
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Production of highly concentrated and hyperpolarized metabolites within seconds in high and low magnetic fields†
Sergey
Korchak
ab,
Meike
Emondts
cd,
Salvatore
Mamone
ab,
Bernhard
Blümich
 d and
Stefan
Glöggler
*ab
d and
Stefan
Glöggler
*ab
aNMR Signal Enhancement Group Max-Planck-Institute for Biophysical Chemistry, Am Faßberg 11, 37077 Göttingen, Germany. E-mail: stefan.gloeggler@mpibpc.mpg.de
bCenter for Biostructural Imaging of Neurodegeneration, Von-Siebold-Straße 3A, 37075 Göttingen, Germany
cDWI-Leibniz Institute for Interactive Materials, Forckenbeckstr. 50, D-52056 Aachen, Germany
dInstitut für Technische Chemie und Makromolekulare Chemie, RWTH-Aachen University, Worringerweg 2, 52074 Aachen, Germany
First published on 8th October 2019
Abstract
Hyperpolarized metabolites are very attractive contrast agents for in vivo magnetic resonance imaging studies enabling early diagnosis of cancer, for example. Real-time production of concentrated solutions of metabolites is a desired goal that will enable new applications such as the continuous investigation of metabolic changes. To this end, we are introducing two NMR experiments that allow us to deliver high levels of polarization at high concentrations (50 mM) of an acetate precursor (55% 13C polarization) and acetate (17% 13C polarization) utilizing 83% para-state enriched hydrogen within seconds at high magnetic field (7 T). Furthermore, we have translated these experiments to a portable low-field spectrometer with a permanent magnet operating at 1 T. The presented developments pave the way for a rapid and affordable production of hyperpolarized metabolites that can be implemented in e.g. metabolomics labs and for medical diagnosis.
Introduction
Low-field nuclear magnetic resonance (NMR) devices have become versatile tools for numerous applications including the investigations of materials, chemicals and for the in situ monitoring of chemical reactions.1–8 The main advantage of low-field NMR is that the instruments can be made small for transportation to a location of interest. This has enabled NMR studies of objects that cannot necessarily be moved such as concrete constructions or coatings. With respect to medical applications, low-field devices can be brought to the point of care.Although NMR is excellent for investigating all of the above, its inherent drawback is the low sensitivity. In order to detect analytes in low concentration and to enhance NMR signals, hyperpolarization (HP) techniques have been introduced. Hyperpolarization is a method to increase NMR signals by up to four orders of magnitude.9–45 Among the various methods spin-exchange optical pumping (SEOP),9–12 dynamic nuclear polarization (DNP)13–18 and para-hydrogen-based techniques such as hydrogenative para-hydrogen induced polarization (PHIP) and signal amplification by reversible exchange (SABRE) have drawn the most attention in biochemical, biological and biomedical research.19–45 One particular application concerns the hyperpolarization of metabolites so that metabolomics experiments can be performed with high sensitivity. The most widely pursued application is to utilize hyperpolarized metabolites directly as contrast agents in medical MRI.46 To that end, for example, 13C-enriched pyruvate is signal enhanced and injected in vivo.13–16 In a tumor, pyruvate is converted into lactate which can be observed in a chemical shift imaging experiment by a change in resonance frequency.13
The most prominent technique to achieve this is dissolution DNP.13–18 Although dissolution DNP has been the main driving force in the field of metabolite imaging, it is a rather slow and costly technique that hyperpolarizes substances in tens of minutes to hours and requires a dedicated magnet. para-Hydrogen based techniques promise to generate contrast agents much faster (in seconds) and in a more cost-efficient way.19–41 In the classical PHIP approach, an unsaturated bond is hydrogenated with para-hydrogen.28 The spin order of the para-hydrogen's nuclear singlet state is thereby converted into observable magnetization and can subsequently be transferred to a 13C nucleus in a metabolite.22,23,43,44,47,48 Detecting the polarization in a hetero-nucleus is beneficial because hydrogen background signals are absent. Another aspect is that changes in molecular structure can be observed better due to larger changes in chemical shift. Moreover, hetero-nuclei typically possess longer longitudinal relaxation times T1 than hydrogen. T1 is affected by the molecular dynamics and is a measure of how long the hyperpolarization can be stored. Thus, hyperpolarized hetero-nuclei prolong the time for in vivo tracing. Historically, the preparation of in vivo contrast agents via PHIP has been achieved utilizing field cycling approaches or radio frequency transfer experiments in low field polarizers operating in the millitesla range.20,22,47–50 Polarizing metabolites for PHIP appeared to be challenging for a long time due to limited number of unsaturated metabolite precursors, which targeted succinate and phospholactate.22,49,51,52 In particular succinate was hyperpolarized up to 28% in concentrations of 30 mM.49,53,54 A detailed table on hyperpolarizable molecules via PHIP can be found in ref. 42. The availability of hyperpolarized metabolites has changed with the introduction of PHIP-SAH (PHIP by means of sidearm hydrogenation) for which an unsaturated ester of a metabolite of interest is generated as starting compound.23para-hydrogen is subsequently reacted with the precursor, the polarization transferred to 13C nuclear spins of the metabolite, and the ester cleaved to yield the desired contrast agent. We would like to point out that the hyperpolarization and the polarization transfer are conducted in chloroform in the presence of a homogeneous catalyst.25,55 Cleavage is accomplished by addition of an aqueous base solution that will extract the desired metabolite in the aqueous phase which upon adjusting the pH can be used for preclinical in vivo experiments.25,55 Thereby, most of the metal catalyst is retained in the organic phase. Most recently, cardiac images of pyruvate and its metabolism into lactate where obtained in this way.55 To achieve the polarization transfer, experiments were performed via field cycling limiting the maximal achievable polarization to about 40%.24,56 Pulsed NMR techniques have afterwards been developed that can theoretically yield close to unity polarization.43,44 So far, 60% 13C polarization was achieved experimentally in low concentrations (1 mM) in a super-conducting high field system for the PHIP-SAH approach.44 For metabolic imaging experiments it is desirable that contrast agent concentrations one order of magnitude higher can be generated with high polarization levels (10% polarization is typically perceived as the threshold for in vivo experiments).42
Once the concentrations becomes larger than 1 mM, we have observed nonlinear effects such as radiation damping (RD) caused by the high levels of polarization, that severely reduce the efficiency of transferring para-hydrogen spin order to the 13C-nuclei of interest in a metabolite.57–60 Here, we introduce pulsed NMR experiments counteracting these effects and thereby yielding an average 13C polarization of 55% in an acetate precursor at concentrations of 50 mM at 7 T magnetic field. We have translated our approach into a compact permanent magnet spectrometer at 1 T (in which RD was observed as well) and show that our approach delivers polarization levels at high concentrations relevant to in vivo applications. We believe that the results presented in this article will therefore speed up metabolomics experiments as well as make hyperpolarized contrast agents more accessible to a wider community via mobile benchtop polarizers.
In this study we have investigated at first the production of the hyperpolarized acetate precursor (ethyl acetate-d6 (EA)) as depicted in Scheme 1, and its subsequent cleavage into acetate. Hyperpolarization was transferred from protons to the carbonyl carbon of the acetate moiety employing a recently introduced pulsed transfer method (ESOTHERIC).43,44 EA currently yields the highest 13C polarization and has a desirable spin-coupling network to design precursors for other metabolites such as lactate or pyruvate that will yield most likely similar polarization values in the future.61,62 Acetate itself is an important metabolite and plays a role in the energy metabolism. It has previously been injected into humans in large quantities to observe glial metabolism.63 It has more recently been utilized in hyperpolarization studies on in vivo animal models to e.g. monitor cardiac metabolism and renal clearance.64,65
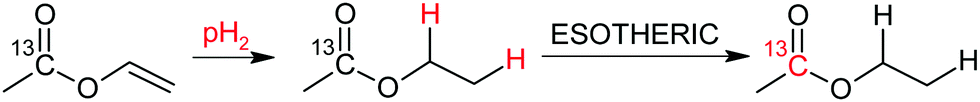 | ||
| Scheme 1 Production of the 13C carbonyl hyperpolarized acetate precursor ethyl acetate-d6 (EA) from perdeuterated vinyl acetate. | ||
Results and discussion
In accordance with the preparation protocol of PHIP-SAH contrast agents for preclinical imaging as described above, we have conducted all of our precursor hyperpolarization studies in chloroform. The main obstacle to achieve high concentrations of HP metabolite is the effect of the huge sample magnetization: 60% 1H-polarization of 4 mM EA is equivalent to a sample of protonated water (55 M) measured in a 300 MHz spectrometer, which is known to induce radiation damping (RD).57–60 Any attempt to bring magnetization into the transverse plane results in generation of a current in the excitation/detection coil that is large enough not only to be detected but in turn to generate a B1 magnetic field which couples to the nuclear magnetization. RD returns transverse magnetization during spin evolution back to the longitudinal direction. The RD effect on the line shape can be seen with the para-hydrogenation of vinyl acetate-d6 at a concentration of as low as 2.5 mM (Fig. 1). The absorption lines become broader from the sampling of the free induction decay (FID) while the emission lines narrow.59,60 At higher concentrations and thus higher resulting magnetization RD is more pronounced. RD is typical for HP systems, where this effect was first detected on helium and xenon.66 As dissolution DNP and SABRE techniques improved, this effect became more severe for protons even leading to spontaneous emission.32,67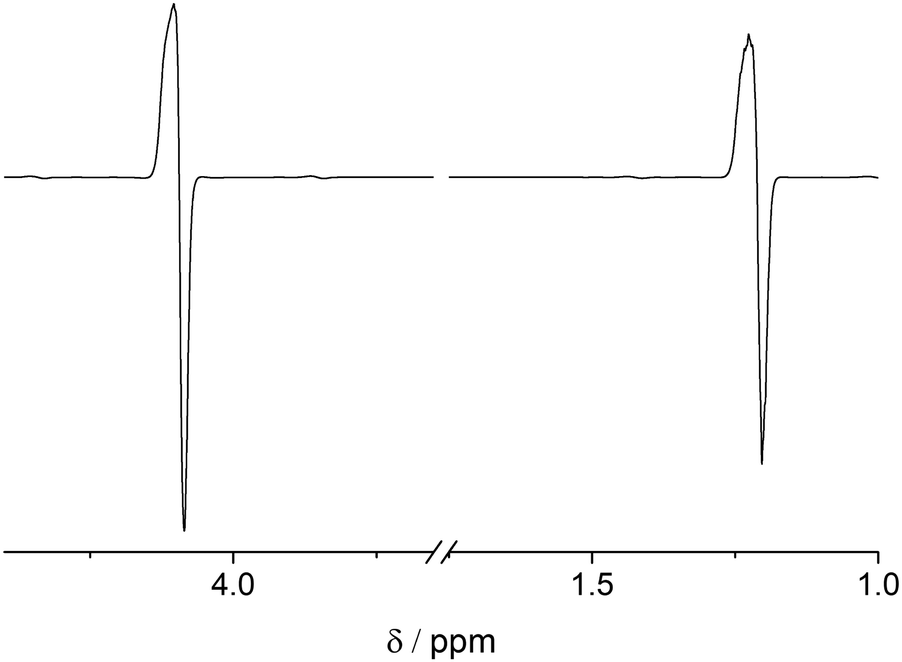 | ||
| Fig. 1 Hyperpolarized 1H spectrum of 2.5 mM EA in chloroform-d (B0 = 7 T) detected following a 45° pulse. One can see effects of radiation damping (RD): the absorption lines are broadened and the emission lines are narrowed. | ||
Despite the fact that the damping field is tiny and thus leads to narrow frequency selectivity exactly on the polarized signals, it appears to be sufficiently effective, as it lasts long enough and is amplified in the resonance circuit of the probe head due to the high quality factor Q of the detection circuit. Considering that the investigated molecule contains 13C in natural abundance, RD is mostly induced by HP protons. However, the 1.1% of 1-13C enriched EA display the same 1H chemical shift thus being subject to uncontrolled B1 fields on protons. As a result, RD spoils the spin dynamics during the proton spin-order transfer to carbon making it inefficient. Moreover, we observed that spoiling processes already start during the para-hydrogenation reaction, transforming the original spin I1zI2z state to some other state, which is inferior to utilizing polarization transfer sequences. RD is characterized by the radiation damping rate 1/τrd, and the most detrimental effects start when τrd is larger than the inverse relaxation time:68

with μ0, η, Q, γ, M0 being the vacuum permeability, the filling factor of the coil, the quality factor of the detection circuit, the nuclear gyromagnetic ratio and the initial magnetization, respectively.
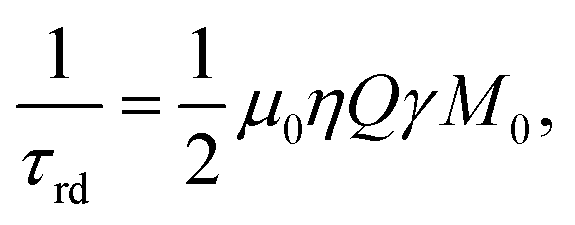
There are several ways to circumvent RD that were developed in response to high solvent concentration.58 Three of them can be used while keeping a high polarization: reduction of the filling factor, the quality factor, or T2*. Reduction of the filling factor by choosing smaller diameter sample tubes in probe heads of 5 or 10 mm also reduces the total magnetization of the sample and shows less RD. However, this approach was not sufficient to completely prevent RD in a commercial spectrometer at high concentrations (see Table 1 for achieved polarization levels) and cannot be used if large substrate quantities are necessary. Another solution would be to employ low Q-factor coils50,69 a Q-switch70 or active electronic feedback71 for the proton channel. But unfortunately, this is not possible in most of the commercial spectrometers. A partial solution that we investigated here is detuning the detection coil, which indeed prevents RD at mildly high concentrations. This measure reduced the Q-factor by a factor of 8 and did not completely prevent RD while prolonging the excitation pulses, hence narrowing the excitation band. In this case, the two protons of EA, which are 870 Hz apart, are excited with angular uniformity and phase shifts that degrade polarization transfer efficiency. A special excitation technique would need to be used that fits the ESOTHERIC pulse sequence. Consequently, we investigated the possibility of suppressing RD by the third method: reducing T2*. Interestingly, the onset of RD depends on the application of 2H-decoupling because the linewidth changes in EA. Decoupling of deuterons narrows the lines in the 1H-spectrum and consequently RD manifests itself at an even lower concentration of ∼0.5 mM. To reduce T2*, a field gradient can be applied, which spreads the frequency range of the signal and thus prevents focusing the energy at one particular frequency.58,72 To control the efficiency of the polarization transfer to the carbon, we have first evaluated the proton polarization utilizing the PHIP-echo pulse sequence (Fig. 2).73–75 The strength of this method is that it provides a net polarization pattern without losses due to overlapping antiphase lines. Largely concentrated solutions of HP molecule are prone to RD as can be seen from Fig. 2b, trace 2. The application of a weak field gradient with 1 ppm line broadening (50 mG cm−1) allowed us to suppress RD in PHIP-echo experiments with concentrations of HP substances up to 50 mM (it was not possible to measure signals larger than this in our spectrometer). In the ESOTHERIC experiments, application of a gradient during para-hydrogenation and polarization transfer allowed us to achieve 50% polarization on carbonyl carbon of EA at 10 mM concentration in a tuned high-field spectrometer inside a 5 mm NMR tube. Higher substrate concentration of ∼50 mM in the same tube resulted in degradation of the polarization level down to 36%, although the conversion of substrate was confirmed to be complete under the reaction conditions. Increasing the gradient strength further did not improve the situation. It appears that a gradient larger than 50 mG cm−1 influences the efficiency of ESOTHERIC transfer sequence (see Fig. S1, ESI†). This is likely due to molecule diffusion when the time period under a gradient is relatively long although the gradient strength is weak.76 However, combining a moderate gradient of 35–70 mG cm−1 over small samples (2 and 3 mm NMR tubes in a 10 mm probe head) allowed us to get 40% and 42% 13C polarization for substrate concentrations of up to 50 mM.
| Conditions | c/mM | 1H PHIP-echo | 13C ESOTHERIC |
|---|---|---|---|
| High field | 2 | 30% and 30% | 56% |
| 3 mm tube in | 9.5 | 27% and 32% | 56 ± 2% |
| 10 mm probe | 50 | 25% and 26% | 42% (35 mG cm−1) |
| High field | 50 | 27.0 ± 0.9% | 40 ± 1% (70 mG cm−1) |
| 2 mm tube in | 27.4 ± 0.2% | ||
| 10 mm probe | |||
| High field | 0.8 | 30% and 30% | 60% |
| 5 mm tube in | 10 | 30% and 30% | 50% (10 mG cm−1) |
| 5 mm probe | 50 | 26% and 23% (50 mG cm−1) | 36% (50 mG cm−1) |
| 54 ± 1% (BPFG) | |||
| 55 ± 1% (50 mG cm−1 Ref) | |||
| 20% (BPFG) | |||
| 93 | n/a, overload | 18% (50 mG cm−1 Ref) | |
| Low field | 1 | 26 ± 1% and 29 ± 1% | 35 ± 2% |
| 5 mm tube in | 10 | 30% and 24% (7 mG cm−1) | 37 ± 1% (7 mG cm−1) |
| 5 mm probe | 15 ± 1% (7 mG cm−1) | ||
| 50 | 10% and 10% (23 mG cm−1) | 23 ± 1% (7 mG cm−1 Ref) | |
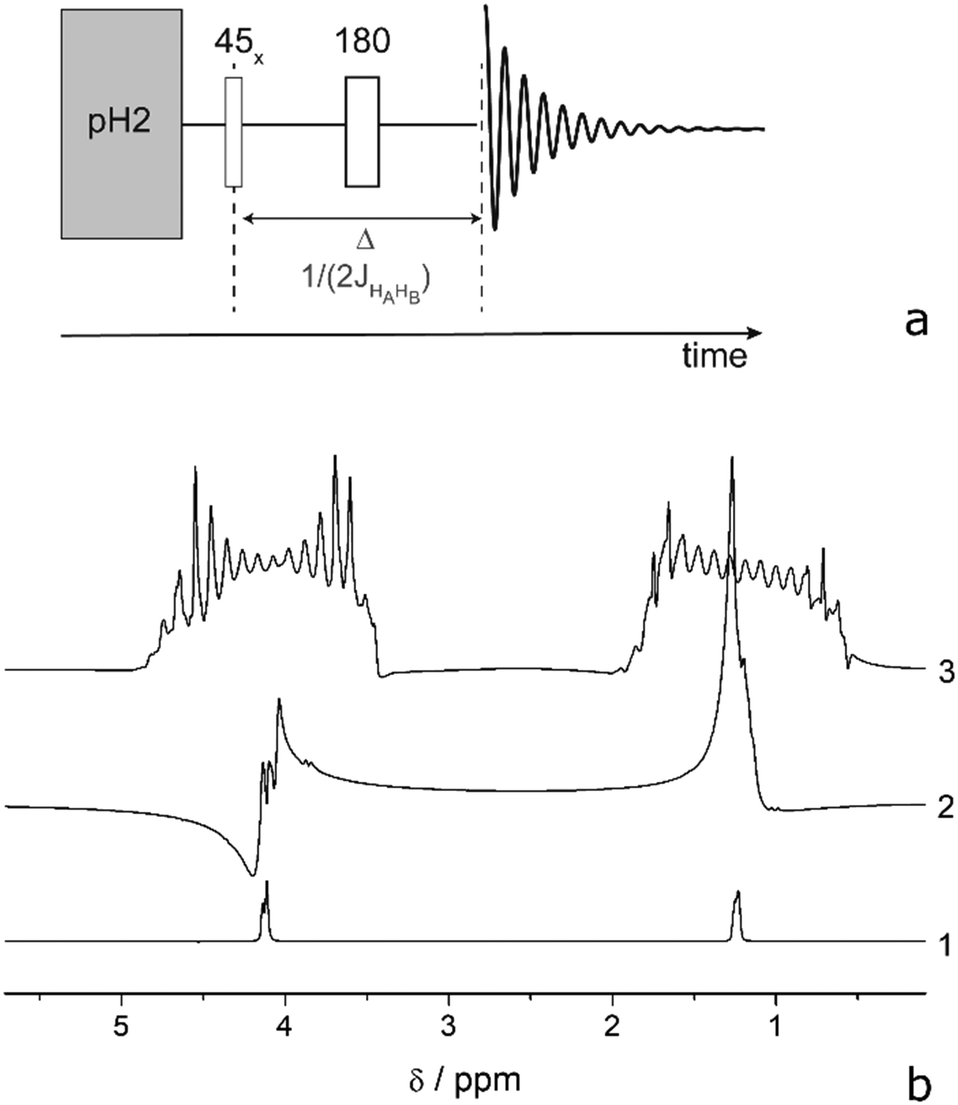 | ||
| Fig. 2 (a) PHIP-echo pulse sequence. (b) Hyperpolarized 1H spectra of EA in chloroform-d at 320 K under various conditions: (1) PHIP-echo of 2 mM substrate in a 3 mm tube resulting in 30% and 30% 1H-polarization; (2) PHIP-echo of 50 mM substrate in a 5 mm tube: RD completely spoils the polarization pattern; (3) PHIP-echo of 50 mM in a 5 mm tube under a 50 mG cm−1 gradient resulting in 26% and 23% polarization of the proton spins. | ||
To further reduce RD suppression without influence of diffusion effects, we have incorporated bipolar pulsed field gradients (BPFG)72,77 in our sequence. In this way, the transverse magnetization is refocused several times during the evolution between RF pulses. Thus, the effect of diffusion under the gradient is reduced because the magnetization stays defocused for short periods only.77Fig. 3(a) shows a modified ESOTHERIC pulse sequence using BPFG (ESOTHERIC-Grad). A constant field gradient is applied during the para-hydrogenation reaction to prevent RD at this stage. Here, no BPFG is necessary because the spin state does not contain a transverse component. After the first 90° pulse the large transverse magnetization is quickly defocused by BPFGs. The magnetization is defocused and refocused 25 times before application of the next RF pulse. Using this method, we were able to obtain up to 54% carbon polarization of a 50 mM solution EA in a 5 mm tube within a 5 mm probe head.
 | ||
| Fig. 3 (a) ESOTHERIC pulse sequence together with bipolar pulsed field gradients (ESOTHERIC-Grad). (b) ESOTHERIC pulse sequence together with several refocusing composite 180° pulses (ESOTHERIC-Ref). (c) 13C polarization of EA carbonyl as a result of the ESOTHERIC pulse sequence after para-hydrogenation of 50 mM of vinyl acetate-d6 in chloroform-d at 320 K: (1) ESOTHERIC without any gradient P = 1%; (2) ESOTHERIC-Ref under 5 mG cm−1P = 5%; (3) ESOTHERIC-Grad 50 mG cm−1 gradient during bubbling and 0.5 G cm−1 during transfer P = 22%; (4) ESOTHERIC under 50 mG cm−1 gradient P = 36%; (5) ESOTHERIC-Grad under 0.15 G cm−1 gradient during bubbling and 2.5 G cm−1 during transfer P = 54%; (6) ESOTHERIC-Ref under 50 mG cm−1 gradient during bubbling and transfer P = 56%. The inset shows a signal of the 13C carbonyl of product of hydrogenation (EA) with thermal polarization at B0 = 7 T (100 mM, 1100 scans, 1H- and 2H-dec). | ||
BPFG is an elaborated technique that may not be available or adds additional costs to spectrometers. We therefore investigated the possibility of refocusing magnetization under a constant gradient utilizing 180° pulses. Thereby, instead of changing the gradient direction in order to reverse magnetization rotation, one can flip magnetization and continue to rotate the magnetization in the same direction until it refocuses. The alternative sequence is shown in Fig. 3b (ESOTHERIC-Ref). Here, several refocusing periods are applied to reduce the defocusing/refocusing times and thus suppress the negative diffusion effect. It was found that composite 90°x180°y90°x pulses were beneficial for the transfer efficiency. 55% 13C polarization was achieved which is as good as with the BPFG-method. All polarization levels recorded under the various conditions are summarized in Table 1.
These promising results motivated us to translate the experiments to a 1 T low-field spectrometer (Magritek Spinsolve-43). Since no BPFGs were available in the utilized system, we have performed experiments with 180°-refocusing pulses only. In 5 mm tubes, it was possible to obtain ∼35% polarization on the carbonyl carbon of EA for a low concentration of 1 mM. As the spectrometer maintains 25 °C inside its body case the sample was preheated in a water bath to 50 °C to facilitate the hydrogenation reaction. For concentrations higher than 1 mM RD was observed when the sample was perfectly shimmed. To prevent RD, a constant gradient of 7 mG cm−1 was applied via the z1-shim along the z-direction (1 ppm). Under these conditions the 13C-polarization reached 37% for 10 mM and only around 15% polarization for 50 mM. Using ESOTHERIC-Ref with non-composite 180°-refocusing pulses allowed us to increase the polarization to 23%. At smaller concentrations (1 mM and 10 mM) we achieved 35% and 37% 13C polarization which is also smaller than at high magnetic fields. We mainly attribute this discrepancy to uncontrolled temperature drops in the low field experiments. The utilized low-field spectrometer runs at room temperature and samples are preheated in an adjacent water bath before they are inserted into the spectrometer to supply para-hydrogen and initiate the polarization transfer. Consequently, the sample cools while it rests in the spectrometer and during the supply of para-hydrogen, leading to a slowed down reaction and conversion rate. As a result, lower proton polarization is generated that can be transferred to 13C. This issue may, however, be circumventable in the future if a low-field spectrometer is utilized that maintains a higher, stable temperature at the site of the sample during bubbling and polarization transfer. Nevertheless, the achieved polarization level is above the in vivo threshold.42
Lastly, we have investigated the production of the hyperpolarized acetate metabolite in high concentrations in high and low-magnetic field in methanol-d4. We have chosen methanol as a solvent to directly demonstrate the cleavage within the spectrometer. Implementation of a phase extraction procedure in accordance to ref. 55 for obtaining preclinical imaging contrast agents is currently under investigation in our lab. Fig. 4 shows the hydrolysis of the hyperpolarized EA in mixtures of methanol-d4 and 1 M NaOD in D2O. Room temperature base is added and mixed in the 5 mm NMR tube while it rests inside a spectrometer after polarization transfer. We would like to note that using methanol as a solvent lead in our experiments to a reduction in initial 13C-polarization (6.6% in low field and 42% in high field) probably because of a slower reaction rate compared to the hydrogenation in chloroform. The discrepancy compared to the high-field case is due to the fact that no elevated temperature was available in the utilized 1 T system and methanol appears to be particular prone to this circumstances. The sample was preheated in a water bath and the temperature dropped during the para-hydrogen supply inside the low-field spectrometer. This will however be circumventable in the future with heated benchtop devices. After cleavage we determined the polarization levels of the metabolite to be 2.2% (low field) and 17% (high field). Noteworthy is the very long T1 of free acetate, which we found to be 147 s at 7 T even in presence of the catalyst (Fig. S14, ESI†). We therefore attribute the loss in polarization mainly to the fact that not all of the sample is excited during the pulsed transfer based on inhomogeneities in B1 generated by the NMR coil and incomplete B1 coverage. Once the base is added, non-irradiated regions, hence unpolarised sample will mix with hyperpolarized metabolites and give an overall reduction in polarization. In future studies we are aiming at elucidating the phenomenon, nevertheless high levels of metabolite polarization are already attainable. Based on the presented results we are now aiming to improve the polarization setup by constructing a low field system with stable temperature, larger and homogeneous B1 field and the possibility to apply field gradients or composite pulses. In addition, implementing an optimized cleavage and phase extraction protocol from chloroform that includes the use of heated base as published by Cavallari et al. will lead to higher degrees of polarization of the desired metabolite in water.25
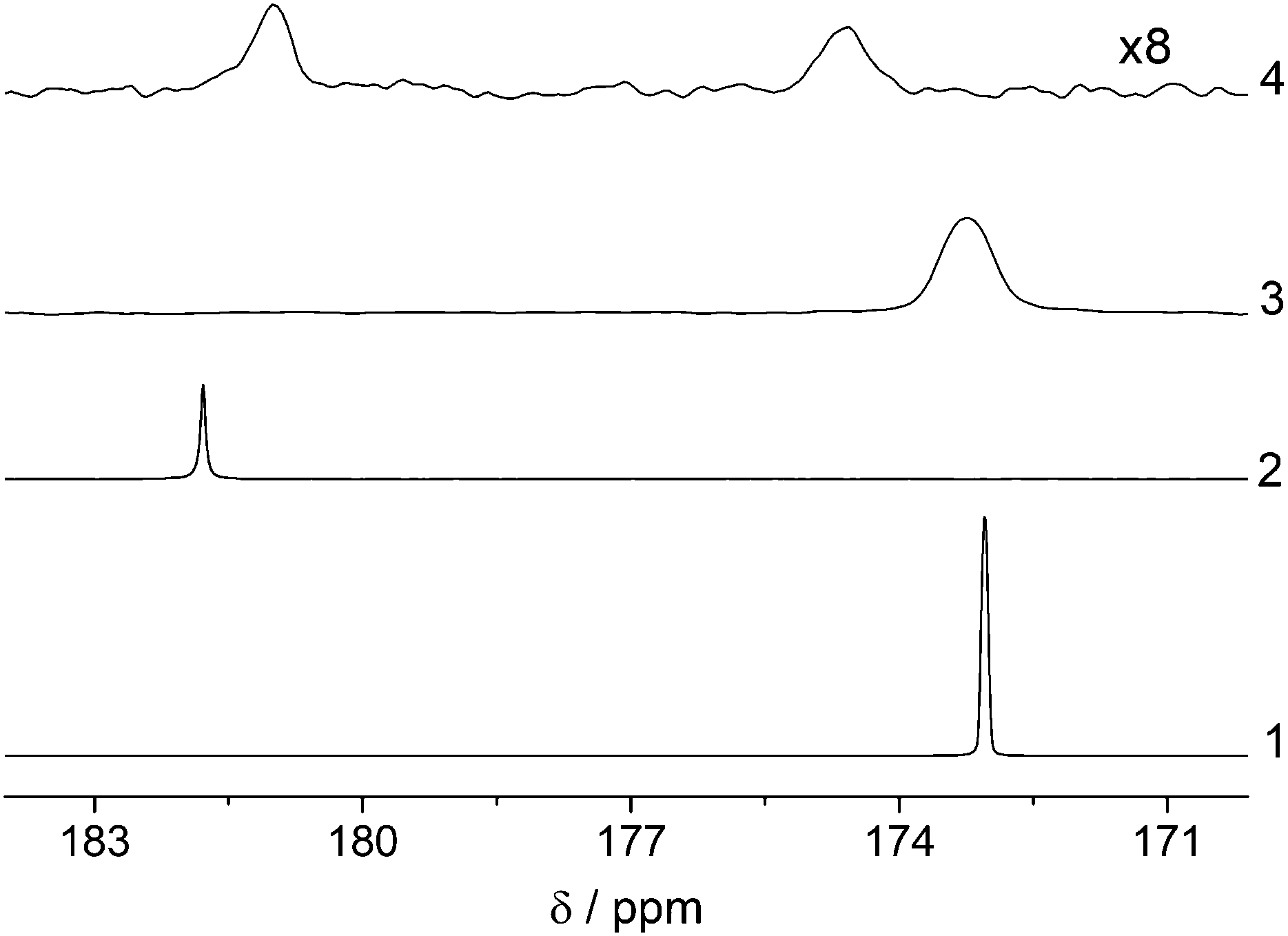 | ||
| Fig. 4 13C spectra of carbonyl carbon of EA hydrolysis. para-Hydrogenation and polarization transfer was done in methanol-d4. Hydrolysis was induced by addition of same volume of 1 M NaOD in D2O. High field experiments were done with 50 mM vinyl acetate-d6 using ESOTHERIC-Ref as polarization transfer sequence to 13C of acetate. At low field ESOTHERIC without gradient was used in reaction of 10 mM vinyl acetate-d6 for better resolution that, however, resulted in smaller polarization levels. At the end of the polarization transfer, longitudinal magnetization was formed by an additional 90° pulse for storage before reaction. 13C spectra were detected with a single pulse: (1) EA with ∼173 ppm and P = 42% before reaction and (2) acetate anion with P = 17% and ∼182 ppm after ∼40 s hydrolysis; in low field: (3) EA with P = 6.6% and ∼173 ppm before reaction and (2) mixture of EA and acetate with P = 2.2% after ∼10 s hydrolysis. Incomplete cleavage is due to the low temperature in the low field system. Chemical shift between high and low field conditions may vary because of internal and external lock in two spectrometers respectively. | ||
Conclusions
In conclusion, we have introduced two pulsed NMR polarization transfer schemes that can produce highly polarized (>50%) 13C-spins in the metabolite precursor EA and the cleaved metabolite (>17%) and at high concentrations of 50 mM. Furthermore, we were able to translate the experiments to a compact low-field spectrometer, achieving high degrees of polarization and concentrations. The obtained quantities (hundreds of microliters) are typically sufficient for preclinical in vivo experiments on rodents.49 This development paves the way for a cost-efficient and fast (seconds) production of hyperpolarized metabolites and their application e.g. in metabolomics labs or for biomedical imaging. We furthermore believe that another advantage of the presented approach is that it can easily be implemented in many labs with minimal efforts. All that is required is a two-channel spectrometer with > 1 T field strength and para-hydrogen supply which is available in a cost-effective way when produced at 77 K (50% para-state enrichment). Moreover, we have reported the observation of non-linear effects in addition reactions with para-hydrogen as a result of the large polarization. Although further investigations are in progress, we postulate that e.g. masing effects in connection with PHIP exist, which can be detected over a large range of magnetic fields and will foster high precision experiments in physics in the future.32Conflicts of interest
BB is on the board of directors of Magritek.Acknowledgements
The authors acknowledge generous funding from the Max-Planck-Society and from the German Research Foundation (grant number DFG 418416679). Open Access funding provided by the Max Planck Society.Notes and references
- F. Presciutti, J. Perlo, F. Casanova, S. Glöggler, C. Miliani, B. Blümich, B. G. Brunetti and A. Sgamellotti, Appl. Phys. Lett., 2008, 93, 033505 CrossRef.
- P. J. McDonald, J. Mitchell, M. Mulheron, P. S. Aptaker, J.-P. Korb and L. Monteilhet, Cem. Concr. Res., 2007, 37, 303–309 CrossRef CAS.
- J. N. King, A. Fallorina, J. Yu, G. Zhang, V.-V. Telkki, C. Hilty and T. Meldrum, Chem. Sci., 2018, 9, 6143–6149 RSC.
- B. Blümich, F. Casanova and S. Appelt, Chem. Phys. Lett., 2009, 477, 231–240 CrossRef.
- M. E. Halse, A. Coy, R. Dykstra, C. Eccles, M. Hunter, R. Ward and P. T. Callaghan, J. Magn. Reson., 2006, 182, 75–83 CrossRef CAS PubMed.
- H. Lee, E. Sun, D. Ham and R. Weissleder, Nat. Med., 2008, 14, 869 CrossRef CAS.
- S. Appelt, H. Kühn, F. W. Häsing and B. Blümich, Nat. Phys., 2006, 2, 105–109 Search PubMed.
- B. Blümich and K. Singh, Angew. Chem., Int. Ed., 2018, 57, 6996–7010 CrossRef PubMed.
- T. G. Walker and W. Happer, Rev. Mod. Phys., 1997, 69, 629–642 CrossRef CAS.
- S. Appelt, A. B.-A. Baranga, C. J. Erickson, M. V. Romalis, A. R. Young and W. Happer, Phys. Rev. A: At., Mol., Opt. Phys., 1998, 58, 1412–1439 CrossRef CAS.
- H. E. Möller, X. J. Chen, B. Saam, K. D. Hagspiel, G. A. Johnson, T. A. Altes, E. E. de Lange and H.-U. Kauczor, Magn. Reson. Med., 2002, 47, 1029–1051 CrossRef PubMed.
- L. Schröder, T. J. Lowery, C. Hilty, D. E. Wemmer and A. Pines, Science, 2006, 314, 446–449 CrossRef PubMed.
- K. Golman, R. I. T. Zandt and M. Thaning, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 11270–11275 CrossRef CAS PubMed.
- S. E. Day, M. I. Kettunen, F. A. Gallagher, D.-E. Hu, M. Lerche, J. Wolber, K. Golman, J. H. Ardenkjaer-Larsen and K. M. Brindle, Nat. Med., 2007, 13, 1382 CrossRef CAS PubMed.
- F. A. Gallagher, M. I. Kettunen, S. E. Day, D.-E. Hu, J. H. Ardenkjær-Larsen, R. I. T. Zandt, P. R. Jensen, M. Karlsson, K. Golman, M. H. Lerche and K. M. Brindle, Nature, 2008, 453, 940 CrossRef CAS PubMed.
- S. J. Nelson, J. Kurhanewicz, D. B. Vigneron, P. E. Z. Larson, A. L. Harzstark, M. Ferrone, M. van Criekinge, J. W. Chang, R. Bok, I. Park, G. Reed, L. Carvajal, E. J. Small, P. Munster, V. K. Weinberg, J. H. Ardenkjaer-Larsen, A. P. Chen, R. E. Hurd, L.-I. Odegardstuen, F. J. Robb, J. Tropp and J. A. Murray, Sci. Transl. Med., 2013, 5, 198ra108 Search PubMed.
- J. Kurhanewicz, D. B. Vigneron, K. Brindle, E. Y. Chekmenev, A. Comment, C. H. Cunningham, R. J. DeBerardinis, G. G. Green, M. O. Leach, S. S. Rajan, R. R. Rizi, B. D. Ross, W. S. Warren and C. R. Malloy, Neoplasia, 2011, 13, 81–97 CrossRef CAS PubMed.
- J. H. Ardenkjær-Larsen, B. Fridlund, A. Gram, G. Hansson, L. Hansson, M. H. Lerche, R. Servin, M. Thaning and K. Golman, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 10158–10163 CrossRef PubMed.
- A. B. Schmidt, S. Berner, W. Schimpf, C. Müller, T. Lickert, N. Schwaderlapp, S. Knecht, J. G. Skinner, A. Dost, P. Rovedo, J. Hennig, D. von Elverfeldt and J. B. Hövener, Nat. Commun., 2017, 8, 14535 CrossRef CAS PubMed.
- P. Bhattacharya, E. Y. Chekmenev, W. H. Perman, K. C. Harris, A. P. Lin, V. A. Norton, C. T. Tan, B. D. Ross and D. P. Weitekamp, J. Magn. Reson., 2007, 186, 150–155 CrossRef CAS PubMed.
- P. Bhattacharya, E. Y. Chekmenev, W. F. Reynolds, S. Wagner, N. Zacharias, H. R. Chan, R. Bünger and B. D. Ross, NMR Biomed., 2011, 24, 1023–1028 CrossRef CAS PubMed.
- M. Goldman, H. Jóhannesson, O. Axelsson and M. Karlsson, C. R. Chim, 2006, 9, 357–363 CrossRef CAS.
- F. Reineri, T. Boi and S. Aime, Nat. Commun., 2015, 6, 5858 CrossRef CAS PubMed.
- E. Cavallari, C. Carrera, T. Boi, S. Aime and F. Reineri, J. Phys. Chem. B, 2015, 119, 10035–10041 CrossRef CAS PubMed.
- E. Cavallari, C. Carrera, S. Aime and F. Reineri, J. Magn. Reson., 2018, 289, 12–17 CrossRef CAS PubMed.
- M. Haake, J. Natterer and J. Bargon, J. Am. Chem. Soc., 1996, 118, 8688–8691 CrossRef CAS.
- J. Natterer and J. Bargon, Prog. Nucl. Magn. Reson. Spectrosc., 1997, 31, 293–315 CrossRef.
- C. R. Bowers and D. P. Weitekamp, Phys. Rev. Lett., 1986, 57, 2645–2648 CrossRef CAS PubMed.
- R. W. Adams, J. A. Aguilar, K. D. Atkinson, M. J. Cowley, P. I. P. Elliott, S. B. Duckett, G. G. R. Green, I. G. Khazal, J. López-Serrano and D. C. Williamson, Science, 2009, 323, 1708–1711 CrossRef CAS PubMed.
- P. J. Rayner, M. J. Burns, A. M. Olaru, P. Norcott, M. Fekete, G. G. R. Green, L. A. R. Highton, R. E. Mewis and S. B. Duckett, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E3188–E3194 CrossRef CAS PubMed.
- D. A. Barskiy, K. V. Kovtunov, I. V. Koptyug, P. He, K. A. Groome, Q. A. Best, F. Shi, B. M. Goodson, R. V. Shchepin, A. M. Coffey, K. W. Waddell and E. Y. Chekmenev, J. Am. Chem. Soc., 2014, 136, 3322–3325 CrossRef CAS PubMed.
- M. Suefke, S. Lehmkuhl, A. Liebisch, B. Blümich and S. Appelt, Nat. Phys., 2017, 13, 568 Search PubMed.
- N. Eshuis, N. Hermkens, B. J. A. van Weerdenburg, M. C. Feiters, F. P. J. T. Rutjes, S. S. Wijmenga and M. Tessari, J. Am. Chem. Soc., 2014, 136, 2695–2698 CrossRef CAS PubMed.
- T. Theis, M. L. Truong, A. M. Coffey, R. V. Shchepin, K. W. Waddell, F. Shi, B. M. Goodson, W. S. Warren and E. Y. Chekmenev, J. Am. Chem. Soc., 2015, 137, 1404–1407 CrossRef CAS PubMed.
- P. Spannring, I. Reile, M. Emondts, P. P. M. Schleker, N. K. J. Hermkens, N. G. J. V. D. Zwaluw, B. J. A. V. Weerdenburg, P. Tinnemans, M. Tessari, B. Blümich, F. P. J. T. Rutjes and M. C. Feiters, Chem. – Eur. J., 2016, 22, 9277–9282 CrossRef CAS PubMed.
- K. V. Kovtunov, M. L. Truong, D. A. Barskiy, O. G. Salnikov, V. I. Bukhtiyarov, A. M. Coffey, K. W. Waddell, I. V. Koptyug and E. Y. Chekmenev, J. Phys. Chem. C, 2014, 118, 28234–28243 CrossRef CAS PubMed.
- W. Iali, A. M. Olaru, G. G. R. Green and S. B. Duckett, Chem. – Eur. J., 2017, 23, 10491–10495 CrossRef CAS PubMed.
- J. F. P. Colell, M. Emondts, A. W. J. Logan, K. Shen, J. Bae, R. V. Shchepin, G. X. Ortiz, P. Spannring, Q. Wang, S. J. Malcolmson, E. Y. Chekmenev, M. C. Feiters, F. P. J. T. Rutjes, B. Blümich, T. Theis and W. S. Warren, J. Am. Chem. Soc., 2017, 139, 7761–7767 CrossRef CAS PubMed.
- T. Trantzschel, J. Bernarding, M. Plaumann, D. Lego, T. Gutmann, T. Ratajczyk, S. Dillenberger, G. Buntkowsky, J. Bargon and U. Bommerich, Phys. Chem. Chem. Phys., 2012, 14, 5601–5604 RSC.
- O. G. Salnikov, K. V. Kovtunov and I. V. Koptyug, Sci. Rep., 2015, 5, 13930 CrossRef PubMed.
- A. S. Kiryutin, G. Sauer, S. Hadjiali, A. V. Yurkovskaya, H. Breitzke and G. Buntkowsky, J. Magn. Reson., 2017, 285, 26–36 CrossRef CAS PubMed.
- J.-B. Hövener, A. N. Pravdivtsev, B. Kidd, C. R. Bowers, S. Glöggler, K. V. Kovtunov, M. Plaumann, R. Katz-Brull, K. Buckenmaier, A. Jerschow, F. Reineri, T. Theis, R. V. Shchepin, S. Wagner, P. Bhattacharya, N. M. Zacharias and E. Y. Chekmenev, Angew. Chem., Int. Ed., 2018, 57, 11140–11162 CrossRef PubMed.
- S. Korchak, S. Yang, S. Mamone and S. Glöggler, ChemistryOpen, 2018, 7, 344–348 CrossRef CAS PubMed.
- S. Korchak, S. Mamone and S. Glöggler, ChemistryOpen, 2018, 7, 672–676 CrossRef CAS PubMed.
- J. McCormick, S. Korchak, S. Mamone, Y. N. Ertas, Z. Liu, L. Verlinsky, S. Wagner, S. Glöggler and L.-S. Bouchard, Angew. Chem., Int. Ed., 2018, 57, 10692–10696 CrossRef CAS PubMed.
- L. E. Olsson, C.-M. Chai, O. Axelsson, M. Karlsson, K. Golman and J. S. Petersson, Magn. Reson. Med., 2006, 55, 731–737 CrossRef PubMed.
- K. Golman, O. Axelsson, H. Johannesson, S. Mansson, C. Olofsson and J. S. Petersson, Magn. Reson. Med., 2001, 46, 1–5 CrossRef CAS PubMed.
- M. Goldman, H. Johannesson, O. Axelsson and M. Karlsson, Magn. Reson. Imaging, 2005, 23, 153–157 CrossRef CAS PubMed.
- A. M. Coffey, R. V. Shchepin, M. L. Truong, K. Wilkens, W. Pharm and E. Y. Chekmenev, Anal. Chem., 2016, 88, 8279–8288 CrossRef CAS PubMed.
- S. Kadlecek, V. Vahdat, T. Nakayama, D. Ng, K. Emami and R. Rizi, NMR Biomed., 2011, 24, 933–942 CrossRef CAS PubMed.
- N. M. Zacharias, H. R. Chan, N. Sailasuta, B. D. Ross and P. Bhattacharya, J. Am. Chem. Soc., 2012, 134, 934–943 CrossRef CAS PubMed.
- R. V. Shchepin, A. M. Coffey, K. W. Waddell and E. Y. Chekmenev, Anal. Chem., 2014, 86, 5601–5605 CrossRef CAS PubMed.
- N. M. Zacharias, C. R. McCullogh, S. Wagner, N. Sailasuta, H. R. Chen, Y. Lee, J. Hu, W. H. Perman, C. Henneberg, B. D. Ross and P. Bhattacharya, J. Mol. Imaging Dyn., 2016, 6, 123 Search PubMed.
- A. M. Coffey, M. A. Feldman, R. V. Shchepin, D. A. Barskiy, M. L. Truong, W. Pham and E. Y. Chekmenev, J. Magn. Reson., 2017, 281, 246–252 CrossRef CAS PubMed.
- E. Cavallari, C. Carrera, M. Sorge, G. Bonne, A. Muchir, S. Aime and F. Reineri, Sci. Rep., 2018, 8, 8366 CrossRef PubMed.
- N. J. Stewart, H. Kumeta, M. Tomohiro, T. Hashimoto, N. Hatae and S. Matsumoto, J. Magn. Reson., 2018, 296, 85–92 CrossRef CAS PubMed.
- H. Desvaux, Prog. Nucl. Magn. Reson. Spectrosc., 2013, 70, 50–71 CrossRef CAS PubMed.
- V. V. Krishnan and N. Murali, Prog. Nucl. Magn. Reson. Spectrosc., 2013, 68, 41–57 CrossRef CAS PubMed.
- H. Barjat, G. P. Chadwick, G. A. Morris and A. G. Swanson, J. Magn. Reson., Ser. A, 1995, 117, 109–112 CrossRef CAS.
- X.-A. Mao and C.-H. Ye, Concepts Magn. Reson., 1997, 9, 173–187 CrossRef CAS.
- O. G. Salnikov, N. V. Chukanov, R. V. Shchepin, I. V. Manzanera Esteve, K. V. Kovtunov, I. V. Koptyug and E. Y. Chekmenev, J. Phys. Chem. C, 2019, 123, 12827–12840 CAS.
- N. V. Chukanov, O. G. Salnikov, R. V. Shchepin, K. V. Kovtunov, I. V. Koptyug and E. Y. Chekmenev, ACS Omega, 2018, 3, 6673–6682 CrossRef CAS PubMed.
- S. Blümel, A. Moreno-Torres, F. Shic, C.-H. Nguy and B. D. Ross, NMR Biomed., 2002, 15, 1–5 CrossRef PubMed.
- A. Flori, M. Liserani, F. Frijia, G. Giovannetti, V. Lionetti, V. Casieri, V. Positano, G. D. Aquaro, F. A. Recchia, M. F. Santarelli, L. Landini, J. H. Ardenkjaer-Larsen and L. Menichetti, Contrast Media Mol. Imaging, 2005, 10, 194–202 CrossRef PubMed.
- E. F. R. Mikkelsen, C. Ø. Mariager, T. Nørlinger, H. Qi, R. F. Schulte, S. Jakobsen, J. Frøkiær, M. Pedersen, H. Stødkilde-Jørgensen and C. Laustsen, Sci. Rep., 2017, 7, 16002 CrossRef PubMed.
- D. J. Y. Marion, G. Huber, P. Berthault and H. Desvaux, ChemPhysChem, 2008, 9, 1395–1401 CrossRef CAS PubMed.
- H.-Y. Chen, Y. Lee, S. Bowen and C. Hilty, J. Magn. Reson., 2011, 208, 204–209 CrossRef CAS PubMed.
- A. Vlassenbroek, J. Jeener and P. Broekaert, J. Chem. Phys., 1995, 103, 5886–5897 CrossRef CAS.
- J.-B. Hövener, E. Y. Chekmenev, K. C. Harris, W. Perman, L. Robertson, B. D. Ross and P. Bhattacharya, Magn. Reson. Mater. Phys., Biol. Med., 2009, 22, 111–121 CrossRef PubMed.
- W. E. Maas, F. H. Laukien and D. G. Cory, J. Magn. Reson., Ser. A, 1995, 113, 274–277 CrossRef CAS.
- P. Broekaert and J. Jeener, J. Magn. Reson., Ser. A, 1995, 113, 60–64 CrossRef CAS.
- V. Sklenar, J. Magn. Reson., Ser. A, 1995, 114, 132–135 CrossRef CAS.
- T. Ratajczyk, T. Gutmann, S. Dillenberger, S. Abdulhussaein, J. Frydel, H. Breitzke, U. Bommerich, T. Trantzschel, J. Bernarding, P. C. M. M. Magusin and G. Buntkowsky, Solid State Nucl. Magn. Reson., 2012, 43–44, 14–21 CrossRef CAS PubMed.
- D. A. Barskiy, K. V. Kovtunov, E. Y. Gerasimov, M. A. Phipps, O. G. Salnikov, A. M. Coffey, L. M. Kovtunova, I. P. Prosvirin, V. I. Bukhtiyarov, I. V. Koptyug and E. Y. Chekmenev, J. Phys. Chem. C, 2017, 121, 10038–10046 CrossRef CAS.
- A. S. Kiryutin, G. Sauer, D. Tietze, M. Brodrecht, S. Knecht, A. V. Yurkovskaya, K. L. Ivanov, O. Avrutina, H. Kolmar and G. Buntkowsky, Chem. – Eur. J., 2019, 25, 4025–4030 CrossRef CAS PubMed.
- E. O. Stejskal and J. E. Tanner, J. Chem. Phys., 1965, 42, 288–292 CrossRef CAS.
- S. Zhang and D. G. Gorenstein, J. Magn. Reson., Ser. A, 1996, 118, 291–294 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Material and methods, general setup and reaction conditions, experiments in low field, relaxation times. See DOI: 10.1039/c9cp05227e |
| This journal is © the Owner Societies 2019 |