
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolecular methods for assessment of non-covalent metallodrug–DNA interactions
Andrew
Kellett
 *a,
Zara
Molphy
a,
Creina
Slator
a,
Vickie
McKee
ab and
Nicholas P.
Farrell
*c
*a,
Zara
Molphy
a,
Creina
Slator
a,
Vickie
McKee
ab and
Nicholas P.
Farrell
*c
aSchool of Chemical Sciences and the National Institute for Cellular Biotechnology, Dublin City University, Glasnevin, Dublin 9, Ireland. E-mail: andrew.kellett@dcu.ie; Tel: +353 1 7005461
bDepartment of Physics, Chemistry and Pharmacy, University of Southern Denmark, Campusvej 55, 5230 Odense M, Denmark
cDepartment of Chemistry, Virginia Commonwealth University, Richmond, VA 23284-2006, USA. E-mail: npfarrell@vcu.edu; Tel: +1 804 8286320
First published on 4th February 2019
Abstract
The binding of small molecule metallodrugs to discrete regions of nucleic acids is an important branch of medicinal chemistry and the nature of these interactions, allied with sequence selectivity, forms part of the backbone of modern medicinal inorganic chemistry research. In this tutorial review we describe a range of molecular methods currently employed within our laboratories to explore novel metallodrug–DNA interactions. At the outset, an introduction to DNA from a structural perspective is provided along with descriptions of non-covalent DNA recognition focusing on intercalation, insertion, and phosphate binding. Molecular methods, described from a non-expert perspective, to identify non-covalent and pre-associative nucleic acid recognition are then demonstrated using a variety of techniques including direct (non-optical) and indirect (optical) methods. Direct methods include: X-ray crystallography; NMR spectroscopy; mass spectrometry; and viscosity while indirect approaches detail: competitive inhibition experiments; fluorescence and absorbance spectroscopy; circular dichroism; and electrophoresis-based techniques. For each method described we provide an overview of the technique, a detailed examination of results obtained and relevant follow-on of advanced biophysical/analytical techniques. To achieve this, a selection of relevant copper(II) and platinum(II) complexes developed within our laboratories are discussed and are compared, where possible, to classical DNA binding agents. Applying these molecular methods enables us to determine structure–activity factors important to rational metallodrug design. In many cases, combinations of molecular methods are required to comprehensively elucidate new metallodrug–DNA interactions and, from a drug discovery perspective, coupling this data with cellular responses helps to inform understanding of how metallodrug–DNA binding interactions manifest cytotoxic action.
Dr Andrew Kellett is a graduate of Maynooth University and Technological University Dublin. He is currently Associate Professor of Inorganic and Medicinal Chemistry at Dublin City University (DCU). His research intersects bioinorganic and nucleic acids research fields and his work is supported by Science Foundation Ireland (SFI), the Synthesis and Solid State Pharmaceutical Centre (SSPC), CÚRAM, the Irish Research Council (IRC), and the European Union's Horizon 2020 programme. The current focus of his group is centred on artificial gene editing and therapeutic nucleic acids. |
 From left to right: Dr Zara Molphy, Dr Andrew Kellett, Prof. Nicholas P. Farrell, Dr Creina Slator, and Prof. Vickie McKee | Dr Zara Molphy completed her BSc in Chemical and Pharmaceutical Sciences at DCU, graduating with a first class honours degree in 2012. She was awarded an Irish Research Council (IRC) Postgraduate Scholarship in 2013 and received her PhD in the area of bioinorganic and nucleic acid chemistry under the supervision of Prof. Andrew Kellett in 2017. She is currently a postdoctoral researcher in the Science Foundation Ireland (SFI) funded polynuclear platinum biomaterials project with expertise in drug design, molecular biology, and DNA damage/repair. |
Dr Creina Slator obtained her BSc in Chemical and Pharmaceutical Sciences (Hons) in 2012 at Dublin City University (DCU). She received her PhD in bioinorganic and medicinal chemistry in 2017 under the supervision of Prof. Andrew Kellett and was a recipient of a Distinguished Scholarship from DCU and also an Irish Research Council Scholarship. She is now a postdoctoral research fellow at DCU working in Science Foundation Ireland (SFI) funded research projects dedicated to platinum and copper biomaterials development. |
Professor Vickie McKee obtained a PhD in macrocyclic chemistry from Queen's University, Belfast. She is currently Adjunkt Professor in Syddansk Universitet, Odense, Denmark and Adjunct Professor in Dublin City University. Her research interests are primarily in bioinorganic coordination chemistry and crystallography. |
Professor Nicholas P. Farrell is a graduate of University College Dublin and Sussex University. He is currently Professor of Chemistry at Virginia Commonwealth University (VCU) where he was Distinguished Research Scholar for 2003–2004. Professor Farrell's activities are both highly interdisciplinary and international in scope. He and his collaborators have received over sixty patents world-wide from his inventions. He was Chair of the first Gordon Research Conference on Metals in Medicine in 2002. He is a 2010–2015 Jefferson Science Fellow and a Corresponding Member of the Brazilian Academy of Sciences. |
Key learning points• Essentials of nucleic acid structure.• Principles of nucleic acid–metallodrug recognition. • Application of mass spectrometry, nuclear magnetic resonance spectroscopy and X-ray crystallography to probe selective metallodrug–DNA binding sites. • Quantification of metallodrug–DNA interactions using high-throughput optical techniques based on fluorescence and absorbance spectroscopy. • Electrophoretic techniques and the use of on-chip microfluidic assays to identify drug–DNA damage. |
1. An introduction to DNA structure and function.
1.1. DNA structure
Nucleic acids are a critical molecular target for candidate antitumoral metallodrugs. The electron-rich phosphate backbone, donor heteroatoms in the nucleobases, and intricate secondary and tertiary structures combine to create potential binding environments for both “free” metal ions and discrete complexes. From a medicinal chemistry perspective, the ability to probe metal complex coordination to DNA is essential for developing new architectures that selectively target oligonucleotides and specific oligonucleotide conformations. Furthermore, given the essentiality of the DNA double-helix for the storage of genetic information and thus mediation of faithful cell replication, the interruption of biogenesis at this juncture provides a basic target for metallodrug discovery.1The biologically predominant B-form of duplex DNA comprises a right-handed double helix containing two antiparallel sugar-phosphate chains (Fig. 1A). The heteroaromatic bases lie in the centre of the helix and hydrogen bonding between them effectively binds the two helical chains together. This structure is supported by stacking interactions that orient the base pairs perpendicular to the helical axis. DNA bases follow Chargaff's rules whereby a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (purine:pyrimidine) relationship exists between A and T as well as G and C (Fig. 1B).2 The angle between the glycosidic bonds and the hydrogen bonds results in unequal grooves in the helical structure referred to as the minor and major grooves. Structural characteristics of B-DNA can thus be exploited for molecular recognition as the major groove (10.5 Å) is wider than the minor groove (4.8 Å), although their depths are generally identical.3 Between the base pairs of B-DNA there is an axial rise of 3.4 Å (0.34 nm) and a 34.5° twist angle associated with every residue rotation while the helix diameter is 20 Å (Fig. 1C). In B-DNA the 2′-deoxyribose ring exists in the C2′-endo twist conformation, in contrast to the C3′-endo twist conformation found within both A and Z-DNA (Fig. 1D).4,5 Along with Watson–Crick (WC) base pairing, π–π stacking interactions provide additional stability to the double helix; the overlap between successive 5′ → 3′ bases differs significantly however as evidenced by the overlap of TA/TA and AT/AT steps shown in Fig. 1E.
:1 (purine:pyrimidine) relationship exists between A and T as well as G and C (Fig. 1B).2 The angle between the glycosidic bonds and the hydrogen bonds results in unequal grooves in the helical structure referred to as the minor and major grooves. Structural characteristics of B-DNA can thus be exploited for molecular recognition as the major groove (10.5 Å) is wider than the minor groove (4.8 Å), although their depths are generally identical.3 Between the base pairs of B-DNA there is an axial rise of 3.4 Å (0.34 nm) and a 34.5° twist angle associated with every residue rotation while the helix diameter is 20 Å (Fig. 1C). In B-DNA the 2′-deoxyribose ring exists in the C2′-endo twist conformation, in contrast to the C3′-endo twist conformation found within both A and Z-DNA (Fig. 1D).4,5 Along with Watson–Crick (WC) base pairing, π–π stacking interactions provide additional stability to the double helix; the overlap between successive 5′ → 3′ bases differs significantly however as evidenced by the overlap of TA/TA and AT/AT steps shown in Fig. 1E.
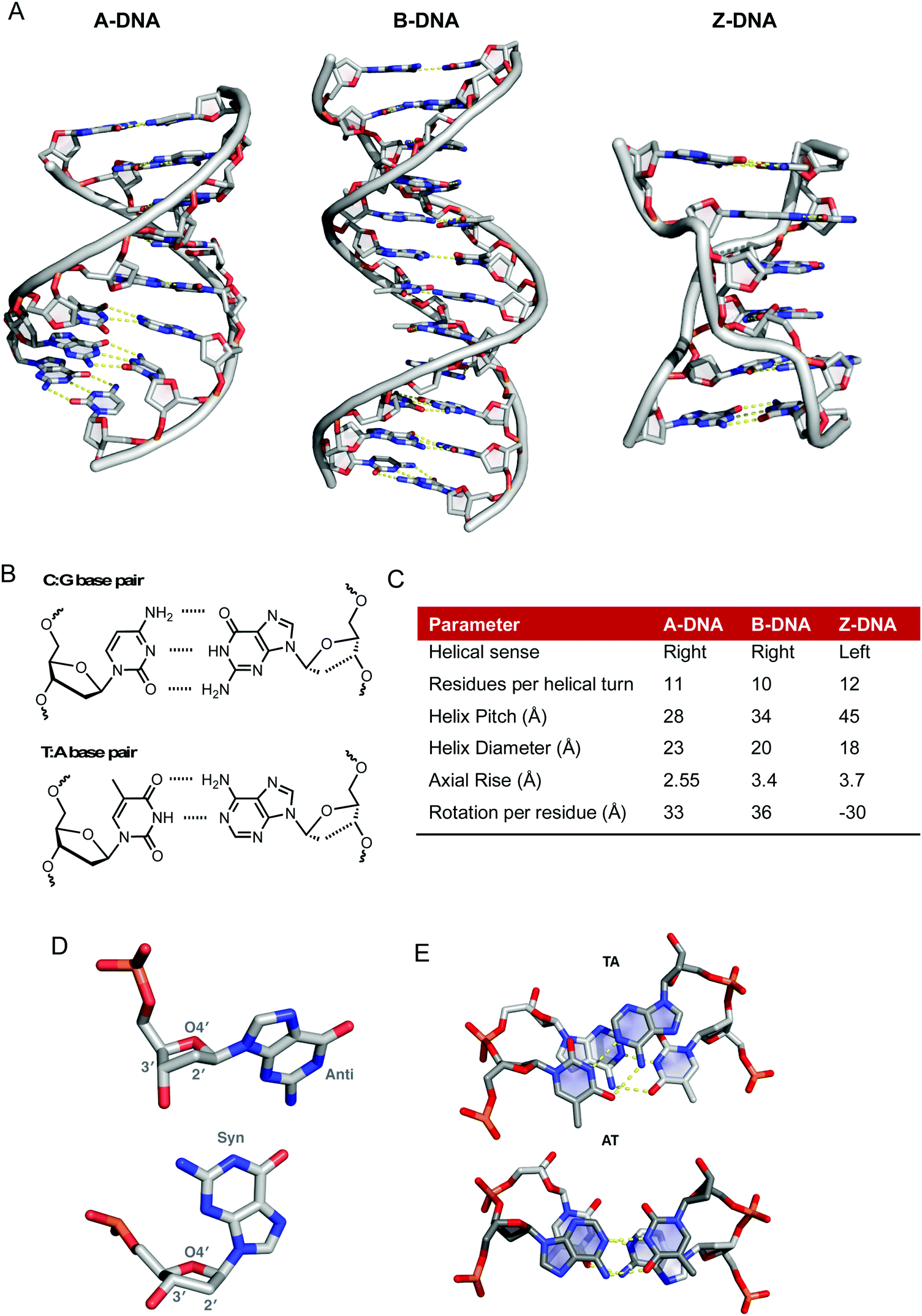 | ||
| Fig. 1 (A) X-ray structures of A-, B- and Z-DNA from PDB files 1VJ4, 1BNA and 2DCG respectively, (B) hydrogen bonding between C–G and T–A base pairs, (C) summary of structural differences between A-, B- and Z-DNA;6 (D) conformational preferences of the 2′-deoxyribose rings of DNA taken from 1BNA and 2DGC with respective anti- and syn-conformations of guanine nucleobase and (E) top down view of stacking interactions in TA and AT steps from PDB 167D (d(CCATTAATGG)2). Hydrogen bonds are shown as dashed yellow lines; the angle between the vectors representing the two N3(T) → N1(A) hydrogen bonds is 118° for the TA step and 160° for the AT step. | ||
The flexibility of DNA lends itself to the formation of many alternative conformations generated under the influence of solvent conditions and small molecules. A-DNA is the dehydrated form of B-DNA and has a similar but more rigid and compacted structure consisting of 11 base pairs per helical turn with a axial rise of 2.55 Å between bases pairs (Fig. 1A and C).6 Z-DNA is formed during the transcription process in vivo due to the torsional strains generated as negative supercoils are created by RNA polymerase moving along the sequence of the DNA double helix. There is a radical difference between Z- and B-DNA as the helical sense flips from right- to left-handed; this conformational change is due to alternating syn- and anti-conformations of purine and pyrimidine bases, respectively. Structurally, Z-DNA is elongated and narrow with a diameter of 18 Å and is composed of a single narrow groove analogous to that of the minor groove of B-DNA resulting in a zigzag arrangement of the backbone (Fig. 1A).5 Besides these “canonical forms” of DNA other forms of DNA such as triplexes, G-quadruplexes and I-motifs are important for gene-specific drug targeting.7,8
Significant effort has been expended in recent years to determine how metallodrug molecules interact with nucleic acids and a number of complementary molecular methods have been developed to probe reversible and non-reversible interactions. In the absence of high-resolution structural data from X-ray diffraction or NMR studies, the mode of binding must be inferred indirectly from molecular and biophysical solution studies. A wealth of information regarding the binding mode and sequence specificity of these interactions can be determined and in this tutorial review we provide an overview of selected molecular methods to identify metallodrug–DNA interactions with particular emphasis on copper(II) and platinum(II) complexes of current interest to our research programmes (cf.Fig. 2A–D). While much evidence has been accumulated on covalently bound adducts, the potential for sequence specificity is in some ways linked to the pre-association or “non-covalent” interactions occurring prior to covalent bond formation and these aspects are emphasized herein.
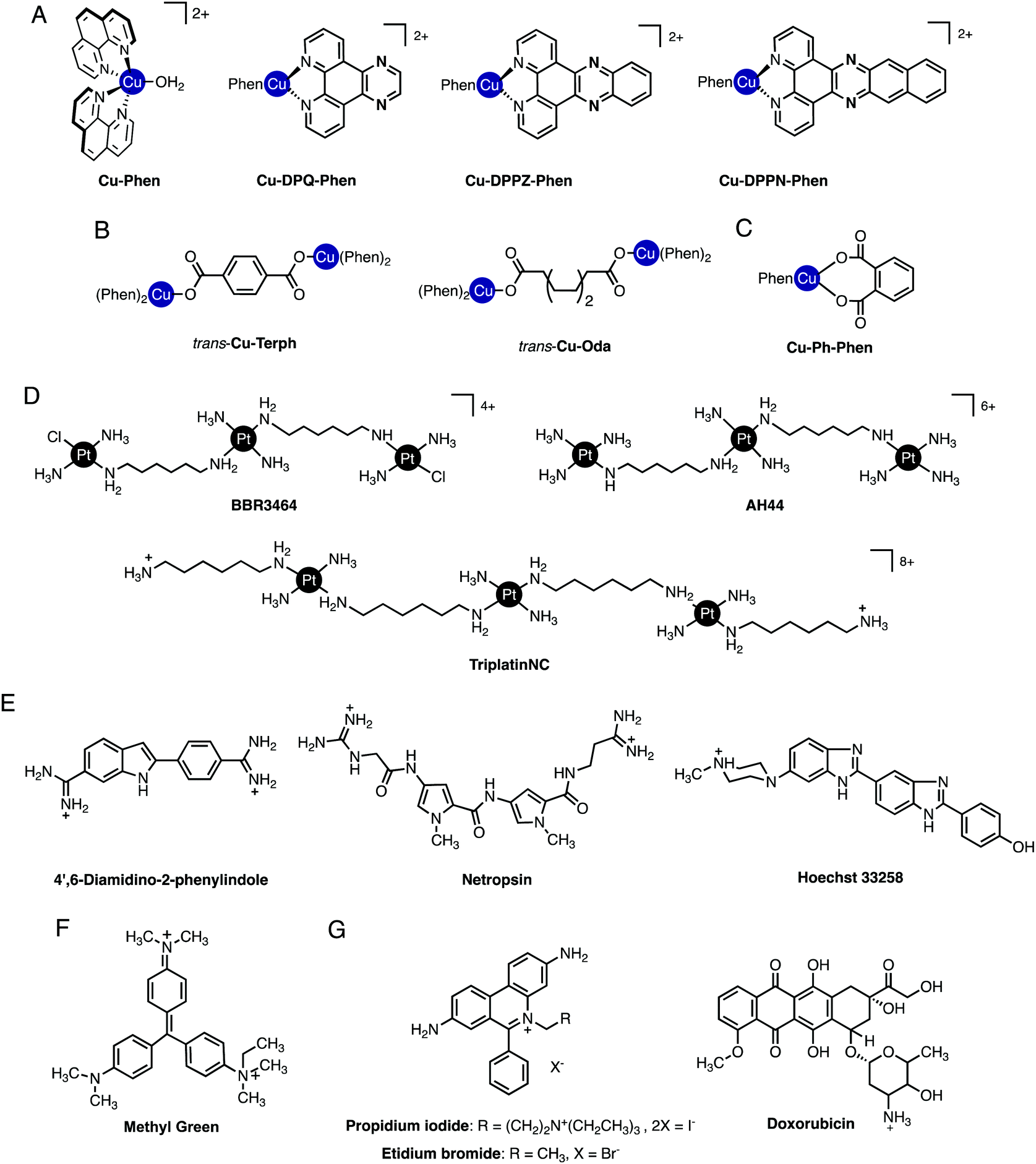 | ||
| Fig. 2 (A) Cu-Phen and ternary copper(II) phenanthrene complexes Cu-DPQ-Phen, Cu-DPPZ-Phen and Cu-DPPN-Phen; (B) di-nuclear copper(II) terephthalate (Cu-Terph) and octanedioate (Cu-Oda) complexes; (C) square planar copper(II) complex Cu-Ph-Phen incorporating o-phthalate and 1,10-phenanthroline; (D) polynuclear platinum complexes (PPCs) [{transPtCl(NH3)2}2-μ-{trans-Pt(NH3)2(NH2(CH2)6NH2)2}]4+ (Triplatin, BBR3464), [{Pt(NH3)3}2-μ-{trans-Pt(NH3)2(NH2(CH2)6NH2)2}]6+ (AH44), and [{transPt(NH3)2(NH2(CH2)6NH3)}2-μ-({trans-Pt(NH3)2(NH2(CH2)6NH2)2})]8+ (TriplatinNC); and (E) Minor groove binding agents 4′,6-diamidino-2-phenylindole (DAPI), netropsin (Net), Hoechst 33258; (F) major groove binding methyl green (MG); and (G). planar heterocyclic intercalators propidium iodide (PI), ethidium bromide (EtBr) and doxorubicin (Dox). | ||
1.2. Nucleic acids as metallodrug targets. Modes of binding by coordination and classical compounds
Non-covalent modes by which metal complexes bind to DNA include intercalation,9,10 insertion,11,12 groove binding,13,14 and by discrete phosphate coordination such as the ‘phosphate clamp’.15 In all cases, structural factors based on the shape and charge of the complex underpin these binding modes and considerable effort is focused to uncover new inorganic scaffolds with selective and robust binding capability. The structural aspects of DNA are, of course, essential to the recognition process since they provide the template to which metal complexes bind. From a classical perspective, crescent-shaped organic molecules containing protonated terminal amines (e.g. netropsin and distamycin, Fig. 2E) are well-matched to the minor groove curvature and, once bound, give rise to helical contraction due to secondary non-covalent phosphate backbone-amine interactions.16 Organic-based major groove binders, on the other hand, contain non-coplanar aromatic propeller-like conformations (e.g. methyl green, Fig. 2F) that bind due to hydrophobic interactions with the major groove floor with cationic amines located on the molecular periphery likely providing ancillary stabilization.17 In contrast to both major and minor groove binders, DNA intercalators (e.g. ethidium bromide) contain planar heteroaromatic structures, often with extended π-backbones, to facilitate penetration into the DNA backbone and subsequent van der Waals contacts between WC pairs (Fig. 2G).18 The significance of these classical interactions to the discovery of new metallodrug–DNA binding modes and in proposing new metal complex-nucleic acid adducts is important to consider. Since classical binding modes are selective and in many cases structurally determined by primary techniques including X-ray structural analysis, comparisons using direct and indirect methods can be made between suspect metallodrug–DNA binding agents and suitable organic controls. In this tutorial review we take advantage of this approach and describe how classical agents can assist in uncovering drug–DNA interactions of novel agents.2. Primary techniques
2.1. X-ray crystallography
Definitive visualization of coordination binding modes comes firstly from X-ray crystallography. Much of our current knowledge of detailed structural information is derived from X-ray crystallographic studies on single crystals of DNA, or oligonucleotides and their complexes with drug molecules. From the initial Franklin photographs demonstrating the helical nature of B-DNA in 1953,19 through the conformational effects of cisplatin binding,20 to more recent studies on intercalation, metalloinsertion and the phosphate clamp, crystallographic studies underpin our understanding of metallodrug–DNA structural chemistry. In recent years most biological macromolecular crystallography has been carried out at 3G synchrotron facilities where the availability of tunable, high intensity X-ray sources and fast, sensitive detectors has enabled study of ever smaller crystals at ever higher resolution.21,22 At the same time, developments in laboratory instrumentation, notably microsource X-ray tubes and CCD or solid state detectors are arguably bringing small macromolecules such as nucleotide oligomers within range of laboratory sources.Good quality data reveal a wealth of detailed, atomic-level structural information (and moderate quality data may also be very revealing). Essentially the experiment yields a 3D map of electron density from which the atomic positions are determined. Once the positions of the atoms are known, geometric data such as bond lengths and angles can be readily calculated and the structure can be explored and illustrated using graphics programs. For example, crystallographic data allow analysis of the hydrogen bonding responsible for the backbone-tracking phosphate clamp behaviour of TriplatinNC as shown in Fig. 2D as well as the stacking and hydrogen bonding involved in Δ-α-[Rh{(R,R)-Me2trien}phi]3+ intercalation (Fig. 3 and 4A).12
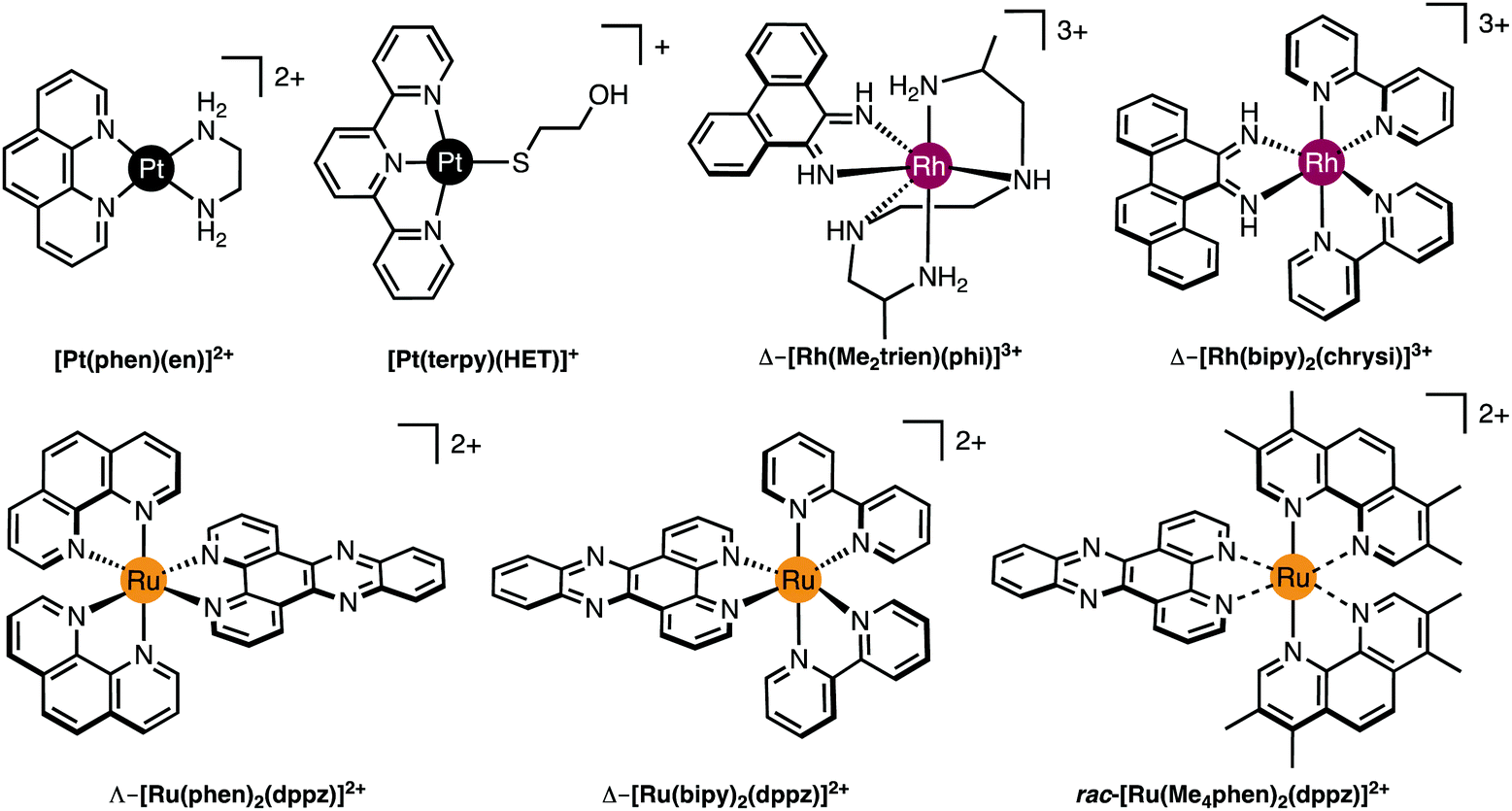 | ||
| Fig. 3 Molecular structures of selected Pt(II), Rh(III) and Ru(II) intercalating and insertion complexes. | ||
The main difficulty in using crystallography to study drug interactions with DNA or oligonucleotides is growing a good quality crystal of reasonable size. For this, a significant quantity of homogeneous, pure material is required and it may be necessary to test a wide range of conditions before a suitable set is found, so it may also be an expensive undertaking in terms of oligonucleotide materials.
Since only the planar section of the ligand can intercalate, it needs to be large enough to achieve significant π-overlap, without bringing the remainder of the complex into unfavourable steric or electronic interaction. Ligands such as 1,10-phenanthroline (phen) and terpyridine (terpy) in square planar Pt complexes intercalate effectively but large ligands such as 9,10-phenanthrenequione diime (phi) or dipyridophenazine (dppz) are more effective for 6-coordinate complexes.12 Recent examples of intercalation involving Ru(II) coordinated phen and TAP (1,4,5,8-tetra-aza-phenanthrene) ligands show partial intercalation that gives rise to DNA ‘kinking’ (Fig. 4D and PDB 4YMC).10 The orientation of the intercalator in the “slot” between two base pairs may be symmetrical, or laterally offset relative to the principal axis of the DNA helix, presumably reflecting the best accessible set of stacking and other interactions, hence dependent on the detailed electronic structures of the intercalating ligand and the base pairs lining the intercalation site. There are ten possible intercalation sites,25 differing in existing π-interactions between base pairs and their affinity for a specific intercalator. For example, in 1979 Lippard and co-workers showed that [Pt(phen)(en)]2+ and [Pt(terpy)(HET)]+ (where HET = 2-hydroxyethanethiolate) exhibit GC selectivity,26 while [Ru(phen)2(dppz)]2+ (Fig. 3) displays preferential intercalation for poly-d(AT) over poly-d(GC).12 The same complex intercalates symmetrically at the TA/TA step in d(CCGGTACCGG)2 (Fig. 4C) but not the AT/AT site in d(CCGGATCCGG)2.10 In fact, many metallointercalators bind preferentially to DNA at specific sites, and this function can be amplified by interaction of the ancillary ligands with the DNA duplex. In an early example, the photoactive complex Δ-α-[Rh{(R,R)-Me2trien}phi]3+ (where Me2trien = 2,9-diamino-4,7-diazadecane, Fig. 3) was found to specifically cleave the sequence 5′-TGCA-3′. The structural basis for this specificity was established by the X-ray structure of the complex bound to 5′-G(5|U)TGCAAC-3′ (Fig. 4A) in the major groove specifically at the 5′-TG|CA-3′ site (where | indicates phi insertion).12 The intercalative π-stacking is supported by H-bonds from the amines of the Me2trien ancillary ligands to the guanine-O6 acceptors as well as to some ordered water molecules.
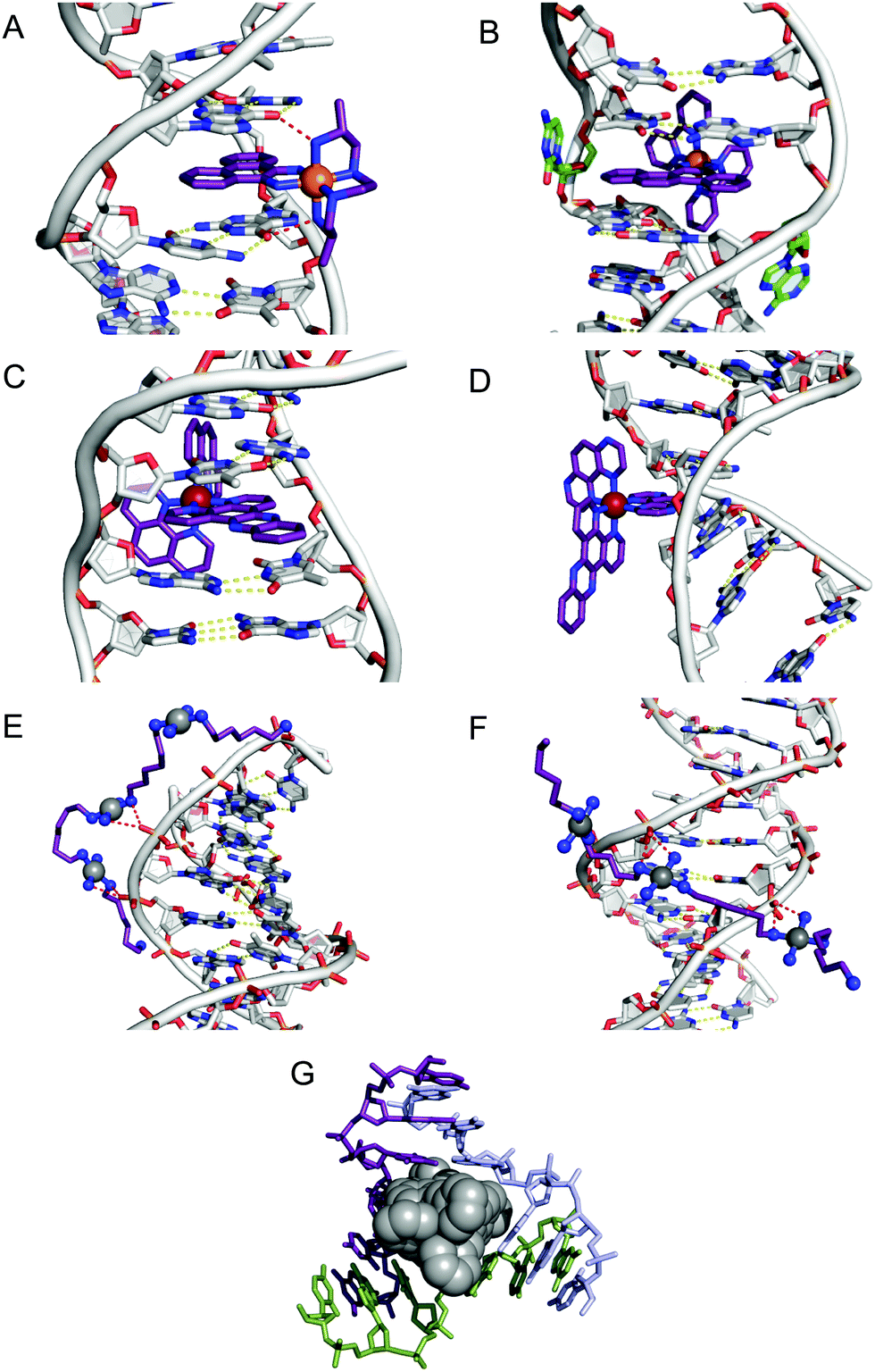 | ||
Fig. 4 Intercalation and insertion. (A) Δ-α-[Rh{(R,R)-Me2trien}phi]3+ intercalated into 5′-G(5|U)TGCAAC-3′ with additional stabilisation by H-bonds from the ancillary ligand (PDB 454d), black lines indicate hydrogen bonds and (B) Δ-[Rh(bpy)2(chrysi)]3+ inserted into (5′-CGGAAATTCCCG-3′), displacing a mismatched AC pair shown in green (PDB 2O1I) b; (C) intercalation of Λ-[Ru(phen)2(dppz)]2+ into d(CCGGTACCGG)2 (PDB 3U38); (D) Λ-[Ru(TAP)2(dppz)]2+ bound to d(CCGGATCCGG)2 by semi-intercalation (PDB 4YMC); TriplatinNC bound to Dickerson–Drew dodecamer (B-DNA) through backbone tracking (E) and groove spanning (F) interactions. (N–H⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) P hydrogen bonds shown as dashed red lines); and (G) three-way DNA junction recognition by a metallosupramolecular helicate (Fe2L3)4+ where L = C25H20N4 (PDB 2ET0). P hydrogen bonds shown as dashed red lines); and (G) three-way DNA junction recognition by a metallosupramolecular helicate (Fe2L3)4+ where L = C25H20N4 (PDB 2ET0). | ||
Metalloinsertion is closely related to intercalation, also involving incorporation of a ligand into the base pair stack. The main difference is that in metalloinsertion one base pair is ejected and replaced by the incoming ligand. This interaction was predicted by Lerman but first structurally characterized in a metal complex only in 2007 for Δ-[Rh(bpy)2(chrysi)]3+ (where chrysi = 5,6-chrysenequinone diimine, Fig. 3 and 4B),12 extended to a family of chrysi derivatives and more recently for both Δ-[Ru(bipy)2(dppz)]2+ and Λ-[Ru(phen)2(dppz)]2+ (where bipy = 2,2′-bipyridine, Fig. 3), in all three cases the insertion is at a mis-matched base pair and from the minor groove (in contrast to intercalation).10,12 The dppz complexes are capable of both intercalation and insertion and it may be that other known intercalators are also capable of insertion in the presence of base-pair mismatches and perhaps matched base-pairs but evidence for this has yet to be established. Insertion involves less modification to the helical structure of DNA than intercalation. There is no significant extension or change in helicity although the “flipped out” bases are free to interact in other ways with the DNA helix or the inserting complex. The stabilisations due to hydrogen bonding of the displaced base pair and its stacking interactions are both lost and need to be replaced by the new π-stacking interactions (plus any new interactions involving the flipped out base pair). The relative contributions of base-stacking and base-pairing to DNA duplex stability depend on the particular sequence considered and, in some cases, the loss of the H-bonding component might not be expected to have a high cost. This is consistent with the observation that metalloinsertion can be specific for mismatched base pairs, especially those where the resulting base-pairing is energetically disfavoured (CC and CA). For example, Δ-[Rh(bipy)2(chrysi)]2+ (Fig. 3) can promote specific cleavage at a single mismatch site in a 2725 base pair linearized plasmid heteroduplex. This specificity is useful in detection of mismatches and is potentially diagnostic for cells with impaired mismatch repair (MMR) mechanisms; notably, a series of Rh-chrysi complexes that showed selective cytotoxicity for MMR-deficient cancer cells.12
As for intercalation, interaction of the ancillary ligands with DNA can be used to tune the binding properties of the inserting complex. For example, [Ru(bipy)2dppz]2+ binds to DNA via both intercalation and by insertion at mismatch sites but [Ru(Me4phen)2(dppz)]2+ (where Me4phen = 3,4,7,8-tetramethyl-1,10-phenanthroline, Fig. 3) is a mismatch-specific metalloinsertor where the methyl groups disfavour intercalation (due to steric interaction with the backbone) and also control the depth of dppz insertion.12 As might be expected, the metalloinsertion correlates with the stability of the mismatch site, strongest for CC and CA (less stable), less striking for GG AA (more stable).
P) hydrogen bonding directed through cis-oriented NH3 (ammine) and -RNH2 (amine) ligands.27 The discrete mechanism of phosphate clamping gives rise to distinctive binding motifs: phosphate tracking and groove spanning (Fig. 4E and F). The groove spanning mode is dependent on helical topology localised to the minor groove and base sequence composition. These structures have been recently reviewed.15
| Method summary | DNA X-ray crystallography (direct) |
|---|---|
| Requirements | 3G synchrotron facilities/microsource X-ray diffractometer. |
| Results | Definitive visualization of metallodrug–DNA binding interactions. |
| Advantages | Detailed structural data. |
| Limitations | Solid-state interactions only and transient/dynamic/pre-associative interactions may be precluded. Crystals of suitable quality required. |
| Complementary and advanced techniques | NMR and MS (direct) (Sections 2.2 and 2.3). CD/fluorescence/absorbance spectroscopy (indirect) (Sections 3.1 and 3.2). |
| Advanced complementary methods include analysis by optical tweezers. |
2.2. NMR studies
NMR studies complement the solid-state structures and give more information to the conformational variability in solution. NMR spectroscopy has benefitted significantly from advances in field strength, magnetic shielding and cryogenic probes. The most useful isotopes for NMR studies of platinum anticancer agents are 1H, 15N and 195Pt. The latter is extremely sensitive to the nature of the ligands attached and can be used for speciation and kinetic studies. The use of {1H,15N} HMQC/HSQC NMR spectroscopy greatly enhances sensitivity and is especially useful in kinetic studies with biological molecules.29,30 Briefly, transfer of magnetization from the proton to a second heteronucleus such as 15N gives a 2-dimensional spectrum with one axis for proton (1H) and the other for 15N, thus producing a spectrum with a peak for each unique proton attached to 15N with the unique advantage of overcoming the inherent insensitivity of the 15N nucleus. The sensitivity of the chemical shift and coupling constants (e.g.1J{15N–195Pt}) to the nature of the trans ligands, and coupled to the fact that the only protons observed are those bound to the 15N nucleus makes the technique of great practical use.30 Pre-association and strong non-covalent binding can be observed as well as kinetics of DNA binding.Cisplatin is a bifunctional DNA-binding agent which preferentially binds to N7 of guanine and adenine and typically results in 1,2-GG/AG intrastrand crosslinks with structural distortion through helical unwinding by 13°, and helical bending of 30–40° towards the major groove. It is generally accepted that aquation of cisplatin to give the more substitution-labile aquated species is necessary prior to covalent binding to DNA. Using {1H,15N} HSQC NMR the stepwise aquation, monofunctional and bifunctional nucleobase binding can be monitored and their respective rates of formation and equilibrium constants can be determined.30
When complexes with higher positive charge are concerned, pre-association with the biomolecule, and the effects of such pre-association on conformational preferences, can be observed. Triplatin (BBR3464, Fig. 2D) was the first multi-nuclear platinum complex to enter human clinical trials. The complex forms crosslinked bi-functional DNA adducts distinct from those of mononuclear species such as cisplatin. More specifically, the compound produces long-range inter- and intra-strand DNA platination and this behaviour is distinct from cisplatin's short range platination. A 3-fold slowing of the aquation of BBR3464 occurs in the presence of dsDNA but not ssDNA. This feature may account for the kinetic binding preference and may also be relevant in stabilization of G-quadruplex structures using BBR3464.15 The results emphasize how the alteration of chemical properties of small molecules in the presence of large host interactions is dependent on the conformation and nature of that host and these examples show the utility of NMR techniques in probing these subtle interactions. Two examples are relevant to this tutorial:
(i) A unique feature of long-range {Pt,Pt} interstrand crosslinks is the occurrence of directional 5′ → 5′ and 3′ → 3′ isomers – the existence of the unusual 3′ → 3′ linkage isomer in the sequence was also confirmed by 2D NMR spectroscopy.15,30 The initial orientation of BBR3464 on DNA mediated by electrostatic interactions transiently engaging with phosphate groups in the minor groove is an important feature which dictates not only isomer directionality but also the final conformation of covalently-bound interstrand crosslinks (1,4 versus 1,6) because in some cases the covalent adduct can be formed directly but in others, diffusion off the DNA is necessary for covalent binding to occur. These subtleties can be followed by 2D {1H,15N} HSQC NMR.30,31
(ii) Using fully 15N-labelled TriplatinNC, the presence of the phosphate clamp in solution with the Dickerson–Drew Duplex (DDD) was confirmed by observation of large chemical shift differences of the δ(NH3) and δ(–NH2R) in both the 1H and 15N dimensions.32 The 2D {1H,15N} HSQC NMR spectrum of 15N-labeled TriplatinNC shows only two cross-peaks and a weak peak due to the dangling amine (Fig. 5). In the presence of DDD at pH 6, dramatic downfield 15N shifts of approximately 20 ppm are observed, emphasising the dependency of shift on hybridization. The coupling constant changes are also consistent with formation of the phosphate clamp.15
| Method summary | NMR analysis (direct) |
|---|---|
| Requirements | Good solubility. Labelled compounds (e.g.15N) best although natural abundance can be used in favourable circumstances. |
| Results | Conformation of metallodrug–DNA solution interactions. |
| Advantages | Modification of electronic properties of complex upon binding to oligonucleotide; kinetics of bond formation upon initial pre-association. |
| Limitations | Need NMR active nuclei and mainly diamagnetic compounds. |
| Complementary and advanced techniques | X-ray and MS (direct) (Sections 2.1 and 2.3). |
| HSQC NMR can be combined with 3-D NOESY-HSQC or TOCSY-HSQC. | |
| Saturation transfer difference (STD) NMR to study transient complex-biomolecule interactions. |
 | ||
| Fig. 5 {1H,15N} HSQC NMR of TriplatinNC (left) and Dickerson–Drew Duplex (DDD, right). Satellites from 1J(15N–195Pt) are clearly visible. Adapted with permission from Qu et al.32 Reproduced from ref. 32 with permission from the Royal Society of Chemistry, copyright 2015. | ||
2.3. Mass spectrometry
Advances in technologies and ionization techniques now make Electrospray Ionization-Mass Spectrometry (ESI-MS) an indispensable tool for probing drug-nucleic acid interactions with several advantages; including the need for only a small sample, speed of use, and ease of analysis.33 Covalent binding with biomolecules in general is easily observed and with appropriate digestion can pinpoint stoichiometry and binding sites of metallodrugs. It is in the study of non-covalent interactions where most interest resides because, if strong enough, the canonical non-covalent binding modes of hydrogen-bonding, electrostatic interactions, and intercalation can be transferred to the gas phase without disruption. Using appropriate non-denaturing conditions and carefully controlled instrumental optimization, electrospray ionization is able to transfer non-covalent complexes into the gas phase of the mass spectrometer without dissociation. Single-stranded, double-stranded and G-quadruplex DNA have all been studied and primary spectra combined with MS–MS techniques can give information on strength and sites of binding.33ESI-MS has also proven useful in probing relative binding affinities of metal complexes to duplex and quadruplex DNA. Differences in binding within a series of octahedral metallointercalators based on [Ru(phen)3]2+ and [Ru(phen)2(dppz)]2+ and square-planar analogs such as [Pt(en)(phen)]2+ demonstrated that the binding affinity in general towards quadruplex DNA is significantly less than that towards dsDNA.33
DNA as a template affects kinetics of substitution reactions occurring within its domain. Mass spectrometric studies using short 18-mer oligos showed a kinetic preference for binding of BBR3464 to ssDNA over dsDNA. In this case, electrospray ionization coupled with Fourier transform ion cyclotron resonance mass spectrometry (ESI-FTICR-MS) is sufficiently sensitive to observe the ‘pre-association’ of the covalently binding molecule prior to Pt–DNA bond formation. For single-stranded DNA, the site of binding of the substitution-inert TriplatinNC and AH44 on a 18-mer ssDNA (5′-TCTCCCAGCGTGCGCCAT-3′) was ascertained using Tandem MS–MS of the 1:1 adducts (Fig. 6).34 The binding is sufficiently strong that the fragment ion pattern is distinctly different and upon MS–MS there is no drug–DNA dissociation, only cleavage of the oligonucleotide backbone.
 | ||
| Fig. 6 ESI-MS/MS schematic of free (top) and PPC (either TriplatinNC or AH44) adducted (bottom) 5′-d(TCTCCCAGCGTGCGCCAT) at 100 and 120 V of collisional energy, respectively.34 Fragmentation of the glycosidic bonds is prevalent throughout the free, with the region of enhanced stability in teal. The associated fragment ions (w82−, w92−, and a9–a12 using standard McLuckey nomenclature) are absent in the adduct indicating the area of PPC binding. | ||
Full scan ESI-MS spectra of dsDNA AT duplex {5′-TAGCGCTTTTTCCGTA-3′}–{5′-TACGCGAAAAAGCGCTA-3′} complexed with substitution-inert PPCs also confirmed that the non-bonding interaction is strong enough to be transferred from solution to the gas phase. The CID spectra showed duplex unzipping into single strands with again no loss of PPC–DNA binding and duplex stabilization correlates with increasing charge and hydrogen bonding character of the complex. Upon increasing the collisional energy the single-stranded DNA formed dissociates as above.29
| Method summary | Mass spectrometry (direct) |
|---|---|
| Requirements | Transfer to gas phase without bond cleavage. |
| Results | Covalent metallodrug–DNA binding interactions/binding stoichiometry/non-covalent binding (gas phase). |
| Advantages | Very small quantities an advantage. |
| Limitation | Gas-phase results may not always translate to solution. |
| Complementary and advanced techniques | X-ray and NMR (direct) (Sections 2.1 and 2.2). |
| High resolution CZE-MS, LA-ICP MS, and MALDI. Conformational preferences from ion-mobility studies. |
2.4. Viscosity
Viscosity is a direct method where drug–DNA interactions are studied as a function of hydrodynamic changes induced by the binding agent. Experiments are carried out by introducing increasing ratios of drug to a solution containing a fixed concentration of DNA and observing how the velocity of DNA sedimentation influences a change in centipoise (cP). Relative viscosity data can be represented as (η/ηo) versus the [compound]/[DNA] ratio r, where η is the viscosity of complex treated DNA and ηo is the viscosity of untreated DNA. The technique is sensitive to changes in DNA chain length and individual modes of binding can be distinguished effectively since covalent and non-covalent binding modes display different hydrodynamic characteristics. Intercalators induce extension and unwinding of the DNA backbone due to separation of base pairs in order to accommodate the bound ligand. This results in the lengthening of the DNA molecule in proportion to the amount of drug bound (Fig. 7). The opposite effect can be noted for non-covalent major and minor groove binding agents since they cause little or no distortion to the phosphate backbone of DNA. In contrast, condensing agents such as the highly cationic TriplatinNC are capable of compacting and precipitating DNA that is indicated by a decreasing trend in viscosity relative to titrated complex (Fig. 7).35 Cisplatin is also known to reduce viscosity since upon covalent binding it kinks the backbone which shortens the axis length of the DNA helix, thereby decreasing the relative viscosity. Since long-range coiling can impact viscosity measurements, high molecular weight DNA fibers can be sheared and examined in parallel to native experiments. We recently applied this approach with salmon testes DNA fibres and the Triplatin series to show sheared DNA fragments aggregate more efficiently due to increasing intramolecular phosphate clamping interactions on smaller nucleotide fragments.| Method summary | Viscosity (direct) |
|---|---|
| Requirements | Digital viscometer and polymeric nucleic acid sample (typically salmon testes/calf thymus DNA). |
| Results | Hydrodynamic (solution) phase modification to nucleic acids. |
| Advantages | Easy of use. Low cost. Suitable for bulk analysis. |
| Limitations | Compounds must result in changes to viscosity of DNA solution; minor groove binding compounds such as netropsin and pentamidine result in no change. Metal ions themselves can compact DNA (charge neutralization). |
| Complementary and advanced techniques | Optical techniques including thermal melting and circular dichroism (Sections 3.1.1 and 3.1.2). Gel based techniques including superhelical DNA unwinding and topoisomerase I-mediated DNA relaxation (Section 4.2). |
| Advanced techniques include turbidity (UV), AFM, and DLS. |
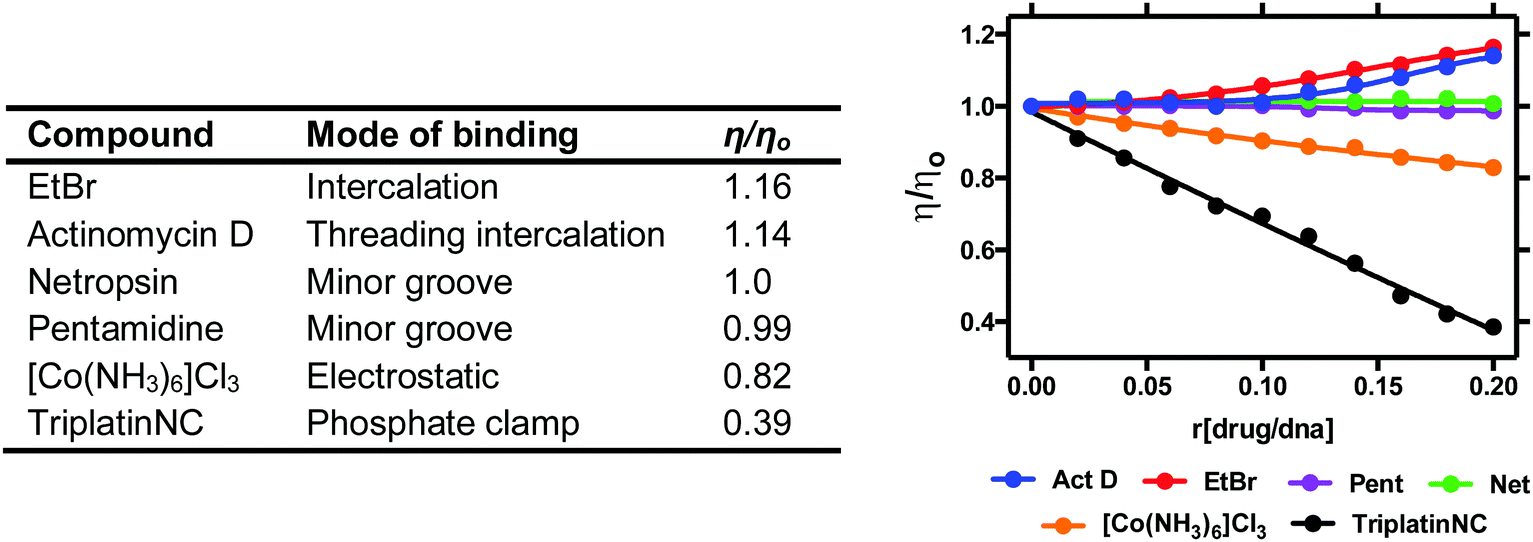 | ||
| Fig. 7 Relative viscosity values of organic and inorganic compounds bound to duplex stDNA. | ||
3. Spectroscopic techniques
3.1. Absorbance based techniques
Since nucleobases have low symmetry and several heteroatom lone pairs, UV-visible spectroscopy of nucleic acids is dominated by their base absorptions. Transitions for individual bases overlap and produce a single broad strong absorption band for the whole nucleic acid polymer, with an absorbance maximum (Amax) between 250–280 nm. The maximal absorbance of nucleic acids (λmax) is dependent on its AT and GC base content. Likewise, the molar extinction coefficient (εmax) of the nucleic acid is dependent on base composition and the preferred secondary structure adopted. A useful resource can be freely accessed on https://www.atdbio.com/tools/oligo-calculator, where the UV and thermodynamic properties of DNA sequences can be calculated.:50 equilibrium exists between helical and single stranded states. Since AT regions contain fewer non-covalent interactions, these hydrogen bonds melt first promoting the initial unwinding of the DNA helix followed by the melting of remaining GC rich regions. The thermal melting process is based on the loss of both hydrophobic interactions and π–π stacking interactions from nearest neighbor interactions due to the denaturation of the double helix and ultimate loss of secondary structure when bases become unstacked. This process is reversible and full renaturation of duplex DNA can occur approximately 25 °C below the denaturation temperature. TM is a powerful technique that relies on the intrinsic extinction coefficient of nucleic acids and is used to probe the thermodynamic parameters involved in metallodrug–DNA binding interactions.36
The differential stability of AT and GC rich regions and splitting of the duplex into single strands, is critical to many cellular processes such as transcription and recombination and is also central to the polymerase chain reaction (PCR). The thermal denaturation of DNA is also strongly influenced by duplex environment including salt and buffered solvent conditions. For example, higher salt concentrations generally result in higher TM values since the negative charge on the phosphate backbone is diffused due to electrostatic stabilisation. The relationship between TM and ionic strength can accordingly be exploited to change TM to a more convenient experimental temperature range.
Thermal denaturation can be easily monitored using a UV-vis spectrophotometer by observing the change in absorbance as a function of temperature. The method offers a useful insight into the strength of drug–DNA interactions as more energy is required to denature the stabilized secondary structure relative to the untreated polynucleotide, the stronger the drug interaction (ΔTM) and vice versa. Since structural factors and base composition impact the affinity of metallodrug–DNA binding this influence can be identified by measuring TM data. At a basic level, classical measurements involving actinomycin D (intercalator) and netropsin (minor groove binder) with co-polymers poly[d(G–C)2] and poly[d(A–T)2] reveal stabilization factors associated with G–C selective intercalation and A–T dependent minor groove binding (Table 1A). UV melting experiments with alternating copolymers have been applied to study Triplatin complexes and, separately, the influence of intercalating phenazine ligands within Cu-DPQ-Phen and Cu-DPPZ-Phen complexes (Table 1A).35,37 While alternating co-polymers, and to some extent calf thymus DNA, provide useful binding data, base-selective interactions are highly desirable to identify. Influenced by Cardin and co-workers structural characterisation of selective Λ-[Ru(phen)2(dppz)]2+ oligonucleotide (ON) binding (Section 2.1.1.), we recently probed the UV-melting of Cu-Oda (Fig. 2B) with palindromic dodecamers containing AT/AT and TA/TA central steps (Table 1B). Sequences with central AT/AT steps were only marginally stabilised whereas the TA/TA step showed significant stabilisation of +8.77 °C and enhanced duplex stability of 4.34 kJ mol−1 that we ascribed to selective intercalation.38
| Method summary | UV thermal melting (indirect) |
|---|---|
| Requirements | Spectrometer equipped with cell holder containing Peltier heat pumps connected to an external temperature controller. Quartz cuvettes with PTFE stoppers (commonly used). |
| Results | Thermal melting of phase transitions/molar enthalpy/association constants of metallodrug–nucleic acid interactions. |
| Advantages | Simple experimental setup. High sensitivity and reproducibility. Suitable for comparing/ranking binding affinity (SAR). Small sample size. |
| Limitation | Metallodrugs stability/optical transparency at elevated temperatures. Binding information generally provided at non-physiological temperatures (>37 °C). |
| Complementary and advanced techniques | NMR and viscosity (Sections 2.2 and 2.4) and optical techniques including circular dichroism spectroscopy, indirect fluorometric assays and topoisomerase I-mediated DNA relaxation (Sections 3.1.2, 3.2.1 and 4.2). Advanced complementary techniques include isothermal titration calorimetry (ITC) and qPCR. |
| (A) | ||
|---|---|---|
| Agent | ΔTM (°C) | |
| Poly[d(A–T)2] | Poly[d(G–C)2] | |
| Netropsin | 12.3 ± 0.8 | 02.8 ± 0.4 |
| Actinomycin D | −0.3 ± 0.3 | 12.1 ± 0.9 |
| Cu-DPQ-Phen | 0.6 ± 0.2 | 11.4 ± 1.1 |
| Cu-DPPZ-Phen | 0.5 ± 0.1 | 10.4 ± 1.1 |
| (B) | ||
|---|---|---|
| Agent | ΔTM (°C) | |
| d(GCCGGTACCGGC)2 | d(GCCGGATCCGGC)2 | |
| Cu-Oda | 8.7 ± 1.5 | 0.8 ± 0.6 |
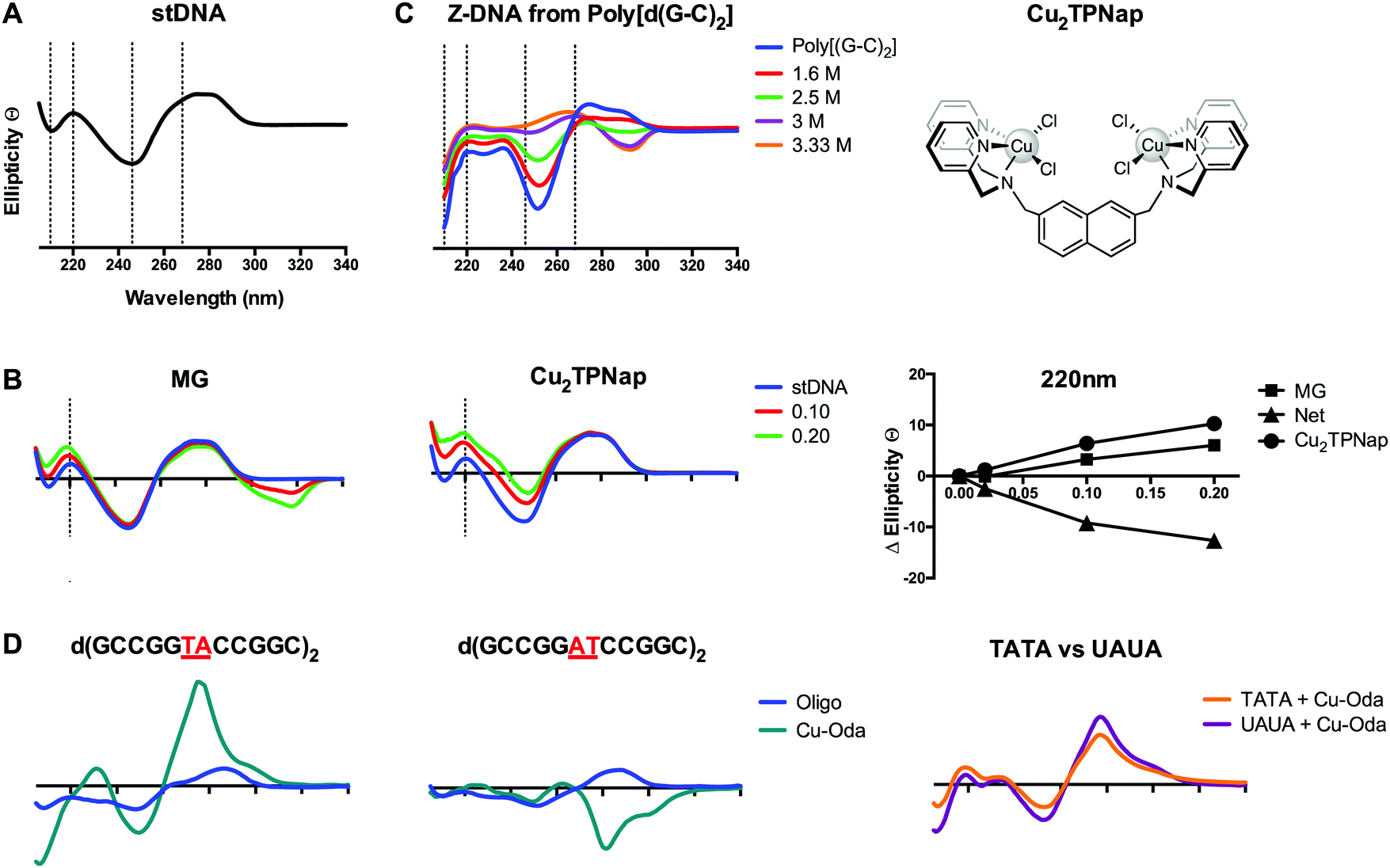 | ||
| Fig. 8 (A) CD profile of B-form salmon testes DNA (stDNA) highlighting wavelengths of interest including 210, 220, 246 and 268 nm; (B) change in ellipticity of stDNA at 220 nm in the presence increasing ratios of methyl green (MG) and di-nuclear Cu2TPNap; (C) B → Z NaCl titration of alternating co-polymer poly[d(G–C)2]; and (D) CD spectra of 12mer sequences d(GCCGGTACCGGC)2, d(GCCGGATCCGGC)2 and overlay of d(GCTTTATAAAGC)2 and d(GCUUUAUAAAGC)2 sequences in the presence of Cu-Oda. | ||
Intercalation of B-DNA typically leads to an increase in the elliptical signal associated with helicity (246 nm) and base pair stacking interactions (268 nm), while minor groove binding agents typically cause a decrease in the elliptical signal associated with hydrogen bonding interactions (220 nm) (Fig. 8B). These interactions can be understood from differences between the action of intercalators where perturbation to the helical backbone is required, and minor groove binders that contract the minor groove due to DNA-adduct H-bonding. The mapping of specific changes to ellipticity as a function of titrated metallo-drug is an important approach and was recently applied to identify major groove binding by a novel di-copper(II) complex (Cu2TPNap); results detailed in Fig. 8B indicate correlation between MG and Cu2TPNap with subsequent biophysical and computational experiments supporting the interaction.14 CD can also identify B → Z and B → A conformational changes and the Z transition in poly[d(G–C)2] is shown in Fig. 8C where NaCl was used to stimulate the inversion (unpublished). Poly[d(G–C)2] and poly[d(G)·d(C)] when treated with mono- and bi-functional platinum complexes (including BBR3464). Helical changes associated with condensation (ψ-DNA) may also be monitored and recent evidence of TriplatinNC-mediated DNA condensation using CD spectroscopy were subsequently corroborated by AFM.15
An elegant function of CD spectroscopy is its ability to probe sequence-specific metallodrug–ON (oligo) interactions. To gain an understanding of DNA sequence-context to the recognition process of Cu-Oda (Fig. 2B) this approach was applied to palindromic dodecamers with varying AT/AT and TA/TA steps, together with TATA repeats and substituted uracil (i.e. UAUA) congeners.38 Site-specific intercalation (ca. 280 nm) at the central TA/TA step in oligo d(GCCGGTACCGGC)2 was identified while a strong Cotton minimum in the same region indicated localized ‘Z-like’ DNA formation at the central AT/AT step in oligo d(GCCGGATCCGGC)2 (Fig. 8D). Intercalation was also observed in two sequences containing the TATA repeat but when thymine (T) was substituted with uracil (U), the elliptical signal at 288 nm increased suggesting a portion of intercalation occurring at the major groove site with the absence of bulky methyl groups in T rendering the major groove accessible to the di-nuclear complex (Fig. 8D).
CD offers many advantages over NMR and X-ray crystallography in the analysis of structural interactions in biological systems as it is inexpensive and requires small amounts of sample allowing for rapid and highly-sensitive analysis. Linear dichroism—another powerful form of polarized-light spectroscopy complementing CD—has been successfully employed to deduce drug–DNA binding geometries and has been extensively reviewed elsewhere.41 An additional application of CD spectroscopy, beyond the scope of this tutorial review, is induced circular dichroism (ICD) where wavelength-specific spectral changes are measured from coupling of electronic transition moments of the ligand (or complex) and DNA.40 Finally, care must be taken in the CD analysis of epigenetic bases (e.g. formyl-cytosine; fC) as oligomers containing these bases can display non-classical Cotton behavior in the B-form.42
| Method summary | Circular dichroism (indirect) |
|---|---|
| Requirements | Circular dichroism spectrometer with continuous nitrogen flow, quartz cuvettes required. |
| Results | Conformational changes to nucleic acid structure upon metallodrug binding. |
| Advantages | Excellent sensitivity and reproducibility. Provides structural information related to specific binding interactions. Small sample size. |
| Limitation | Caution must be taken with chiral metallodrugs. Experimental conditions must carefully maintain DNA structure. Limited solvent choice (UV activity). |
| Complementary and advanced techniques | Direct binding analysis by UV-vis and fluorescence spectroscopy, DNA unwinding experiments (4.2), and viscosity (2.4). |
| Advanced techniques include linear dichroism along with X-ray crystallography, NMR, UV thermal melting (Sections 2.1, 2.2 and 3.1.1) and molecular dynamics. |
3.2. Fluorescence based methods
 | ||
| Fig. 9 (A) Indirect fluorescence displacement assay using ethidium bromide as a reporter molecule to determine apparent binding constants (Kapp) of non-fluorescent DNA-binding agents; (B) FPX-TriPtNC binding assessment assay; and (C) fluorescence quenching of limited bound intercalator (EtBr) to poly[d(G–C)2] and poly[d(A–T)2] upon titration of netropsin and actinomycin D. Reproduced from ref. 37 with permission from the American Chemical Society, copyright 2014. | ||
| Method summary | Fluorescence reporting (indirect) |
|---|---|
| Requirements | Fluorescence spectrophotometer and quartz cuvette or fluorescence plate reader for high throughput analysis and suitable fluorescent reporter dyes. |
| Results | Indirect evidence of DNA binding when combined with specific reporter dyes. Binding constant can be obtained for compounds lacking a chromophore. |
| Advantages | High-throughput analysis possible when combined with 96 well plates. |
| Limitation | Indirect method with a reliance on signal from a fluorogenic reporter. Quenching of the reporter by metallodrug must be examined prior to analysis. |
| Complementary and advanced techniques | NMR, mass spectrometry and viscosity (Sections 2.2, 2.3, and 2.4) and optical techniques including thermal melting and circular dichroism (Sections 3.1.1 and 3.1.2). Advanced techniques include surface plasmon resonance (SPR) and molecular dynamics. |
4. Electrophoretic-based techniques
4.1. An introduction to gel mobility shift assays
Electrophoretic gel mobility shift assays are a well-established analytical technique used to analyze, separate and purify nucleic acid samples. Gel electrophoresis involves the movement of DNA through a solid-phase sieve-like medium such as agarose or polyacrylamide, under the influence of an electric potential difference. Due to its inherent negative charge, DNA moves in the electric field as an anion, from the cathode to the anode. The rate of DNA migration through the gel is dependent on several factors including the length of the DNA sequence and its conformation. Thus, long DNA fragments migrate slower than shorter fragments through an agarose medium as they experience greater resistance when travelling through a matrix of small pores within the gel. A number of external parameters can be controlled in order to maximize the resolution of a gel including the agarose content, its physical length, and the voltage applied allied with running time. Another important aspect of gel electrophoresis is the study of plasmid DNA vectors. These vectors are isolated from bacteria and are generally found in a supercoiled (SC) state. Manipulation of SC plasmid DNA by endonucleases or damaging agents gives rise to open-circular (OC) and linear (L) isoforms (Fig. 10A). Since supercoiled DNA is tightly compacted and has a small mass-to-charge ratio, it migrates with ease through an agarose medium. Once the plasmid becomes nicked on a single strand (i.e. single strand break (SSB) induction), supercoils become released resulting in the formation of open-circular DNA which experiences more resistance due to its larger size in the agarose matrix. Linear DNA results from double strand breaks (DSBs) and clustered DNA damage and this form experiences less resistance than open-circular DNA during its migration through an agarose medium (Fig. 10A). Metallodrug–DNA damage can therefore be identified using plasmid DNA and the rate of conversion between SSB and DSBs estimated by band densitometry.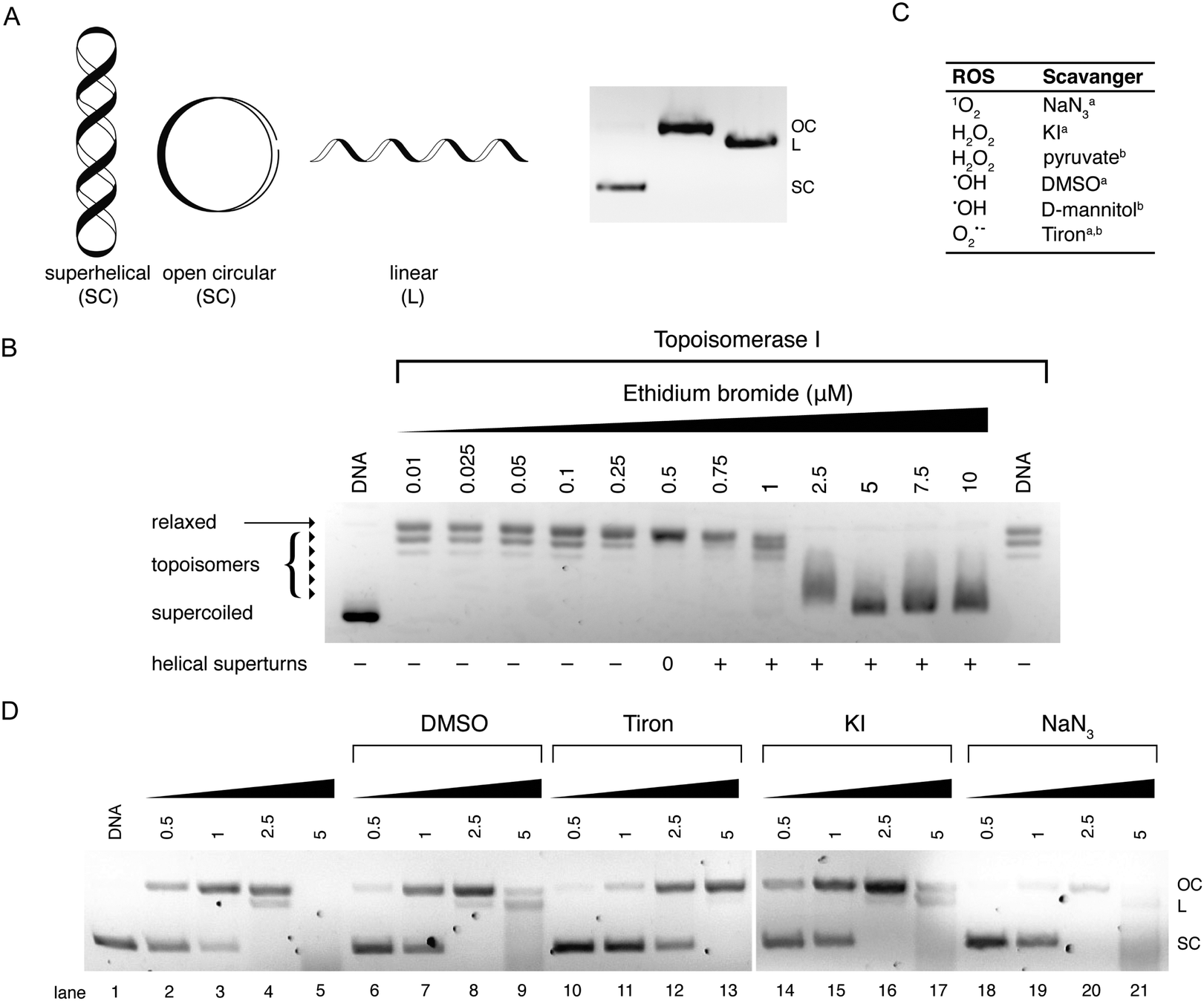 | ||
| Fig. 10 (A) Cartoon illustration of superhelical (SC), open circular (OC) and linear (L) DNA and agarose gel electrophoresis representation of respective DNA forms; (B) topoisomerase I mediated DNA relaxation assay in the presence of intercalating agent EtBr; (C) table of selected free radical scavengersa and intracellular antioxidantsb; and (D) gel profile of plasmid DNA treated with Cu-Ph-Phen in the absence (lanes 2–5) and presence of radical-specific antioxidants and trapping agents (lanes 6–21). Reproduced from ref. 50 with permission from the American Chemical Society, copyright 2016. | ||
4.2. Superhelical DNA unwinding and topoisomerase I-mediated relaxation
Electrophoretic mobility shift assays can be employed to determine the influence of covalent drug interactions on DNA supercoiling. The terms that need to be considered when discussing topological constraint stem from the formula Lk = T + W, where Lk = linking number, Tw = twist (number of helical turns of the DNA duplex) and Wr = writhe (number of supercoils). This technique has previously been used to investigate a series of structurally related Pt(II) complexes that differed in their covalent co-ordination mode (mono-functional/bi-functional).44 In the presence of increasing concentrations of a covalent binding small molecule, the migration rate of SC–DNA is monitored. When all supercoils are removed from the plasmid—due to crosslinking or intercalation—the migration pattern of DNA is identical to that of OC–DNA. The bound drug-to-nucleotide ratio at this point can be visualised in the agarose gel and referred to as the coalescence point. When the drug loading ratio is further increased beyond this point, positive supercoiling can be detected as the plasmid becomes wound in the opposite direction under the influence of excess metallodrug/intercalator.Another method to reliably identify DNA intercalation is via topoisomerase I-mediated DNA relaxation. Topoisomerases (Topo) are a specialised class of nuclear enzymes that catalyse the transient cleavage, manipulation and resealing of either a single strand (Topo I) or double strand (Topo II) of DNA. This activity allows the relaxation of chain intertwinement and the release of superhelical tension to permit topological changes required during replication, transcription, and DNA repair.45 Topo I-mediated relaxation is a robust assay to identify intercalative properties of suspect DNA binding metallodrugs. Complexes with intercalating moieties, typically constructed from planar aromatic ligands, unwind and elongate the helical structure inhibiting topoisomerase enzymes from binding to DNA, or alternatively stabilising the enzyme–DNA complex.
Topo I isolated from E. coli relaxes negatively coiled superhelical plasmid DNA giving a distinct topological pattern of negative, or right handed, topoisomers (Fig. 10B). In the presence of intercalating agent EtBr, plasmid DNA becomes unwound and transitions from its negatively wound topological pattern to fully relaxed OC DNA, and then to positively (left-handed) SC plasmid as the plasmid becomes wound in the opposite direction.46 This unwinding effect is evident in lanes 2–14 of Fig. 10B where EtBr induces helical unwinding by 26°, with positively wound topology of scDNA becoming visible after 0.5 μM exposure. The distinct “up and down” migration pattern evident in Fig. 10B is a typical trend observed when SC plasmid DNA is treated with an intercalating agent. As the concentration of the bound intercalant increases, negatively charged SC DNA becomes unwound forming relaxed OC DNA which migrates slower through the agarose medium than tightly packed SC DNA. When the concentration of the intercalant is further increased, OC DNA is wound tightly in the opposite direction forming positively charged SC DNA that migrates at a similar rate to negatively charged SC DNA.
4.3. Oxidative DNA damage detection DNA damage
Our efforts to develop this metallodrug class have focused on the incorporation of designer DNA intercalating ligands such as DPPZ and also on the role of nuclearity. One recent study involved the isolation of ternary Cu(II) complexes incorporating both 1,10-phenathroline and phenazine ligands; through the systematic extension of the ligated phenazine ligand in the Cu2+ bis-phenanthroline model, it was possible to enhance the chemical nuclease profile of the series relative to Cu-Phen where the activity trend Cu-DPQ-Phen ≈ Cu-DPPZ-Phen > CuPhen (Fig. 2) was observed.37 Reactive oxygen species (ROS) responsible for oxidative DNA damage by this series were later investigated using superhelical plasmid DNA with a selection of ROS-specific antioxidants and stabilisers as follows: NaN3 for singlet oxygen (1O2); KI for hydrogen peroxide (H2O2); DMSO for the hydroxyl radical (•OH); and D2O for stabilising singlet oxygen (1O2) (Fig. 10C). Experiments revealed the hydroxyl radical as chiefly responsible for degradation.49 Recently, this method was extended to examine the in vitro and intracellular cleavage efficacy of the developmental therapeutic [Cu(o-phthalate)(1,10-phenanthroline)] (Cu-Ph-Phen, Fig. 2C) and results are shown in Fig. 10D.50 Experiments here revealed superoxide (O2˙−) as a prominent species responsible for DNA oxidation as co-incubation with 4,5-dihydroxy-1,3-benzenedisulfonic acid (tiron) significantly impeded DNA damage and prevented double strand damage (lane 10–13). The presence of DMSO diminished activity to a lesser extent while DNA damage was only marginally altered by KI (lane 14–17) and NaN3 (lane 18–21). Significantly, in the same study ovarian adenocarcinoma cancer cells (SKOV3) pretreated with tiron and subsequently exposed to an LD50 concentration of Cu-Ph-Phen displayed enhanced survival by ∼26%.50 A further example showing correlation between ROS mediators of in vitro DNA damage and cell culture cytotoxicity can be found in ref. 38.
Oxidative DNA damage is known to be dependent on a number of factors including and not limited to: DNA plasmid type and conformation, presence/absence of exogenous oxidant and reductant, presence of chelating agents and dependence on hydrogen peroxide. The presence of non-covalent recognition elements such as minor groove (netropsin), major groove (methyl green) and electrostatic agents ([Co(NH3)6]Cl3) can influence accessibility for DNA damaging drugs. Analysis of plasmid DNA cleavage involving the copper(II) phenazine series (Cu-Phen-DPQ; Cu-Phen-DPPZ; and Cu-Phen-DPPN) with pre-incubation of the major groove binding agent methyl green (Fig. 2F) resulted in enhanced chemical nuclease activity, indicative of drug–DNA interactions being directed towards the minor groove, while the presence of the minor groove binding agent netropsin was found to reduce the oxidative DNA damage profile of the same complex series.49
4.4. Melphalan protection assay
To establish the minor groove as a preferred drug–DNA binding site the aromatic nitrogen mustard melphalan can be employed. When in solution, the bis(2-chloroethyl)amino side chain of the molecule can cyclize to form the aziridinium ion which can add to a nucleophilic site on DNA forming a mono-alkylated covalent DNA adduct. This process can be repeated to form a di-alkylated adduct resulting in intra- or inter-strand crosslinking between two complementary strands of DNA. Subsequent heat-treatment with piperidine induces thermolabile DNA cleavage at guanine N7 (major groove) and adenine N3 (minor groove) sites (Fig. 11B). When DNA is pretreated with cationic minor groove binding agents such as netropsin or distamycin, these molecules afford protection to DNA from the alkylation process by occupying the narrow AT-rich minor groove and prevent thermolabile scission from occurring. Interestingly, the same protective effects can be seen in the case of PPCs and classical minor groove binders, where melphalan is prevented from accessing A-rich regions in the minor groove and this protective effect is outlined in Fig. 11C.14,29| Method summary | Agarose gel electrophoresis (indirect) |
|---|---|
| Requirements | Gel electrophoresis tank, power pack, transilluminator, agarose, plasmid DNA and DNA specific stain. DNA/RNA ladders necessary for accurate sizing. |
| Results | DNA damage and stepwise conversion of SC → OC → LC. Also possible to determine conformational changes to nucleic acid structure upon metallodrug binding. |
| Advantages | Simple experimental setup, cost effective, small sample size. Fast analysis time. |
| Limitation | Quantitation of DNA damage/modification (with densitometry, for example) relies on reporters (indirect) that bind conformers at different ratios. |
| Complementary and advanced techniques | Polyacrylamide gel electrophoresis (PAGE), Mass spectrometry and on-chip microfluidics (Sections 2.3 and 4.5). Advanced techniques include qPCR and LC-MS/MS stable isotope dilution detection for DNA damage, and 8-oxo-dG ELISA detection. |
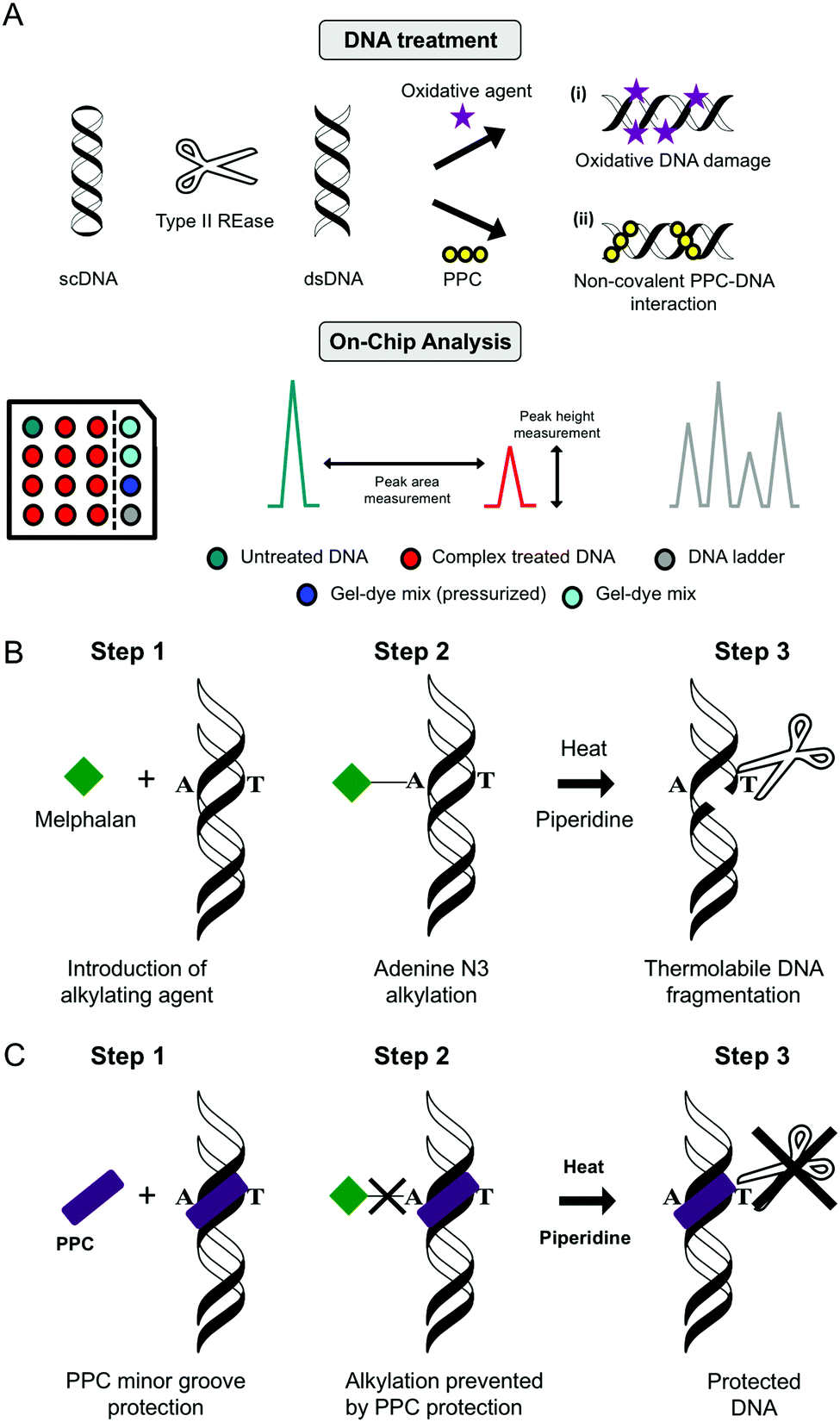 | ||
| Fig. 11 (A) Double stranded DNA “on-chip” protocol developed within our research laboratories used to assess (i) oxidative DNA damage and (ii) PPC non-covalent binding interactions using the Agilent BioAnalyser 2100 platform; (B) melphalan alkylation assay using piperidine and heat to induce thermolabbile scission and (C) minor groove protection assay polynuclear platinum complexes (PPC). | ||
4.5. On-chip microfluidics
Lab on chip technology (e.g. the Agilent Bioanalyser 2100) has provided a platform to conduct high-throughput gel electrophoretic experiments on a microfluidic chip with up to 12 DNA, RNA or protein samples being analyzed and processed sequentially. There are many advantages to the method including broad sample size range (25–12000 bp), minimal sample consumption and high sizing resolution with accuracy normalised to internal reference markers. Data can be processed and generated with output including electrograms, peak height, peak area and digital electropherograms. Many applications for nucleic acids have been found for this technique including determination of sample size, quality and concentration (Fig. 11A).
Recently, we employed this technique to determine drug–DNA damage and selective binding interactions with double stranded DNA sequences. In the first case, an assay was devised to compare the oxidative degradation profiles of a family of structurally related bis-chelate Cu(II) complexes of Cu-DPQ-Phen, Cu-DPPZ-Phen, and Cu-DPPN-Phen on restricted pUC19. By employing both peak height and area it was possible to rank the fragmentation activity of the complex series with Cu-DPQ-Phen identified as the most active agent.37 In another application of this technique, cationic Triplatin complexes of varying linker length [{trans-Pt(NH3)2(NH2(CH2)nNH3)}2-μ-({trans-Pt(NH3)2(NH2(CH2)nNH2)2})](NO3)8, where n = 5 (AH78P), 6 (AH78 TriplatinNC) and 7 (AH78H) were determined to inhibit site-selective excision by type II restriction endonucleases—a consequence of which might indicate their ability to negate native DNA excision repair processes. The experimental setup involved pre-incubating DNA with PPC complexes followed by endonuclease exposure to BamHI, EcoRI, and SalI. Concentration dependent inhibition was identified in many cases, with high intensities of the native pUC19 band becoming visible.35 Additionally, the technique identified how PPC linker chain length modulates endonuclease access to the restriction sites. In this work, the pentanediamine complex (n = 5) protected or inhibited digestion in the nanomolar range, while a 10-fold increase was required to block restriction activity in the hexane- (n = 6) and heptane-bridged (n = 7) congeners.
| Method summary | On-chip microfluidics (indirect) |
|---|---|
| Requirements | Commercially available microfluidic systems such as the Agilent Bio-Analyser and preferred chip types (e.g. DNA 1000, DNA 7500). |
| Results | Output from this technique includes electropherograms, electrograms, and advanced analysis including %DNA degradation. |
| Advantages | Quantitative method with high sizing resolution. Accuracy of individual chips is normalised to internal reference markers. Experiments can be optimised using inexpensive agarose methods. |
| Limitation | Expense, scaling between DNA sizes requires change of microfluidic chip, can be analytically challenging. |
| Complementary techniques | Mass spectrometry analysis (Section 2.3). |
5. Conclusion
DNA is a well-established pharmacological target for metallodrugs and developmental anticancer complexes continue to be rationally designed for potential clinical use against this target. The elucidation of non-covalent binding modes, along with helical and groove residency is a crucial area of study in this field. This tutorial review has focused on selected molecular methods for investigating metallodrug–DNA interactions using gel electrophoretic, electronic and fluorescent spectroscopic, NMR spectroscopic, X-ray crystallographic and mass spectrometric techniques. Significantly, when combinations of complementary molecular methods are employed, the full picture of solution binding properties can be defined, which ultimately broaden our understanding of complex-DNA binding. Recent work on probing the nucleic acid binding mode by ‘phosphate clamping’ Triplatin complexes elegantly reveals how these molecular techniques function synergistically.35 Using a family of cationic tri-platinum(II) complexes of varying aliphatic linker length [{trans-Pt(NH3)2(NH2(CH2)nNH3)}2-μ-({trans-Pt(NH3)2(NH2(CH2)nNH2)2})](NO3)8, where n = 5 (AH78P), 6 (AH78; TriplatinNC) and 7 (AH78H), high-affinity PPC–DNA interactions were uncovered using ethidium bromide fluorescence quenching, while cooperative fluorescence binding of Hoechst 33258 was observed at the minor groove. Conformational changes on long DNA were then identified using viscosity, electrophoretic, and CD spectroscopic methods where aggregation/condensation of nucleic acids was evidenced in tandem with conversion from B → Z-DNA. 2D-1H NMR experiments, in conjunction with several other molecular methods, then indicated two limiting modes of phosphate clamping—backbone tracking (GC dependent) and groove spanning (AT dependent)—could be distinguished and implied DNA condensation was driven, primarily, by minor-groove spanning. Further application of electrophoresis (including ‘on-chip’ microfluidics) showed Triplatin–DNA binding prevented endonuclease activity by type II restriction enzymes. Subsequent work by Farrell and co-workers identified nucleolar condensation in colorectal cancer cells using confocal microscopy with atomic force microscopy (AFM) studies showing this class to condense both tRNA and duplex DNA structures.15 Other examples on the application of molecular methods have been described in this review and, with this in mind, the overlap and versatility of complementary techniques can augment our understanding of non-covalent metallodrug–DNA interactions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Science Foundation Ireland (15/CDA/3648). AK acknowledges support from the Marie Skłodowska-Curie Innovative Training Network (ITN) ClickGene (H2020-MSCA-ITN-2014-642023). NF acknowledges support by NIH RO1CA78754.References
- N. P. E. Barry and P. J. Sadler, Chem. Commun., 2013, 49, 5106–5131 RSC.
- E. Chargaff, Science, 1971, 172, 637–642 CrossRef CAS.
- R. Wing, H. Drew, T. Takano, C. Broka, S. Tanaka, K. Itakura and R. E. Dickerson, Nature, 1980, 287, 755–758 CrossRef CAS.
- A. Rich and S. Zhang, Nat. Rev. Genet., 2003, 4, 566–572 CrossRef CAS PubMed.
- H. R. Drew and R. E. Dickerson, J. Mol. Biol., 1981, 152, 723–736 CrossRef CAS.
- R. E. Dickerson, H. R. Drew, B. N. Conner, R. M. Wing, A. V. Fratini and M. L. Kopka, Science, New Series, 1982, 216, 475–485 CAS.
- M. Zeraati, D. B. Langley, P. Schofield, A. L. Moye, R. Rouet, W. E. Hughes, T. M. Bryan, M. E. Dinger and D. Christ, Nat. Chem., 2018, 10, 631–637 CrossRef CAS PubMed.
- G. Biffi, D. Tannahill, J. McCafferty and S. Balasubramanian, Nat. Chem., 2013, 5, 182–186 CrossRef CAS PubMed.
- C. Hiort, P. Lincoln and B. Nordén, J. Am. Chem. Soc., 1993, 115, 3448–3454 CrossRef CAS.
- C. J. Cardin and J. P. Hall, in DNA-targeting Molecules as Therapeutic Agents, ed. M. J. Waring, Royal Society of Chemistry, Cambridge, 2018, pp. 198–227 Search PubMed.
- H. Song, J. T. Kaiser and J. K. Barton, Nat. Chem., 2012, 4, 615–620 CrossRef CAS PubMed.
- J. K. Barton, A. N. Boynton and K. M. Boyle, in DNA-targeting Molecules as Therapeutic Agents, ed. M. J. Waring, Royal Society of Chemistry, Cambridge, 2018, pp. 367–390 Search PubMed.
- A. Oleksi, A. G. Blanco, R. Boer, I. Usón, J. Aymamí, A. Rodger, M. J. Hannon and M. Coll, Angew. Chem., Int. Ed., 2006, 118, 1249–1253 CrossRef.
- Z. Molphy, D. Montagner, S. S. Bhat, C. Slator, C. Long, A. Erxleben and A. Kellett, Nucleic Acids Res., 2018, 46, 9918–9931 CrossRef PubMed.
- N. P. Farrell, Chem. Soc. Rev., 2015, 44, 8773–8785 RSC.
- T. C. Jenkins, in Drug–DNA Interaction Protocols, ed. K. R. Fox, Totowa, NJ, 1st edn, 1997, vol. 90, pp. 195–218 Search PubMed.
- S. K. Kim and B. Nordén, FEBS Lett., 1993, 315, 61–64 CrossRef CAS PubMed.
- P. Lincoln, L. M. Wilhelmsson and B. Nordén, in DNA-targeting Molecules as Therapeutic Agents, ed. M. J. Waring, Royal Society of Chemistry, Cambridge, 2018, pp. 45–73 Search PubMed.
- R. E. Franklin and R. G. Gosling, Nature, 1953, 171, 740–741 CrossRef CAS PubMed.
- P. M. Takahara, A. C. Rosenzweig, C. A. Frederick and S. J. Lippard, Nature, 1995, 377, 649–652 CrossRef CAS PubMed.
- E. F. Garman, Science, 2014, 343, 1102–1108 CrossRef CAS PubMed.
- J. R. Helliwell and E. P. Mitchell, IUCrJ, 2015, 2, 283–291 CrossRef CAS PubMed.
- L. S. Lerman, J. Mol. Biol., 1961, 3, 18–30 CrossRef CAS PubMed.
- R. Galindo-Murillo, J. C. García-Ramos, L. Ruiz-Azuara, T. E. Cheatham and F. Cortés-Guzmán, Nucleic Acids Res., 2015, 43, 5364–5376 CrossRef CAS PubMed.
- P. J. Bond, R. Langridge, K. W. Jennette and S. J. Lippard, PNAS, 1975, 72, 4825–4829 CrossRef CAS.
- M. Howe-Grant and S. J. Lippard, Biochemistry, 1979, 18, 5762–5769 CrossRef CAS PubMed.
- S. Komeda, T. Moulaei, K. K. Woods, M. Chikuma, N. P. Farrell and L. D. Williams, J. Am. Chem. Soc., 2006, 128, 16092–16103 CrossRef CAS PubMed.
- M. J. Hannon, Chem. Soc. Rev., 2007, 36, 280–295 RSC.
- S. Komeda, Y. Qu, J. B. Mangrum, A. Hegmans, L. D. Williams and N. P. Farrell, Inorg. Chim. Acta, 2016, 452, 25–33 CrossRef CAS.
- S. J. Berners-Price, L. Ronconi and P. J. Sadler, Prog. Nucl. Magn. Reson. Spectrosc., 2006, 49, 65–98 CrossRef CAS.
- J. J. Moniodis, D. S. Thomas, M. S. Davies, S. J. Berners-Price and N. P. Farrell, Dalton Trans., 2015, 44, 3583–3593 RSC.
- Y. Qu, R. G. Kipping and N. P. Farrell, Dalton Trans., 2015, 44, 3563–3572 RSC.
- T. Urathamakul, D. J. Waller, J. L. Beck, J. R. Aldrich-Wright and S. F. Ralph, Inorg. Chem., 2008, 47, 6621–6632 CrossRef CAS PubMed.
- J. B. Mangrum and N. P. Farrell, Chem. Commun., 2010, 46, 6640–6650 RSC.
- A. Prisecaru, Z. Molphy, R. G. Kipping, E. J. Peterson, Y. Qu, A. Kellett and N. P. Farrell, Nucleic Acids Res., 2014, 42, 13474–13487 CrossRef CAS PubMed.
- J. B. Chaires, in DNA-targeting Molecules as Therapeutic Agents, ed. M. J. Waring, Royal Society of Chemistry, Cambridge, 2018, pp. 74–95 Search PubMed.
- Z. Molphy, A. Prisecaru, C. Slator, N. Barron, M. McCann, J. Colleran, D. Chandran, N. Gathergood and A. Kellett, Inorg. Chem., 2014, 53, 5392–5404 CrossRef CAS.
- C. Slator, Z. Molphy, V. McKee, C. Long, T. Brown and A. Kellett, Nucleic Acids Res., 2018, 46, 2733–2750 CrossRef PubMed.
- G. R. Bishop and J. B. Chaires, Curr. Protoc. Nucleic Acid Chem., 2002, 11, 7.11.1–7.11.8 Search PubMed.
- N. C. Garbett, P. A. Ragazzon and J. B. Chaires, Nat. Protoc., 2007, 2, 3166–3172 CrossRef CAS.
- B. Nordén, F. Tjerneld and E. Palm, Biophys. Chem., 1978, 8, 1–15 CrossRef.
- J. S. Hardwick, D. Ptchelkine, A. H. El-Sagheer, I. Tear, D. Singleton, S. E. V. Phillips, A. N. Lane and T. Brown, Nat. Struct. Mol. Biol., 2017, 24, 544–552 CrossRef CAS PubMed.
- A. R. Morgan, J. S. Lee, D. E. Pulleyblank, N. L. Murray and D. H. Evans, Nucleic Acids Res., 1979, 7, 547–569 CrossRef CAS PubMed.
- M. V. Keck and S. J. Lippard, J. Am. Chem. Soc., 1992, 114, 3386–3390 CrossRef CAS.
- J. C. Wang, Nat. Rev. Mol. Cell Biol., 2002, 3, 430–440 CrossRef CAS.
- P. Peixoto, C. Bailly and M.-H. David-Cordonnier, in Drug–DNA Interaction Protocols, ed. K. R. Fox, Humana Press, 2nd edn, 2010, vol. 613, pp. 235–256 Search PubMed.
- M. Pitié and G. Pratviel, Chem. Rev., 2010, 110, 1018–1059 CrossRef.
- D. S. Sigman, A. Mazumder and D. M. Perrin, Chem. Rev., 1993, 93, 2295–2316 CrossRef CAS.
- Z. Molphy, C. Slator, C. Chatgilialoglu and A. Kellett, Front. Chem., 2015, 3, 1–9 CAS.
- C. Slator, N. Barron, O. Howe and A. Kellett, ACS Chem. Biol., 2016, 11, 159–171 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2019 |