoto and T. Ogawa, Carbohydr. Res., 1994, 263, C1–C6 CrossRef CAS , and N-tetrachlorophthaloyl (only O-4 reacted), see L. Lay, L. Manzoni, R. R. Schmidt, Carbohydr. Res., 1998, 310, 157–171, both with galactopyranose donors. The N-tetrachlorophthaloyl protected glucosamine diol reacted with an l-fucosyl donor with slight preference for O-3 (O-3/O-4, 2:1).
- J. D. C. Codée, L. J. van den Bos, A.-R. de Jong, J. Dinkelaar, G. Lodder, H. S. Overkleeft and G. A. van der Marel, J. Org. Chem., 2009, 74, 38–47 CrossRef.
- S. Masamune, W. Choy, J. S. Petersen and L. R. Sita, Angew. Chem., Int. Ed. Engl., 1985, 24, 1–30 CrossRef.
- N. M. Spijker and C. A. A. van Boeckel, Angew. Chem., Int. Ed. Engl., 1991, 30, 180–183 CrossRef.
- D. Lafont, P. Boullanger and B. Fenet, J. Carbohydr. Chem., 1994, 13, 565–583 CrossRef CAS.
- C. A. A. van Boeckel and M. Petitou, Angew. Chem., Int. Ed. Engl., 1993, 32, 1671–1690 CrossRef.
- M. Petitou and B. C. A. A. van, Pure Appl. Chem., 2009, 69, 1839–1846 Search PubMed.
- H. A. Orgueira, A. Bartolozzi, P. Schell and P. H. Seeberger, Angew. Chem., Int. Ed., 2002, 41, 2128–2131 CrossRef CAS PubMed.
- D. Magaud, R. Dolmazon, D. Anker, A. Doutheau, Y. L. Dory and P. Deslongchamps, Org. Lett., 2000, 2, 2275–2277 CrossRef CAS PubMed.
- J. C. Castro-Palomino, Y. E. Tsvetkov, R. Schneider and R. R. Schmidt, Tetrahedron Lett., 1997, 38, 6837–6840 CrossRef CAS.
- Thioethers at the reducing end of acceptors may change the reactivity of the acceptor (for example see: K. Zegelaar-Jaarsveld, S. A. W. Smits, G. A. van der Marel and J. H. van Boom, Bioorg. Med. Chem., 1996, 4, 1819–1832 CrossRef CAS and S.-T. Liew, A. Wei, Carbohydr. Res., 2002, 337, 1319–1324.). Thioethers can also participate to the extent of aglycon transfer when the thiofunction is more reactive than the alcohol in the acceptor (for example see: A.-R de Jong, B. Hagen, V. van der Ark, H. S. Overkleeft, J. D. C. Codée, G. A. Van der Marel, J. Org. Chem., 2012, 77, 108–125, Z. Li, J. C. Gildersleeve, Tetrahedron Lett., 2007, 48, 559–562 and H. M. Christensen, S. Oscarson, H. H. Jensen, Carbohydr. Res., 2015, 408, 51–95.). Such a migration may be inhibited by bulky anomeric thio ethers (R. Geurtsen, G.-J. Boons, Tetrahedron Lett., 2002, 43, 9429–9431).
- Q. Zhang, E. R. van Rijssel, M. T. C. Walvoort, H. S. Overkleeft, G. A. van der Marel and J. D. C. Codée, Angew. Chem., Int. Ed., 2015, 54, 7670–7673 CrossRef CAS PubMed.
- M. T. C. Walvoort, G. Lodder, J. Mazurek, H. S. Overkleeft, J. D. C. Codée and G. A. van der Marel, J. Am. Chem. Soc., 2009, 131, 12080–12081 CrossRef CAS PubMed.
- M. T. C. Walvoort, W. de Witte, J. van Dijk, J. Dinkelaar, G. Lodder, H. S. Overkleeft, J. D. C. Codée and G. A. van der Marel, Org. Lett., 2011, 13, 4360–4363 CrossRef CAS PubMed.
- G. Anilkumar, M. R. Gilbert and B. Fraser-Reid, Tetrahedron, 2000, 56, 1993–1997 CrossRef CAS.
- G. Anilkumar, L. G. Nair and B. Fraser-Reid, Org. Lett., 2000, 2, 2587–2589 CrossRef CAS.
- C. J. J. Elie, R. Verduyn, C. E. Dreef, D. M. Brounts, G. A. van der Marel and J. H. van Boom, Tetrahedron, 1990, 46, 8243–8254 CrossRef CAS.
- C. J. J. Elie, R. Verduyn, C. E. Dreef, G. A. van der Marel and J. H. van Boom, J. Carbohydr. Chem., 1992, 11, 715–739 CrossRef CAS.
- H. Paulsen, in Selectivity – a Goal for synthetic efficiency, ed. W. Bartmann and B. M. Trost, Verlag Chemie, Weinheim, 1984, pp. 169–190 Search PubMed.
- H. Paulsen, M. Paal, D. Hadamczyk and K.-M. Steiger, Carbohydr. Res., 1984, 131, C1–C5 CrossRef CAS.
- H. Paulsen, D. Hadamczyk, W. Kutschker and A. Bünch, Eur. J. Org. Chem., 1985, 129–141 CAS.
- L. Bohé and D. Crich, Trends Glycosci. Glycotechnol., 2010, 22, 1–15 CrossRef.
- B. Fraser-Reid, J. C. Lopez, K. V. Radhakrishnan, M. Mach, U. Schlueter, A. Gomez and C. Uriel, Can. J. Chem., 2002, 80, 1075–1087 CrossRef CAS.
- B. Fraser-Reid, J. C. López, K. V. Radhakrishnan, M. V. Nandakumar, A. M. Gómez and C. Uriel, Chem. Commun., 2002, 2104–2105 RSC.
- C. Uriel, A. M. Gómez, J. C. López and B. Fraser-Reid, Synlett, 2003, 2203–2207 CAS.
- B. Fraser-Reid, J. C. López, A. M. Gómez and C. Uriel, Eur. J. Org. Chem., 2004, 1387–1395 CrossRef CAS.
- M. Guillemineau and F.-I. Auzanneau, Carbohydr. Res., 2012, 357, 132–138 CrossRef CAS PubMed.
- A. Forman and F.-I. Auzanneau, Carbohydr. Res., 2016, 425, 10–21 CrossRef CAS.
- B. Fraser-Reid and J. C. López, in Reactivity Tuning in Oligosaccharide Assembly, ed. B. Fraser-Reid and J. Cristóbal López, Springer Berlin Heidelberg, Berlin, Heidelberg, 2011, pp. 1–29 Search PubMed.
- F. Della Felice, E. A. Rúveda, C. A. Stortz and M. I. Colombo, Carbohydr. Res., 2013, 380, 167–173 CrossRef CAS PubMed.
- The group of Fraser-Reid has explained the regioselective preference of the different donors by the ability of participating groups to offer a more ‘diffuse’ anomeric charge (via an dioxo- or trioxolenium ion) compared to non-participating groups. See ref. 40 and G. Anilkumar, Z. J. Jia, R. Kraehmer and B. Fraser-Reid, J. Chem. Soc., Perkin Trans. 1, 1999, 3591–3596 RSC.
- D. Crich and W. Cai, J. Org. Chem., 1999, 64, 4926–4930 CrossRef CAS PubMed.
- J. R. Krumper, W. A. Salamant and K. A. Woerpel, Org. Lett., 2008, 10, 4907–4910 CrossRef CAS PubMed.
- J. R. Krumper, W. A. Salamant and K. A. Woerpel, J. Org. Chem., 2009, 74, 8039–8050 CrossRef CAS PubMed.
- M. G. Beaver and K. A. Woerpel, J. Org. Chem., 2010, 75, 1107–1118 CrossRef CAS PubMed.
- T. G. Minehan and Y. Kishi, Tetrahedron Lett., 1997, 38, 6815–6818 CrossRef CAS.
- R. J. Hinkle, Y. Lian, N. D. Litvinas, A. T. Jenkins and D. C. Burnette, Tetrahedron, 2005, 61, 11679–11685 CrossRef CAS.
- H. Mayr and A. R. Ofial, Acc. Chem. Res., 2016, 49, 952–965 CrossRef CAS.
- J. Ammer and H. Mayr, J. Phys. Org. Chem., 2013, 26, 59–63 CrossRef CAS.
- S. Minegishi, S. Kobayashi and H. Mayr, J. Am. Chem. Soc., 2004, 126, 5174–5181 CrossRef CAS PubMed.
- A. D. Dilman and H. Mayr, Eur. J. Org. Chem., 2005, 1760–1764 CrossRef CAS.
- J. Ammer, C. Nolte and H. Mayr, J. Am. Chem. Soc., 2012, 134, 13902–13911 CrossRef CAS PubMed.
- H. Mayr, B. Kempf and A. R. Ofial, Acc. Chem. Res., 2003, 36, 66–77 CrossRef CAS PubMed.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- C. G. Swain and E. C. Lupton, J. Am. Chem. Soc., 1968, 90, 4328–4337 CrossRef CAS.
- P. J. Garegg, P. Konradsson, I. Kvarnström, T. Norberg, S. C. T. Svensson and B. Wigilius, Acta Chem. Scand., Ser. B, 1985, 39, 569–577 CrossRef.
- T. Bowden, P. J. Garegg, J.-L. Maloisel and P. Konradsson, Isr. J. Chem., 2000, 40, 271–277 CrossRef CAS.
- B. Schumann, S. G. Parameswarappa, M. P. Lisboa, N. Kottari, F. Guidetti, C. L. Pereira and P. H. Seeberger, Angew. Chem., Int. Ed., 2016, 55, 14431–14434 CrossRef CAS PubMed.
- S. Chatterjee, S. Moon, F. Hentschel, K. Gilmore and P. H. Seeberger, J. Am. Chem. Soc., 2018, 140, 11942–11953 CrossRef CAS PubMed.
- K. Le Mai Hoang and X.-W. Liu, Nat. Commun., 2014, 5, 5051 CrossRef PubMed.
- In a different study by the same group, the stereochemical outcome of palladium catalyzed glycosylations of glycal donors, was also shown to critically depend on the nucleophilicity of the acceptor. Weak nucleophiles, such as phenol or trifluoroethanol, solely provided α-products, whereas stronger nucleophiles, and moderately nucleophilic carbohydrate acceptors all predominantly gave the β-product. S. Xiang, K. L. M. Hoang, J. He, Y. J. Tan and X.-W. Liu, Angew. Chem., Int. Ed., 2015, 54, 604–607 CrossRef CAS PubMed.
- S. van der Vorm, T. Hansen, H. S. Overkleeft, G. A. van der Marel and J. D. C. Codee, Chem. Sci., 2017, 8, 1867–1875 RSC.
- For selected examples, see ref. 59 and (a) D. Crich and S. Sun, J. Org. Chem., 1996, 61, 4506–4507 CrossRef CAS; (b) D. Crich and S. Sun, J. Org. Chem., 1997, 62, 1198–1199 CrossRef CAS; (c) D. Crich and M. Smith, Org. Lett., 2000, 2, 4067–4069 CrossRef CAS; (d) D. Crich and M. Smith, J. Am. Chem. Soc., 2001, 123, 9015–9020 CrossRef CAS; (e) K. Sasaki and K. Tohda, Tetrahedron Lett., 2018, 59, 496–503 CrossRef CAS.
- For selected examples, see ref. 40, 41 and (a) J. Dinkelaar, A. R. de Jong, R. van Meer, M. Somers, G. Lodder, H. S. Overkleeft, J. D. C. Codée and G. A. van der Marel, J. Org. Chem., 2009, 74, 4982–4991 CrossRef CAS; (b) J. D. C. Codée, L. J. van den Bos, A.-R. de Jong, J. Dinkelaar, G. Lodder, H. S. Overkleeft and G. A. van der Marel, J. Org. Chem., 2009, 74, 38–47 CrossRef; (c) M. T. C. Walvoort, H. van den Elst, O. J. Plante, L. Kröck, P. H. Seeberger, H. S. Overkleeft, G. A. van der Marel and J. D. C. Codée, Angew. Chem., Int. Ed., 2012, 51, 4393–4396 CrossRef CAS.
- B. Hagen, S. van der Vorm, T. Hansen, G. A. van der Marel and J. D. C. Codée, Selective Glycosylations: Synthetic Methods and Catalysts, Wiley-VCH Verlag GmbH & Co. KGaA, 2017, pp. 1–28 Search PubMed.
- D. M. Whitfield, Carbohydr. Res., 2007, 342, 1726–1740 CrossRef CAS.
- T. Hosoya, P. Kosma and T. Rosenau, Carbohydr. Res., 2015, 411, 64–69 CrossRef CAS.
- B. Hagen, S. Ali, H. S. Overkleeft, G. A. van der Marel and J. D. C. Codée, J. Org. Chem., 2017, 82, 848–868 CrossRef CAS PubMed.
- B. Hagen, J. H. M. van Dijk, Q. Zhang, H. S. Overkleeft, G. A. van der Marel and J. D. C. Codée, Org. Lett., 2017, 19, 2514–2517 CrossRef CAS.
- S. van der Vorm, J. M. A. van Hengst, M. Bakker, H. S. Overkleeft, G. A. van der Marel and J. D. C. Codée, Angew. Chem., Int. Ed., 2018, 57, 8240–8244 CrossRef CAS.
- S. Kaeothip, S. J. Akins and A. V. Demchenko, Carbohydr. Res., 2010, 345, 2146–2150 CrossRef CAS.
- J. Kalikanda and Z. Li, Carbohydr. Res., 2011, 346, 2380–2383 CrossRef CAS.
- J. Kalikanda and Z. Li, Tetrahedron Lett., 2010, 51, 1550–1553 CrossRef CAS.
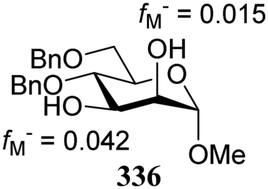
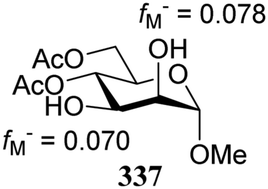
- The numerical values can provide a measure of regioselectivity within the same molecule. Comparing these values between different molecules does not seem to be valid. A 4,6-dimethyl substituted mannose diol acceptor gave values fM− = 0.050 for O-2 and fM− = 0.078 for O-3, which differ significantly from the ones reported for the dibenylated acceptor 336, and are closer to the diacetyl acceptor 337.
- M. I. Colombo, E. A. Rúveda and C. A. Stortz, Org. Biomol. Chem., 2011, 9, 3020–3025 RSC.
- M. B. Cid, I. Alonso, F. Alfonso, J. B. Bonilla, J. López-Prados and M. Martín-Lomas, Eur. J. Org. Chem., 2006, 3947–3959 CrossRef CAS.
- M. B. Cid, F. Alfonso, I. Alonso and M. Martín-Lomas, Org. Biomol. Chem., 2009, 7, 1471–1481 RSC.
- S. Inouye, Chem. Pharm. Bull., 1968, 16, 1134–1137 CrossRef CAS.
- C. M. Pedersen, J. Olsen, A. B. Brka and M. Bols, Chem. – Eur. J., 2011, 17, 7080–7086 CrossRef CAS.
- S. S. Kulkarni, in Selective Glycosylations: Synthetic Methods and Catalysts, ed. C. S. Bennett, Wiley-VCH Verlag GmbH & Co. KGaA, 2017, pp. 255–276 Search PubMed.
- M. S. Taylor, in Selective Glycosylations: Synthetic Methods and Catalysts, ed. C. S. Bennett, Wiley-VCH Verlag GmbH & Co. KGaA, 2017, pp. 231–253 Search PubMed.
- J. Lawandi, S. Rocheleau and N. Moitessier, Tetrahedron, 2016, 72, 6283–6319 CrossRef CAS.
- The order of reactivity roughly corresponds with calculated enthalpies of hydroxyl deprotonation (M. E. Brewster, M. Huang, E. Pop, J. Pitha, M. J. S. Dewar, J. J. Kaminski, N. Bodor, Carbohydr. Res., 1993, 242, 53–67), however, measured dissociation constants suggest a reversed order of reactivity (M. Matwiejuk, J. Thiem, Eur. J. Org. Chem., 2012, 2180–2187). Regardless of both these studies, the reactivity of an acceptor glycoside is better estimated by its ability to accept positive charge than to lose a hydroxyl proton.
- J.-C. Lee, W. A. Greenberg and C.-H. Wong, Nat. Protoc., 2007, 1, 3143–3152 CrossRef.
- C.-H. Hsu, S.-C. Hung, C.-Y. Wu and C.-H. Wong, Angew. Chem., Int. Ed., 2011, 50, 11872–11923 CrossRef CAS.
- M. Huang, P. Retailleau, L. Bohé and D. Crich, J. Am. Chem. Soc., 2012, 134, 14746–14749 CrossRef CAS.
- P. O. Adero, T. Furukawa, M. Huang, D. Mukherjee, P. Retailleau, L. Bohé and D. Crich, J. Am. Chem. Soc., 2015, 137, 10336–10345 CrossRef CAS.
- M. Huang, T. Furukawa, P. Retailleau, D. Crich and L. Bohé, Carbohydr. Res., 2016, 427, 21–28 CrossRef CAS.
- M. Huang, G. E. Garrett, N. Birlirakis, L. Bohé, D. A. Pratt and D. Crich, Nat. Chem., 2012, 4, 663–667 CrossRef CAS.
- D. Crich and N. S. Chandrasekera, Angew. Chem., Int. Ed., 2004, 43, 5386–5389 CrossRef CAS.
- R. A. Sneen, Acc. Chem. Res., 1973, 6, 46–53 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2019 |