
Recent advances in nucleophile-triggered CO2-incorporated cyclization leading to heterocycles
Sheng
Wang
a and
Chanjuan
Xi
 *ab
*ab
aMOE Key Laboratory of Bioorganic Phosphorus Chemistry & Chemical Biology, Department of Chemistry, Tsinghua University, Beijing 100084, China. E-mail: cjxi@tsinghua.edu.cn
bState Key Laboratory of Elemento-Organic Chemistry, Nankai University, Tianjin 300071, China
First published on 27th November 2018
Abstract
Carbon dioxide (CO2) has emerged as a sustainable, feasible, abundant one-carbon synthon and displays great potential in the synthesis of heterocycles such as lactones, lactams, and 2-oxazolidinones, which are privileged motifs in pharmaceutical chemistry demonstrating bioactivities. Although the fixation of CO2 is restricted due to its thermodynamic stability and kinetic inertness, multiple breakthroughs have been realized in annulation chemistry. This review concentrates on the advances made in the last five years in CO2-incorporated cyclization triggered by N-, O-, and C-nucleophiles. Three transformation modes of CO2 including carboxylative cyclization, carbonylative cyclization, and reductive cyclization have been summarized. Moreover, typical mechanisms and significant applications of these reactions are also described.
 Sheng Wang | Sheng Wang obtained his BSc degree in Chemistry from Shandong University in 2014. He is currently a PhD candidate under the supervision of Professor Chanjuan Xi at Department of Chemistry, Tsinghua University. His research interests include CO2 activation and its application in synthetic chemistry. |
 Chanjuan Xi | Chanjuan Xi is a Professor of Department of Chemistry, Tsinghua University. She graduated from Lanzhou University in 1986. She worked at Lanzhou Institute of Chemical Physics, CAS (1986–1995). In the course of her work, she earned a Master's degree in Chemistry in 1994. In 1996, she joined Hokkaido University and earned her PhD in Pharmaceutical Sciences in 1999, and spent one year as a postdoctoral fellow in Catalysis Research Center. In 2000, she joined Tsinghua University as Associate Professor and then she was promoted to a full Professor in 2005. Her research interests include organometallics and sustainable organic chemistry. |
1. Introduction
The utilization of CO2 as a one-carbon source to prepare value-added chemicals has received considerable attention.1 Such interests initially arose as a consequence of fossil fuel depletion and the continuously increasing emission of CO2 caused by human behavior. In this context, CO2 has been regarded as a promising one-carbon source to release the environmental pressure and moreover provide various benefits in organic synthesis such as safety, effectiveness, low toxicity, abundance, and sustainability. Despite these merits, recycling CO2 into valuable molecules effectively is restricted due to its thermodynamic stability and kinetic inertness, which makes the majority of CO2 lost as waste. Therefore, investigations concentrating on the utilization of CO2 in chemical transformations are worthwhile and highly desirable.Up to now, several strategies to overcome the thermodynamic stability and kinetic inertness of CO2 have been developed. It is well known that CO2 is a non-polar molecule with a delocalized π bond containing three centers and four electrons, which stabilizes the CO2 molecule. To realize CO2 transformation, the utilization of high-energy substrates such as organometallic reagents and 3-membered ring compounds is a straightforward method, while the addition of high-energy additives to trigger the reaction and provide energy for a catalytic cycle is also effective. Moreover, the application of catalysts, which could activate CO2 or starting materials and accelerate the reaction by lowering the energy barrier of the transition state, is also key to achieve a thermodynamically favored reaction.
Based on the strategy mentioned above, diverse reactions of CO2 have been reported. The central carbon serves as a weak electrophilic site due to the polar C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O double bond and displays excellent affinity towards strong nucleophiles, while the Lewis basic oxygen can donate electrons. As a result, reducing CO2 in a straightforward manner to prepare formic acid, formaldehyde, methanol or methane by 2-electron to 8-electron reduction processes using suitable reductants has been developed. Both homogeneous metal catalysts with different ligands2 and heterogeneous catalysts with controlled nano-structures3 realize reduction smoothly. More importantly, value-added carbonates, carbamates, carboxylic acids, and various sophisticated heterocycles containing “CO2”, “CO”, “CH2” or “CH” moieties are obtained in the presence of CO2 and different substrates with amino groups, hydroxyl groups, and carbon nucleophiles. Among the diverse products, heterocycles are of key importance due to their synthetic applications and biological properties as significant motifs (Scheme 1).4 Consequently, CO2-incorporated cyclization triggered by nucleophiles has been explored continuously in recent years as an effective and green approach to heterocycles.
O double bond and displays excellent affinity towards strong nucleophiles, while the Lewis basic oxygen can donate electrons. As a result, reducing CO2 in a straightforward manner to prepare formic acid, formaldehyde, methanol or methane by 2-electron to 8-electron reduction processes using suitable reductants has been developed. Both homogeneous metal catalysts with different ligands2 and heterogeneous catalysts with controlled nano-structures3 realize reduction smoothly. More importantly, value-added carbonates, carbamates, carboxylic acids, and various sophisticated heterocycles containing “CO2”, “CO”, “CH2” or “CH” moieties are obtained in the presence of CO2 and different substrates with amino groups, hydroxyl groups, and carbon nucleophiles. Among the diverse products, heterocycles are of key importance due to their synthetic applications and biological properties as significant motifs (Scheme 1).4 Consequently, CO2-incorporated cyclization triggered by nucleophiles has been explored continuously in recent years as an effective and green approach to heterocycles.
 | ||
| Scheme 1 Selected biologically active heterocycles possessing different one-carbon moieties. | ||
According to reported literature, CO2-incorporated cyclization could be divided into three modes (Scheme 2): (I) nucleophilic attack on CO2 followed by intramolecular cyclization to afford carboxylative cycles; (II) sequential nucleophilic attack on CO2 with substrates containing two nucleophilic sites to give cyclic carbonyl derivatives; (III) nucleophilic attack on the reduced CO2 followed by cyclization to generate cyclic derivatives. Nucleophiles such as amines, alcohols, and some carbon nucleophiles could be carboxylated by CO2 in the presence of base, but the problem is that these unstable carboxylated products would decarboxylate and return back to starting materials after workup. Therefore, the key to success lies in fixation of CO2, for instance, transformation of unstable carboxylated intermediates to a stable product or reducing stable CO2 to more applicable one-carbon sources. To solve the problem, the substrate in mode I provides an electrophilic site to accept excess electrons from oxygen while mode II offers another nucleophile to attack the CO2. In these two modes, the valence of central carbon is invariant. In mode III, depending on the electron acceptor properties of the central carbon, CO2 could be switched to a more flexible and applicable one-carbon synthon serving as “CH”, “CO” or “CH2” in the construction of heterocycles. Based on these reaction modes, various well-designed substrates and catalysis systems have achieved transformations of CO2 into molecules as one-carbon building blocks to generate different heterocycles. This review endeavors to illustrate different elegant CO2-incorporated cyclizations in the last 5 years initiated by appropriate nucleophiles to yield various heterocycles: (1) nucleophile-triggered CO2-incorporated carboxylative cyclization; (2) nucleophile-triggered CO2-incorporated carbonylative cyclization; (3) CO2-incorporated reductive cyclization. The cyclization of aziridines and epoxides with CO2 is not included as it is discussed in detail in previous works.5
 | ||
| Scheme 2 Reaction modes of CO2-incorporated annulation triggered by nucleophiles. | ||
2. Nucleophile-triggered CO2-incorporated carboxylative cyclization
As an electrophile, CO2 could be easily trapped by different strong nucleophiles with the formation of C–N, C–O or C–C bonds with the negative charge being transferred to the O atom of CO2. Taking advantage of the resulting nucleophilic oxygen anions, well-designed substrates undergo tandem cyclization assisted by a catalyst or base to give various valuable chemicals such as 2-oxazolidinones, cyclic carbonates, and lactones.2.1 N-Nucleophile-triggered carboxylative cyclization
Amines bearing unsaturated bonds such as allylamines and propargylamines are significant substrates for CO2-incorporated synthesis of attractive 2-oxazolidinones.6 Although great progress has been made in the preparation of 2-oxazolidinones, the synthesis of functionalized 2-oxazolidinones is still attractive considering their versatility in pharmacy and materials science. To facilitate the annulation, strategies involving activation of unsaturated bonds and CO2 have been fully applied.In 2016, Yu and co-workers developed copper-catalyzed oxytrifluoromethylation of allylamine 1 with CO2 (1 atm) and Togni's reagent (Scheme 3).7 CF3-Containing 2-oxazolidinones 2 were prepared chemo-, regio-, and diastereoselectively with moderate to good yield. Notably, bulky trisubstituted alkenyl amines could also undergo oxytrifluoromethylation, which was extremely challenging. Although azotrifluoromethylation of allylamine was competitive, the great affinity between CO2 and the amine controlled the reactivity of the amine and avoided undesirable byproduct formation. The addition of radical scavengers such as TEMPO (2,2,6,6-tetramethylpiperidine-1-oxyl) and BHT (butylated hydroxytoluene) generated TEMPO-CF3 and BHT-CF3, which proved the existence of ˙CF3. Based on experiments and DFT calculations,8 a mechanism involving Cu(I)–Cu(II) catalytic cyclization was proposed. Initially, the amine coordinates with Cu(I) followed by deprotonation with 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU). Subsequently, intermediate I-2 attacks CO2 to afford copper carboxylated complex I-3. Then, the I-CF3 bond undergoes homolysis to generate a ˙CF3 radical and a Cu(II) species followed by the addition of a ˙CF3 radical to the C–C double bond. Finally, intramolecular radical–radical cross-coupling takes place diastereoselectively to deliver spirocyclic product 3 with the release of the Cu(I) catalyst.
 | ||
| Scheme 3 Cu-Catalyzed carboxylative cyclization of allylamines with CO2. | ||
Recently, Yu and co-workers further developed copper-catalyzed trifluoromethylative dearomatization of indoles 4 and furans 6 with Togni's reagent and CO2 (1 atm) in the presence of 1,5-diazobicyclo[4.3.0]non-5-ene (DBN) (Scheme 4).9 A variety of CF3-containing spirocyclic indolines 5 and spiroacetals 7 were constructed. The observation of TEMPO-CF3 in the 19F NMR spectrum after the addition of TEMPO as a radical scavenger indicated that the radicals ˙CF3 played an important role in the reaction. Supported by DFT calculations, the reaction starts with the addition of an amine–copper complex to CO2 in the presence of base followed by radical addition of ˙CF3 to the CC double bond.
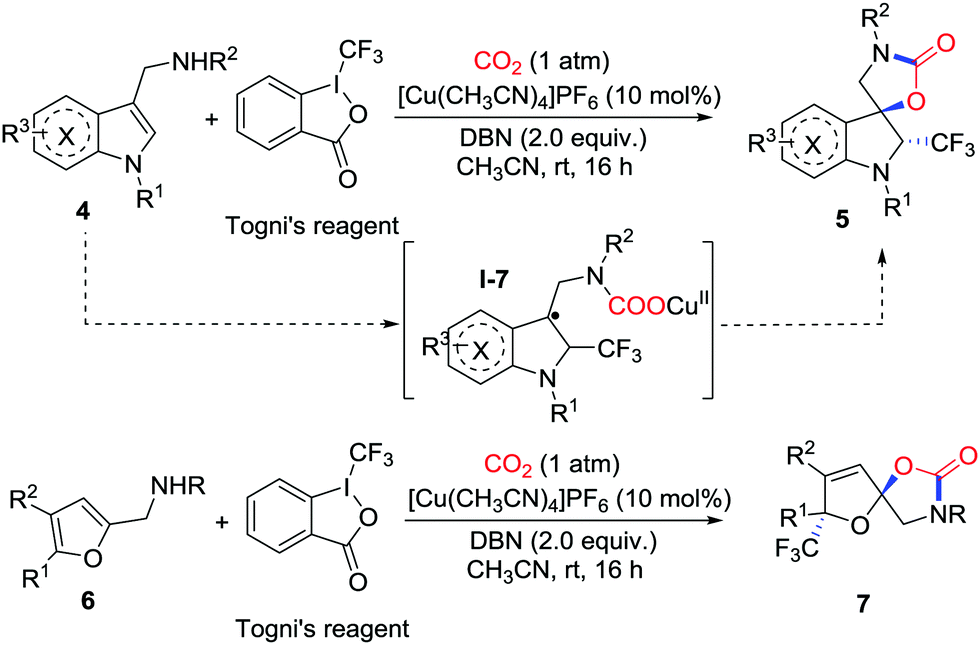 | ||
| Scheme 4 Copper-catalyzed carboxylative cyclization of indoles and furans with CO2. | ||
Light-driven reactions have been developed in recent years due to their high efficiency and mild conditions. He and co-workers disclosed photo-initiated perfluoroalkylation of allylamines 8 with CO2 (1 atm) and perfluoroalkyl iodides at room temperature to afford oxazolidinones 9 in good yield (Scheme 5).10 Controlled experiments and 1H NMR spectroscopy suggested a possible mechanism as shown in Scheme 5. The allylamine initially attacks CO2 with the assistance of 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD). The perfluoroalkyl iodide undergoes homolytic cleavage to generate perfluoroalkyl radicals. Subsequently, addition of the perfluoroalkyl radicals to the alkene gives secondary carbon radicals I-7, which could deliver iodoperfluoroalkylated carbamate I-8via coupling with radical I˙ (path I). However, path II involving radical propagation of I-7 and another molecule of perfluoroalkyl iodide is feasible as well.
 | ||
| Scheme 5 Photo-initiated carboxylative cyclization of allylamine with CO2. | ||
More recently, Yu and co-workers realized visible-light photoredox-catalyzed oxy-difluoroalkylation of allylamines 10 with CO2 (1 atm) to afford difluoroalkylated 2-oxazolidones 11 (Scheme 6).11 This reaction showed good tolerance to functional groups and steric hindrance. Apart from bromodifluoroacetates, a variety of difluoroalkyl reagents such as bromodifluoroacetamides, triisopropylsilyl-protected gem-difluoropropargyl bromides and phosphonyldifluoromethyl bromides gave the corresponding product under suitable conditions (longer time or iridium catalyst). Radical inhibition experiments and radical-clock experiments were conducted, which indicated the existence of ˙CF2CO2Et in the cyclization step. A possible mechanism was proposed as shown in Scheme 6. Initially, 1,4-diazabicyclo[2.2.2]octane (DABCO) reductively quenches the excited ruthenium catalyst to give the vital Ru(I) species, which reduces BrCF2COOEt to afford the radical ˙CF2COOEt I-11. Then, radical addition of the intermediate I-11 to carbamate I-10 generates the benzyl radical I-12 followed by oxidation of excited Ru(II) and intramolecular cyclization to yield the product 11.
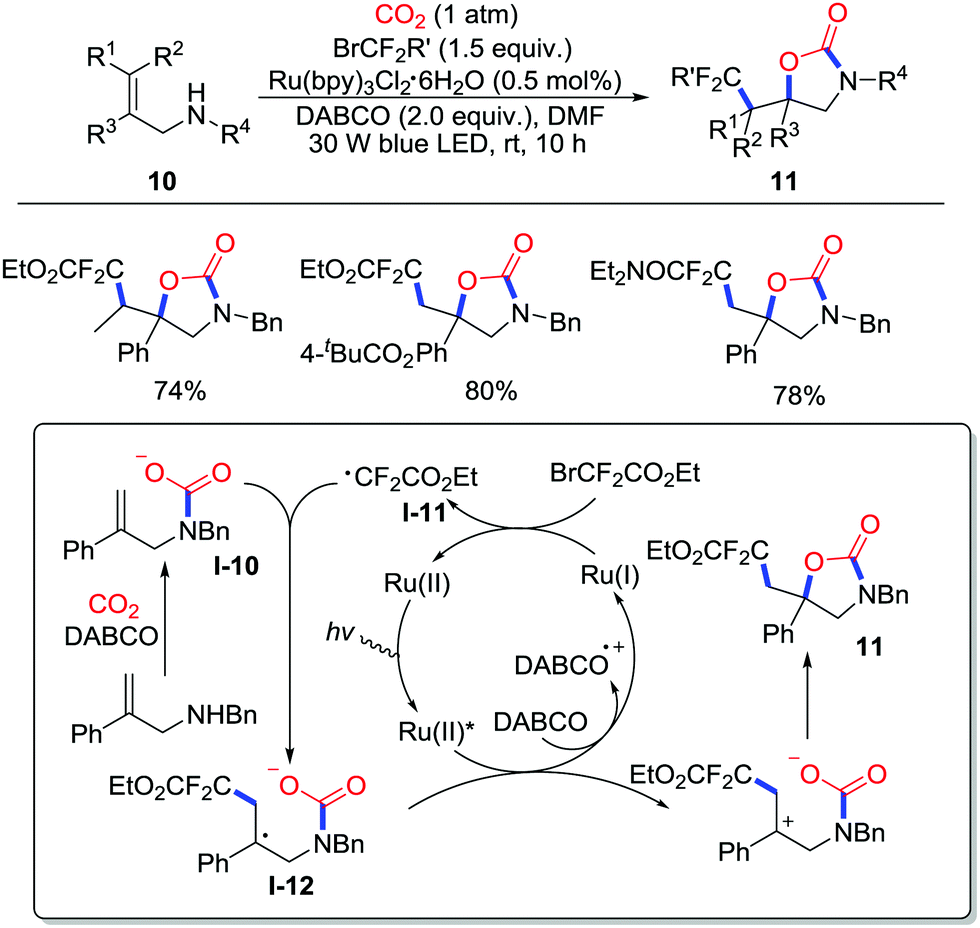 | ||
| Scheme 6 Visible-light photoredox-catalyzed oxy-difluoroalkylation of allylamines with CO2. | ||
Yu and co-workers further realized visible-light-driven oxy-alkylation of allylamines 12 with CO2 (1 atm) and unactivated alkyl bromides 13 (Scheme 7).12 Various functional groups could be tolerated in this process except some electron-withdrawing groups such as Bz, Boc, and Ts groups. A variety of primary, secondary, and tertiary alkyl bromides were applied in this reaction as well. Radical inhibition experiments and radical clock experiments proved the existence of alkyl radicals which were also detected by radical trapping experiments with TEMPO. A plausible mechanism was proposed as shown in Scheme 7. First, the Pd(0) is excited by photons and undergoes a single electron transfer (SET) process with alkyl bromide to generate Pd(I) and an alkyl radical. Then, radical addition to the carbamate takes place and gives benzylic radical I-13, which subsequently regenerates Pd(0) and delivers the stabilized cation I-14via SET. Intramolecular cyclization rapidly occurs to give the corresponding 2-oxazolidinone 14.
 | ||
| Scheme 7 Visible-light photoredox-catalyzed oxy-alkylation of allylamines with CO2. | ||
Although the prolinol scaffold is attractive in medical chemistry as a valuable heterocycle, the synthetic methods are limited for protected allylamines due to the undesirable reaction between free amines and oxidants. In 2017, our group demonstrated a concise iodine-mediated oxidative bicyclization of pent-4-en-1-amines 15 with CO2 (1 atm) via the oxyamination process to prepare prolinol carbamates 16, which could be easily transformed to various functionalized prolinols (Scheme 8).13 A mechanism was proposed as shown in Scheme 8 based on controlled reactions. The amine first adds CO2 with the formation of carbamate salts I-15. In the second step, the alkene is activated by I2 and attacked by a CO2− protected amine. The resulting iodo-carbamic acid I-16 affords the oxyamination product 16 after intramolecular substitution.
 | ||
| Scheme 8 I2-Mediated carboxylative cyclization of pent-4-en-1-amines with CO2. | ||
Cyclization of propargylamines to prepare 2-oxazolidinones with CO2 has been reported with various catalysts such as metal catalysts,14a–e organic catalysts,14f,g ionic liquids,14h CO2-adduct catalysts,14i,j and metal–organic frameworks (MOFs).14k,l
Moreover, atom-economical and step-economical multicomponent reactions to afford highly functionalized 2-oxazolidinones are still desirable. Nevada and co-workers disclosed a palladium-catalyzed multicomponent reaction with propargylamines 17, aryl halides and CO2 (1 atm) (Scheme 9).15 In the presence of atmospheric pressure CO2, highly functionalized 5-methylene-1,3-oxazolidin-2-ones 18 were generated stereoselectively in high efficiency. In this reaction, Pd(0) is first oxidized by aryl iodide to Pd(II), which activates the alkyne to facilitate cyclization giving vinyl palladium complex I-17 by π-activation. Subsequently reductive elimination delivers the product 18. In the case of a terminal alkyne, with the addition of excess aryl iodide, CuI (5 mol%), and DABCO (1,4-diazabicyclo[2.2.2]octane) as a base, Sonogashira reaction would occur before cyclization. The palladium catalyst serves as not only a π-Lewis acid but also a bridge connecting the carboxylative cyclization with cross coupling, which provides an approach to functionalized 2-oxazolidinones 20.
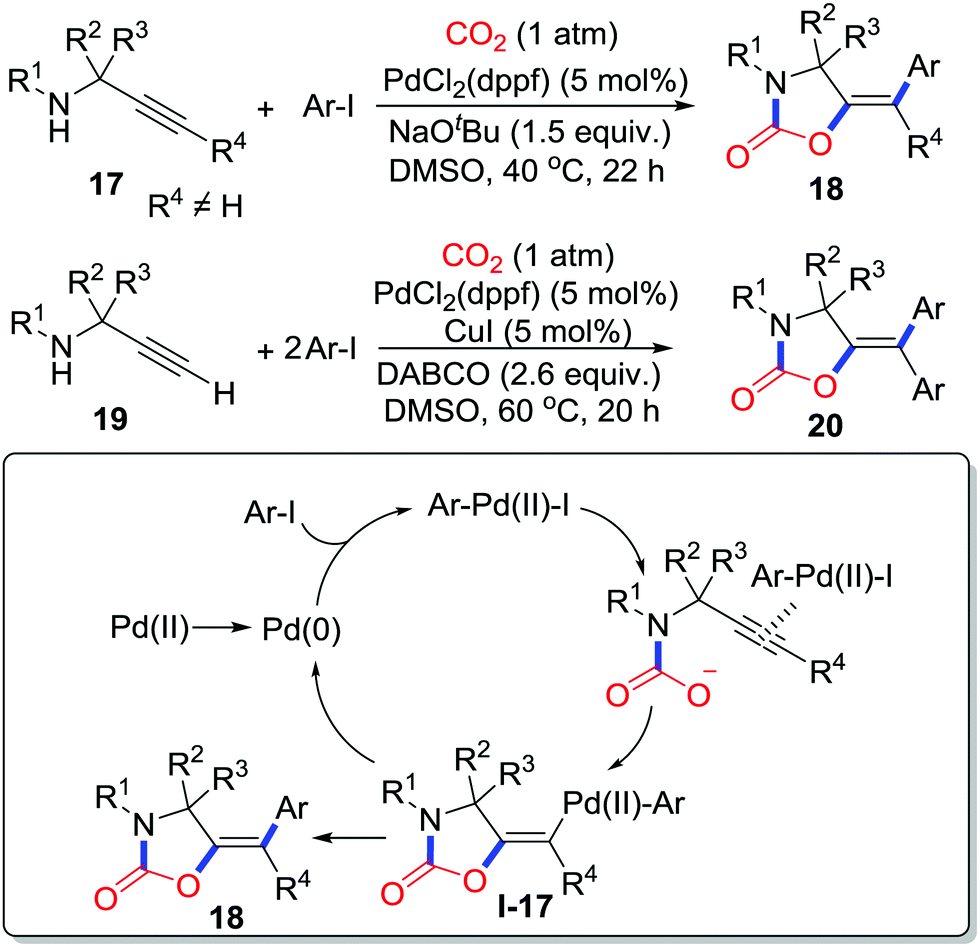 | ||
| Scheme 9 Palladium-catalyzed carboxylative cyclization of propargylamines with CO2. | ||
Li and co-workers reported an efficient copper-catalyzed four-component coupling with terminal alkynes, aldehydes, amines, and CO2 (1 atm) to prepare 2-oxazolidinones 21 bearing exocyclic alkenes.16 Moreover, Zhou and co-workers realized the asymmetric synthesis of 2-oxazolidinones 21 in a similar pathway through the tandem asymmetric coupling carboxylative cyclization (Scheme 10).17 Copper in the presence of the chiral PYBOX ligand L1 catalyzes the three-component coupling reaction to provide chiral N-aryl propargylaniline I-18. Downstream cyclization catalyzed by AgOBz/DPG (1,3-diphenylguanidine) affords 2-oxazolidinones 21. Notably, copper was reused as a catalyst in the cyclization step.
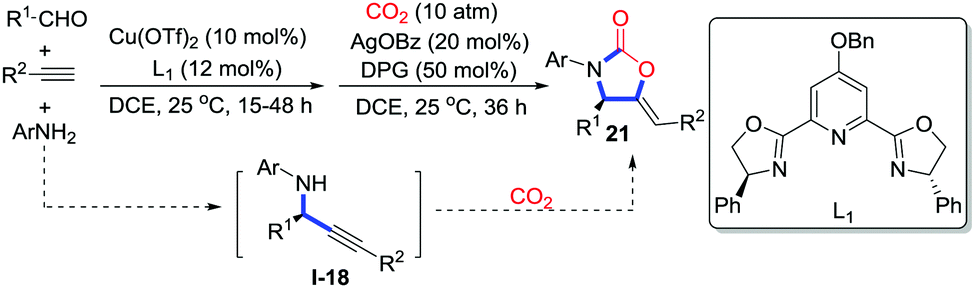 | ||
| Scheme 10 Tandem asymmetric carboxylative cyclization with alkynes, aldehydes, aryl amines and CO2. | ||
Besides propargylamines and allylamines, 2,3-allenyl amines were also well-investigated in CO2 chemistry. In 2013, Ma and co-workers reported a stereoselective palladium-catalyzed synthesis of 5-vinyloxazolidin-2-ones 23 from CO2 (1 atm), aryl iodides or alkenyl iodides, and 2,3-allenyl amines 22 (Scheme 11).18 Furthermore, inspired by previous palladium-catalyzed cyclization, Ikariya and co-workers demonstrated a carboxylative cyclization of 2,3-allenyl amines 24 with CO2 (10–70 atm) and Ag(OAc)(IPr) as a catalyst to form product 25 in 32 to 78% yields (Scheme 11).19 Owing to the silver catalyst, only trace amounts of intramolecular hydroamination products or 6-membered ring cyclic products were observed. Detailed comparisons with silver catalyst and gold catalyst were made by experiments and DFT calculations. The alkenyl carbon in the alkenyl-silver complex possesses more natural atomic charges than the alkenylgold complex as shown by DFT calculations. In the 13C NMR spectra, the chemical shift of the alkenyl carbon adjacent to the Au(I) is larger than Ag(I). According to theoretical and experimental results, the better performance of the silver catalyst might be attributed to the more polarized metal–carbon bond in the alkenyl–metal complex.
 | ||
| Scheme 11 Palladium-catalyzed and silver-catalyzed carboxylative cyclization of 2,3-allenyl amines with CO2. | ||
When allylamines or propargylamines were used, 5-membered heterocycles were obtained in most cases via a 5-exo-dig or 5-exo-trig process. Longer carbochains between amino groups and unsaturated bonds might give larger rings in the presence of CO2. Triggered by amino groups, less nucleophilic anilines were also utilized to prepare diverse attractive heterocycles with CO2 undergoing the 6-exo-trig process. In 2013, Yamada and co-workers described the silver-catalyzed synthesis of benzoxazin-2-one 27 derivatives from o-alkynylanilines 26 and CO2 (10 atm) assisted by DBU (Scheme 12).20 The silver catalyst serves as a Lewis acid to catalyze the cyclization and gives only the Z-type product via a 6-exo-dig process. Primary anilines as well as secondary anilines also gave the corresponding product smoothly.
 | ||
| Scheme 12 Silver-catalyzed carboxylative cyclization of o-alkynylanilines with CO2. | ||
Apart from the addition to the unsaturated bond, the substitution reaction has displayed application in fixing the CO2 into heterocycles. Oxazolidinedione is a versatile scaffold in diverse molecules demonstrating biological activities such as anti-bacterial and anti-cancer. In 2015, Kim and co-workers reported a copper/Cs2CO3-catalyzed reaction of 2-bromo-3-phenylacrylic acids 28, amines, and CO2 (1 atm) to afford various oxazolidinediones 29 (Scheme 13).21 The hypothesized mechanism is presented in Scheme 13. First, insertion of CO2 to amine affords the carbamic acid intermediate I-20. Then, 2-bromo-3-phenylacrylic acid coordinates with Cu(I) followed by the intramolecular oxidative addition with the C–Br bond. In the third step, the copper complex was coordinated by carbamic salt I-20 to form the new complex I-21. Subsequently reductive elimination generates the C–O bond. Finally, intramolecular nucleophilic attack of the amino group yields oxazolidinedione releasing the free copper catalyst. Isotope-labelling experiments with C18O2 also support this mechanism.
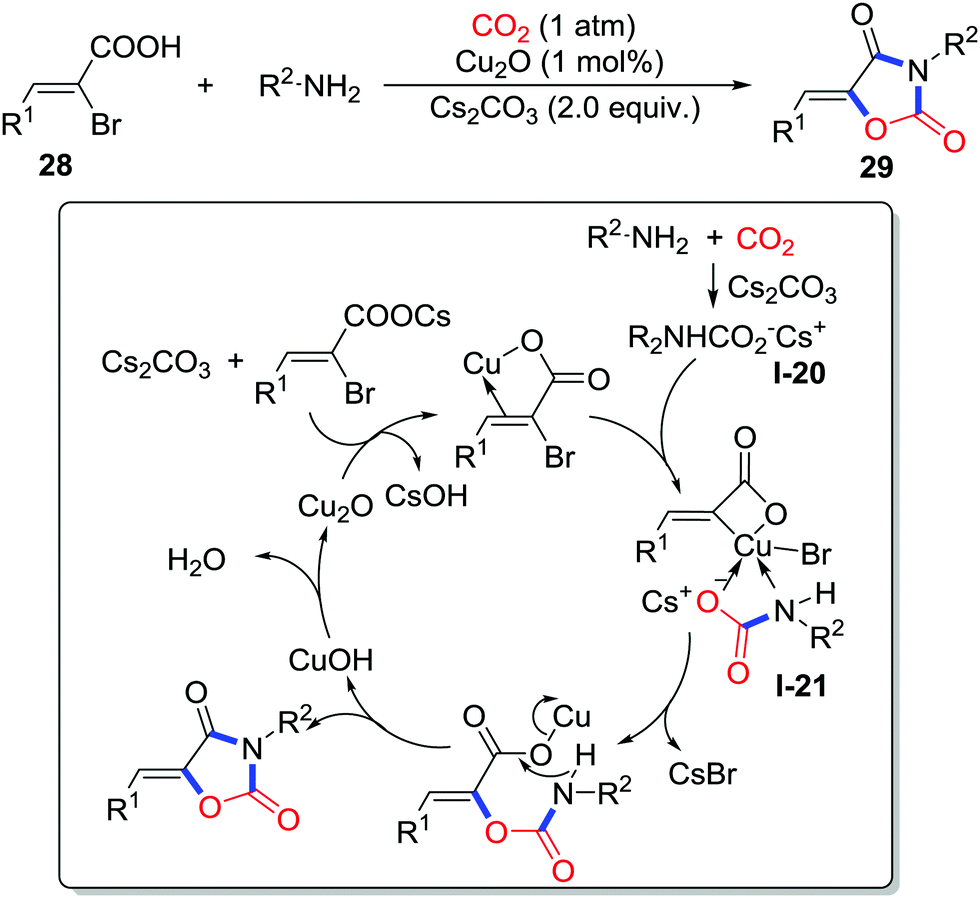 | ||
| Scheme 13 Copper-catalyzed carboxylative cyclization of 2-bromo-3-phenylacrylic acids and amines with CO2. | ||
1,3,4-Oxadiazole-2(3H)-one is a privileged skeleton widely existing in numerous bioactive molecules. Previous facile approaches to this structure mostly involve toxic CO or phosgene, while CO2 as an environmental-friendly feedstock exhibited superiority. Lu and co-workers disclosed a CsF/18-crown-6-mediated 1,3-dipolar cycloaddition of nitrile imine precursors 30 to synthesize 1,3,4-oxadiazole-2(3H)-ones 31 under CO2 (20 atm) (Scheme 14).24 Some variations in the substituents were made as shown in Scheme 14 and further application in synthesizing potential drugs was conducted. Controlled experiments and NMR detection indicated that 18-crown-6 promotes the formation of the nitrile imine intermediate and CsF/18-crown-6 enhances the reactivity of CO2 as a 1,3-dipolarophile.
 | ||
| Scheme 14 CsF/18-crown-6-promoted carboxylative cyclization of nitrile imines with CO2. | ||
Another approach to oxazolidinediones with CO2 (1 atm) was reported by Lu and co-workers in 2015 (Scheme 15).23 The α-ketoester 29 reacts with P(NMe2)3via Kukhtin–Ramirez addition reaction affording the electrophilic α-ketoester-tris(dimethylamino)phosphine adduct I-22. Then, the adduct traps the carbamic acid generated in situ to yield the carbamic ester intermediate I-23, which gives the corresponding product 30 in the presence of NaOMe.
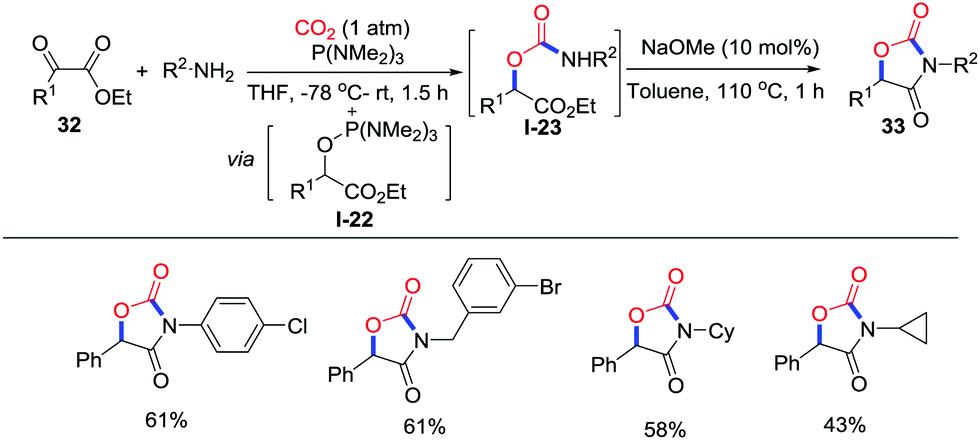 | ||
| Scheme 15 Phosphorus-mediated carboxylative cyclization of α-ketoesters and amines with CO2. | ||
More recently, a similar strategy using an additive to enhance the ability of the functional group to leave was utilized by Repo and co-workers to synthesize the cyclic carbamates 35 from amino alcohols 33 with the assistance of p-toluenesulfonyl chloride (TsCl) as an activating reagent (Scheme 16).22 5-Membered and 6-membered rings could be obtained with the corresponding amino alcohols while attempts to prepare 7-membered or 8-membered rings failed. Moreover, chiral substrates inverted the configuration at the stereocenter to deliver the product stereoselectively. DFT calculations suggested that the ring closure step was an intramolecular SN2 step with leaving of the TsO− group, which is in accordance with conversion of configuration when chiral substrates were used.
 | ||
| Scheme 16 TsCl-promoted carboxylative cyclization of amino alcohols with CO2. | ||
Besides addition and substitution, other miscellaneous methods to accept electrons from oxygen could realize carboxylative cyclization as well. In 2017, Zhang and co-workers disclosed palladium-catalyzed carboxylative annulation of o-iodoanilines 36 with CO2 (10 atm) and CO (5 atm) to generate isatoic anhydrides 37 (Scheme 17), which possess great potential in heterocycle synthesis and RNA structure probing.25 The reaction is compatible with both electron-donating groups and electron-withdrawing groups from moderate to good yield. Experiments utilizing labeled CO2 and CO revealed a possible reaction pathway. CO inserts into a C–Pd(II) bond generated by oxidative addition of ArX to give acyl-palladium intermediate I-24, which was transformed to the 7-membered acyl-palladium carbamate I-25 after addition of CO2. Then, reductive elimination affords the corresponding isatoic anhydrides with the release of Pd(0). Another possible route involving the formation of a 6-membered palladium carbamate I-26 from CO2 insertion cannot be excluded.
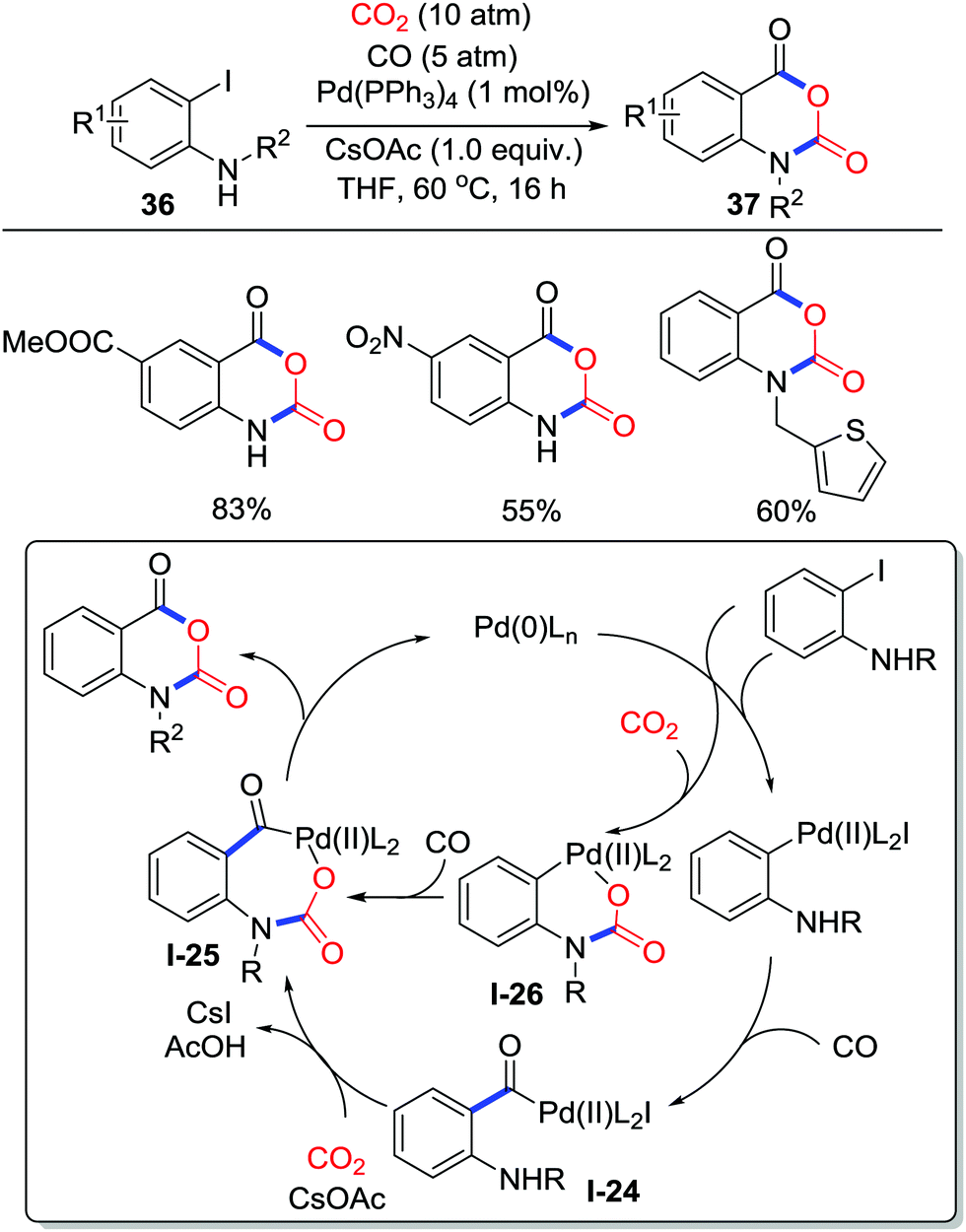 | ||
| Scheme 17 Palladium-catalyzed carboxylative cyclization of o-iodoanilines with CO2 and CO. | ||
2.2 O-Nucleophile-triggered carboxylative cyclization
O-Nucleophiles such as alcohols are capable of inserting CO2 with the formation of carbonic acid species, and the subsequent intramolecular addition or substitution would fix CO2 into the heterocycles. Alcohols containing unsaturated bonds or leaving groups could undergo carboxylative cyclization and afford useful cyclic carbonates. Methods utilizing metal catalysts,26a,b NHC-carbenes,26c organic bases,26d,e and ionic liquids26f have been well-investigated, which provide multiple ways to synthesize promising cyclic carbonates. However, highly functionalized cyclic carbonates are rarely reported.Alcohols containing C–C double bonds are reactive in the carboxylative cyclization via the addition process. Minakata and co-workers achieved tBuOI-mediated cyclization of allyl alcohols and homoallylic alcohols to afford cyclic carbonates bearing an iodomethyl group.6c However, the synthesis of chiral cyclic carbonates is still challenging. In 2016, Johnston and co-workers applied the dual Brønsted acid/chiral base organocatalyst to construct chiral centers and succeeded in preparing the 6-membered iodo-substituted cyclic carbonate 39 (Scheme 18).27 In the presence of the catalyst, I+ generated from the interaction of alkene and NIS (N-iodosuccinimide) induced the cyclization of carbonic acid, which is generated from the alcohols and CO2. Moreover, the resulting iodocarbonate was a versatile starting material and further transformations such as reduction and hydrolysis were demonstrated. Water exhibited a potent effect on the yield of the product due to the reversible combination with CO2, which is a competitive reaction to addition of alcohols to CO2. It is believed that the high enantioselectivity derives from the hydrogen bond between the transient carbonic acid intermediates and the dual Brønsted acid/chiral base catalysts.
 | ||
| Scheme 18 NIS-mediated carboxylative cyclization of homoallylic alcohols with CO2. | ||
Carboxylative cyclization involving propargylic alcohols and CO2 (1 atm) has been studied thoroughly. Recently, Cheng and co-workers realized a palladium-catalyzed one-pot carboxylative cyclization of propargylic alcohols 40, organic halides and CO2 (1 atm) under mild conditions (Scheme 19) via Pd(II) species I-27.28 This reaction features excellent step-economy, a broad substrate scope, good functional group tolerance, and high stereoselectivity, affording various functionalized α-alkylidene cyclic carbonates.
 | ||
| Scheme 19 Palladium-catalyzed carboxylative cyclization of propargylic alcohols and organic halides with CO2. | ||
In most cases, epoxides are initiated by external nucleophiles to capture CO2 and give the corresponding cyclic carbonates via an SN2 mechanism. Different from conventional mechanisms, Kleij and co-workers realized CO2-incorporated carboxylative cyclization of epoxy alcohols and amines 42 in MEK (methyl ethyl ketone) via an SNi mechanism (Scheme 20).29 The nucleophilic oxygen of in situ generated carbonates I-28 attacked epoxides in the presence of aluminum catalyst to generate cyclic carbonate 43 in a 5-exo-tet process. Moreover, this reaction was employed in organic synthesis, and some natural products containing an epoxy alcohol scaffold could give different carbonates with high chemoselectivity in different modes by the regulation of reaction conditions.
 | ||
| Scheme 20 Aluminum-catalyzed carboxylative cyclization of epoxy alcohols with CO2. | ||
In 2016, the NHC-copper-catalyzed multicomponent coupling of CO2 (5 atm), bis(pinacolato)diborons, and aldehydes was disclosed by Hou and co-workers (Scheme 21).30 A wide range of lithium boracarbonate ion pairs bearing different functional groups were prepared efficiently, which possess great potential as electrolytes in lithium ion batteries. Based on the isolated intermediates, a multi-component reaction mechanism was proposed. The reaction starts with ligand exchange between LiOtBu and [(SIMes)CuCl] to generate the copper alkoxide [(SIMes)Cu(OtBu)] followed by its reaction with B2(pin)2 to afford the copper complex [(SIMes)Cu(Bpin)]. Then, addition of Cu–B to the aldehyde occurs generating a new copper complex I-29, which subsequently attacks CO2 to afford the cyclic boracarbonate derivative I-30 with the formation of a B–O bond. Transmetalation between LiOtBu and the copper complex provides the final product releasing the copper catalyst.
 | ||
| Scheme 21 Copper-catalyzed carboxylative cyclization of aldehydes and bis(pinacolato)diborons with CO2. | ||
2.3 C-Nucleophile-triggered carboxylative cyclization
Strong C-nucleophiles, such as Grignard reagents and organic lithium reagents, are able to insert CO2 with the formation of C–C bonds to give the corresponding carboxylic acids. However, weak C-nucleophiles could only attack CO2 reversibly to generate unstable carboxylates, which tend to be decarboxylated. To fix CO2 into the molecules, further intramolecular addition to the unsaturated bond is necessary. Additionally, the resulting lactones are vital scaffolds in pharmacy.Obviously, enolates possess sufficient nucleophilicity to capture CO2, but the corresponding β-ketocarboxylic acids would revert to starting materials due to their thermodynamic instability. In 2012, Yamada's group realized the silver-catalyzed intramolecular cyclization of β-ketocarboxylic acids to construct 5-membered lactones.31 Furthermore, in 2017, they applied a similar strategy for the synthesis of tetronic acids 46 in good yields. The MTBD (7-methyl-1,5,7-triazabicyclo[4.4.0]dec-5-ene)/AgOAc system achieved the capture of CO2 with conjugated ynones 45via a 5-exo-dig cyclization selectively (Scheme 22).32 Application of this reaction in the total synthesis of Aspulvinone E exhibited enormous potential in synthetic chemistry.
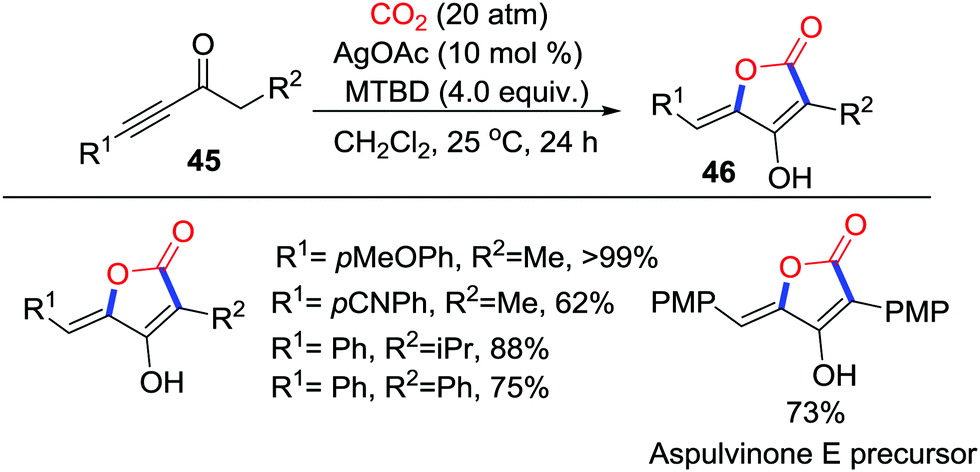 | ||
| Scheme 22 Silver-catalyzed carboxylative cyclization of conjugated ynones with CO2. | ||
Organosilane compounds are versatile tools to construct C–C bonds as potential nucleophiles. In the presence of F− or Lewis acids, carboxylation of the C–Si bond with CO2 (10 atm) to yield carboxylic acids or esters has been investigated. In 2015, Yamada and co-workers reported the silver-catalyzed and CsF-promoted carboxylative cyclization of trimethyl(2-methylenebut-3-yn-1-yl)silane derivatives 47 with CO2 (10 atm) to form the 2-furanones 48 and 2-pyrones 49 in satisfactory yield regioselectively (Scheme 23).33 When aryl-substituted alkynes were used, the 2-furanone derivatives 48 were obtained via a 5-exo-dig cyclization; single crystal X-ray diffraction performed on the products showed that they exist as Z-isomers. However, when alkyl-substituted alkynes were used, 2-pyrones 49 were formed as the main products.
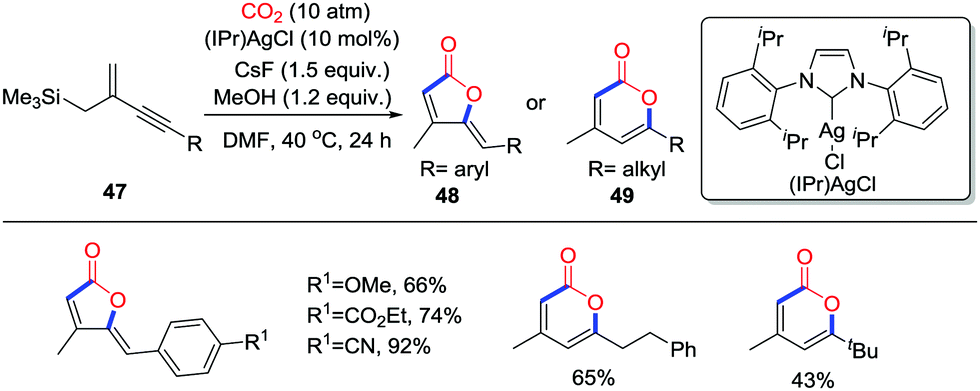 | ||
| Scheme 23 Silver-catalyzed and CsF-promoted carboxylative cyclization of trimethyl(2-methylenebut-3-yn-1-yl)silane derivatives with CO2. | ||
The C3-position of indoles is a promising C-nucleophile to react with electrophiles, and carboxylation has been established.34 In 2015, Skrydstrup and co-workers described the 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD)-catalyzed carboxylation of alkynyl indoles 50 at 100 °C (Scheme 24).35 30 mol% TBD catalyzed the cyclization efficiently when R2 was an aryl group, and 1.0 equivalent of TBD was necessary when R2 was an alkyl. This work provides an effective path to prepare tricyclic indole-containing rings 51 bearing different functional groups. A regular mechanism involving the indole-C3 position attacking the TBD-CO2 adduct to form the carboxylate and then delivering the lactone by a 6-endo-dig process was proposed. However, more recently, DFT calculation36 conducted by Wong and co-workers revealed that the TBD might not only activate CO2 to facilitate carboxylation of indole-C3 position but also stabilize the carboxylate to assist cyclization. This theoretical calculation did not contradict the demonstrated experimental result.
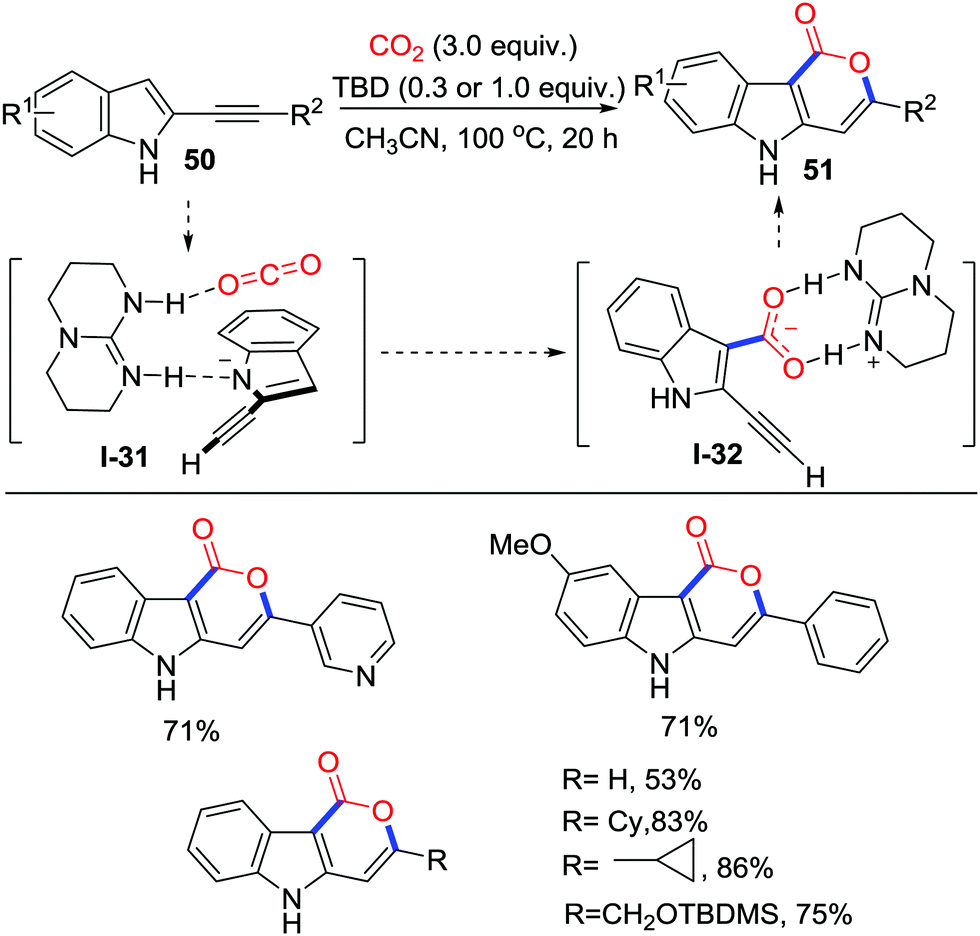 | ||
| Scheme 24 TBD-catalyzed carboxylative cyclization of alkynyl indoles with CO2. | ||
Benzyne is a highly reactive intermediate with great electrophilicity. The addition of nucleophiles to benzynes could generate C-nucleophiles to trap electrophiles, which provide an access to 1,2-disubtituted arenes. In 2014, Kobayashi and co-workers demonstrated the copper-catalyzed tandem cyclization reaction of terminal alkynes, CO2 (15 atm) and benzynes to prepare a series of functionalized isocoumarins 53 (Scheme 25).37 Assisted by NHC-copper catalysts and three equivalents of Cs2CO3, isocoumarins were obtained in moderate to good yield bearing electron-withdrawing and electron-donating functional groups. The mechanism proposed is shown in Scheme 25. The terminal alkyne is basified and generates an alkyne copper complex in the presence of copper catalyst. Assisted by CsF, 2-(trimethylsilyl)phenyl triflate 52 is converted to benzyne, which subsequently reacts with the alkynyl copper species to give o-alkynyl aryl copper intermediate I-33. Then, reversible nucleophilic addition to CO2 occurs and a subsequent 6-endo-dig cyclization produces isocoumarin copper complex I-34. Transmetalation regenerates the copper catalyst. Finally, the resulting cesium salt I-35 gives the product 53 after hydrolysis, which is also supported by controlled experiments with deuterium-enriched alkynes.
 | ||
| Scheme 25 Copper-catalyzed carboxylative cyclization of benzynes and terminal alkynes with CO2. | ||
The addition to alkynes is also an effective route to generate C-nucleophiles. Elegant substrate design might lead to different heterocycles. In 2018, Wu and co-workers disclosed the first visible-light-driven carbocarboxylation of alkynes 54 with the Ir/Co dual catalyst system and CO2 (1 atm) to prepare 2-benzoxepinones 55 (Scheme 26).38 When ortho-ester substituted aryl alkyne 54 was applied, an unprecedented cobalt-carboxylation/acyl migration occurred to deliver γ-hydroxybutenolides efficiently. Initially, the Co(II) oxidizes Ir(II) which was reduced by iPr2NEt and visible-light to give vital Co(I) species. Alkyne 54 reacts with CO2 and the cobalt catalyst to generate a 5-membered cobaltacycle intermediate I-36. Addition to the ester group occurs to generate carboxylate I-37. Then, the cobalt complex I-37 undergoes photoredox-catalyzed reduction and transmetalation with the photocatalyst and ZnBr2 to release the Co(I) species and carboxylate I-38. Tautomerization spontaneously takes place to deliver γ-keto acryliczinc complex I-39, which is followed by intramolecular cyclization to generate γ-hydroxybutenolide 55. When terminal alkynes were used, 2-pyrones were obtained (Scheme 27). The terminal alkyne inserts 5-membered cobaltacycle intermediate I-40 to give the 7-membered cobaltacycle I-41. Further reductive elimination releases the 2-pyrones and regenerates the Co(I) species.
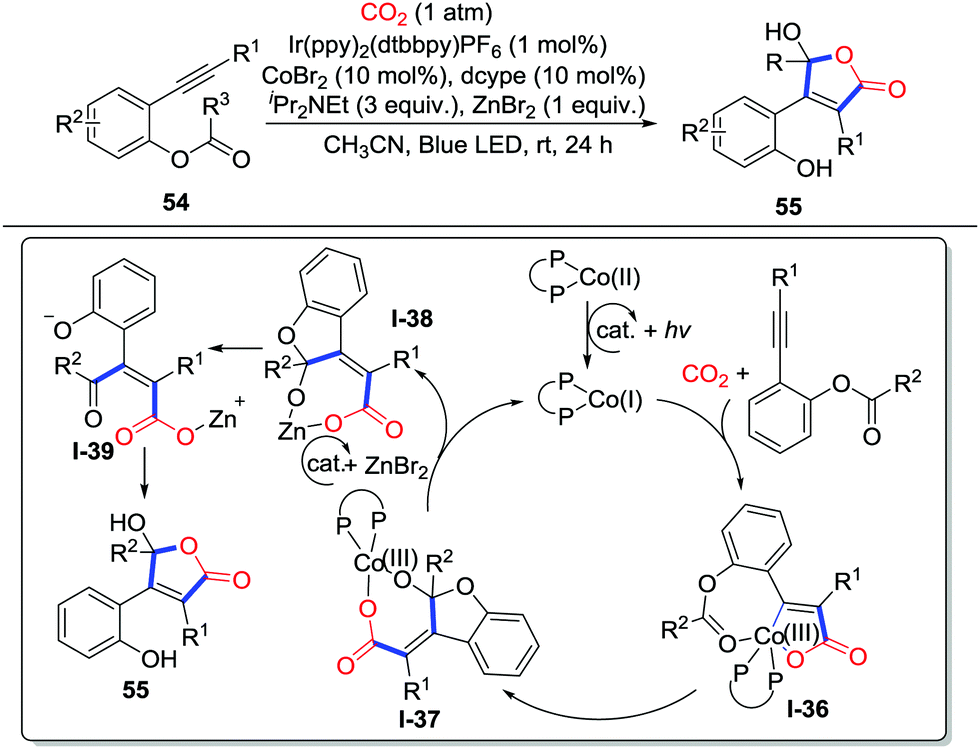 | ||
| Scheme 26 Visible-light-driven cobalt-catalyzed carboxylative cyclization of ortho-ester substituted aryl alkynes with CO2. | ||
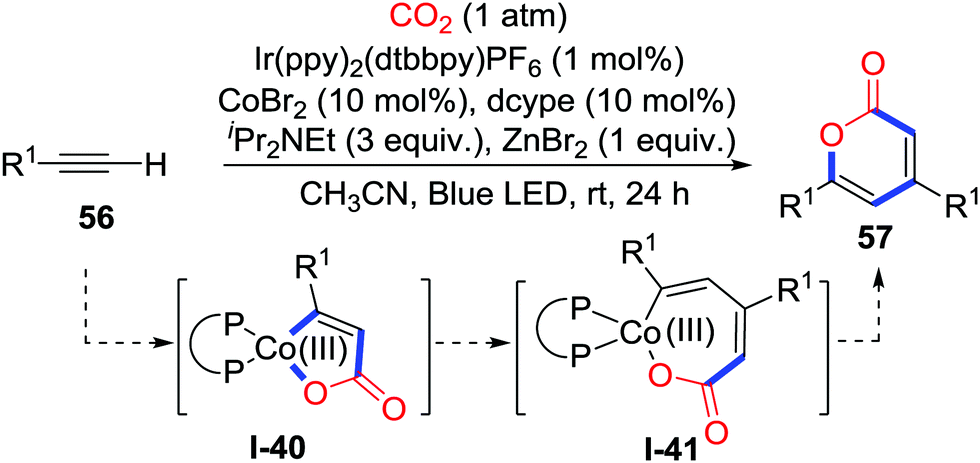 | ||
| Scheme 27 Visible-light-driven cobalt-catalyzed carboxylative cyclization of terminal alkynes with CO2. | ||
Apart from lactones, other heterocycles such as maleic anhydrides can be easily prepared with CO2 (1 atm) by miscellaneous methods. Tsuji and co-workers demonstrated the nickel-catalyzed double carboxylation of internal alkyne 58 at room temperature with Zn as a reducing reagent and MgBr2 as an additive (Scheme 28).39 Controlled experiments and DFT calculations were conducted to elucidate the mechanism. Initially, Ni(II) is reduced by Zn to afford Ni(0), which is followed by the oxidative cycloaddition with an internal alkyne and CO2 to generate Ni(II) metallacycle I-42. Then, the Ni(I) species I-43 is formed from the Ni(II) species with the assistance of Zn and MgBr2via a single electron transfer. Addition of the C–Ni bond to CO2 takes place with the formation of the dicarboxylated acid I-44 that leads to maleic anhydride after acidification.
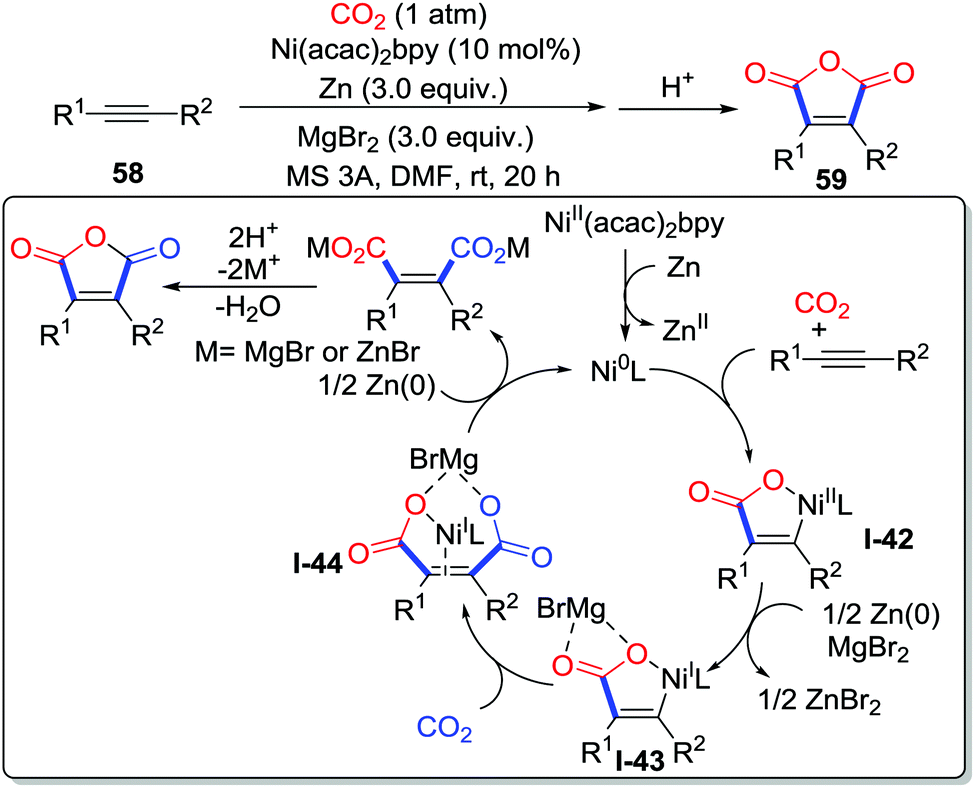 | ||
| Scheme 28 Nickel-catalyzed carboxylative cyclization of internal alkynes with CO2. | ||
The CO2-incorporated carboxylative cyclization has been developed to afford a diversity of heterocycles such as 2-oxazolidinones, cyclic carbonates, 1,3,4-oxadiazole-2(3H)-ones, and lactones based on different nucleophiles and electrophilic sites. Among these transformations, substrate design is a dominant factor. The nucleophilic site of the substrate traps CO2 while its electrophilic site fixes CO2 as part of a heterocycle. Transition metal catalysts often play a vital role in cyclization to enhance the nucleophilicity of nucleophiles or electrophilicity of electrophiles.
3. Nucleophile-triggered CO2-incorporated carbonylative cyclization
In contrast to the rapid development of carboxylative cyclization, carbonylative cyclization utilizing CO2 has been rarely reported until recently. Compared with traditional carbonyl sources such as CO40 and phosgene, CO2 is undoubtedly more promising due to it being non-toxic, renewable, and safe. Theoretically, there are three pathways to incorporate CO2 as a carbonyl source into a heterocycle. To reduce CO2 to CO with a reductant represents an effective means to exploit CO2 as a CO surrogate.41 However, when it comes to carbonylative cyclization, the employment of a reductant makes the transformation less atom-economical. Obviously, a more straightforward method is the carbonylative cyclization with CO2 in substrates containing two nucleophilic sites in a redox-neutral process. This reaction is initiated by the addition to CO2 followed by a C–O bond cleavage to deliver the carbonylative product and oxygen-containing species. The main difficulty identified is the poor leaving ability of oxygen after the addition to CO2. Additionally, the rearrangement of unstable carbamate cycles deriving from CO2 is also a possible mode. Although various challenges exist, the displacement of toxic CO and phosgene with CO2 is still promising, which may extend the application of CO2 as “CO” and “O” synthon in a redox-neutral pathway.3.1 N-Nucleophile-triggered carbonylative cyclization
Carbonylative cyclization triggered by N-nucleophiles with CO2 may produce multiform heterocycles such as lactams and imidazolidin-2-ones, which depend on the substitution in the second step forming the new bonds.Oxindoles are important lactams widely existing as skeletons in natural products and medicinal chemistry. In 2015, Hirao and co-workers described the CO2-incorporated carbonylative cyclization with ketoamines 60 to prepare 3-aryl-3-hydroxy-2-oxindoles 61, which are one of the most important compounds in oxindole derivatives (Scheme 29).42 Magnesium served as a reductant to reduce the carbonyl group. Moreover, in the absence of Me3SiCl, alcohols were the main product and no desired products were observed, which suggested the assistance of Me3SiCl in the CO2 transformation. Notably, the bromo and ester group could survive in this cyclization. In addition, N-methyl substrates also produced the corresponding lactams smoothly. However, the mechanism remains unclear up to now. Single-electron reduction of the ketone to attack CO2 was proposed as a reasonable path. However, a two-electron reduction process cannot be excluded.
 | ||
| Scheme 29 Me3SiCl-promoted carbonylative cyclization of ketoamines with CO2. | ||
In 2016, Yu and co-workers developed the NaOtBu promoted carbonylation of o-alkenyl- or o-arylanilines 62 with the incorporation of CO2 (1 atm), which provided a useful method for the formation of important 2-quinolinones and polyheterocycles 63via the lactamization of alkenyl or heteroaryl C–H bonds (Scheme 30).43 This reaction features a broad substrate scope, scalability, and environmental friendliness and presents a feasible approach to various significant lactam derivatives such as the Tipifarnib precursor. Moreover, compared with CO, application of CO2 in the carbonylation avoided the addition of oxidants and realized the carbonylation of sp2 C–H bonds in redox-neutral conditions. Experiments concentrating on the mechanism showed that isocyanates I-45 deriving from CO2 and anilines might be vital intermediates to give the corresponding lactam after cyclization.
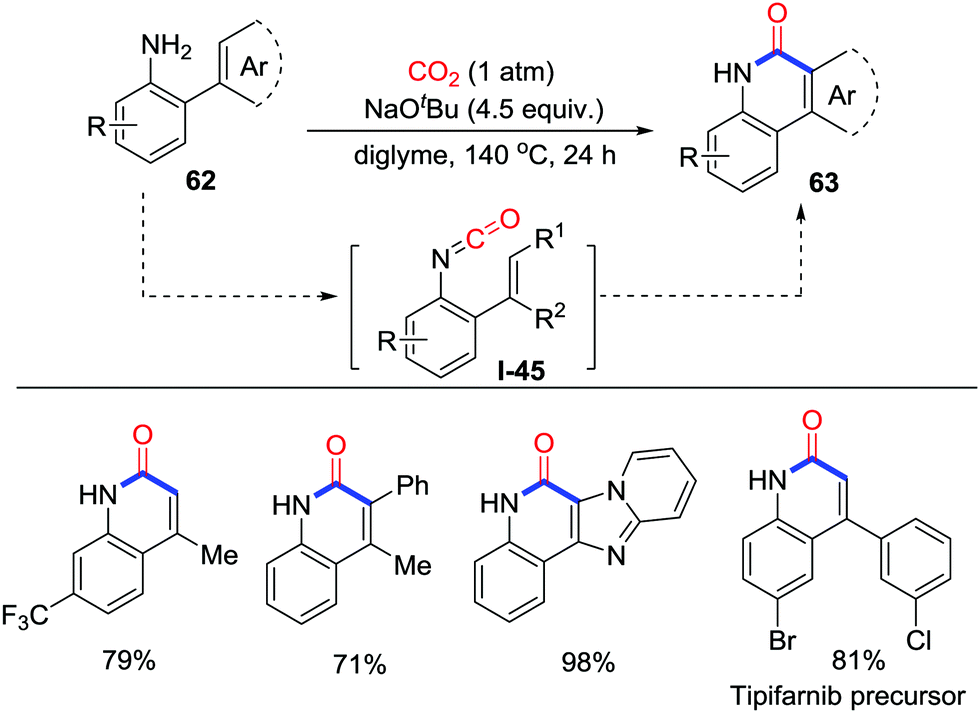 | ||
| Scheme 30 NaOtBu-mediated carbonylative cyclization of o-alkenyl- or o-arylanilines with CO2. | ||
Our group developed MeOTf and TBD mediated carbonylative cyclization of o-arylanilines 64 leading to phenanthridinones 65 with CO2 (1 atm) in o-DCB (1,2-dichlorobenzene) at 140 °C (Scheme 31).44 Although o-alkenylanilines failed to be transformed to the corresponding lactams in this reaction, the conversion of o-arylanilines exhibited good performance owing to the great reactivity of MeOTf, which provided a complementary route to that shown in Yu's work.43 The electronic effect was obvious and the electron-donating functional group enhanced the yield. The isolation of methyl carbamate and the observation of isocyanate in high temperature demonstrated the possible mechanism. First, aniline reacts with the TBD-CO2 adduct to give methyl carbamate I-46 with the aid of MeOTf. In the second step, after losing HOTf at high temperature, the methyl carbamate intermediate is converted to isocyanate I-47, which is induced by MeOTf to generate 6-methoxyphenanthridine I-48. Finally, phenanthridinone is obtained after acidification.
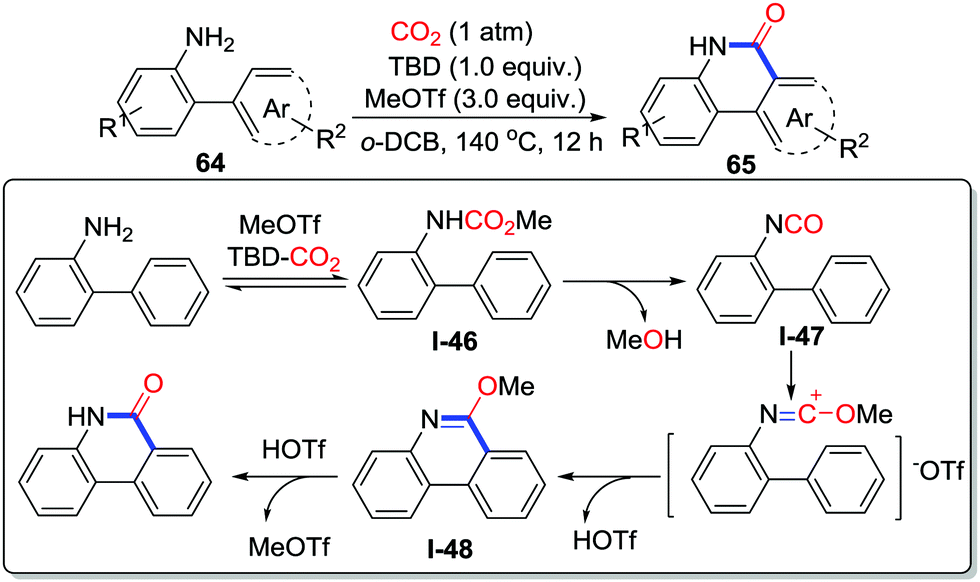 | ||
| Scheme 31 TBD and MeOTf-mediated carbonylative cyclization of o-arylanilines with CO2. | ||
Recently, Cheng's group reported a palladium-catalyzed multicomponent tandem reaction involving N-tosylhydrazones 66, o-iodoanilines 67, and CO2 (1 atm) to prepare a series of 4-aryl-2-quinolinones 68 (Scheme 32).45 The reaction might start with palladium-catalyzed cross-coupling of N-tosylhydrazones and o-iodoanilines followed by the carbonylation of CO2. However, a pathway generating 1-iodo-2-isocyanatobenzene before cross-coupling cannot be excluded.
 | ||
| Scheme 32 Palladium-catalyzed carbonylative cyclization of N-tosylhydrazones and o-iodoanilines with CO2. | ||
Compared with lactamization mentioned above, carbonylation with stronger nucleophiles forming C–N or C–O bonds is less challenging. In 2015, Suen and co-workers accomplished KOH-promoted carbonylation of hydrazides 70 with CO2 (bubble) (Scheme 33).46 The hydrazides 70 could be prepared from acid chlorides 69 and hydrazine monohydrate in high efficiency affording biologically significant 5-substituted-3H-[1,3,4]-oxadizole-2-one 71 under mild conditions.
 | ||
| Scheme 33 KOH-promoted carbonylation of hydrazides with CO2. | ||
2-Benzimidazolone is an attractive heterocycle for application in pharmaceutical chemistry. Liu and co-workers displayed DBU-based ionic-liquid-catalyzed carbonylative cyclization with the formation of two C–N bonds utilizing CO2 (1 atm) at 120 °C (Scheme 34).47 When 2-aminothiophenols were used, the corresponding benzothiazolones could also be obtained efficiently. Notably, [DBUH][OAc] is a dual-active catalyst. [DBUH]+ activates CO2 with tertiary nitrogen, while [OAc]− activates o-phenylenediamine through hydrogen bonding. Furthermore, the azole-anion-based aprotic ionic liquids were also applied in this transformation and delivered 2-benzimidazolones in a similar process.48
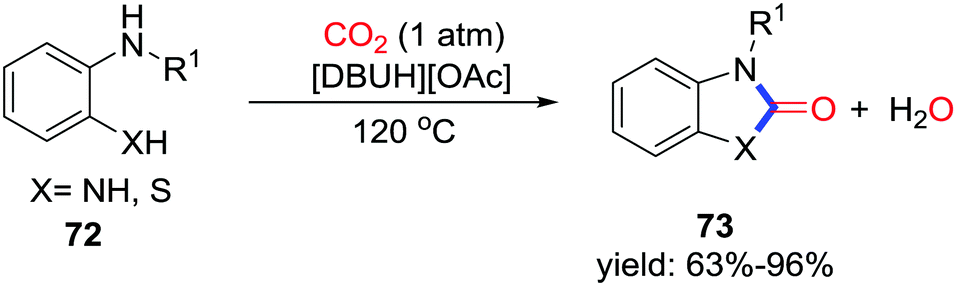 | ||
| Scheme 34 [DBUH][OAc]-catalyzed carbonylative cyclization of phenylenediamines with CO2. | ||
In 2017, Cheng's group reported the NaOtBu-mediated dearomative carbonylation of N-2-pyridylamidines 74 with the formation of two C–N bonds under 1 atm of CO2 (Scheme 35).49 This annulation provided a range of pyrido[1,2-a]-1,3,5-triazin-4-ones 75 bearing different functional groups, and the iodo-substituted product was extended to synthesize artificial nucleosides in a straightforward manner. The 6π-electron cyclic reaction from isocyanate intermediates I-49 was described as a possible process.
 | ||
| Scheme 35 NaOtBu-mediated carbonylative cyclization of N-2-pyridylamidines with CO2. | ||
Although heteroatoms possess sufficient nucleophilicity, some carbonylation using CO2 is not thermodynamically favorable. For instance, the reaction between 2-aminoethanols and CO2 to yield 2-oxazolidinones is restricted thermodynamically. In 2015, He and co-workers provided an approach to overcome this difficulty by adding additives to trigger the reaction. With the addition of propargylic alcohols 77 and silver catalyst to avoid the conventional dehydration step, 2-aminoethanol derivatives 77 were transformed to a wide range of 2-oxazolidinones 78 with CO2 (10 atm) (Scheme 36).50 Controlled experiments and DFT calculations were conducted to elucidate the mechanism, and a key intermediate I-50 was also isolated when the reaction was conducted over two hours. Based on the evidence, a plausible mechanism was proposed. Initially, the propargylic alcohol attacks CO2 to afford cyclic carbonate. The resulting carbonate reacts with 2-aminoethanol via the ring-opening reaction to afford carbamate intermediate I-50,51 which affords the corresponding 2-oxazolidinone 78 and α-hydroxyl ketone 79 through intramolecular nucleophilic substitution. Furthermore, a similar strategy was also applied in the synthesis of cyclic carbonates.52
 | ||
| Scheme 36 Silver-catalyzed carbonylation of 2-aminoethanol derivatives with CO2. | ||
Carbonylation of inert C–H bonds utilizing CO2 is still the most attractive approach due to the atom-economy and high efficiency. In 2018, Yu and co-workers revealed the palladium-catalyzed carbonylation of unactivated C–H bonds in benzamides 80 with CO2 (1 atm) to give the phthalimides 81 under redox-neutral conditions (Scheme 37).53 The nature of substituents on the nitrogen of the benzamides was crucial, with only a methyl group giving good yield which might arise from the less steric hindrance and better directing ability. It is noteworthy that the carbonylation only occurred at the less-hindered ortho position when meta-substituted substrates were used. The kinetic isotope effect (KIE) value suggested that the C–H activation step might be the rate-determining-step. Although the precise mechanism of the carbonylation step was not clear, the palladacycle was believed as the significant intermediate for detection of I-51 in ESI-HRMS when N,N,N′,N′-tetramethylethylenediamine (TMEDA) was applied as the ligand.
 | ||
| Scheme 37 Palladium-catalyzed carbonylation of benzamide derivatives with CO2. | ||
Isotope labeling has demonstrated remarkable application in early diagnosis of diseases and pharmaceuticals. In 2018, Audisio and co-workers applied the Staudinger/aza-Wittig reaction in the late-stage carbon labelling with o-azidoanilines 82 or 3-azidopropan-1-amines 84 and labelled CO2 (1 equiv.) replacing conventional toxic labelled phosgene or CO (Scheme 38).54 All isotopes of carbon (11C, 13C, 14C) could be utilized in this method. Moreover, this reaction features excellent tolerance to different functional groups and practicability. Various drug candidates such as Domperidone 86 and even an unprotected peptide could be labelled utilizing this method.
 | ||
| Scheme 38 Carbonylation of o-azidoaniline derivatives with labelled CO2. | ||
Utilizing CO2 as the carbonyl source via rearrangement reactions has also been well established.55 2-Aminobenzonitriles 88 are typical substrates that undergo carboxylative cyclization to afford quinazoline-2,4(1H,3H)-diones 89 after rearrangement (Scheme 39). Various strategies were applied to realize efficient transformation via similar rearrangement processes. The amino group reacts with CO2 and the resulting carbamic acid attacks the C–N triple bond to give a carbamate I-54. Subsequently, the carbamate undergoes C–O bond cleavage with the formation of isocyanate intermediate I-55, which rearranges to quinazoline-2,4(1H,3H)-diones 89 by the nucleophilic addition due to its thermodynamic instability.
 | ||
| Scheme 39 NaOtBu-mediated carbonylative cyclization of N-2-pyridylamidines with CO2. | ||
When primary o-alkynylaniline 90 was utilized in the silver-catalyzed carboxylative cyclization, Yamada's group observed 4-hydroxyquinolin-2(1H)-one 91 as a by-product, which was identified by 1H NMR, 13C NMR, and single crystal X-ray diffraction. Isotopic labelling experiments with C18O2 and in situ IR measurements strongly suggested the possible mechanism as shown in Scheme 40.56 First, carboxylative cyclization occurs to generate benzoxazin-2-one I-56 as in previous work.20 Assisted by DBU, deprotonation followed by C–O bond cleavage delivers isocyanate I-57, which is subsequently transformed to the corresponding product by a nucleophilic addition of the enolate to the C–O double bond. They further extended this intramolecular rearrangement to propargylic amines with CO2 (1 atm) and a variety of tetramic acid derivatives were obtained in good yield.57 In 2014, Lu and co-workers found similar rearrangement using CuI as catalyst (Scheme 41).58
 | ||
| Scheme 40 Silver-catalyzed carbonylative cyclization of o-alkynylanilines with CO2. | ||
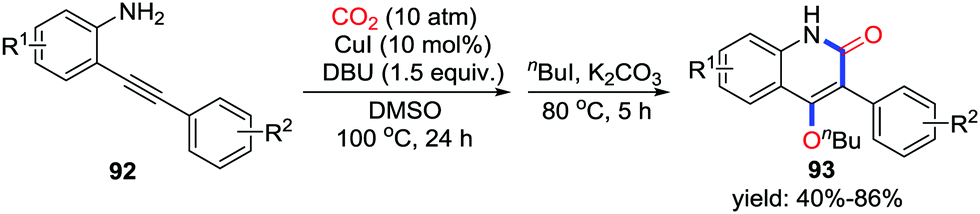 | ||
| Scheme 41 Copper-catalyzed carbonylative cyclization of o-alkynylanilines with CO2. | ||
Furthermore, this strategy was extended to multicomponent reactions incorporating CO2 into heterocycles as a carbonyl source. In 2017, Cheng and co-workers developed a palladium-catalyzed three-component reaction between o-alkynylanilines 94, aryl iodides, and CO2 (1 atm), which provided a sustainable method to construct 3,3-diaryl 2,4-quinolinediones 95 (Scheme 42).59 The mechanism is shown in Scheme 42. Firstly, o-alkynylaniline captures CO2 to give carbamate. Then, ArPdX produced by oxidative addition of ArI and Pd(0) activates the carbamate, which undergoes a trans-oxopalladation to produce intermediate I-58. Subsequently, reductive elimination of the intermediate I-58 takes place to afford heterocycles I-59 with the regeneration of Pd(0). Finally, deprotonation followed by rearrangement delivers the corresponding product 95.
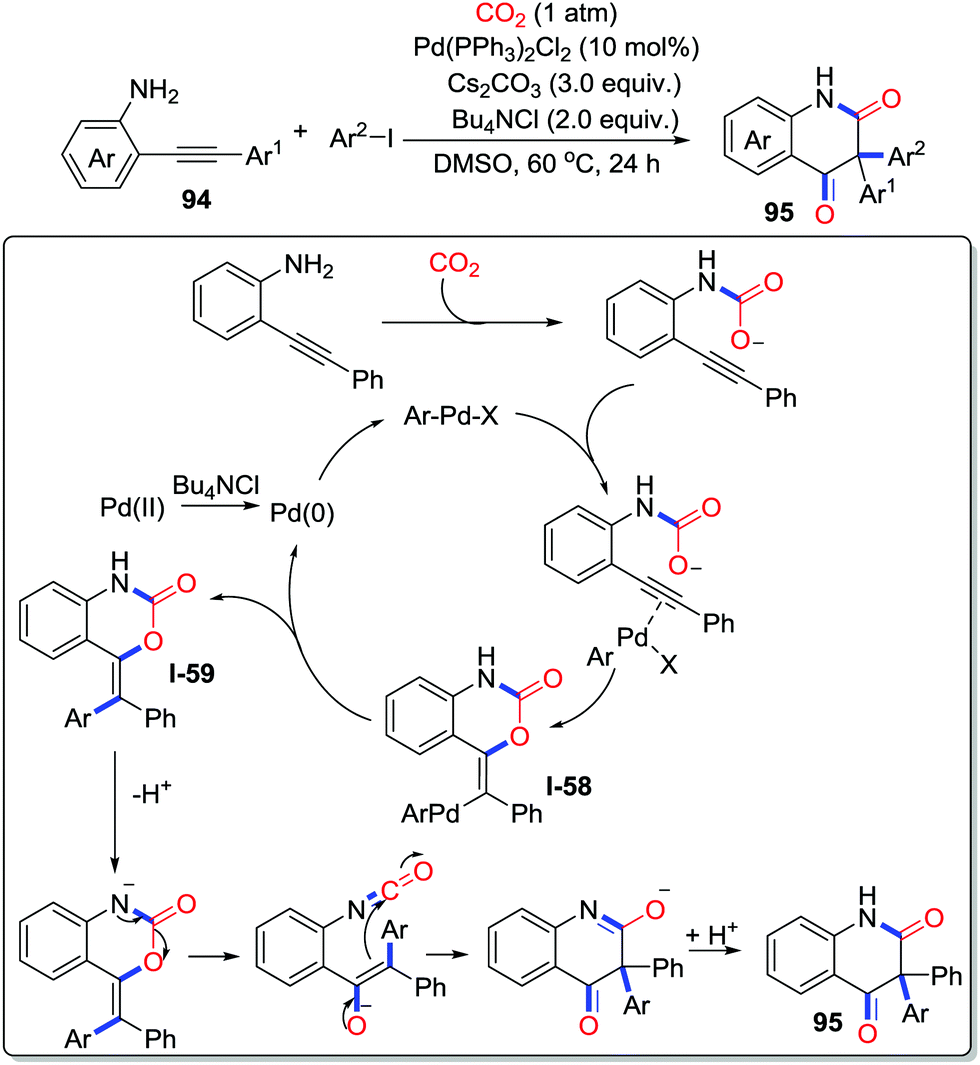 | ||
| Scheme 42 Palladium-catalyzed carbonylative cyclization of o-alkynylanilines and aryl iodides with CO2. | ||
The synthesis of N3-substituted quinazoline-2,4(1H,3H)-diones has attracted great attention due to their wide existence in bioactive molecules such as ketanserin and nitraquazone as well as their utilization in synthetic chemistry. In 2017, Beller and co-workers realized the three-component reaction involving o-bromoanilines 96, isocyanides 97, and CO2 (10 atm) in the catalysis of Pd(OAc)2 and BuPAd2 (butyldi-1-adamantylphosphine) (Scheme 43).60 This reaction demonstrated good chemoselectivity, regioselectivity, and tolerance to tertiary, secondary, and primary isocyanides. The post-functionalization of the product could be conducted conveniently. Moreover, as an economical 13C-carbonylating reactant, 13CO2 could also deliver the desired heterocycles under optimized reaction conditions, which exhibits application in preparing selective-labelled products. When N-benzyl-2-bromoaniline was used, N-tert-butyl-N′-benzyl-2-amino-benzamide was obtained in 68% yield instead of the formation of the desired heterocycle, which strongly suggested the existence of isocyanate intermediates in the rearrangement. A possible reaction mechanism is shown in Scheme 43. Intermediate I-60 was formed by the oxidative addition of Pd(0) with o-bromoaniline followed by the migratory insertion of isocyanide to a C–Pd(II) bond. In the presence of Cs2CO3, a nucleophilic addition to CO2 followed by reductive elimination generated intermediate I-61 with the release of Pd(0). Finally, cyclic carbamate intermediate I-61 is treated with base to afford quinazoline-2,4(1H,3H) diones 98via isocyanate intermediate I-62. Notably, Ji and Lu's group also independently realized a similar palladium-catalyzed reaction with o-iodoanilines or o-bromoanilines.61
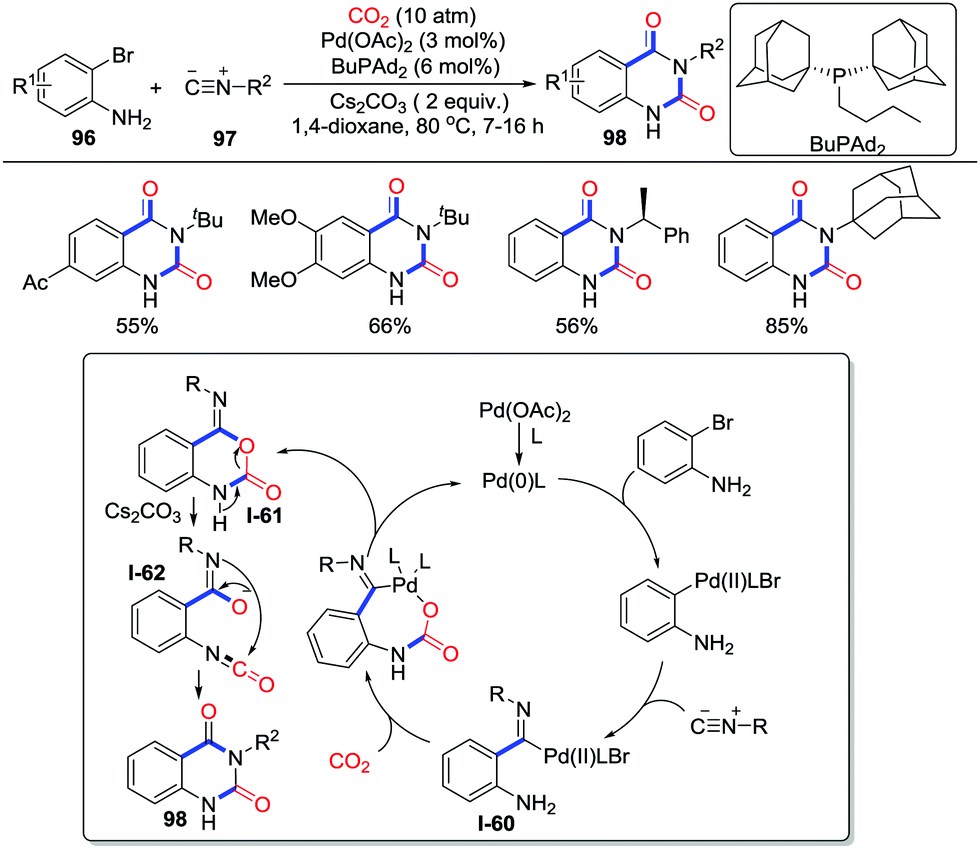 | ||
| Scheme 43 Palladium-catalyzed carbonylative cyclization of o-bromoanilines and isocyanides with CO2. | ||
3.2 O-Nucleophile-triggered carbonylative cyclization
As potent nucleophiles, O-nucleophiles are expected to trap CO2 and generate lactones after consecutive intramolecular substitution with one C–O bond formation and one C–O bond cleavage. When the o-heteroarylphenols 99 were employed, important coumarin derivatives were obtained utilizing CO2 (1 atm) (Scheme 44).62 Based on controlled experiments, a possible mechanism involving addition of carbonate I-63 to CO2 followed by substitution of nucleophilic sp2 C–H was proposed as shown in Scheme 44. In addition, treatment of o-alkenylphenols 101 with CO2 also afforded compounds 102 (Scheme 45). | ||
| Scheme 44 KOtBu-mediated carbonylative cyclization of o-heteroyl phenols and o-alkenylphenols with CO2. | ||
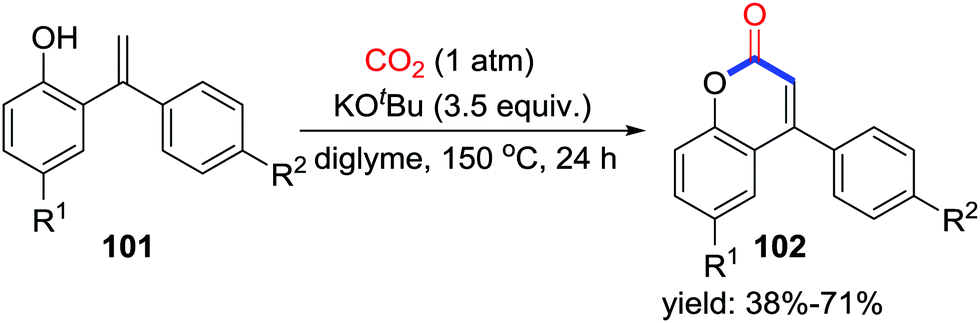 | ||
| Scheme 45 KOtBu-mediated carbonylative cyclization of o-alkenylphenols with CO2. | ||
Although direct carboxylation of C–H bonds with CO2 is difficult due to the low reactivity of CO2, the C–H bond with a directing group possesses sufficient nucleophilicity to many electrophiles. Yu and co-workers applied this property in the lactonization of tertiary benzylic alcohols 103 with CO2 (1 atm) to prepare important phthalides 104 (Scheme 46).63 Primary and secondary benzylic alcohols failed due to β-H elimination. When a 18O-labeled substrate was used, 18O was fully retained in the product which supported the possible carbonylative process. DFT calculations were conducted to elucidate the mechanism. The reaction starts with coordination of the benzylic alcohol with the palladium catalyst in the presence of LiOtBu, which subsequently generates the palladacycle intermediate I-64via a concerted-metalation–deprotonation (CMD) process. Then, the CO2 insertion to the Pd–O bond occurs successively to generate 9-membered palladacycle intermediate I-65. Subsequently, intramolecular nucleophilic addition gives the intermediate I-66. Finally, decarbonization releases lactone and the palladium catalyst.
 | ||
| Scheme 46 Palladium-catalyzed carbonylative cyclization of tertiary benzylic alcohols with CO2. | ||
Apart from the straightforward sequential addition to CO2, rearrangement gave the carbonyl as well when O-nucleophiles were applied. In 2018, Yu and co-workers developed the practical synthesis of appealing tetronic acids 106 with propargylic alcohols 105 and CO2 (1 atm) in 1,3-dimethyl-2-imidazolidinone (DMI) (Scheme 47).64 Substrates with electron-withdrawing groups exhibited better reactivity which might be explained by the higher electrophilicity of the C–C triple in the cyclization step. Carbonate I-67 was considered as an intermediate based on transformation from the carbonate to the product without CO2. A mechanism involving rearrangement of the cyclic carbonates was proposed. Propargylic alcohol is basified to attack CO2 and then undergoes intramolecular cyclization to give cyclic carbonate. Nucleophilic attack of the Cs2CO3 to the carbonyl promotes the ring-opening process and delivers the intermediate I-68. Then, the Dieckmann-type condensation generates Cs2CO3 and the re-cyclized product 106 which could be transformed to tetronic acid.
 | ||
| Scheme 47 Cs2CO3-mediated carbonylative cyclization of propargylic alcohols with CO2. | ||
3.3 C-Nucleophile-triggered carbonylative cyclization
Unlike the N-nucleophiles and O-nucleophiles, C-nucleophiles only exist in a specific position, which are unfavorable to attack CO2. Furthermore, considering the difficulty of unactivated C–H carboxylation with CO2 up to now, C–H carbonylative cyclization is still challenging. In 2013, Iwasawa and co-workers disclosed the direct carboxylation of an alkenyl C–H bond in catalysis using palladium with CO2 (1 atm) to afford a variety of coumarins 108 in good yields (Scheme 48).65 Importantly, an alkenyl palladium complex coordinated by cesium salt was isolated and confirmed by single crystal X-ray diffraction, which indicated that C–H bond cleavage had occurred. Additionally, the carboxylation step turns out to be reversible, which displays a tendency to decarboxylation side. Coumarins were obtained by the reaction of I-70 with another molecule o-alkenylphenol 107. | ||
| Scheme 48 Palladium-catalyzed carbonylative cyclization of o-alkenylphenols with CO2. | ||
Triggered by various nucleophiles, benzynes could be transformed to 1,3-zwitterionic intermediates. In 2014, Biji and co-workers demonstrated the isocyanide-triggered cyclization of benzynes with CO2 (1 atm) (Scheme 49).66 Transformation of I-71 to the product 111 could be realized in the presence of F−. Consequently, CsF turns out to be crucial not only in the activation of benzyne precursors but also in the rearrangement step.
 | ||
| Scheme 49 CsF-mediated carbonylative cyclization of benzynes and isocyanides with CO2. | ||
In 2015, Lu and co-workers reported the carbonylation of o-hydroxyacetophenones 112 and o-aminoacetophenones 114via the carboxylation of enolates with DBU (Scheme 50).67 The formation of stable heterocycles is the driving force that promotes reversible reaction between enolates and CO2 towards the carboxylation.
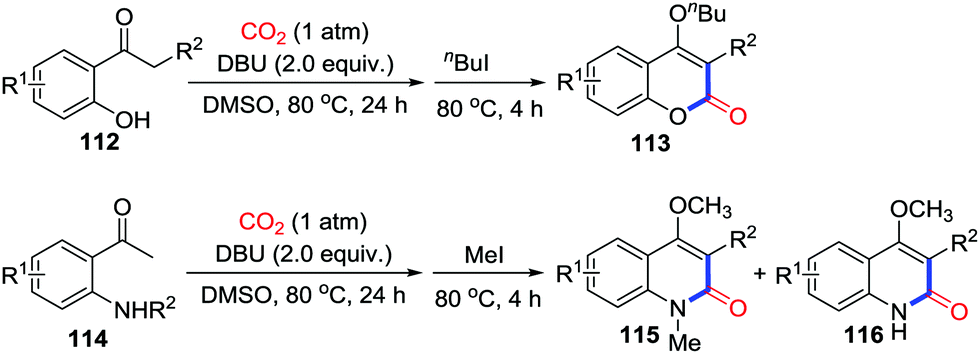 | ||
| Scheme 50 DBU-mediated carbonylative cyclization of o-hydroxy- and o-acetamido-acetophenones with CO2. | ||
This practical protocol was further applied by Lu's group in 2016 (Scheme 51).68 CsF-promoted carbonylation of propenyl ketones 117 with CO2 (30 atm) at 100 °C was shown to afford α-pyrones 118, which possessed a significant biological activity. It was assumed that the high pressure of CO2 and low concentration of reaction inhibited dimerization of substrates. C18O2 was also utilized in this reaction to gain more insight into the mechanism and to demonstrate that CO2 only acts as a CO moiety in this transformation. The reaction might be initialized by carboxylation followed by intramolecular substitution while another mechanism similar to Yu's work62 that was triggered by O-nucleophiles could not be excluded.
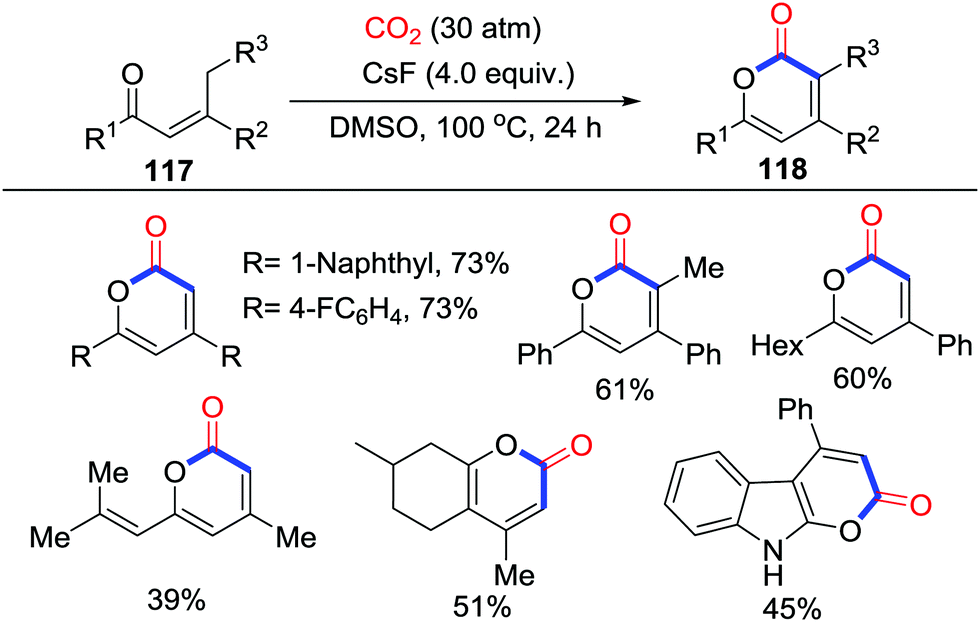 | ||
| Scheme 51 CsF-mediated carbonylative cyclization of propenyl ketones with CO2. | ||
Recently, Yu and co-workers reported a one-pot transition-metal-free lactonization of enamides 119 to prepare 1,3-oxazin-6-ones 120 (Scheme 52).69 EtI was used to quench the reaction and the corresponding esters were obtained. N-Methyl substituted enamides failed to be carboxylated in the presence of base. Moreover, the utilization of 13CO2 gave the selective-labelled product. On the basis of these experiments, it is believed that the reaction is initiated by the carboxylation of electron-rich alkenes.
 | ||
| Scheme 52 NaOtBu-promoted carbonylative cyclization of enamides with CO2. | ||
Transition-metal catalyzed C–H carbonylation with CO2via C–H bond activation has rarely been reported since Iwasawa's work.65 The main challenge lies in the fact that the acidic and oxidative condition for C–H activation is incompatible with basic and reductive condition for CO2 activation. In 2018, Li and co-workers displayed an exciting redox-neutral rhodium(II) catalyzed carbonylation of 2-arylphenols with CO2 (1 atm) (Scheme 53).70 Phosphine ligands were extremely important to enhance the reactivity of the rhodium catalyst and to avoid the well-known Kolbe–Schmitt type reaction. KOtBu was considered to enhance the concentration of CO2 in the solution by strong interactions with CO2. Due to steric hindrance, the carboxylation tends to occur at the less hindered C–H bond. The novel complex A prepared was characterized by X-ray crystallography, which could be used as a catalyst to realize the carbonylation of 2-arylphenols efficiently. A tentative mechanism was proposed as shown in Scheme 53. First, 121 is basified with KOtBu and reacts with complex A to generate complex I-73. Then, assisted by the base ROK (such as KOtBu), the aryl C–H bond activation of I-73, via a proton abstraction process, generates the metallacycle I-74 reversibly. CO2 insertion into the C–Rh bond occurs reversibly to give the 8-membered rhodium carboxylate I-75. Subsequently, lactonization rapidly delivers the product 122 and regenerates complex A. More recently, Li's group extended this reaction to more challenging electron-deficient 2-pyridylphenols by tuning ligands, which provide an access to pyrido-heterocycles.71
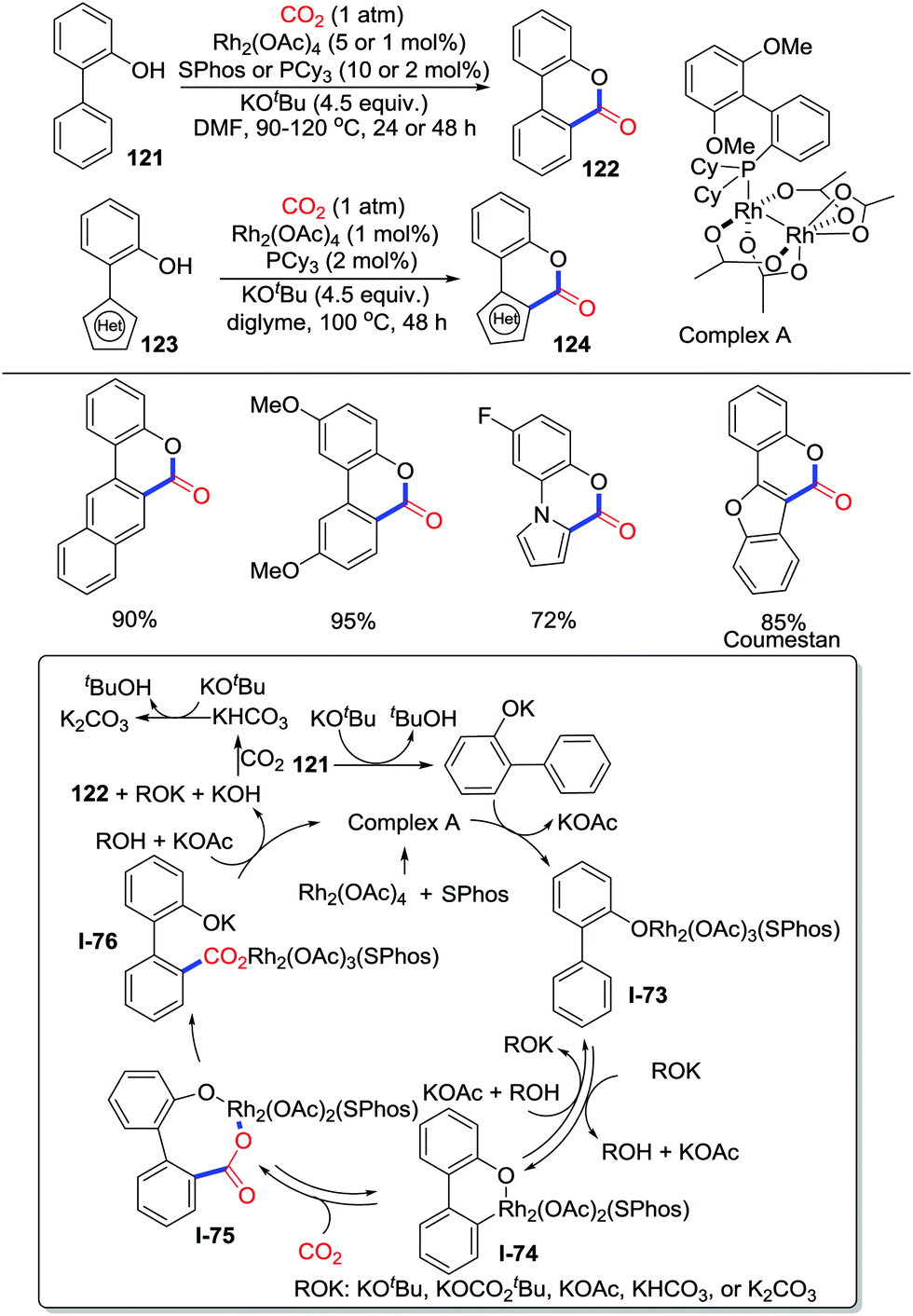 | ||
| Scheme 53 Rhodium-catalyzed carbolyative cyclization of 2-arylphenols with CO2. | ||
Without reductants, there are two pathways to realize the carbonylation reaction utilizing CO2. The first method is the successive nucleophilic attack on CO2 with the substrate possessing two nucleophilic sites. The difficulty of this approach lies in the generation of the small molecule, which makes the reaction thermodynamically unfavorable. As a result, additives or high temperature to provide energy is necessary. Another mode is the rearrangement reaction, which has also been developed with different substrates. The initial cyclization reaction generates an unstable cyclic intermediate, which rearranges immediately to a stable heterocycle.
4. CO2-Incorporated reductive cyclization
Considering that the carbon center in CO2 is at its highest valence state, great effort has been made with various reduction systems involving different catalysts and reductants such as hydrogen, boranes, and silanes. Based on the number of electrons donated by reductants, CO2 could be reduced to different oxidation states to give the corresponding product such as formates and acetals, which could be captured by nucleophiles to afford value-added products.2,72 Substrates with two nucleophilic sites are able to attack reductive intermediates in sequence to deliver diverse heterocycles.4.1 2-Electron reductive cyclization
2-Electron reduction could reduce CO2 to formates, which deliver the amides after formylation with amines.2 In 2013, Cantat and co-workers designed a cascade process utilizing substrates with two nucleophilic sites and combined formylation with condensation to produce various heterocycles with a formamidine structure.73 With the aid of NHC-carbenes, 1,2-diamines, anthranilamides, and aminobenzylamines could conduct the cascade reaction successfully with atmospheric CO2 to generate benzimidazoles, quinazolinones, formamidines and their derivatives in the presence of poly(methylhydrosiloxane) (PMHS) (Scheme 54). This work provided an effective strategy to realize the deoxygenation of CO2 and made CO2 more flexible in the construction of heterocycles. | ||
| Scheme 54 IPr-catalyzed reductive cyclization of 1,2-diamines, anthranilamides, and aminobenzylamines with CO2. | ||
A similar strategy was utilized by Liu and co-workers. In the presence of a mixture of gases (150 atm, CO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2 = 3:2), o-phenylenediamines were catalyzed by RuCl(dppe)2 to give a series of benzimidazoles (Scheme 55).74 Formic acid could react with o-phenylenediamine immediately at 120 °C to provide benzimidazole via formamide I-77. On the basis of this result, a mechanism was proposed. Initially, CO2 is reduced by H2 in the presence of a ruthenium catalyst and o-phenylenediamine as a base. Afterwards, N-formylation occurs followed by condensation to give the desired product.
:H2 = 3:2), o-phenylenediamines were catalyzed by RuCl(dppe)2 to give a series of benzimidazoles (Scheme 55).74 Formic acid could react with o-phenylenediamine immediately at 120 °C to provide benzimidazole via formamide I-77. On the basis of this result, a mechanism was proposed. Initially, CO2 is reduced by H2 in the presence of a ruthenium catalyst and o-phenylenediamine as a base. Afterwards, N-formylation occurs followed by condensation to give the desired product.
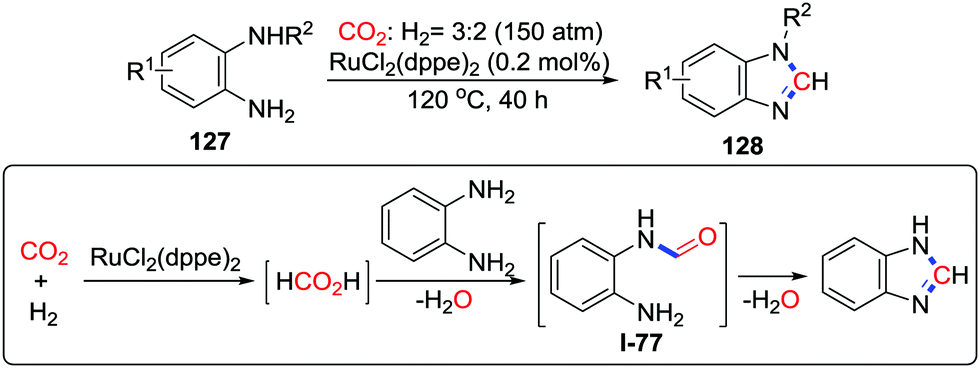 | ||
| Scheme 55 Ruthenium-catalyzed reductive cyclization of o-phenylenediamines with CO2. | ||
In 2015, Liu and co-workers reported that o-aminothiophenols and o-phenylenediamines 129 could be used for the reductive cyclization with CO2 (5 atm) in the presence of an ionic liquid and (EtO)3SiH as reductant (Scheme 56).75 Substrates bearing electron donating groups such as –OMe and –NR2, or electron withdrawing groups such as –F, –Cl, –Br, and –NO2 were accommodated in this reaction. In this reaction, the Si–H bond in (EtO)3SiH was activated by [Bmim]+, while the substrates were activated by [OAc]−via a hydrogen bond. The cyclic products were formed by a successive substitution from a formoxysilane intermediate.
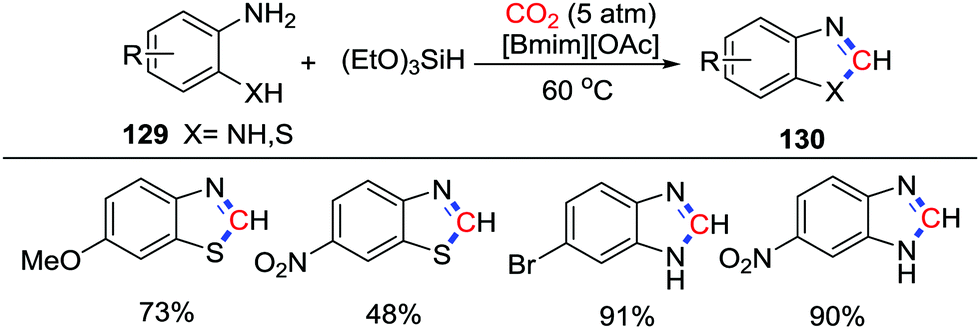 | ||
| Scheme 56 [Bmim][OAc]-catalyzed reductive cyclization of o-aminothiophenols and o-phenylenediamines with CO2. | ||
Frustrated Lewis pairs (FLPs) have demonstrated tremendous potential in activating small molecules. Liu and co-workers applied FLPs as catalysts in the CO2 transformation to realize N-methylation of amines in 2015.76 Subsequently, Sun's group extended FLPs as catalysts to the construction of heterocycles. In the presence of B(C6F5)3, o-phenylenediamines 131 reacted with CO2 (10 atm) and four equivalents of PhSiH3 at 120 °C affording benzimidazoles 132,77 which has attracted attention due to the relevance to natural products and biologically active molecules (Scheme 57). The amine-CO2-B(C6F5)3 adduct was detected by NMR spectroscopy. Based on this evidence and further investigations, a plausible mechanism was proposed as displayed in Scheme 57. Initially, amine-CO2-B(C6F5)3I-78 is formed. Activated by B(C6F5)3, PhSiH3 reduces this adduct via a 2-electron process to release the catalyst and generates the formamide intermediate I-79, which affords the desired product after dehydration.
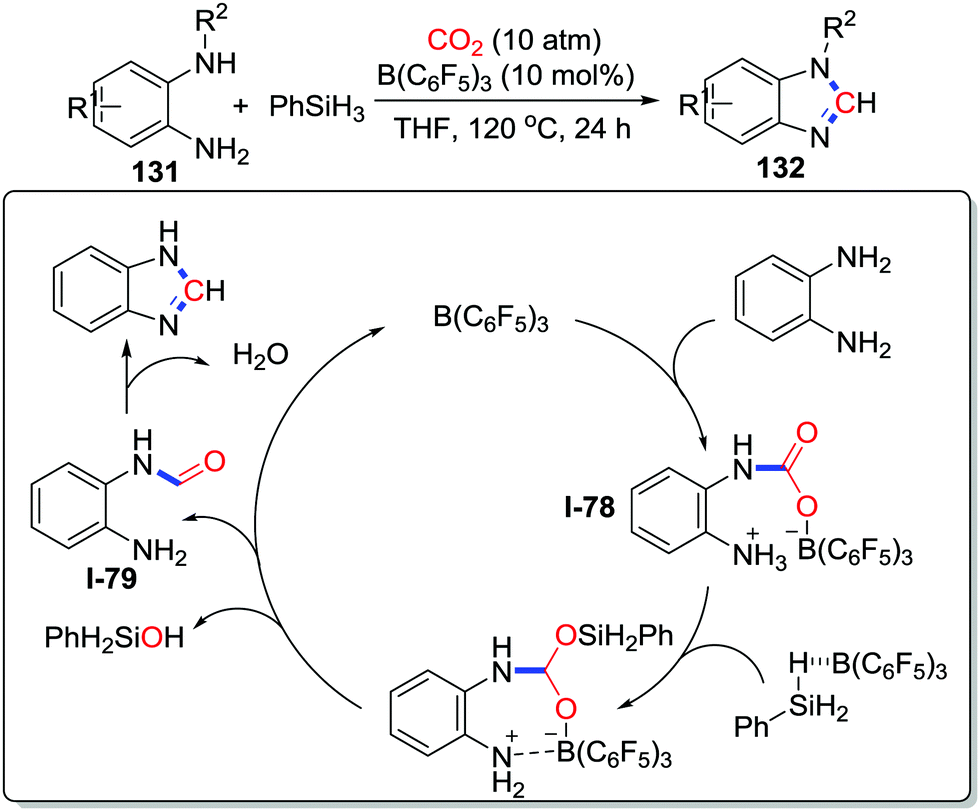 | ||
| Scheme 57 B(C6F5)3-catalyzed reductive cyclization of o-phenylenediamines with CO2. | ||
Recently, our group reported the Lewis base-promoted selective reduction of CO2 into boryl formates using BH3NH3 as a reductant under mild conditions (Scheme 58).78 The in situ generated boryl formates 133 were reacted at 80 °C with benzene-1,2-diamine, 2-aminophenol, and 2-aminobenzenethiol to afford 1H-benzo[d]imidazole (60% yield), 2-aminobenzenethiol (53% yield), and benzo[d]thiazole (40% yield), respectively.
 | ||
| Scheme 58 BH3NH3-mediated reductive cyclization with CO2. | ||
The process of 2-electron reductive cyclization mentioned above by fixing CO2 through reduction, formylation and condensation in sequence is not the only pathway in 2-electron reductive cyclization. In 2017, Cheng and co-workers described the 2-electron reductive carbonylative cyclization of 2-hydrazinopyridines 135 and benzamidrazones 137 to give triazolones in the presence of K2CO3 and CO2 (1 atm) (Scheme 59).79 5 equivalents of HSi(OEt)3 served as the reductant to reduce CO2 in a 2-electron process. To elucidate the mechanism, controlled experiments were conducted. When phenyl hydrazine was used, only the formylated product was obtained under standard conditions, which supported a 2-electron reduction of CO2. Moreover, the utilization of the pre-prepared compound I-80 as a substrate afforded triazolone 136 along with H2 in the absence of base and HSi(OEt)3. This result indicated I-80 as a possible intermediate in the reductive cyclization. Consequently, the reaction might start with formylation of the substrate in the presence of HSi(OEt)3. Then, nucleophilic attack takes place to generate intermediate I-81. Finally, the product is obtained with extrusion of H2.
 | ||
| Scheme 59 Reductive cyclization of 2-hydrazinopyridines and benzamidrazones with CO2. | ||
4.2 4-Electron reductive cyclization
Acetal species can be obtained from CO2via a 4-electron reduction process, which serve as methylene groups after successive nucleophilic attacks. Nevertheless, this process is not common due to further reduction taking place leading to a 6-electron reduction by C–O bond cleavage. In 2013, Cantat and co-workers reported the TBD-catalyzed 4-electron reduction of CO2 to give aminals 140 with the formation of two C–N bonds under atmospheric pressure CO2 (1 atm) (Scheme 60).80 Although this work focused on cross-coupling of CO2 and two amines, the application of diamino substrates also provided the corresponding heterocycles in excellent yield. The reaction starts with reduction of CO2 with PhSiH3 to give a silylacetal species. The formed bis(silyl)acetal undergoes successive nucleophilic attack by amines rapidly to deliver the product. Later, He81 and Han82 achieved the glycine betaine-catalyzed 4-electron reduction of CO2, which was captured by amines, respectively. Although heterocycles were not synthesized in these works, it possesses great potential for 4-electron reductive cyclization. | ||
| Scheme 60 TBD-catalyzed reductive cyclization of diamino substrates with CO2. | ||
Based on a similar process, Bontemps and co-workers also described iron-catalyzed reductive functionalization of CO2 with 9-BBN (9-borabicyclo[3.3.1]nonane) (Scheme 61).83 CO2 was reduced to bis(boryl)acetals and no further reduction product was observed in a short reaction time. Nucleophiles were added to functionalize the resulting intermediates 142in situ. Dihydroimidazoles 143 were obtained after addition of N1,N2-dimethylbenzene-1,2-diamines while addition of NH3 gave hexamethylenetetramines 144 straightforwardly.
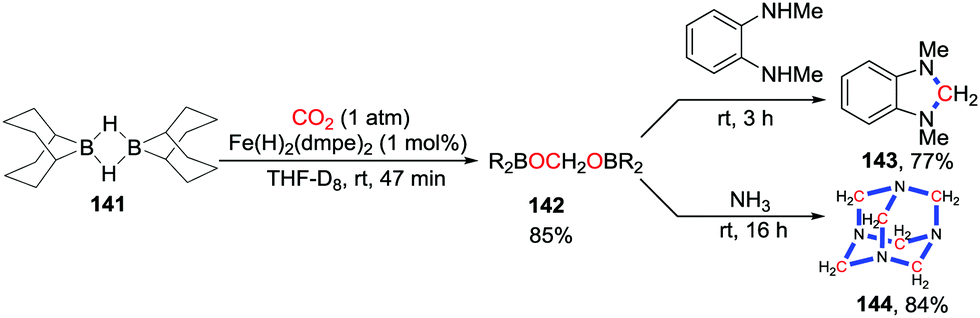 | ||
| Scheme 61 Iron-catalyzed 4-electron reductive cyclization with CO2. | ||
More recently, Xia and co-workers developed reductive CO2 fixation to construct spiroindolepyrrolidines 146via a 4-electron reduction with the formation of C–N and C–C bonds (Scheme 62).84 The mechanistic study revealed a pathway involving CO2 fixation. CO2 is reduced in the presence of PhSiH3 and TBD via a 4-electron process to deliver bis(silyl)acetal I-83. Subsequently, the acetal is captured by the amino group and the C3 carbon in tryptamine 145 with the formation of C–N and C–C bonds to afford spiroindolepyrrolidine 146 following dearomatization.
 | ||
| Scheme 62 TBD-catalyzed reductive cyclization of tryptamines with CO2. | ||
Besides the N atom, other heteroatoms could also attack the acetal species. Recently, Klankermayer's group reported the Ru- and Co-based co-catalyst for the synthesis of dimethoxymethane with CO2 as a methylene source.85 Even though the S atom possesses excellent nucleophilicity, 4-electron and 6-electron reductive fixations on the S atom were not reported. In 2018, our group developed a 4-electron reduction of S-nucleophiles to prepare dithioacetals from thiophenols or thiols using the NaBH4/I2 system with CO2 (1 atm) (Scheme 63).86 When propane-1,3-dithiols 147 were used, 1,3-dithianes 148 could be easily obtained in 60%. NaBH4 is activated by I2 to give the reactive borane and reduce CO2.
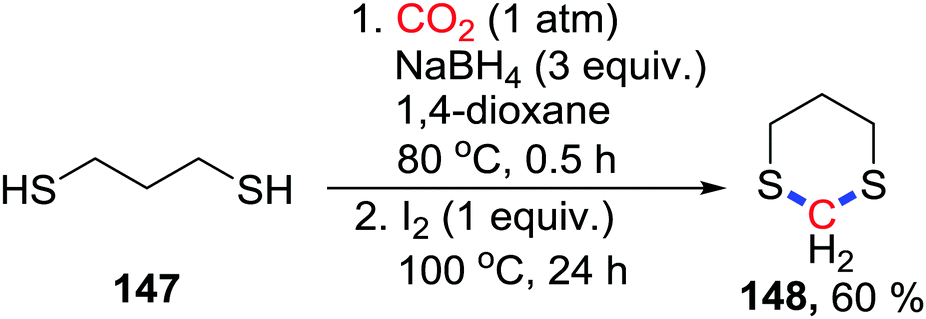 | ||
| Scheme 63 NaBH4/I2 mediated reductive cyclization of propane-1,3-dithiols with CO2. | ||
Research involving CO2-incorporated reductive cyclization concentrates on catalyst systems, which could reduce CO2 to different oxidation states via 2-electron or 4-electron reduction. The corresponding intermediates could be captured by substrates containing two nucleophilic sites. Although the reduction of CO2 has been well investigated, the combination of CO2 reduction and cyclization still remains undeveloped.
5. Conclusions and perspectives
From the perspective of reaction mode, the core idea in carboxylative, carbonylative, and reductive cyclization involves a nucleophilic attack on CO2 and transformation of the carboxylate to a stable compound. Based on this idea, and as a result of deeper understanding of the properties of CO2 along with the development of catalytic systems, three reaction modes have been highlighted that have shown exciting growth.In the past, CO2-incorporated carboxylative cyclization was restricted to only a few substrates such as epoxides and aziridines. However, substrates have been extended to transform CO2 elegantly in recent years due to imaginative design strategies developed by chemists. Consequently, a variety of heterocycles have been prepared such as functionalized 2-oxazolidinones, benzoxazin-2-ones, oxazolidinediones, 1,3,4-oxadiazole-2(3H)-ones, cyclic carbonates, isocoumarins, and even maleic anhydrides. The application of nucleophilic NHC-carbene ligands and organic bases also facilitates these types of cyclization. Although catalysts tend to activate substrates rather than CO2, the reaction is feasible to provide an approach to highly functionalized heterocycles. Carbonylation utilizing CO2 remained dormant until these years. Compared with the traditional toxic carbonyl source CO and phosgene, CO2 is certainly a greener carbon source. Carbonylation conducted with sequential nucleophilic attack or rearrangement generates lactones and lactams. It is noteworthy that carbonylation involving inert sp2 C–H bonds was realized, which shows the great potential of CO2 in synthetic chemistry. In contrast to development in CO2 reduction, few works have been accomplished in CO2 reductive cyclization. More substrates with two nucleophilic sites to construct heterocycles are still lacking. The oxidation state of the central carbon is decisive in this transformation, which depends on the reducibility imparted by the hydrogen sources and catalysts. Although efficient catalysts such as FLPs, ionic liquids, and organic superbases have been utilized in reductive cyclization with accessible hydrogen sources, deeper exploration with different catalysts, ligands, and hydrogen sources is required.
In spite of the rapid development in CO2-incorporated cyclization to prepare multifarious heterocycles in recent years, there still exist large challenges and potential for further CO2 utilization. Besides the elegant design of more efficient and cheaper catalysts with higher TON and TOF, some points include
(1) Light- and electro-driven CO2 transformation: light- and electro-chemistry has exhibited wide application, while light- or electro-triggered CO2-incorporated organic synthesis has been rarely reported. The utilization of light and electricity in CO2 transformation not only makes the transformations greener, but also broadens the substrate scope.
(2) Oxidation: besides a one-carbon source, CO2 is a mild oxidant as well. Although carbon was not utilized in the oxidation, the by-products deriving from the deoxygenation of CO2 are also valuable carbon sources for further functionalization.51,52
(3) Protecting group: as an electrophile, CO2 is able to react with amines to give carbamates, which weaken the nucleophilicity and reducibility of amines to facilitate downstream reaction.13 This property suggested a possible exploitation of CO2 as a protecting group like Boc (t-butyloxy carbonyl) in a traceless path.87
(4) Lewis acid: the central carbon possesses weak Lewis acidity. The addition of CO2 to substrates could make the substrates more electron-deficient and reactive to facilitate the reactions such as cross-coupling88a,b and oxidation.88c
(5) CO2 as a carbon source in the synthesis of important smaller and larger heterocycles: 4-membered rings such as β-lactams and larger heterocycles such as macrolides are of significance in medicinal chemistry, while CO2-incorporated cyclization only focused on building 5-membered and 6-membered rings.
(6) The incorporation of rational substrate design and heterogeneous catalysts such as MOFs14k,l and nanostructured materials3 may exhibit potential in CO2 utilization.
We believe that further developments in this field will promote CO2 as an ideal feedstock capable of turning waste into wealth.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21472106, 91645120, and 21871163). We thank Bo Zhang and Wei Hang from Tsinghua University for the advice.References
-
(a) T. Sakakura, J. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 CrossRef CAS PubMed
; (b) M. Cokoja, C. Bruckmeier, B. Rieger, W. A. Herrmann and F. E. Kühn, Angew. Chem., Int. Ed., 2011, 50, 8510–8537 CrossRef CAS PubMed
- K. Dong, R. Razzaq, Y. Hu and K. Ding, Top. Curr. Chem., 2017, 375, 23 CrossRef PubMed
-
(a) S. Xie, Q. Zhang, G. Liu and Y. Wang, Chem. Commun., 2016, 52, 35–59 RSC
-
(a) M. Patel, S. S. Ko, R. J. McHugh, J. A. Markwalder, A. S. Srivastava, B. C. Cordova, R. M. Klabe, S. Erickson-Viitanen, G. L. Trainor and S. P. Seitz, Bioorg. Med. Chem. Lett., 1999, 9, 2805–2810 CrossRef CAS PubMed
-
(a) X. Lu and D. J. Darensbourg, Chem. Soc. Rev., 2012, 41, 1462–1484 RSC
-
(a) V. A. Peshkov, O. P. Pereshivko, A. A. Nechaev, A. A. Peshkov and E. V. Van der Eycken, Chem. Soc. Rev., 2018, 47, 3861–3898 RSC
- J. Ye, L. Song, W. Zhou, T. Ju, Z. Yin, S. Yan, Z. Zhang, J. Li and D. Yu, Angew. Chem., Int. Ed., 2016, 55, 10022–10026 CrossRef CAS PubMed
- L. Zhu, J. Ye, M. Duan, X. Qi, D. Yu, R. Bai and Y. Lan, Org. Chem. Front., 2018, 5, 633–639 RSC
- J. Ye, L. Zhu, S. Yan, M. Miao, X. Zhang, W. Zhou, J. Li, Y. Lan and D. Yu, ACS Catal., 2017, 7, 8324–8330 CrossRef CAS
- M. Wang, Y. Cao, X. Liu, N. Wang, L. He and S. Li, Green Chem., 2017, 19, 1240–1244 RSC
- Z. Yin, J. Ye, W. Zhou, Y. Zhang, L. Ding, Y. Gui, S. Yan, J. Li and D. Yu, Org. Lett., 2018, 20, 190–193 CrossRef CAS PubMed
- L. Sun, J. Ye, W. Zhou, X. Zeng and D. Yu, Org. Lett., 2018, 20, 3049–3052 CrossRef CAS PubMed
- S. Wang, X. Zhang, C. Cao, C. Chen and C. Xi, Green Chem., 2017, 19, 4515–4519 RSC
-
(a) S. Hase, Y. Kayaki and T. Ikariya, Organometallics, 2013, 32, 5285–5288 CrossRef CAS
- P. García-Domínguez, L. Fehr, G. Rusconi and C. Nevado, Chem. Sci., 2016, 7, 3914–3918 RSC
- W. J. Yoo and C. Li, Adv. Synth. Catal., 2008, 350, 1503–1506 CrossRef CAS
- X. Gao, C. Gan, S. Liu, F. Zhou, H. Wu and J. Zhou, ACS Catal., 2017, 7, 8588–8593 CrossRef CAS
- S. Li, J. Ye, W. Yuan and S. Ma, Tetrahedron, 2013, 69, 10450–10456 CrossRef CAS
- K. Yamashita, S. Hase, Y. Kayaki and T. Ikariya, Org. Lett., 2015, 17, 2334–2337 CrossRef CAS PubMed
- T. Ishida, S. Kikuchi, T. Tsubo and T. Yamada, Org. Lett., 2013, 15, 848–851 CrossRef CAS PubMed
- S. Sharma, A. K. Singh, D. Singh and D. Kim, Green Chem., 2015, 17, 1404–1407 RSC
- T. Niemi, I. Fernandez, B. Steadman, J. K. Mannisto and T. Repo, Chem. Commun., 2018, 54, 3166–3169 RSC
- W. Zhang, T. Xia, X. Yang and X. Lu, Chem. Commun., 2015, 51, 6175–6178 RSC
- C. Guo, W. Zhang, N. Zhang and X. Lu, J. Org. Chem., 2017, 82, 7637–7642 CrossRef CAS PubMed
- W. Zhang, N. Zhang, Y. Sun, Y. Ding and X. Lu, ACS Catal., 2017, 7, 8072–8076 CrossRef CAS
-
(a) X. Tang, C. Qi, H. He, H. Jiang, Y. Ren and G. Yuan, Adv. Synth. Catal., 2013, 355, 2019–2028 CrossRef CAS
- B. A. Vara, T. J. Struble, W. Wang, M. C. Dobish and J. N. Johnston, J. Am. Chem. Soc., 2015, 137, 7302–7305 CrossRef CAS PubMed
- S. Sun, B. Wang, N. Gu, J. Yu and J. Cheng, Org. Lett., 2017, 19, 1088–1091 CrossRef CAS PubMed
- J. Rintjema, R. Epping, G. Fiorani, E. Martín, E. C. Escudero-Adán and A. W. Kleij, Angew. Chem., Int. Ed., 2016, 55, 3972–3976 CrossRef CAS PubMed
- B. Carry, L. Zhang, M. Nishiura and Z. Hou, Angew. Chem., Int. Ed., 2016, 55, 6257–6260 CrossRef CAS PubMed
- S. Kikuchi, K. Sekine, T. Ishida and T. Yamada, Angew. Chem., Int. Ed., 2012, 51, 6989–6992 CrossRef CAS PubMed
- Y. Sadamitsu, K. Komatsuki, K. Saito and T. Yamada, Org. Lett., 2017, 19, 3191–3194 CrossRef CAS PubMed
- K. Sekine, Y. Sadamitsu and T. Yamada, Org. Lett., 2015, 17, 5706–5709 CrossRef CAS PubMed
- W. Yoo, M. G. Capdevila, X. Du and S. Kobayashi, Org. Lett., 2012, 14, 5326–5329 CrossRef CAS PubMed
- Z. Xin, C. Lescot, S. D. Friis, K. Daasbjerg and T. Skrydstrup, Angew. Chem., Int. Ed., 2015, 54, 6862–6866 CrossRef CAS PubMed
- C. W. Kee, K. Q. E. Peh and M. W. Wong, Chem. – Asian J., 2017, 12, 1780–1789 CrossRef CAS PubMed
- W. Yoo, T. V. Q. Nguyen and S. Kobayashi, Angew. Chem., Int. Ed., 2014, 53, 10213–10217 CrossRef CAS PubMed
- J. Hou, A. Ee, W. Feng, J. Xu, Y. Zhao and J. Wu, J. Am. Chem. Soc., 2018, 140, 5257–5263 CrossRef CAS PubMed
- T. Fujihara, Y. Horimoto, T. Mizoe, F. B. Sayyed, Y. Tani, J. Terao, S. Sakaki and Y. Tsuji, Org. Lett., 2014, 16, 4960–4963 CrossRef CAS PubMed
- Y. Li, Y. Hu and X. Wu, Chem. Soc. Rev., 2018, 47, 172–194 RSC
- C. Lescot, D. U. Nielsen, I. S. Makarov, A. T. Lindhardt, K. Daasbjerg and T. Skrydstrup, J. Am. Chem. Soc., 2014, 136, 6142–6147 CrossRef CAS PubMed
- T. Amaya, I. Kurata and T. Hirao, Org. Chem. Front., 2016, 3, 929–933 RSC
- Z. Zhang, L. Liao, S. Yan, L. Wang, Y. He, J. Ye, J. Li, Y. Zhi and D. Yu, Angew. Chem., Int. Ed., 2016, 55, 7068–7072 CrossRef CAS PubMed
- S. Wang, P. Shao, G. Du and C. Xi, J. Org. Chem., 2016, 81, 6672–6676 CrossRef CAS PubMed
- S. Sun, W. Hu, N. Gu and J. Cheng, Chem. – Eur. J., 2016, 22, 18729–18732 CrossRef CAS PubMed
- M. Brahmayya, S. A. Dai and S. Y. Suen, RSC Adv., 2015, 5, 65351–65357 RSC
- B. Yu, H. Zhang, Y. Zhao, S. Chen, J. Xu, L. Hao and Z. Liu, ACS Catal., 2013, 3, 2076–2082 CrossRef CAS
- Y. Zhao, Y. Wu, G. Yuan, L. Hao, X. Gao, Z. Yang, B. Yu, H. Zhang and Z. Liu, Chem. – Asian J., 2016, 11, 2735–2740 CrossRef CAS PubMed
- M. Xia, W. Hu, S. Sun, J. Yu and J. Cheng, Org. Biomol. Chem., 2017, 15, 4064–4067 RSC
- Q. Song, Z. Zhou, M. Wang, K. Zhang, P. Liu, J. Xun and L. He, ChemSusChem, 2016, 9, 2054–2058 CrossRef CAS PubMed
- Q. Song, Z. Zhou, H. Yin and L. He, ChemSusChem, 2015, 8, 3967–3972 CrossRef CAS PubMed
- Z. Zhou, Q. Song and L. He, ACS Omega, 2017, 2, 337–345 CrossRef CAS
- L. Song, G. Cao, W. Zhou, J. Ye, Z. Zhang, X. Tian, J. Li and D. Yu, Org. Chem. Front., 2018, 5, 2086–2090 RSC
- A. Del Vecchio, F. Caille, A. Chevalier, O. Loreau, K. Horkka, C. Halldin, M. Schou, N. Camus, P. Kessler, B. Kuhnast, F. Taran and D. Audisio, Angew. Chem., Int. Ed., 2018, 57, 9744–9748 CrossRef CAS PubMed
-
(a) J. Ma, B. Han, J. Song, J. Hu, W. Lu, D. Yang, Z. Zhang, T. Jiang and M. Hou, Green Chem., 2013, 15, 1485–1489 RSC
- T. Ishida, S. Kikuchi and T. Yamada, Org. Lett., 2013, 15, 3710–3713 CrossRef CAS PubMed
- T. Ishida, R. Kobayashi and T. Yamada, Org. Lett., 2014, 16, 2430–2433 CrossRef CAS PubMed
- C. Guo, W. Zhang, S. Liu and X. Lu, Catal. Sci. Technol., 2014, 4, 1570–1577 RSC
- B. Wang, S. Sun, J. Yu, Y. Jiang and J. Cheng, Org. Lett., 2017, 19, 4319–4322 CrossRef CAS PubMed
- P. Mampuys, H. Neumann, S. Sergeyev, R. V. A. Orru, H. Jiao, A. Spannenberg, B. U. W. Maes and M. Beller, ACS Catal., 2017, 7, 5549–5556 CrossRef CAS
- P. Xu, F. Wang, T. Wei, L. Yin, S. Wang and S. Ji, Org. Lett., 2017, 19, 4484–4487 CrossRef CAS PubMed
- Z. Zhang, T. Ju, M. Miao, J. Han, Y. Zhang, X. Zhu, J. Ye, D. Yu and Y. Zhi, Org. Lett., 2017, 19, 396–399 CrossRef CAS PubMed
- L. Song, L. Zhu, Z. Zhang, J. Ye, S. Yan, J. Han, Z. Yin, Y. Lan and D. Yu, Org. Lett., 2018, 20, 3776–3779 CrossRef CAS PubMed
- G. Shen, W. Zhou, X. Zhang, G. Cao, Z. Zhang, J. Ye, L. Liao, J. Li and D. Yu, Chem. Commun., 2018, 54, 5610–5613 RSC
- K. Sasano, J. Takaya and N. Iwasawa, J. Am. Chem. Soc., 2013, 135, 10954–10957 CrossRef CAS PubMed
- T. Kaicharla, M. Thangaraj and A. T. Biju, Org. Lett., 2014, 16, 1728–1731 CrossRef CAS PubMed
- W. Zhang, S. Liu and X. Lu, Beilstein J. Org. Chem., 2015, 11, 906–912 CrossRef CAS PubMed
- W. Zhang, M. Yang and X. Lu, Green Chem., 2016, 18, 4181–4184 RSC
- Z. Zhang, C. Zhu, M. Miao, J. Han, T. Ju, L. Song, J. Ye, J. Li and D. Yu, Chin. J. Chem., 2018, 36, 430–436 CrossRef CAS
- L. Fu, S. Li, Z. Cai, Y. Ding, X. Guo, L. Zhou, D. Yuan, Q. Sun and G. Li, Nat. Catal., 2018, 1, 469–478 CrossRef
- Z. Cai, S. Li, Y. Gao and G. Li, Adv. Synth. Catal., 2018, 360, 4005–4011 CrossRef CAS
- A. Tlili, E. Blondiaux, X. Frogneux and T. Cantat, Green Chem., 2015, 17, 157–168 RSC
- O. Jacquet, C. Das Neves Gomes, M. Ephritikhine and T. Cantat, ChemCatChem, 2013, 5, 117–120 CrossRef CAS
- B. Yu, H. Zhang, Y. Zhao, S. Chen, J. Xu, C. Huang and Z. Liu, Green Chem., 2013, 15, 95–99 RSC
- X. Gao, B. Yu, Z. Yang, Y. Zhao, H. Zhang, L. Hao, B. Han and Z. Liu, ACS Catal., 2015, 5, 6648–6652 CrossRef CAS
- Z. Yang, B. Yu, H. Zhang, Y. Zhao, G. Ji, Z. Ma, X. Gao and Z. Liu, Green Chem., 2015, 17, 4189–4193 RSC
- Z. Zhang, Q. Sun, C. Xia and W. Sun, Org. Lett., 2016, 18, 6316–6319 CrossRef CAS PubMed
- B. Zhang, G. Du, W. Hang, S. Wang and C. Xi, Eur. J. Org. Chem., 2018, 1739–1743 CrossRef CAS
- X. Wu, S. Sun, B. Wang and J. Cheng, Adv. Synth. Catal., 2017, 359, 3855–3859 CrossRef CAS
- X. Frogneux, E. Blondiaux, P. Thuéry and T. Cantat, ACS Catal., 2015, 5, 3983–3987 CrossRef CAS
- X. Liu, X. Li, C. Qiao, H. Fu and L. He, Angew. Chem., Int. Ed., 2017, 56, 7425–7429 CrossRef CAS PubMed
- C. Xie, J. Song, H. Wu, B. Zhou, C. Wu and B. Han, ACS Sustainable Chem. Eng., 2017, 5, 7086–7092 CrossRef CAS
- G. Jin, C. G. Werncke, Y. Escudie, S. Sabo-Etienne and S. Bontemps, J. Am. Chem. Soc., 2015, 137, 9563–9566 CrossRef CAS PubMed
- D. Zhu, L. Fang, H. Han, Y. Wang and J. Xia, Org. Lett., 2017, 19, 4259–4262 CrossRef CAS PubMed
-
(a) K. Thenert, K. Beydoun, J. Wiesenthal, W. Leitner and J. Klankermayer, Angew. Chem., Int. Ed., 2016, 55, 12266–12269 CrossRef CAS PubMed
- Z. Guo, B. Zhang, X. Wei and C. Xi, Org. Lett., 2018, 20, 6678–6681 CrossRef CAS PubMed
-
(a) J. Ye, I. Kalvet, F. Schoenebeck and T. Rovis, Nat. Chem., 2018, 10, 1037–1041 CrossRef CAS PubMed
-
(a) S. B. Lang, T. M. Locascio and J. A. Tunge, Org. Lett., 2014, 16, 4308–4311 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2019 |