
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Micro-spectroscopy of HKUST-1 metal–organic framework crystals loaded with tetracyanoquinodimethane: effects of water on host–guest chemistry and electrical conductivity†
Miguel
Rivera-Torrente
a,
Matthias
Filez
a,
Christian
Schneider
 b,
Ewout C.
van der Feltz
a,
Konrad
Wolkersdörfer
c,
Dereje H.
Taffa
c,
Michael
Wark
c,
Roland A.
Fischer
b and
Bert M.
Weckhuysen
*a
b,
Ewout C.
van der Feltz
a,
Konrad
Wolkersdörfer
c,
Dereje H.
Taffa
c,
Michael
Wark
c,
Roland A.
Fischer
b and
Bert M.
Weckhuysen
*a
aInorganic Chemistry and Catalysis, Debye Institute for Nanomaterials Science, Utrecht University, Universiteitsweg 99, 3584 CG Utrecht, The Netherlands. E-mail: b.m.weckhuysen@uu.nl
bDepartment of Chemistry, Technische Universität München, Lichtenbergstraße 4, 85748 Garching, Germany
cInstitute of Chemistry, Chemical Technology 1, Carl von Ossietzky Universität Oldenburg, Carl-von-Ossietzky Straße 9-11, 26129 Oldenburg, Germany
First published on 6th November 2019
Abstract
Composite materials, consisting of a metal–organic framework (MOF) and a guest molecule, further denoted as guest@MOF composites, have gained strong interest due to the guest-induced tunability of the host properties, for example in sensing or electroconductivity applications. However, decoding the complexity of these guest@MOF composites and extracting structure-performance relationships are far from trivial and require the use of a gamut of characterization tools. In this work, we use various micro-spectroscopic tools both under static (ex situ) and dynamic (in situ) conditions to map the properties and diffusion behavior of TCNQ (7,7,8,8-tetracyanoquinodimethane) as a guest molecule in single HKUST-1 crystals as the host. Raman micro-spectroscopy allowed us to map the spatial distribution of TCNQ within HKUST-1 single crystals, thereby revealing a heterogeneous distribution of TCNQ after initial TCNQ-infiltration, concentrated at the crystal edges, and a homogeneous redistribution upon water vapor treatment. These insights are correlated to I–V scans at different temperatures and to electrochemical impedance spectroscopy (EIS), which allowed us to verify the different contributions to conductivity. These data showed changes in electrical conductivity after exposing the sample to moisture and air. In situ FT-IR micro-spectroscopy during treatment with moisturized nitrogen gas suggests lower transient diffusion rates for water inside TCNQ@HKUST-1 relative to pristine HKUST-1, likely due to steric hindrance of the pore-filling TCNQ molecules in the HKUST-1 framework. The application of micro-spectroscopic techniques is crucial to uncovering MOF intracrystal heterogeneities and yielding rationally-derived instructions for the improved design of guest@MOF systems.
Introduction
Metal–organic frameworks (MOFs) have emerged as a class of porous crystalline materials with high potential in a manifold of applications, including gas sorption, catalysis, biomedicine and electronic devices.1,2 One of their main advantages is their high porosity that can allow them to act as hosts for metal nanoparticles and enzyme guests, or to incorporate functional guest molecules that can tune the MOF properties. The performance of these functionalized host–guest materials, further denoted as guest@MOFs, critically depends on the precise location of these guest species within the host material (i.e., the MOF crystal). More particularly, microscopic heterogeneities in the intracrystal distribution of guest molecules might affect both the bulk properties and the performance of the guest@MOF materials. In this sense, spatially resolved techniques are extremely powerful tools to shed light on the precise location of guest molecules within guest@MOF systems, under both ex situ and in situ conditions.In contrast to other classes of porous functional materials, such as zeolites (e.g. FAU, BEA and MOR), mesoporous structured materials (e.g. MCM-41 and SBA-15) and (pillared) clay minerals,3–7 the number of studies using spatially-resolved spectroscopies is very limited in the field of MOFs. Notable examples include the linker distribution mapping in multivariate MTV-MOFs or defect imaging in HKUST-1, as recently addressed by vibrational and fluorescence micro-spectroscopies.8–12 In addition, infrared micro-spectroscopy has been applied by the Kärger group to extract the transient diffusion behaviour of probe molecules within ZIF-8 crystals.13–17 However, to the best of our knowledge, no micro-spectroscopic studies reporting on charting the spatial location, distribution and chemical properties of functional guest molecules in MOFs have been published. Typically, for example, mapping nanoparticle distributions in MOFs is achieved by Transmission Electron Microscopy (TEM).18 However, the probing volume of TEM is limited to the nm3-range, which typically hinders imaging of the distribution of guest entities over an entire MOF crystal. Also, TEM (unless coupled to Energy Dispersive X-ray (EDX) analysis, which usually requires high voltages and intense electron beams, and therefore, could result in beam damage) does not provide chemical information, but rather structural properties of the sample. The application of micro-spectroscopic tools to locate and speciate guest molecules in MOFs can thus offer chemical information with spatial resolution across micrometre-sized MOF crystals in a non-destructive way under both ex situ and in situ conditions.
Among guest@MOF materials, HKUST-1 loaded with TCNQ (TCNQ = 7,7,8,8-tetracyanoquinodimethane) has been one of the most interesting yet challenging systems to be studied. Initially introduced by Allendorf et al.,19 this guest@MOF composite material has attracted the interest of many researchers for its strong increase in electronic conductivity upon infiltration with TCNQ as a guest molecule.20–22 Electrically conductive MOFs have been intensively researched in recent years,23,24 and in this context, the post-synthetic introduction of redox-active species into intrinsically insulating MOF materials represents a promising strategy to combine porosity and electrical conductivity in one material.25 For a precise elucidation of the underlying electrical conductivity mechanism, structural information of the guest molecule is of utmost importance. While theory predicted the coordination of TCNQ to two adjacent metal nodes of HKUST-1,19 just recently, some of us found crystallographic evidence for this preferential adsorption, when TCNQ was introduced via the vapor phase.26 Given the complexity of the system, elaborate characterization techniques are necessary to identify the necessary structure–property relationships and to study how moisture affects the distribution of TCNQ within the MOF and impacts the electroconductivity of the host–guest material.
Here, we have used Raman microscopy to map the spatial distribution of TCNQ as the guest molecule within large HKUST-1 crystals before and after dosing water vapour. Spectral mapping was done under controlled conditions within a sealed chamber, avoiding contact with air, water therein or contaminants. Advanced data processing, by means of Principal Component Analysis (PCA), was used to improve data quality and related spectral interpretation. In addition, in situ FT-IR micro-spectroscopy has been used to compare the diffusion behaviour of TCNQ-loaded and pristine HKUST-1 crystals upon water vapor dosing in the chamber. The obtained results are compared with water uptake isotherms, X-ray photoelectron spectroscopy (XPS) data and current (I)–voltage (V) curves, as obtained from I–V scans. By doing so, we show that the multi-technique approach allows us to further shed light on the complex host–guest chemistry of the TCNQ@HKUST-1 system.
Experimental section
Synthesis of HKUST-1 single crystals
Large HKUST-1 crystals were prepared by following a procedure of Tovar et al.27 In brief, Cu(NO3)2·3H2O (0.49 g, 2.0 mmol, 99%, Acros Organics) was dissolved in 3 mL of deionized water in a glass vial. In another vial, benzene-1,3,5-tricarboxylic acid (0.24 g, 1.1 mmol, H3BTC, 98%, Sigma-Aldrich) was dissolved in 3 mL of ethanol (VWR International, 96%). Then, the Cu solution was mixed with 3 mL of N,N-dimethylformamide (DMF, anhydrous, 99.8%, Sigma-Aldrich) in a 20 mL glass vial. The linker solution and 12 mL of glacial acetic acid (99%, Sigma-Aldrich) were added to the mixture and the vial was placed in a preheated oven at 328 K for 72 h. The crystals were collected and immersed in fresh ethanol and CH2Cl2 (99%, Sigma-Aldrich) for 1 week, respectively, before activation at p ≈ 10−3 mbar and 453 K for 24 h. Activated HKUST-1 was stored in an Ar BraunTM glovebox (O2 < 1 ppm, H2O < 1 ppm) until further use. 7,7,8,8-Tetracyanoquinodimethane (TCNQ, 98%, acbr GmbH, Germany) was recrystallized three times from acetonitrile (anhydrous, 99%, Sigma-Aldrich) under inert conditions to yield molecular, crystalline TCNQ in high purity.Dry HKUST-1 single crystals were loaded into a glass ampoule with an excess of recrystallized TCNQ (TCNQ, approx. 2 eq. of TCNQ per HKUST-1) adapted from the protocol reported in the literature.26
Routine characterization
Ex situ X-ray diffraction (XRD) was done in a 2θ range of 5–50° in steps of 0.0016413° (2θ) with a PANalytical Empyrean instrument equipped with a Cu source (λKα = 1.54184 Å) at 45 kV and 40 mA. The samples were loaded in borosilicate capillaries of 0.7 mm diameter. The radiation was focused onto the sample through a focusing X-ray beam mirror equipped with a 1/8° divergence slit and 0.02 radian Soller slits. The diffracted X-ray beam was detected by a PIXcel1D detector in receiving slit mode equipped with a 1/8° anti-scatter slit and 0.02 radian Soller slits. In situ X-ray diffraction (XRD) measurements were carried out in a 2θ range of 5–30° in steps of 0.05° (2θ), and 1 s per step time, on a Bruker Advance D8 diffractometer in Bragg–Brentano geometry equipped with a Co K1,2 source (λ = 1.79026 Å), a VANTEC-1 detector and 1/8° divergence slits. Scanning Electron Microscopy (SEM) micrographs were recorded with a FEI Helios nanolab 600 DualBeam microscope operated at 2.00 kV voltage and 25 pA current. All images were formed by secondary electrons with a 3 μs dwell time. Images of the large HKUST-1 crystals were recorded on a PhenomPro X microscope, equipped with a CeB6 detector and operated at 10 kV voltage. The single-crystals were supported on C tape deposited over Al stabs (FEI stabs) and inserted in the vacuum chamber without any Au or Pt coatings. X-ray Photoelectron Spectroscopy (XPS) measurements were performed using a Thermo Fisher Scientific ESCALAB 250Xi (UK) instrument equipped with a monochromatic Al Kα radiation source (1486.6 eV). The powder samples were placed into an aluminium sample holder and pressed using a custom-made steel presser and degassed until the base pressure reaches ca. 5 × 10−7 mbar. Core level XPS spectra of Cu 2p, N 1s, O 1s and C 1s were recorded with 10 eV pass energy and 0.01 eV step size and 5 spectra were averaged. The XPS spectra were deconvoluted and fitted with a smart background employing mixed Gaussian–Lorentzian line shapes using the Avantage software (Version 5.986).Conductivity and water uptake isotherms
Water sorption isotherms were collected with a Quantachrome Hydrosorb 1000 instrument controlled with the software HydroWin v5. In a N2-filled glovebox, ca. 50 mg of pre-activated 1.0TCNQ@HKUST-1 was transferred to a sample cell. Five adsorption–desorption cycles were performed at 298 K. Before every cycle, the sample was evacuated at 298 K until constant pressure was achieved. The temperature of the sample cell was controlled by a water bath set at 298 K. Electrical conductivity measurements were conducted on compressed powder pellets. After the sample preparation in a glove box, the previously described custom-made measuring cell was connected to a Zahner-Zennium electrochemical workstation.28 Electrochemical impedance spectroscopy (EIS) measurements were performed and an alternating voltage of 10 mV was employed in the frequency range from 1 Hz to 1 MHz at 0 V DC voltage. Ion current (I)–voltage (V)-curves were collected in the voltage range from −1 to 1 V with a slow rate of 100 mV s−1.Micro-spectroscopy and data analysis
Raman micro-spectroscopy was performed with a RenishawTM inVia microscope, using a 532 nm diode excitation laser with a 20× magnification objective, a 1200 lines mm−1 grating and a Charge Coupled Device (CCD) detector. The objective has a Numerical Aperture (NA) of 0.4, resulting in an 815.5 × 815.5 μm2 diameter spot size. All measurements were performed with a total 0.44 mW power output (power density: 1.27 × 10−5 W cm−2) in the 1500–2300 cm−1 range. In a glovebox, the samples were transferred to a sealed Linkam cell equipped with CaF2 windows and filled with 1 bar of Ar (99.999% Linde AG). Data analysis of the Raman microscopy maps was performed using WiRE 3.4© (Renishaw) and TXM Wizard29 software packages. Principal Component Analysis (PCA) of the signal-to-baseline or single wavelength intensity maps was performed with Matlab R2018a (Mathworks). PCA was done by K-means clustering of pixels with similar spectral features, resulting in Voronoi cells grouped in four different clusters. In order to define the representative components (eigenvectors) of the PCA analysis, the Scree test and Cumulative Variance Explained (CVE) tests are shown (Fig. S1, ESI†). It is clear that with three to four components (Fig. S2, ESI†), a representative part of the data set can be analysed without losing relevant information. Deconvolution of the bands of the average spectra obtained from cluster analysis was carried out using the Fityk 1.3.1© package (Nelder–Mead simplex).Fourier-Transform Infra-Red (FT-IR) micro-spectroscopy was carried out using a PerkinElmer Frontier MIR/FIR spectrometer coupled to a PerkinElmer Spotlight 400 FT-IR microscope, with a Mercury–Cadmium–Telluride (MCT) detector cooled with liquid N2, in a 16 × 16 array of 6.25 × 6.25 μm pixels. The samples were kept in sealed cells equipped with CaF2 windows, under inert conditions. Then, a flow of 60 mL min−1 nitrogen gas (99.99%, Linde AG) saturated with demineralized water (at 298 K) was introduced into the sample cell for 5 min, stopping every 30 s to collect FT-IR microscopy maps of the HKUST-1 crystals.
Results and discussion
The two materials under study, namely large crystals of HKUST-1 and TCNQ@HKUST-1, were obtained by a method described in a previous report by Schneider et al.,26 in which their crystallinity, morphology and porosity are reported. TCNQ was introduced by vapour phase infiltration in an evacuated glass ampoule. During this treatment, TCNQ sublimes and diffuses into the large pores of HKUST-1, in which only half contain uncoordinated Cu sites pointing to the centre of the pore. As shown by Schneider et al., TCNQ chemically binds via its two terminal nitrile groups to two neighbouring, i.e. in opposite sides of the pore cage, metal nodes, as evidenced by the increasing intensity of the (111) reflection in the X-ray diffraction data.26 In addition, physisorbed TCNQ fills the remaining pore space and weakly interacts with the MOF. This leads to loadings reaching up to two TCNQ molecules per large pore, while maintaining pore space accessible for small molecules, as shown by sorption experiments with N2 and H2O.To study the spatial distribution of TCNQ molecules within a HKUST-1 crystal after vapour phase infiltration, Raman micro-spectroscopy was used as the characterization method, as illustrated in Fig. 1. Raman spectroscopy has been extensively used for characterizing TCNQ and Cu-TCNQ compounds, due to the fact that the Raman intensity is highly sensitive to charge transfer processes, such as the binding to metal substrates or electrical switching.30–32 By applying Raman spectroscopy in a spatially resolved manner with a resolution of ∼1 μm, chemical 3-D imaging of the HKUST-1 crystals can be achieved by focussing an incident laser beam on the sample, in combination with pinhole filtering of out-of-plane contributions and signal detection using a charge coupled device (CCD) array detector. If an objective with a long working distance is used, a sealed Linkam cell with CaF2 windows can be placed at the focal point of the beam, which allows us to monitor the dynamics of the MOF material under in situ conditions.33,34 After data collection, Principal Component Analysis (PCA) and Cluster Analysis (CA) can be used to extract the variance of the dataset and to classify the Raman spectra into different groups with similar spectral features.35,36
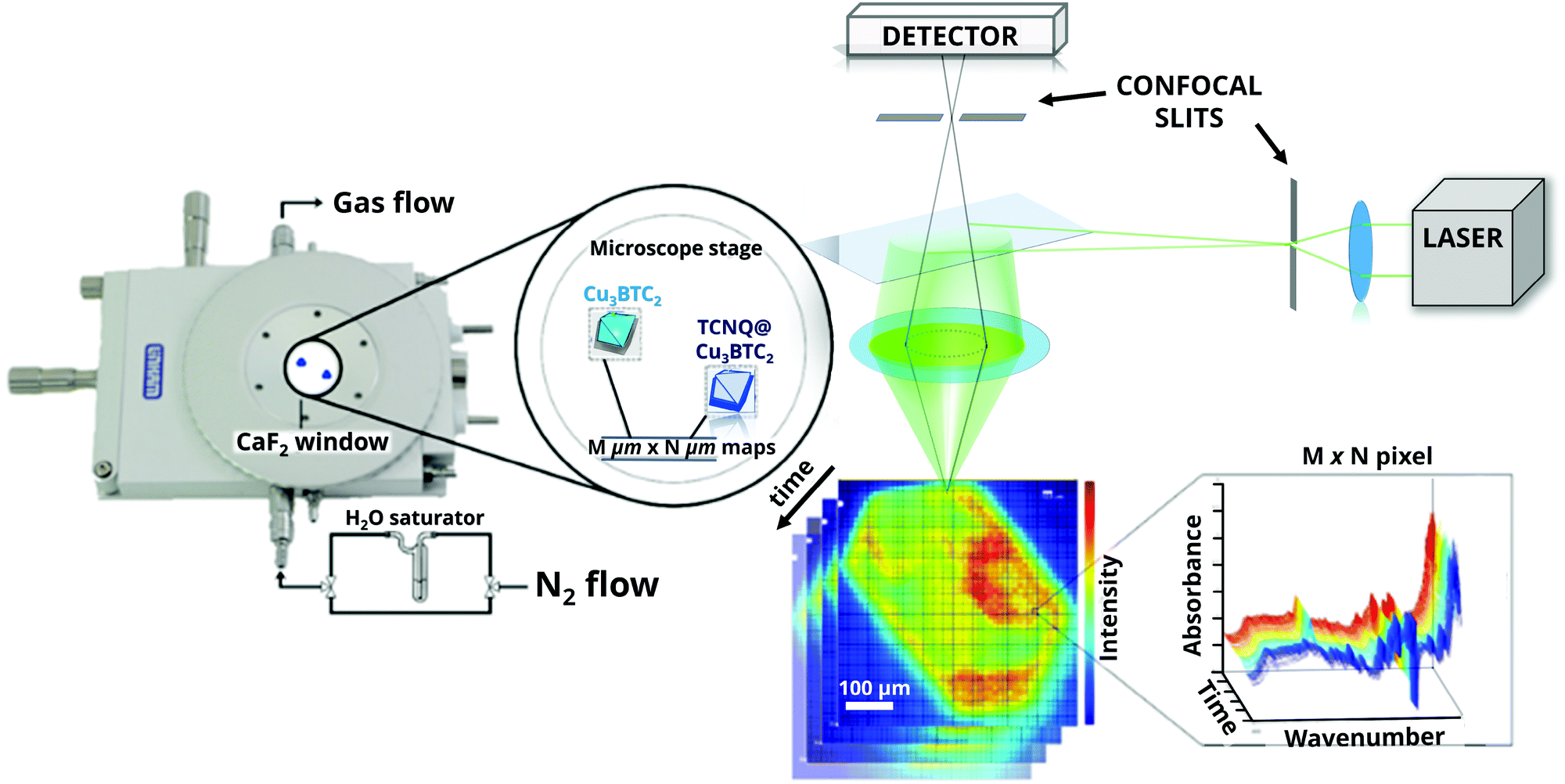 | ||
| Fig. 1 Micro-spectroscopic approach used to investigate the influence of H2O in large crystals of HKUST-1 and TCNQ@HKUST-1 materials. Confocal Raman microscopy allows 3-D in situ chemical imaging of MOF crystals under dynamic conditions by focussing the laser beam into selected (X,Y) areas of the crystal. A saturator can be connected to an air-sealed cell in order to dose water vapour. | ||
Raman micro-spectroscopy maps of a HKUST-1 crystal, loaded with TCNQ, are given in Fig. 2a and b. The Raman intensity map in Fig. 2a displays the aromatic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching band (νCC) at 1605 cm−1 and visually shows where the HKUST-1 crystal and TCNQ are relatively located, since this Raman band is present in the BTC linker of the host material and in TCNQ as guest molecule (Fig. S3, ESI†). In Fig. 2b, the Raman map of the 2226 cm−1 band, associated with stretching of TCNQ C
C stretching band (νCC) at 1605 cm−1 and visually shows where the HKUST-1 crystal and TCNQ are relatively located, since this Raman band is present in the BTC linker of the host material and in TCNQ as guest molecule (Fig. S3, ESI†). In Fig. 2b, the Raman map of the 2226 cm−1 band, associated with stretching of TCNQ C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N nitrile groups (νCN) alone, is also shown. In order to identify and group the Raman spectra within the maps into different classes, PCA and CA have been performed using all the recorded Raman spectra within the map. Aside from extracting the variance of the dataset, the analysis minimizes the influence of scattering, out-of-focus contributions, cosmic rays or impurities that may affect the signal and obscure the interpretation. In the current case, four different clusters are obtained, exhibiting fingerprint spectra that mainly differ in their 1605 (νCC) and 2226 cm−1 (νCN) band intensity and thus their abundance of TCNQ (Fig. 2c and d).
N nitrile groups (νCN) alone, is also shown. In order to identify and group the Raman spectra within the maps into different classes, PCA and CA have been performed using all the recorded Raman spectra within the map. Aside from extracting the variance of the dataset, the analysis minimizes the influence of scattering, out-of-focus contributions, cosmic rays or impurities that may affect the signal and obscure the interpretation. In the current case, four different clusters are obtained, exhibiting fingerprint spectra that mainly differ in their 1605 (νCC) and 2226 cm−1 (νCN) band intensity and thus their abundance of TCNQ (Fig. 2c and d).
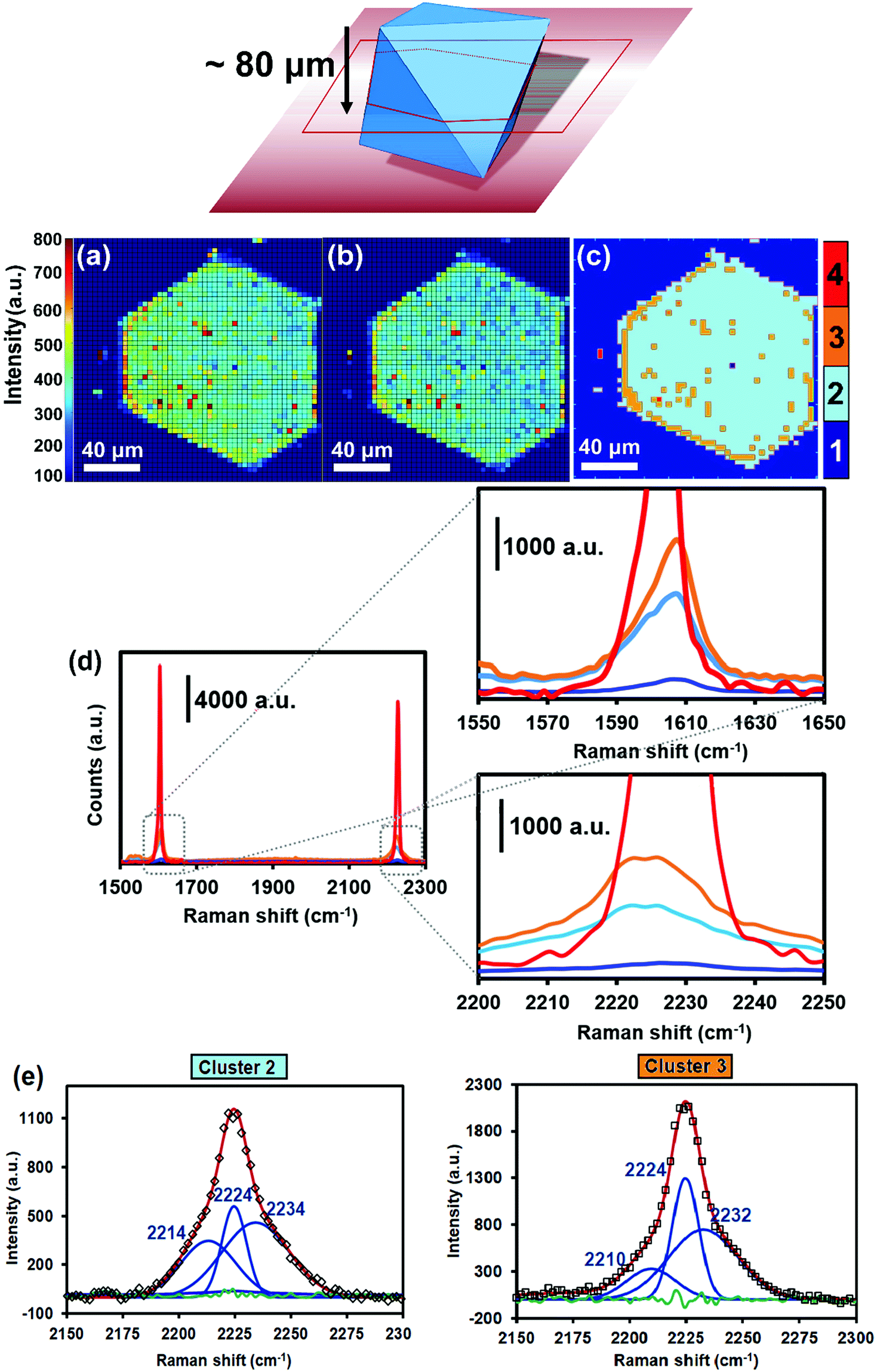 | ||
| Fig. 2 (Top) Scheme showing the cross-section of two intertwined, octahedral HKUST-1 crystals, loaded with TCNQ, and probed with the Raman laser at a depth of z ∼ 80 μm. Signal-to-baseline ratio maps of the Raman bands at (a) 1605 cm−1 (corresponding to the CC aromatic stretching vibration) and at (b) 2226 cm−1 (corresponding to the CN stretching vibration) of a HKUST-1 crystal, loaded with TCNQ. (c) Different spectral clusters obtained from Principal Component Analysis (PCA) of the 1500–2300 cm−1 region, showing different intensities throughout the TCNQ@HKUST-1 crystals. (d) Raman spectra corresponding to each cluster of the map in (c). Scale bars indicate 40 μm in all cases. (e) Deconvolution of the nitrile stretch band in the average spectra of clusters 2 and 3 seen in (d). The green line corresponds to the residual of the Nelder–Mead simplex algorithm used. | ||
Cluster 1 in Fig. 2c represents the spectra containing limited TCNQ Raman intensity. These spectra represent regions located outside the TCNQ@HKUST-1 crystal, i.e. where no TCNQ is present. Cluster 2 contains fingerprint spectra that exhibit moderate TCNQ abundance, mostly populating the crystal interior. Cluster 3 contains spectra that expose increased TCNQ intensity relative to the moderately intense spectra in cluster 2. Pixels classified in cluster 3 mainly situate at the crystal edges, implying that the TCNQ abundance increases radially toward the outside of the HKUST-1 crystal and therefore resembles a core–shell structure. In addition, small, non-uniformly dispersed islands of cluster 3 are observed across the HKUST-1 crystal, implying fluctuations in the TCNQ concentration after initial infiltration. Finally, cluster 4 groups spectra with TCNQ concentrations an order of magnitude higher compared to clusters 2 and 3. Cluster 4 pixels are only observed at a spatially remote location outside TCNQ@HKUST-1, suggesting that this area is represented by TCNQ condensates.
Fig. 3 shows the micrographs of the crystals of pristine HKUST-1 and the TCNQ@HKUST-1 host–guest composite material. In line with previous experiments, a Cu-TCNQ coordination polymer26 can be seen at the surface of the TCNQ@HKUST-1 host–guest composite material. The bands corresponding to both materials were present after deconvolution of the average Raman spectra of each cluster (see Fig. 2e): one contribution arising from physically deposited TCNQ, at 2224 cm−1, and a smaller component at 2210–2214 cm−1, assigned to TCNQ coordinated to the Cu paddlewheels. In contrast, no clear signs of the Cu-TCNQ coordination polymer, which typically shows the CN stretch at 2205 cm−1,32,37,38 were observed. It is important to bear in mind that the average spectra contain the spectral contribution of different pixels located at the edge of the crystal and other areas. Further, the pixel size of 1 × 1 μm2 is not small enough to probe nanometric needles of Cu-TCNQ and TCNQ@HKUST-1, preventing us from a clear distinction of species, yet corroborating different compounds within the TCNQ@HKUST-1 crystals. The focal point of the laser was set to a depth of z ∼ 80 μm from the top edge, allowing us to obtain chemical maps of the cross-sections, rather than the top crystal surfaces of the TCNQ@HKUST-1 host–guest composite material. The formation of aggregates on the edges is due to diffusion barriers that form during the vapour-phase infiltration process, in which the TCNQ molecules that enter the pores first coordinate strongly to the Cu2+ Lewis sites and block these pores as diffusion pathways, preventing other TCNQ molecules from entering the MOF crystal. Not only coordination within the pores, but also formation of a thin, nanometric layer of the dense Cu-TCNQ coordination polymer on the external surface of the HKUST-1 crystals might play a role. The defective Cu+ sites on the external crystal surface react with gaseous TCNQ at high temperature, forming the by-phase observed by SEM (Fig. 3). Additional TCNQ entering the MOF material may interact in different manners with the surface of HKUST-1. For instance, it may be physisorbed in the smaller pore cages, without interacting with any Cu sites, as the Raman band at 2225 cm−1 corresponds to neutral, non-reduced TCNQ (i.e., non-coordinated to the Cu centres).39 Alternatively, it could only partially transfer the electron density towards the Cu centres, keeping the nitrile stretches unchanged in the Raman spectrum.
 | ||
| Fig. 3 SEM micrographs of the crystals of (a) pristine HKUST-1 and (b) the TCNQ@HKUST-1 host–guest composite material. Cu-TCNQ needles covering the surface of the crystal are clearly visible at higher magnifications in (d), in comparison to the neat surface observed in (c). | ||
Yet another possibility would be that TCNQ interacts with a defect Cu+ site and undergoes a redox reaction yielding an oxidized Cu2+ site and a reduced TCNQ− molecule. Thus, it is difficult to offer a detailed molecular description of the TCNQ@HKUST-1 system. This layer of condensed TCNQ has a thickness of ca. 5 μm, i.e. 1–2 pixels in the Raman maps, in contrast to the typical nanometric films of the Cu-TCNQ coordination polymer formed on the surface.40 Important to mention here is that despite the formation of these aggregates, no signs of dimeric TCNQ radicals41,42 were present in the Raman spectra. To verify that this finding is consistent, two intertwined, octahedral HKUST-1 crystals were mapped and analysed following the same procedure. The maps and cluster analysis are presented in Fig. S4 (ESI†), and they also show the characteristic aggregation of TCNQ in the inner edges of the HKUST-1 crystals. Some zones in proximity to the surface also showed a few aggregates of TCNQ. Thus, we could corroborate that the formation of this layer of TCNQ is widespread throughout the TCNQ@HKUST-1 sample probed.
It is known that HKUST-1 crystals have a limited stability with respect to moisture and water, having potential consequences ranging from Cu reduction to pore collapse depending on the exposure time and water vapour pressure.43–48 To ensure that no such effects occur during the hydration experiments in this work, in situ XRD experiments were performed by recording diffractograms during a temperature-programmed experiment with dosing of water-saturated He gas (60 mL min−1 He flow through a bubbler filled with demineralized H2O following the temperature program depicted in Fig. S4 (ESI†), at a heating rate of 5 K min−1). The XRD patterns show that the structure is stable at temperatures up to 573 K and all Bragg reflections are highly intense throughout the whole experiment.49,50
Hydrosorption isotherms over 5 cycles (Fig. S6, ESI†) showed that the total water sorption capacity decreases slightly after cycling. This suggests that although no major structural losses occur, a certain fraction of the surface is lost. This may be ascribed to pore collapse, or to irreversible chemisorption leading to a loss of surface sites. In order to further study the effect of pore blockage on the HKUST-1 crystal edges, caused by TCNQ aggregates, in situ FT-IR microscopy of the materials under study was performed (Fig. 4). However, no signs of framework decomposition were detected, as the XRD peaks showed the same intensity before and after high-temperature vapor dosing. These findings are in line with those of Giovine et al.,51 where decomposition of HKUST-1 in air or moisture does not occur on short timescales, comparable to the exposure times in our experiments. This confirms that the HKUST-1 framework is stable under the described experimental conditions and allows for its in situ micro-spectroscopic study from room temperature up to 573 K in H2O-saturated He or N2 flows. More specifically, we have studied the process of hydration by monitoring the evolution of the FT-IR band at 3400 cm−1, which corresponds to the O–H stretching mode of water, of both pristine HKUST-1 and TCNQ@HKUST-1 crystals. Fig. 4 shows the evolution of two different crystals (parent HKUST-1, on top; and TCNQ@HKUST-1, below) under a stream of N2 saturated with water for 5 min at 298 K. The maps show the absorbance of the IR band at 3400 cm−1 over time for each pixel present in the crystal, where water penetrates the pores from the right side (incoming flow in the cell). At different time steps, from t = 0 to t = 420 s, the absorbance increases steadily on different parts of both crystals: first edges, and then the core of the MOF, indicating that unlike TCNQ, it can flow through the pores with lower diffusional barriers.
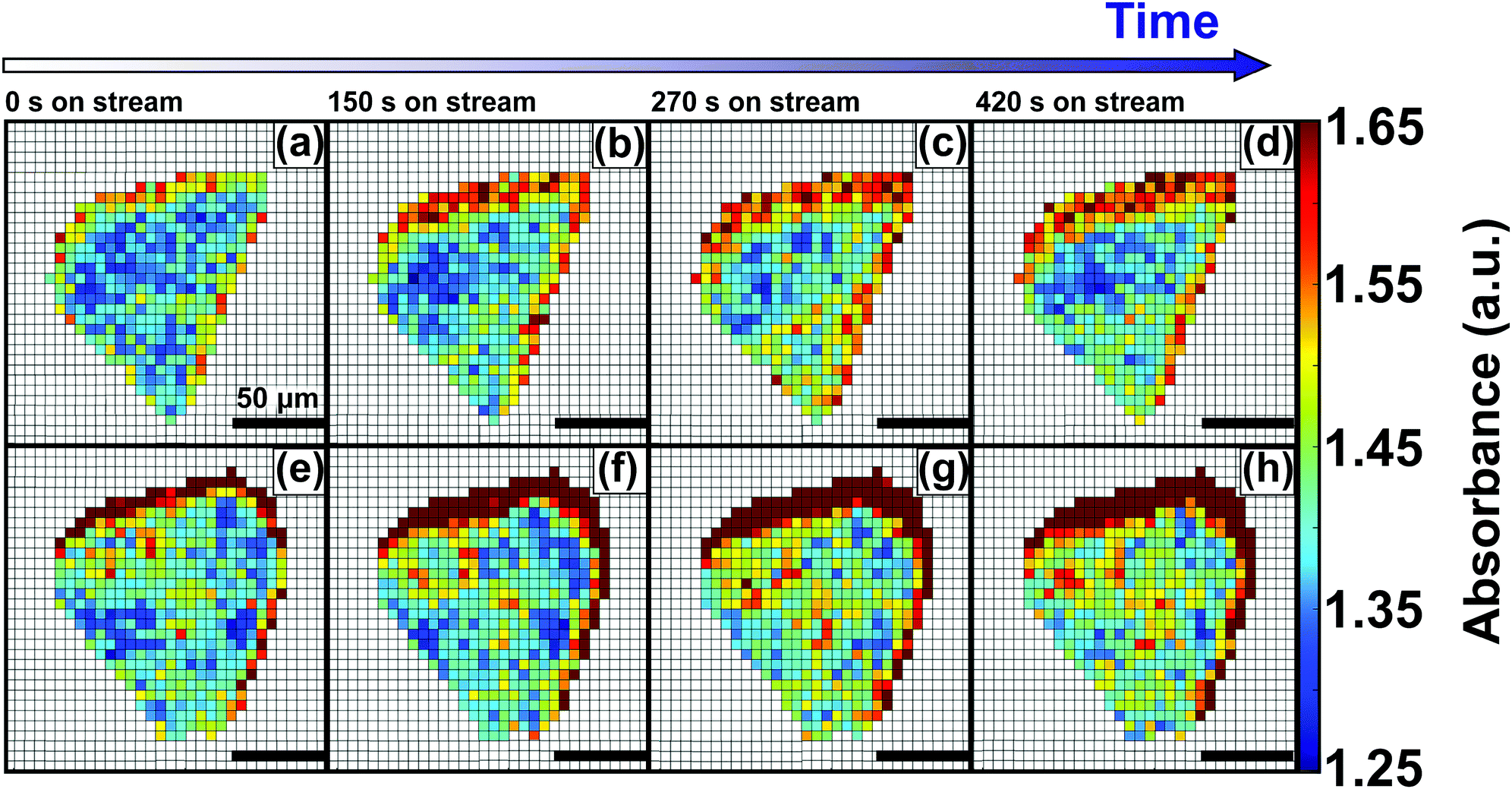 | ||
| Fig. 4 In situ FT-IR microscopy maps of the intensity of the band at 3400 cm−1 of (top; a–d) pristine HKUST-1 and (bottom; e–h) TCNQ@HKUST-1 after hydration by water vapor at room temperature for 0, 150, 270 and 420 s. Note that the preferential uptake on the right side is due to the incoming flow from that side of the spectroscopic cell. All scale bars represent 50 μm. | ||
To obtain more quantitative information, we have plotted the evolution of the absorbance of the 3400 cm−1 band over the entire HKUST-1 crystal, in the absence and presence of TCNQ. The increase in the absorbance of the 3400 cm−1 band, averaged for all the pixels corresponding to the materials, shows a gradual saturation after ca. 5 min (300 s) (Fig. 5).
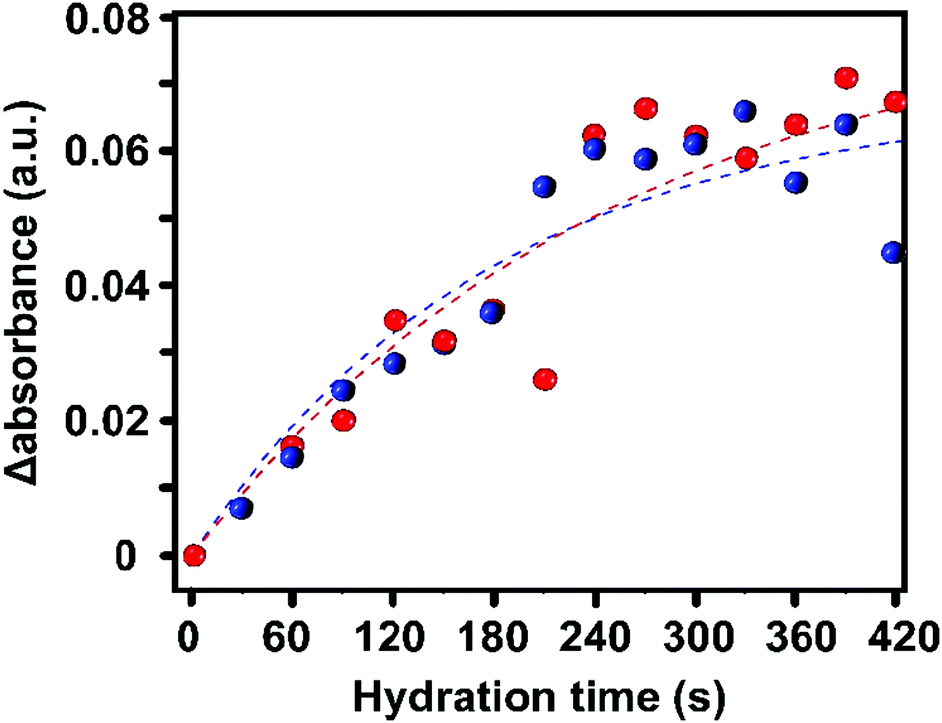 | ||
| Fig. 5 Evolution of the average change in absorbance of all the pixels for each crystal: pristine (blue) and TCNQ@HKUST-1 (red) during the adsorption of water. Note that there is not a significant difference at the bulk scale. | ||
This indicates that the pores are filled with water vapor very shortly after the dosing starts, in line with previous experiments showing the high capacity and kinetics of HKUST-1 for that purpose.52,53 Indeed, water molecules rapidly fill the pores and coordinate to any remaining coordinatively unsaturated Cu2+ sites that may not be occupied by TCNQ. The similar profiles suggest that TCNQ does not play a significant role in hindering the maximum water uptake of HKUST-1 crystals at the bulk scale. As mentioned above, the FT-IR micro-spectroscopy maps, shown in Fig. 4, reveal spatial differences in the water uptake depending on the distance to the centre of the HKUST-1 crystal. In order to better understand the kinetic behaviour of hydration, we have analysed the evolution of the band at 3400 cm−1 with respect to the distance from the centre of the crystal. An algorithm, applied to the FT-IR microscopy maps, recorded during the hydration experiments was used to analyse the average absorbance of the band with an increasing radial distance. The results are given in Fig. 6a, where the red area represents pixels further away from the centre of the HKUST-1 crystal (blue area). The HKUST-1 crystal was divided into 11 different areas from crystal surface to centre, as described in the related schematic. In Fig. 6b and c, the plots show that the average absorbance of the 3400 cm−1 band progressively decays from outer areas of the HKUST-1 crystal, i.e. surface (pixel 1), to inner zones (pixel 11). This observation holds for both the pristine HKUST-1 and TCNQ@HKUST-1 crystals. It is important to note that Fig. 6c shows that the decay observed between pixels 1 to 3 (a radius of around 18.75 μm) is more pronounced in the case of the TCNQ-loaded crystals. This indicates that, indeed, although the guest molecule aggregates do not decrease the total uptake significantly, they do hinder diffusion to some extent within the surface pores towards the inner areas of the HKUST-1 crystal. These different diffusional behaviours can be better seen in Fig. 6d where the apparent rate constant of hydration over time for pixels of increasing distance to the centre of the HKUST-1 crystal was calculated.
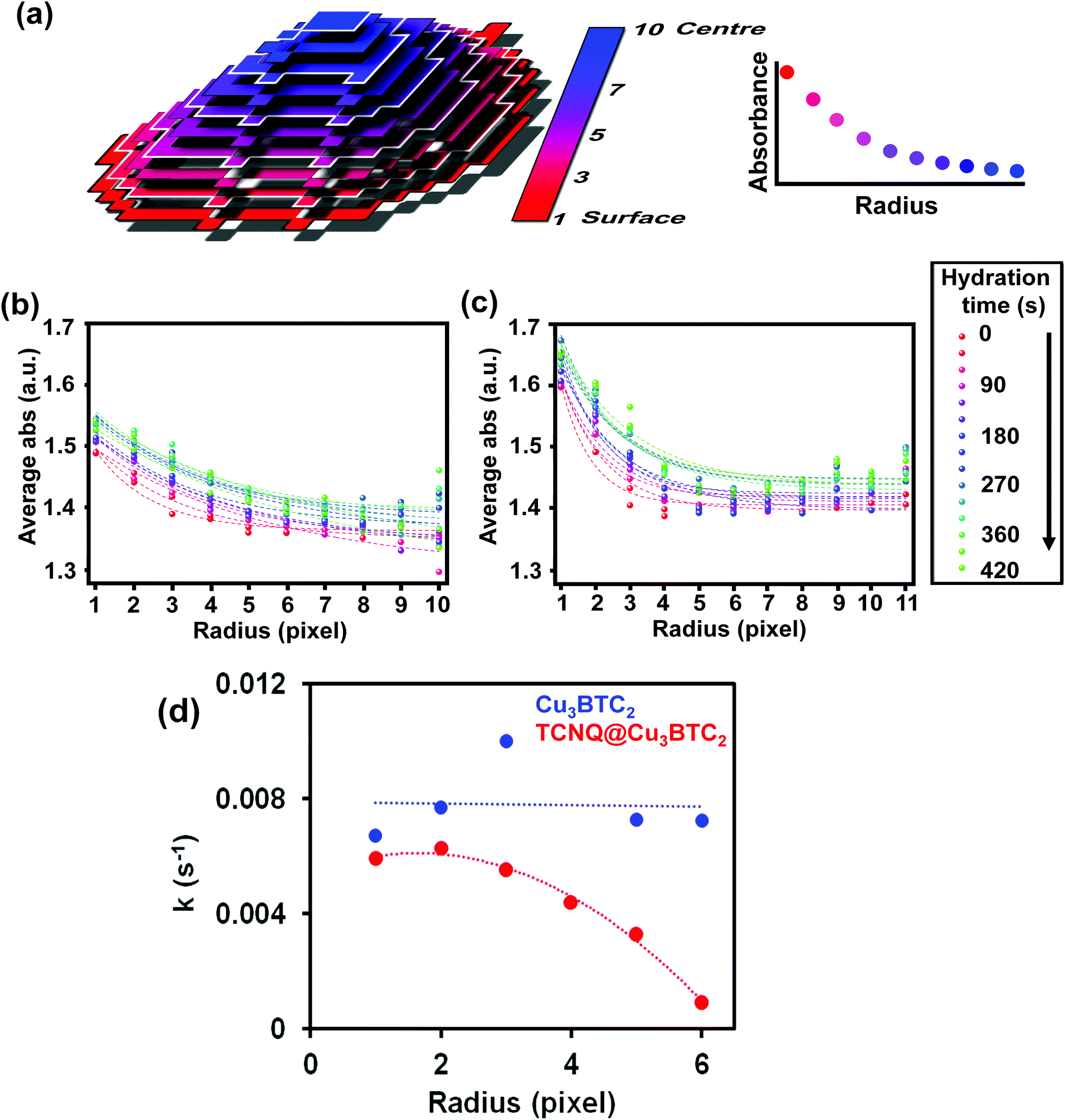 | ||
| Fig. 6 (a) Scheme of the algorithm applied to understand water uptake with respect to the centre of the HKUST-1 crystal. Average absorbance of the water band at 3400 cm−1 for different pixels at increasing distances from the central part of the (b) pristine HKUST-1 and (c) TCNQ@HKUST-1 host–guest composite material. (d) Rate constant (k) for the hydration vs. radius describing the diffusion barrier created by TCNQ at the edges of the HKUST-1 crystals. | ||
The apparent rate constant of increase in absorbance was fitted to a function of the type: ΔAbs = A(1 − e−kt), where A represents the maximum Δ absorbance, t is the hydration time and k is the rate constant of hydration. It is clear that in the pristine HKUST-1 material, hydration occurs at a similar rate throughout the entire MOF crystal. However, the TCNQ-loaded material shows decreasing rates for increasing distance to the surface, indicating that the areas on the outer cores hydrate at similar rates as the pristine HKUST-1, but the areas closer to the centre of the crystal need longer times to hydrate. In other words, water molecules need longer times to reach the inner areas of TCNQ@HKUST-1 and interact with the Cu2+ sites, assuming that the interaction with the MOF occurs much faster than diffusion.
In addition to pore blockage purely due to steric hindrance of water molecules diffusing through, one may as well argue that TCNQ is highly hydrophobic. TCNQ is soluble in organics (THF, CH3CN or dioxane) due to its estimated low dipole moment ![[D with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0044_20d1.gif) ≈ 0.04 D (similar to that of n-hexane); but it is sparingly soluble in water. This may render TCNQ@HKUST-1 more resistant to water absorption and hosting, as previously shown for HKUST-1 etched with fluorohexane.54
≈ 0.04 D (similar to that of n-hexane); but it is sparingly soluble in water. This may render TCNQ@HKUST-1 more resistant to water absorption and hosting, as previously shown for HKUST-1 etched with fluorohexane.54
In order to investigate if the inclusion of water had an impact on the spatial distribution of TCNQ, Raman micro-spectroscopy maps of the band corresponding to the nitrile stretch of TCNQ@HKUST-1 (located at 2226 cm−1) after the hydration experiments were recorded. Importantly, PCA of the Raman spectra obtained reveals that the spatial heterogeneities within the HKUST-1 crystals are not present anymore (see Fig. 7) and consequently a very different picture is observed. Cluster 2 (light blue), with a lower average intensity than Cluster 3 (orange), is present on the crystal edges and outer sections, i.e. the intensity on the surfaces is now lower than in the crystal core and no signs of heterogeneities are observed. This indicates that TCNQ, in contrast to the Raman micro-spectroscopy maps of Fig. 2 and Fig. S3 (ESI†), is more homogeneously distributed throughout the HKUST-1 crystal. Evidently, exposing the HKUST-1 crystals to water vapor results in a redispersion of the TCNQ guest molecules, filling the pore cavities of the MOF material in a more homogeneous manner. A possible explanation is the competition between the excess of water molecules with TCNQ for available Cu unsaturated sites, thereby liberating TCNQ from its strong coordination to the MOF and allowing for diffusion within the pores.
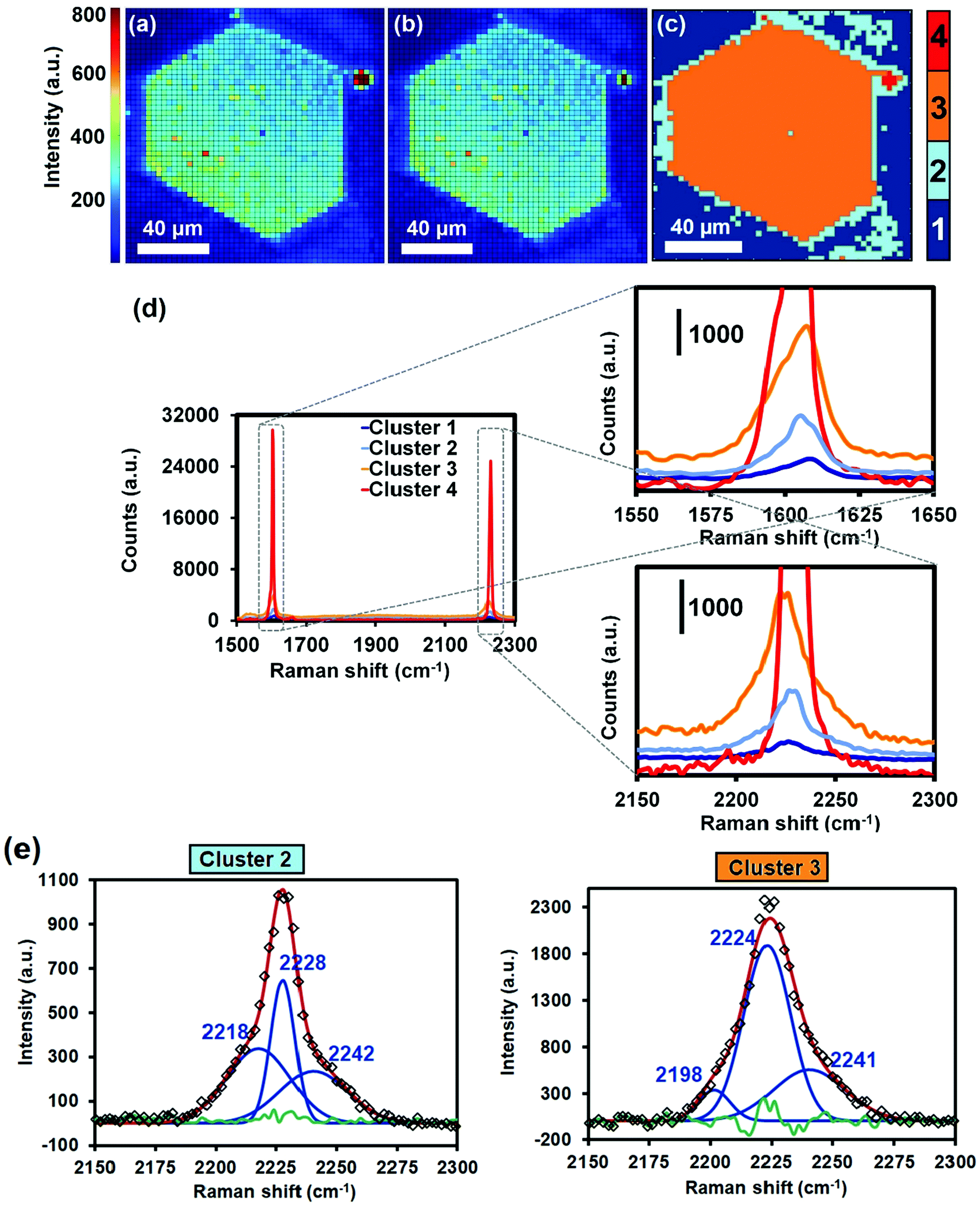 | ||
| Fig. 7 Raman maps of the crystal shown in Fig. 2 after bubbling N2 saturated with deionized water through the sealed cell for 5 min at 298 K. Signal-to-baseline ratio maps of the Raman bands at (a) 1605 cm−1 (corresponding to the CC aromatic stretching vibration) and at (b) 2226 cm−1 (corresponding to the CN stretching vibration) of TCNQ. (c) Different spectral clusters obtained from Principal Component Analysis (PCA) of the 1500–2300 cm−1 region showing different intensities throughout the two adjacent TCNQ@Cu3BTC2 crystals. (d) Raman spectra corresponding to each cluster of the map in (c). (e) Deconvolution of the band corresponding to the nitrile stretch in clusters 2 and 3 of the average spectra shown in (d). The green line corresponds to the residual of the Nelder–Mead simplex algorithm used. | ||
To study the effect of this TCNQ redispersion on the electrical conductivity of the TCNQ@HKUST-1 material, we have measured I–V scans and EIS of the materials under Ar at 298 and 378 K first and after exposure to the atmosphere at those temperatures. It is worth mentioning that the conductivity measurements were carried out using μm-sized Cu3BTC2 powders (10–20 μm octahedra) infiltrated with TCNQ, as previously described in the literature.26 However, we believe that the redispersion phenomenon may also be present in the case of those powdered samples. Previous studies have suggested that TCNQ, aligned within the pores of HKUST-1, leads to a percolation system through which electrons can hop from one pore to the next one. It may be hypothesized that water vapour adsorbed in the pores induces the redispersion of the TCNQ molecules. Competitive coordination of H2O to the Cu cations would then lead to a loss in long range ordering of the TCNQ molecules aligned in the pores that can be observed in the (111) reflection of the XRD patterns. In Fig. S7 (ESI†), it can be seen how the intensity of this reflection at 5.8° strongly decreases upon exposure of the material to air. This suggests that moisture affects the percolation network through which electrons are mobile, with implications for the material's performance as an electrical conductor.
Indeed, using the current–voltage (I–V) graphs of the material at different temperatures (Fig. S8, ESI†), the activation energy for electron conduction was obtained. The Arrhenius plot of the charge transfer of TCNQ@HKUST-1 before hydration reveals a temperature activated conductivity mechanism with an activation energy of 0.18 eV. The exposure of the sample to a low level of relative humidity, i.e. room atmosphere at 298 K, results in a slight decrease of the conductivity (Fig. S9, ESI†). It was found that a high level of relative humidity does not induce a fast and pronounced response of the TCNQ@HKUST-1 material. However, after prolonged exposure (ca. 68 h) to a humidified atmosphere, different unknown electrochemical processes lead to a slight increase in measured current and a slight deviation from its ohmic behaviour, as illustrated in Fig. S9 (ESI†).
In a compressed powder pellet, the interaction between MOF particles plays an important role in the charge transfer. In this manner, it can be assumed that a high loading of TCNQ molecules in the outer shell of the HKUST-1 material is beneficial for the charge transfer. An alternative explanation for electrical conductivity is the presence of the Cu-TCNQ coordination polymer on the crystal surfaces, which would act as a conductive coating over the crystals.40 The lower intensity of the CN vibrational band around the crystal edges in the Raman micro-spectroscopy cluster maps, shown in Fig. 2 and Fig. S3 (ESI†), suggests that this layer is affected by the presence of moisture. This may be an alternative explanation for the drop in conductivity after exposure to water. The absence of a pronounced tail in the EIS measurements (Fig. S10, ESI†) of the humidified samples indicates a low mobility of H2O in the framework. If any, there is just a minor ionic contribution to the overall conductivity of the system, which is in line with a slow diffusion of H2O in TCNQ@HKUST-1.
In order to understand the nature of the species on the HKUST-1 surface, X-ray Photoelectron Spectroscopy (XPS) measurements before and after measuring electrochemical impedance, as well as after the water sorption experiments, were performed (Fig. S11, ESI†). It can be noted that the N 1s spectra of the pristine TCNQ@HKUST-1 material are characterized by two prominent XPS peaks centred at ∼398.91 eV and ∼399.98 eV as well as by an XPS shake-up at ∼401.59 eV. The two main XPS peaks can be assigned to the coordinated and uncoordinated CN groups respectively, and these peak positions are consistent with literature reports.26,55 Upon exposure to moisture (and also after the conductivity measurements), only one prominent XPS peak at ∼399.5 (and ∼399.2 eV, respectively) with an XPS shake-up feature at slightly higher energy is clearly observed. This finding corroborates that water affects the coordination of TCNQ molecules to the Cu centres. Moreover, the Cu 2p3/2 XPS peak can be decomposed into two contributions, assigned to Cu+ and Cu2+ species. The XPS satellite features become more pronounced upon water vapor exposure and the conductivity measurement under a humidified atmosphere, which suggests a slow oxidation of Cu+ to Cu2+. From the literature, it is known that Cu2+ species can be characterized by strong XPS satellite peaks.56 The ratios of Cu+/Cu2+ at the crystallite surface were estimated from the integrals of the XPS peak areas resulting in a value of 2.64 for the pristine sample and values of 1.97 and 1.94 after water vapor adsorption and conductivity measurements, respectively, for the TCNQ@HKUST-1 host–guest composite material. The values obtained for the pristine HKUST-1 sample are in good agreement with the previously reported data.26 Furthermore, the O 1s XPS spectra show two main peaks originating from adsorbed water (531.5–531.9 eV) and from the organic linker BTC molecules (>533 eV). These changes confirm that different processes affect the TCNQ@HKUST-1 host–guest composite material upon dosage of water vapour.
Conclusions
By taking large HKUST-1 MOF crystals as a host material and TCNQ as a guest molecule, we have shown that new physicochemical insights can be obtained into the underlying processes of guest@MOF composites by employing a set of characterization methods. These methods include amongst others Raman micro-spectroscopy, IR micro-spectroscopy, X-ray Photoelectron Spectroscopy (XPS) and Scanning Electron Microscopy (SEM). For example, Raman micro-spectroscopy and subsequent Principal Component Analysis (PCA) of the data obtained showed that large agglomerates of TNCQ are formed on the inner surface of the MOF crystals. In addition to Cu-TCNQ coordination polymer needles visible by SEM, these agglomerates were present within a few microns of the surface.Moreover, in situ IR micro-spectroscopy experiments of the degassed crystals under dynamic hydration conditions showed that the observed TCNQ agglomerates lead to the formation of diffusional barriers, in comparison with pristine HKUST-1 crystals, in which hydration is less hindered by diffusion. Not only is hydration affected, but also the spatial distribution of TCNQ was modified upon exposure of the crystals to nitrogen saturated with water vapour, re-dispersing the guest TCNQ molecules within the host HKUST-1 crystals. Electrical conductivity measurements showed that hydration of the TCNQ@HKUST-1 guest–host composite material results in a slight decrease of conductivity. This study highlights the importance of using a multi-technique approach to tackle the lack of understanding in the functioning of MOFs as functional materials for different applications. The use of this ensemble of characterization tools paves the way for more advanced, spatially-resolved studies of MOF crystals.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Union Horizon 2020 Research and Innovation program under the Marie Sklodowska-Curie grant agreement 641887 (DEFNET) as well as from the priority program SPP 1928 (COORNETs) of the German Research Foundation (DFG). M. F. acknowledges the European Union's Horizon 2020 Research and Innovation program under the Marie Sklodowska-Curie grant agreement (No. 748563). This work is supported by the Netherlands Organization of Scientific Research (NWO) Gravitation program (Netherlands Center for Multiscale Catalytic Energy Conversion, MCEC). C. S. gratefully thanks the German Academic Scholarship Foundation and the German Chemical Industry Fund (FCI) for financial support.References
- H.-C. Zhou, J. R. Long and O. M. Yaghi, Chem. Rev., 2012, 112, 673–674 CrossRef CAS PubMed.
- N. Stock and S. Biswas, Chem. Rev., 2012, 112, 933–969 CrossRef CAS PubMed.
- F. Meirer, D. T. Morris, S. Kalirai, Y. Liu, J. C. Andrews and B. M. Weckhuysen, J. Am. Chem. Soc., 2015, 137, 102–105 CrossRef CAS PubMed.
- D. E. Perea, I. Arslan, J. Liu, Z. Ristanovic, L. Kovarik, B. W. Arey, J. A. Lercher, S. R. Bare and B. M. Weckhuysen, Nat. Commun., 2015, 6, 7589 CrossRef PubMed.
- C. E. Harvey, I. E. I. Petterson, B. M. Weckhuysen, C. Gooijer, F. Ariese and A. J. G. Mank, Appl. Spectrosc., 2012, 66, 1179–1185 CrossRef CAS PubMed.
- I. L. C. Buurmans and B. M. Weckhuysen, Nat. Chem., 2012, 4, 873–886 CrossRef CAS PubMed.
- A. M. Beale, S. D. M. Jacques and B. M. Weckhuysen, Chem. Soc. Rev., 2010, 39, 4656–4672 RSC.
- A. M. Katzenmeyer, J. Canivet, G. Holland, D. Farrusseng and A. Centrone, Angew. Chem., Int. Ed., 2014, 53, 2852–2856 CrossRef CAS PubMed.
- R. Ameloot, F. Vermoortele, J. Hofkens, F. C. De Schryver, D. E. De Vos and M. B. J. Roeffaers, Angew. Chem., Int. Ed., 2013, 52, 401–405 CrossRef CAS PubMed.
- W. Schrimpf, J. Jiang, Z. Ji, P. Hirschle, D. C. Lamb, O. M. Yaghi and S. Wuttke, Nat. Commun., 2018, 9, 1647 CrossRef PubMed.
- K. C. Jayachandrababu, D. S. Sholl and S. Nair, J. Am. Chem. Soc., 2017, 139, 5906–5915 CrossRef CAS PubMed.
- A. Ghosh, P. Mukherjee, S. Deb and R. Bhargava, J. Phys. Chem. Lett., 2017, 8, 5325–5330 CrossRef CAS PubMed.
- C. Chmelik, H. Bux, J. Caro, L. Heinke, F. Hibbe, T. Titze and J. Kärger, Phys. Rev. Lett., 2010, 104, 085902 CrossRef PubMed.
- C. Chmelik, F. Hibbe, D. Tzoulaki, L. Heinke, J. Caro, J. Li and J. Kärger, Microporous Mesoporous Mater., 2010, 129, 340–344 CrossRef CAS.
- C. Chmelik, J. Kärger, M. Wiebcke, J. Caro, J. M. van Baten and R. Krishna, Microporous Mesoporous Mater., 2009, 117, 22–32 CrossRef CAS.
- D. Tzoulaki, L. Heinke, H. Lim, J. Li, D. Olson, J. Caro, R. Krishna, C. Chmelik and J. Kärger, Angew. Chem., Int. Ed., 2009, 48, 3525–3528 CrossRef CAS PubMed.
- F. Hibbe, C. Chmelik, L. Heinke, S. Pramanik, J. Li, D. M. Ruthven, D. Tzoulaki and J. Kärger, J. Am. Chem. Soc., 2011, 133, 2804–2807 CrossRef CAS PubMed.
- C. Wiktor, M. Meledina, S. Turner, O. I. Lebedev and R. A. Fischer, J. Mater. Chem. A, 2017, 5, 14969–14989 RSC.
- A. A. Talin, A. Centrone, A. C. Ford, M. E. Foster, V. Stavila, P. Haney, R. A. Kinney, V. Szalai, F. El Gabaly, H. P. Yoon, F. Léonard and M. D. Allendorf, Science, 2014, 343, 66 CrossRef CAS PubMed.
- K. J. Erickson, F. Léonard, V. Stavila, M. E. Foster, C. D. Spataru, R. E. Jones, B. M. Foley, P. E. Hopkins, M. D. Allendorf and A. A. Talin, Adv. Mater., 2015, 27, 3453–3459 CrossRef CAS PubMed.
- T. Neumann, J. Liu, T. Wächter, P. Friederich, F. Symalla, A. Welle, V. Mugnaini, V. Meded, M. Zharnikov, C. Wöll and W. Wenzel, ACS Nano, 2016, 10, 7085–7093 CrossRef CAS PubMed.
- X. Chen, Z. Wang, P. Lin, K. Zhang, H. Baumgart, E. Redel and C. Wöll, ECS Trans., 2016, 75, 119–126 CrossRef CAS.
- L. Sun, G. Campbell Michael and M. Dincă, Angew. Chem., Int. Ed., 2016, 55, 3566–3579 CrossRef CAS PubMed.
- I. Stassen, N. Burtch, A. Talin, P. Falcaro, M. Allendorf and R. Ameloot, Chem. Soc. Rev., 2017, 46, 3185–3241 RSC.
- M. D. Allendorf, R. Medishetty and R. A. Fischer, MRS Bull., 2016, 41, 865–869 CrossRef.
- C. Schneider, D. Ukaj, R. Koerver, A. A. Talin, G. Kieslich, S. P. Pujari, H. Zuilhof, J. Janek, M. D. Allendorf and R. A. Fischer, Chem. Sci., 2018, 9, 7405–7412 RSC.
- T. M. Tovar, J. Zhao, W. T. Nunn, H. F. Barton, G. W. Peterson, G. N. Parsons and M. D. LeVan, J. Am. Chem. Soc., 2016, 138, 11449–11452 CrossRef CAS PubMed.
- M. Feyand, C. F. Seidler, C. Deiter, A. Rothkirch, A. Lieb, M. Wark and N. Stock, Dalton Trans., 2013, 42, 8761–8770 RSC.
- Y. Liu, F. Meirer, P. A. Williams, J. Wang, J. C. Andrews and P. Pianetta, J. Synchrotron Radiat., 2012, 19, 281–287 CrossRef CAS PubMed.
- G. R. Anderson and J. P. Devlin, J. Phys. Chem., 1975, 79, 1100–1102 CrossRef CAS.
- S. Matsuzaki, R. Kuwata and K. Toyoda, Solid State Commun., 1980, 33, 403–405 CrossRef CAS.
- E. I. Kamitsos and W. M. Risen, J. Chem. Phys., 1983, 79, 5808–5819 CrossRef CAS.
- B. M. Weckhuysen, Chem. Soc. Rev., 2010, 39, 4557–4559 RSC.
- E. Stavitski and B. M. Weckhuysen, Chem. Soc. Rev., 2010, 39, 4615–4625 RSC.
- L. Zhang, M. J. Henson and S. S. Sekulic, Anal. Chim. Acta, 2005, 545, 262–278 CrossRef CAS.
- O. A. Maslova, G. Guimbretière, M. R. Ammar, L. Desgranges, C. Jégou, A. Canizarès and P. Simon, Mater. Charact., 2017, 129, 260–269 CrossRef CAS.
- P. G. Gucciardi, S. Trusso, C. Vasi, S. Patanè and M. Allegrini, Phys. Chem. Chem. Phys., 2002, 4, 2747–2753 RSC.
- P. M. Usov, H. Jiang, H. Chevreau, V. K. Peterson, C. F. Leong and D. M. D’Alessandro, J. Phys. Chem. C, 2017, 121, 26330–26339 CrossRef.
- J. S. Chappell, A. N. Bloch, W. A. Bryden, M. Maxfield, T. O. Poehler and D. O. Cowan, J. Am. Chem. Soc., 1981, 103, 2442–2443 CrossRef CAS.
- K. Thürmer, C. Schneider, V. Stavila, R. W. Friddle, F. Léonard, R. A. Fischer, M. D. Allendorf and A. A. Talin, ACS Appl. Mater. Interfaces, 2018, 10, 39400–39410 CrossRef PubMed.
- S.-i. Terashita, K. Nakatsu, Y. Ozaki and S. Takagi, J. Phys. Chem., 1995, 99, 3618–3628 CrossRef CAS.
- R. Bozio, A. Girlando and C. Pecile, J. Chem. Soc., Faraday Trans. 2, 1975, 71, 1237–1254 RSC.
- M. Todaro, L. Sciortino, F. M. Gelardi and G. Buscarino, J. Phys. Chem. C, 2017, 121, 24853–24860 CrossRef CAS.
- M. Todaro, A. Alessi, L. Sciortino, S. Agnello, M. Cannas, F. M. Gelardi and G. Buscarino, J. Spectrosc., 2016, 8074297, DOI:10.1155/2016/8074297.
- M. Todaro, G. Buscarino, L. Sciortino, A. Alessi, F. Messina, M. Taddei, M. Ranocchiari, M. Cannas and F. M. Gelardi, J. Phys. Chem. C, 2016, 120, 12879–12889 CrossRef CAS.
- N. Drenchev, E. Ivanova, M. Mihaylov and K. Hadjiivanov, Phys. Chem. Chem. Phys., 2010, 12, 6423–6427 RSC.
- L. Alaerts, E. Séguin, H. Poelman, F. Thibault-Starzyk, P. A. Jacobs and D. E. De Vos, Chem. – Eur. J., 2006, 12, 7353–7363 CrossRef CAS PubMed.
- P. St. Petkov, G. N. Vayssilov, J. Liu, O. Shekhah, Y. Wang, C. Wöll and T. Heine, ChemPhysChem, 2012, 13, 2025–2029 CrossRef CAS PubMed.
- Y. Wu, A. Kobayashi, G. J. Halder, V. K. Peterson, K. W. Chapman, N. Lock, P. D. Southon and C. J. Kepert, Angew. Chem., Int. Ed., 2008, 47, 8929–8932 CrossRef CAS PubMed.
- C. Schneider, D. Bodesheim, M. G. Ehrenreich, V. Crocellà, J. Mink, R. A. Fischer, K. T. Butler and G. Kieslich, J. Am. Chem. Soc., 2019, 141, 10504–10509 CrossRef CAS PubMed.
- R. Giovine, F. Pourpoint, S. Duval, O. Lafon, J.-P. Amoureux, T. Loiseau and C. Volkringer, Cryst. Growth Des., 2018, 18, 6681–6693 CrossRef CAS.
- D. Kim and H. Lee, RSC Adv., 2015, 5, 2749–2755 RSC.
- J. M. Castillo, T. J. H. Vlugt and S. Calero, J. Phys. Chem. C, 2008, 112, 15934–15939 CrossRef CAS.
- J. B. Decoste, G. W. Peterson, M. W. Smith, C. A. Stone and C. R. Willis, J. Am. Chem. Soc., 2012, 134, 1486–1489 CrossRef CAS PubMed.
- J. M. Lindquist and J. C. Hemminger, Chem. Mater., 1989, 1, 72–78 CrossRef CAS.
- M. C. Biesinger, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2010, 257, 887–898 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Data analysis of Raman microscopy, IR microscopy, PCA, XPS, water sorption and conductivity measurements. See DOI: 10.1039/c9cp05082e |
| This journal is © the Owner Societies 2019 |