
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAbsorption spectra at the iodine 3d ionisation threshold following the CHxI+ (x = 0–3) cation sequence
Kaja
Schubert†
ab,
Alexander A.
Guda†
 c,
Karolin
Mertens
a,
Jan O.
Schunck
ab,
Stefan
Schippers
d,
Alfred
Müller
e,
Sadia
Bari
b,
Stephan
Klumpp
ab and
Michael
Martins
*a
c,
Karolin
Mertens
a,
Jan O.
Schunck
ab,
Stefan
Schippers
d,
Alfred
Müller
e,
Sadia
Bari
b,
Stephan
Klumpp
ab and
Michael
Martins
*a
aDepartment Physik, Universität Hamburg, Luruper Chaussee 149, 22761 Hamburg, Germany. E-mail: kaja.schubert@desy.de; michael.martins@uni-hamburg.de; Fax: +49 040 8998 5088; Tel: +49 040 8998 5088
bDeutsches Elektronen-Synchrotron DESY, Notkestr. 85, 22607 Hamburg, Germany
cThe Smart Materials Research Institute, Southern Federal University, Sladkova 178/24, 344090 Rostov-on-Don, Russia
dJustus-Liebig-Universität Gießen, I. Physikalisches Institut, Heinrich-Buff-Ring 16, 35392 Gießen, Germany
eJustus-Liebig-Universität Gießen, Institut für Atom- und Molekülphysik, Leihgesterner Weg 217, 35392 Gießen, Germany
First published on 8th November 2019
Abstract
Yields of atomic iodine Iq+ (q ≥ 2) fragments resulting from photoexcitation and photoionisation of the target cations CHxI+ (x = 0–3) have been measured in the photon-energy range 610 eV to 670 eV, which comprises the threshold for iodine 3d ionisation. The measured ion-yield spectra show two strong and broad resonance features due to the excitation of the 3d3/2,5/2 electrons into εf states similar to atomic iodine. In the 3d pre-edge range, electrons are excited into molecular orbitals consisting of iodine, carbon, and hydrogen atomic orbitals. These transitions have been identified by comparison with literature data and by simulations using time-dependent density functional theory (TDDFT) with the KMLYP functional. The ion-yield spectrum for CH3I+ resembles the spectrum of IH+ [Klumpp et al., Phys. Rev. A, 2018, 97, 033401] because the highest occupied molecular orbitals (HOMO) of the H and CH3 fragments both contain a single vacancy, only. For the molecular cations with higher number of vacancies in the valence molecular orbitals CHxI+ (x = 0–2), a stronger hybridisation of the molecular orbitals occurs between the organic fragment and the iodine resulting in a change of bonding from a single σ bond in CH3I+ to a triple bond including two π orbitals in CI+. This is reflected in the resonance energies of the observed absorption lines below the iodine 3d excitation threshold.
Introduction
Inner-shell absorption spectroscopy utilising soft X-rays is an ideal tool to probe the chemical environment of a specific element within a molecule. The power of inner-shell excitation was proven by Siegbahn and co-workers in photoelectron spectroscopy studies of the famous ESCA molecule ethyl trifluoroacetate (see e.g. the recent review by Martensson et al.1) which show a chemical shift of the binding energy of carbon 1s electrons depending on the chemical environments established by the different ligands.In general, to understand the influence of each ligand on a molecule, one approach is to vary the number of the ligands (e.g. by removing them) and to determine the changes in the spectrum. However, these newly formed molecules are often not stable. Here, ionic targets offer a way to prepare various (ionic) molecular species in an ion beam that are not accessible by other methods. The different molecular ions can easily be separated by their mass-to-charge ratio and, thus, purified target beams of molecular fragments (radicals) can be provided for experiments. We have chosen the molecular target ion sequence CHxI+ (x = 0–3) as a model system to study, in a combined experimental and theoretical effort, the dependence of the molecular electronic and geometric structure on the number x of hydrogen atoms bound to the central carbon atom. Previously, we examined the electronic structure of the IH+ cation.2 The nature of most of the observed resonances was dominated by the iodine atom. However, for one observed resonance (peak E in Fig. 6b) the final molecular orbital was identified to result from hybridisation of the atomic iodine 5p and the hydrogen 1s orbital to form a (5pπ) σ* state. By exchanging the bonding partner of the iodine atom from hydrogen in IH+ to carbon in CHxI+ the explicit changes of the electronic structure of the different molecules are studied in the present project.
Neutral iodomethane is intensively studied because it can easily be excited either by electrons,3 by high-power lasers of 193 nm,4–8 266 nm,9–11 or even longer wavelengths,12–18 dissociating the molecule.8,19,20 Photons of these different wavelengths, produced by the Sun,21–24 can dissociate CH3I from natural sources25–27 in Earth's atmosphere forming atomic iodine (ions) which act as catalysts for ozone depletion.28–30 Unravelling the dissociation and excitation mechanisms of the iodomethane molecule is important to understand its role in the chemistry of Earth's atmosphere.31,32
Studies of iodomethane in the soft X-ray regime are limited. Olney et al.33 provide an overview of the work done until 1997. More recent publications in which the valence structure of iodomethane is studied discuss the use of photoelectron spectroscopy with photons in the energy range of up to 30 eV.34–38 The iodine 4d resonance was studied by absorption39 and photoelectron spectroscopy,40–43 by electron excitation techniques,33,44 and is exploited to study charge transfer dynamics using the pump–probe technique with short X-ray pulses produced by free-electron lasers (FELs),45–50 or a combination of optical and XUV pulses from higher harmonic generation (HHG) sources.11,51,52
Inner-shell excitation studies of iodomethane including the carbon 1s53 or the iodine 3d resonance54,55 have been scarce, but a comprehensive study examining the iodine 3d threshold in neutral CH3I was published recently,56 showing photo- and Auger electron spectra in the energy regions of the 3d, 4s, and 4p thresholds.
Experiment
The experiments were carried out at the ion beamline facility PIPE at P04, PETRA III, DESY, using accelerated beams of CHxI+ (x = 0–3) molecular ions. Details of the set-up and operation of PIPE57–59 and of the photon beamline P0460 have been discussed previously. The ion preparation and the beamline parameters for the soft X-ray photons delivered by P04 are chosen as described in the context of the IH+ experiment.2For the production of the target molecular cations, liquid CH3I was evaporated under vacuum conditions. The vapour was leaked into a 10 GHz-ECR61 ion source via an electronically controlled needle valve keeping the gas pressure within the ion source in the order of 1 × 10−4 hPa. Fig. 1 shows the mass spectrum of CHxI+ (x = 0–3) molecular cations measured behind the mass analysing dipole magnet. The resolving power for the ion preparation is sufficient to distinguish each single molecular ion of the sequence without overlapping mass lines. Still, some mixing of molecular ions can occur because of different stable isotopes of the constituent elements. Iodine has only one isotope, but carbon has two. 13C has an abundance of 1%62 and its presence in the experiment is visible at the mass line marked with 13CH3I+. The mass lines of CHxI+ (x = 1–3) can be mixed with ions containing the 13C isotope and one hydrogen atom less, but because of the low abundance of 13C, we consider these contributions to be negligible.
 | ||
| Fig. 1 Mass spectrum of the sequence of CHxI+ (x = 0–3) molecular cations showing the mass peaks to be well separated by our mass-to-charge analysing spectrometer. The red solid line is a fit with 5 Gaussian functions determining the mass resolution of the mass spectrometer with the given entrance- and exit-slit widths to be m/Δm ≈ 710. | ||
To obtain absorption spectra of the CHxI+ (x = 0–3) molecular cations, the merged-beams technique was used.57,63,64 The 6 keV, mass-to-charge selected ion beam from the ECR source was overlaid with the photon beam from the P04 beamline in the approximately 1.5 m long merged beam section of PIPE. To reduce the background from collisions of the chosen molecular ion with residual gas, the background pressure in the merged-beam interaction region of the photons and the ions was kept below 2 × 10−10 mbar.
As a measure for the absorption of photons by the target ions, the atomic fragment-ion channels Iq+ (q = 2–4) were individually recorded as functions of the photon energy (Fig. 2). The maximum count rate in the different ion spectra scales roughly with the available ion current of the target ion for the direct I 3d photoionisation. Because of the low ion current for CI+ and CHI+ target ions, only the fragment channels up to q = 3 were recorded.
 | ||
| Fig. 2 To obtain the ion yield spectra of the different molecular cations CHxI+ (x = 0–3; panels d–a), the different atomic fragment channels Iq+ (q = 2–4/dots, triangles, squares) were measured. Note: for x = 0,1 only q = 2, 3 have been measured. For atomic iodine I+ more fragment channels Iq+ (q = 2–8) can be observed (panel e). The prominent fragment channels here are I4+ (light orange squares), I5+ (blue stars) and I6+ (dark orange hexagons). All other fragment channels of Iq+ are depicted in light red. The upper curve in each panel is the sum of the fragment channels. | ||
The ion fragment channels were separated by means of mass spectrometry by a second dipole magnet and were detected by a channeltron detector capable of counting single ions.57,65 The recorded spectra were normalised to the flux of target ions, measured with a Faraday-cup behind the interaction region inside the fragment-separating dipole magnet, and to the photon flux measured with a calibrated photodiode. Both quantities were recorded in parallel to the ionic fragment counting.
To disperse the soft X-rays of beamline P04, a 400 lines per mm grating was chosen in order to achieve a photon flux of 2 × 1014 photons per s. The exit slit of the P04 monochromator was set to 1500 μm resulting in a bandwidth of the photons of about 1 eV.
Theoretical methods
To model the observed absorption spectra, the molecular geometry, the electronic structure and the transition probabilities were calculated using density functional theory (DFT) with the KMLYP functional in the ADF-2017 software suite.66–68 The total charge for all molecules was set equal to 1+. Relativistic effects were taken into account via the non-collinear spin–orbit approximation, which gives a more accurate prediction of the electron energies for heavy elements than the collinear approach.69 The electron transition energies and oscillator strengths were calculated within the Tamm–Dancoff approximation for the time-dependent DFT (TDDFT)70,71 including the self-consistent calculation of spin–orbit effects. For the TDDFT calculations, no symmetry constraints were used to restrict the wavefunction, while the assignment of the transitions was performed afterwards in terms of non-relativistic symmetric molecular orbitals. The absolute energy values of the calculated spectra were shifted by 0.6 eV using peak A (Fig. 6) in the experimental absorption spectrum of I+ as a reference. This shift is explained by the uncertainty of theoretical DFT calculations for calculating absolute values of the core level energies.72 In general, for deeper core states this error increases. However, the relative chemical energy shifts are more precise within the same computational approach due to cancelling of uncertainties for different spectra originated from the same core level. In our work we have applied a rigid shift of 0.6 eV for all spectra to align them with the experimental data, while relative shifts between calculated spectra for different species were not adjusted. The calculated resonances were convoluted with a 1.0 eV (FWHM) Gaussian matching the experimentally observed width. To describe higher-energy states, the largest available quadruple-zeta basis set with four polarisation functions (QZ4P) was additionally extended with diffuse iodine p-orbitals.2,68Experimental results
For the cations CHxI+ (x = 0–3), different ion fragment channels Iq+ up to q = 4 were observed depicted in Fig. 2, which were summed up to obtain the spectra shown in Fig. 3b. For x = 2, 3 the fragment charge states q = 2–4 and for CHI+ and CI+ the fragment channels q = 2, 3 were measured, as for these two fragments the q = 4 channel was to weak.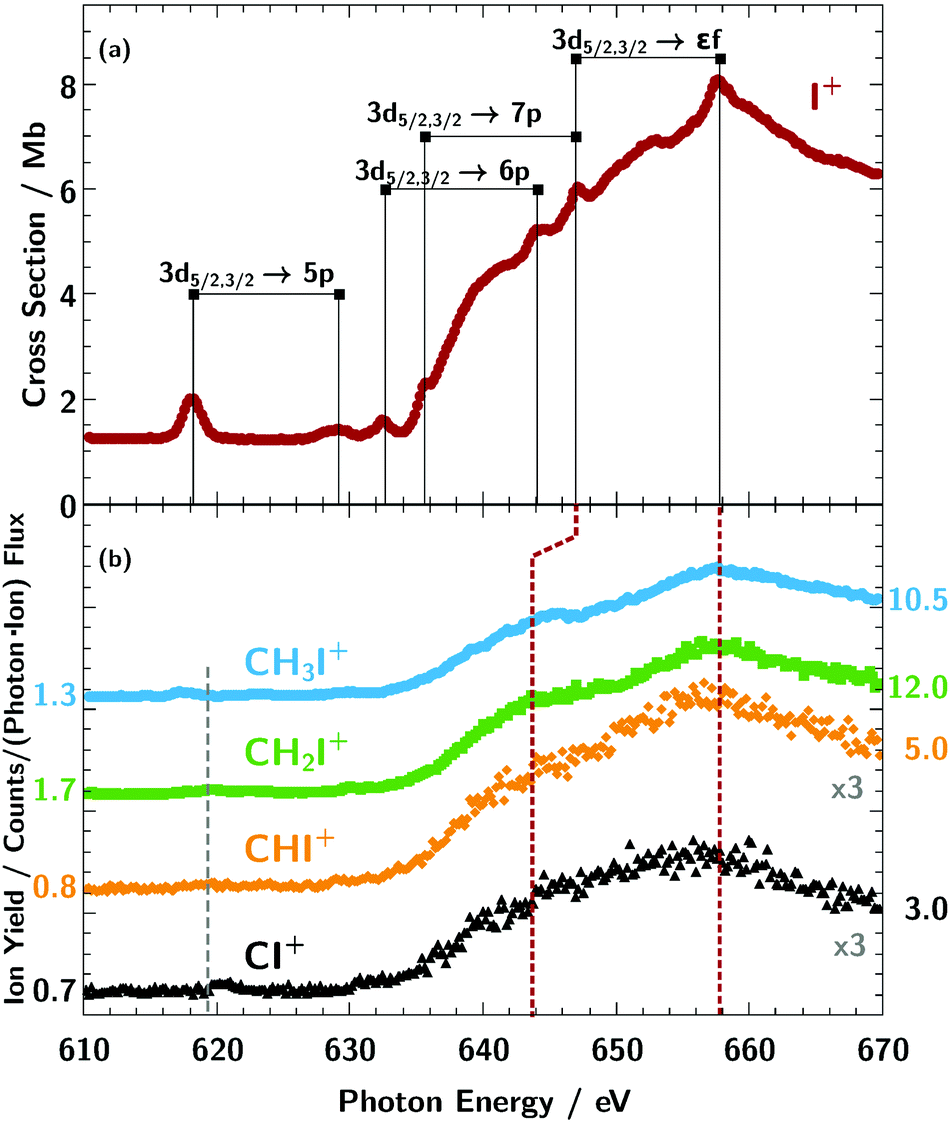 | ||
| Fig. 3 Ion-yield curves for the primary atomic target ion I+ (red dots, panel a) and the primary molecular ion sequence CHxI+ (panel b, light blue dots: x = 3, green squares: x = 2, orange diamonds: x = 1, and black triangles: x = 0) summed over all measured fragment channels Iq+; q = 2–8 for I+, q = 2–4 for x = (2, 3) and q = 2, 3 for x = (0,1). For a better comparison the spectra of the molecular ions were plotted in panel (b) on individual axes with an arbitrary offset and are scaled by a factor shown in the graph. The numbers on the y-axis indicate the yield for each individual curve. The assignment of the observed absorption resonances of I+ is taken from our previous publication.2 For the molecular target ions, the broad 3d → εf features of I+ are visible as well. Below the 3d threshold comparatively weak resonance features can be observed for the molecular target ions in the vicinity of the grey dashed line. | ||
For the IH+ cation we assumed that the hydrogen is ejected as H+ after photoionisation.2 As can be seen in the partial ion-yield absorption spectra in Fig. 2, the dominating fragment channels for the atomic iodine are q = 4–6, especially I5+ (panel e in Fig. 2). For the larger molecular cations CH3I+ and CH2I+, the dominating fragment channel is I2+, while for the smaller cations CHI+ and IC+ the dominating fragment channel is I3+. This indicates, the more constituents a molecule has, especially hydrogen, the more charge can be carried away by its fragments after the charge has been transferred to them.73 This results in a reversal of the I2+/I3+ fragment ratio from CI+ to CH3I+. In the following the sum of the partial ion yields will be discussed which are referred to as absorption spectra. Fig. 3 compares the measured absorption spectra of CHxI+ (x = 0–3) with the spectroscopic features associated with photoproduction of a 3d vacancy in the atomic I+ ion.2
Charged carbon, hydrogen or CHk+ fragments were not observed. The reason is the large kinetic energy release of these light fragments which they acquire during the fragmentation process. With a typical charge q = 3 of the iodine fragment, a singly charged H+ or C+ fragment with a mean C–I or H–I bond length of around 2 Å, the kinetic energy release is approximately 21 eV. This results in a large transverse momentum change of the light fragments, which largely inhibits their detection. The effect reduces also the transmission of the iodine fragments. This transmission must be known for the derivation of absolute cross sections from the measured particle counts rates. In principle, an estimate for the transmission can be derived from detailed ion-optical calculations. However, such simulations are beyond the scope of the present work.
Calculations
To make a line assignment and to understand the contribution of the atomic orbitals to the absorption lines, we performed calculations based on density functional theory. Table 1 shows equilibrium bond length values and Mulliken charges on iodine for the different molecular ion species. The Mulliken charge decreases along with the number x of hydrogen atoms in the series of CHxI+ (x = 0–3) upon filling the anti-bonding orbitals. Similarly, the bond length C–I is larger for CH3I+ compared to CHI+ and CH2I+.| Molecular ion | I–X bond length in Angstrom (X = H or C) | Mulliken charge on I | Molecular symmetry group |
|---|---|---|---|
| IH+ | 1.63 | 0.89 | C ∞v |
| CI+ | 1.88 | 1.00 | C ∞v |
| CHI+ linear (180°) | 1.85 | 0.98 | C ∞v |
| CHI+ bent (152°) | 1.85 | 0.94 | C s |
| CH2I+ | 2.01 | 0.91 | C 2v |
| CH3I+ | 2.10 | 0.81 | C 3v |
The relaxed geometries reported in Table 1 were used further for TDDFT simulations. The TDDFT approach does not account for all many body interactions in the iodine 5d–nf transitions which result in rich multiplet structures. However, we have recently shown2 its applicability to iodine 5d–np transitions. The spectra depicted as solid lines in Fig. 7a–d show the results of the calculations in comparison with the experimentally data. Table 2 lists the final-state configurations of the observed absorption lines according to the theoretical model.
| Line label | Molecular cation | ||
|---|---|---|---|
| IH+ (C∞v) | CH3I+ (C3v) | CH2I+ (C2v) | |
| A | π (I 5px, 5py)/SOB | E (I 5px, 5py)/SOB | B1 (C 2px/I 5px)/SOD |
| B | 78%: π (I 5px, 5py)/SOA | E (I 5px, 5py)/SOA | 52%: B2 (I 6py/C 3py/H 2s, 1s) |
| 22%: π (I 6dxz, 6dyz/I 6px, 6py/H 2px, 2py) | 48%: A1 (C 2pz/I 6pz/H 2s/I 6s) | ||
| C | 72%: π (I 6px, 6py/I 6dxz, 6dyz) | 60%: A1 (I 6pz/H 2s) | 56%: A1 (C 3pz/I 6pz/H 2pz) |
| 17%: π (I 6dxz, 6dyz/I 6px, 6py/H 2px, 2py) | 40%: E (I 6px, 6py/H 2s) | 44%: B2 (I 6py/C 2py/H 1s) | |
| 11%: σ (I 6pz) | |||
| D | 53%: σ (H 2s/H 2pz/I 6dz2) | 51%: E (I 5dxz/H 2pz/I 7px/I 5dx2−y2/H 1s) | 73%: B1 (C 2px/I 5px)/SOA |
| 30%: σ (I 6pz/I 7pz) | 40%: E (C 3px/I 5dxz/H 2s/H 2px) | 27%: B2 (I 7py/I 5dxy) | |
| 17%: σ (I 7Px, 7py) | 9%: A1 (I 6pz) | ||
| E | σ (H 1s/I 5pz, 6dz2) | A1 (I 5pz/C 2pz/I 3dz2) | A1 (I 5pz/C 2pz/I 5dz2) |
| Line label | Molecular cation | ||
|---|---|---|---|
| CHI+ | CI+ (C∞v) | ||
| Linear (C∞v) | Bent (Cs) | ||
| A | π (C 2px, 2py/I 5px, 5py) | A′′ (C 2py/I 5py) | π (C 2px, 2py/I 5px, 5py) |
| B | 62%: σ (I 6pz/H 2pz) | 56%: A′ (I 6pz/H 2pz) | π (I 6px, 6py) |
| 38%: π (I 6px, 6py) | 44%: A′′ (I 6py) | ||
| C | 80%: π (C 3px, 3py/I 6px, 6py) | 43%: A′′ (C 3py/I 5dyz/I 6py) | 57%: σ (I 6pz) |
| 10%: π (I 6px, 6py/I 6dxz, 6dyz/C 3px, 3py) | 31%: A′ (I 6px, 6pz) | 33%: π (I 6dxz, 6dyz/C 6px, 6py/C 3px, 3py) | |
| 10%: π (C 3px, 3py/I 6dxz, 6dyz/I 6px, 6py/H 2px, 2py) | 26%: A′ (I 6dxz/C 3px/I 6px) | 10%: π (I 7px, 7py/I 6dxz, 6dyz/C 3px, 3py) | |
| D | 46%: π (I 7px, 7py) | 31%: A′′ (C 3py/I 5dyz/I 7py) | 59%: σ (C 3pz/I 7pz) |
| 31%: σ (I 7pz) | 22%: A′ (C 3px/I 7pz) | 41%: σ (I 6pz) | |
| 11%: π (C 3px, 3py/I 6dxz, 6dyz/I 7px, 7py/H 2px, 2py) | 19%: A′ (I 7px) | ||
| 6%: σ (I 7pz) | 17%: A′′ (C 3py/I 5dyz/I 7py) | ||
| 6%: π (C 3px, 3py/I 7px, 7py) | 10%: A′ (I 7pz) | ||
| E | σ (I 5pz/I 6dz2/C 2pz) | ||
| E1 | π (C 2px, 2py/I 5px, 5py) | 56%: A′′ (C 2py/I 5py) | |
| 44%: A′ (C 2px/I 5px) | |||
| E2/E3 | σ (I 5pz, 6dz2/C 2s) | A′ (I 5pz, 6dx2−z2/C 2s) | |
The three-dimensional visualisation of the valence molecular orbitals for the molecular cation IH+ and for the cationic sequence CHxI+ (x = 0–3) are shown in Fig. 4.
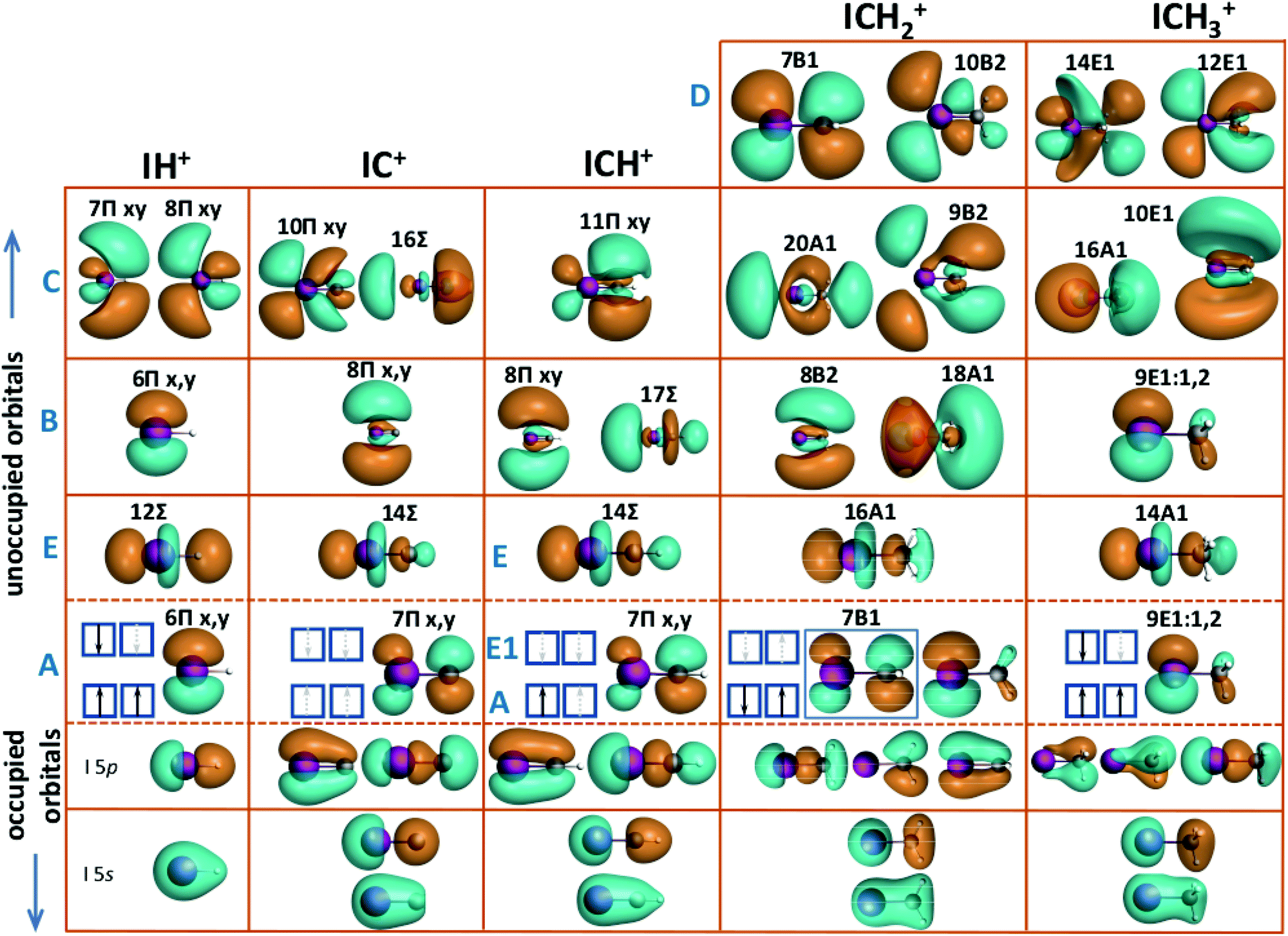 | ||
| Fig. 4 Orbital pictures of the cation IH+ and the cation sequence CHxI+ (x = 0–3). From bottom to top the inner-shell orbitals with s-type symmetry of iodine and the organic constituents are depicted. The HOMO–LUMO orbitals are shown between the dashed lines and above the unoccupied orbitals responsible for the transitions B–E. While the final orbital for transition A is the iodine 5px,y oriented perpendicularly to the I–H or C–I bond, the axial 5pz orbital hybridised with the organic fragment level is responsible for transition E. The detailed line assignments are given in Table 2. For details refer to text. | ||
Fig. 5 shows the molecular orbitals of the neutral constituents H, CH, CH2 and CH3 which form the molecular orbitals after their hybridisation with the iodine 5p states. In terms of electronic configuration, H and CH3 are very similar. As depicted in Fig. 5, both constituents miss one electron to complete their valence shell. The single bond of IH+ is formed by the hybridisation of the valence iodine 5p orbital and the hydrogen 1s orbital.2 Though, CH3 offers a π-type unoccupied orbital oriented along the CI bond which results in its low hybridisation with iodine 5px and 5py orbitals, oriented perpendicularly to the CI bond. In contrast, the empty carbon 2pz orbital of C, CH, and CH2 is suitable for hybridisation. The CI+ molecular ion, like neutral CO, exhibits a triple bond. The valence carbon 2p and the iodine 5p orbitals form three bonding and three anti-bonding orbitals. Two of them of π-type, directed perpendicularly to the CI bond, are degenerate.
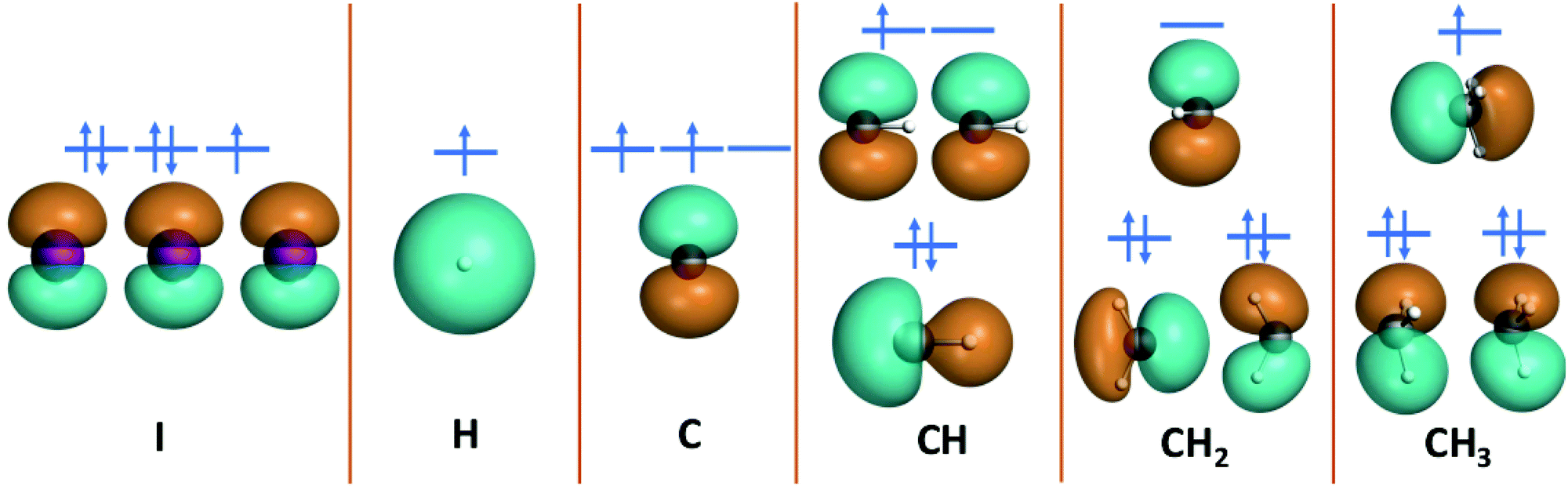 | ||
| Fig. 5 Orbital pictures of the neutral atomic I, H, and C and molecular CH, CH2, and CH3 fragments which are the building blocks forming the molecular (cation) sequence CHxI (x = 0–3). | ||
Discussion
The ionisation thresholds for the 3d5/2 and 3d3/2 levels are located at 619.3 eV and 630.8 eV, respectively, for neutral atomic iodine.74 In the molecular ion spectra shown in Fig. 3b, the broad iodine 3d5/2,3/2 → εf lines are visible above 630 eV, similar to the atomic iodine ion from Fig. 3a. As indicated by the red dashed lines in Fig. 3b, the spin–orbit splitting of the εf lines is apparently larger for the molecular cation sequence and the molecular εf appear to be broader compared to the atomic ones. Further, the position of the εf lines on the energy scale are comparatively stable, slightly shifting to lower photon energies for smaller numbers of hydrogen x in the cation ion sequence CHxI+. This observed small energy shift of the molecular εf lines relative to the I+ resonances at 647 eV and 658 eV can rather be explained by an increase of line broadening with respect to the atomic iodine ion I+ lines than by an increase of spin–orbit splitting of the molecular lines.More distinct spectroscopic features can be seen below the iodine 3d ionisation threshold (Fig. 6 and 7). Obviously, the resonance structure of the cations CHxI+ (x = 0–3) changes with the number of hydrogen atoms in the molecules. This can be easily understood in terms of occupation of molecular orbitals for the different CHx fragments as we show in Fig. 5. A qualitative assignment of the resonances in CH3I+ can be performed by comparison with the absorption spectra2 of I+ and IH+ as shown in Fig. 6.
 | ||
| Fig. 6 Assignment of the resonances of CH3I+ observed below the 3d ionisation threshold: (A) 3d5/2 → 2e4, (B) 3d3/2 → 2e4 or 3d5/2 → 6p, (E) 3d5/2 → 4a1. The assignment follows the results of previous investigations.2,11,33,75 For details see text. | ||
Neutral CH3I is of C3v symmetry. Its electronic ground state can be expressed as:11,33,75
| KIKCLIMINI(1a1)2(2a1)2(1e)4(3a1)2(2e)4 1A1. | (1) |
Accordingly, the ground state of the molecular ion CH3I+ has an electron configuration:76–79
| KIKCLIMINI(1a1)2(2a1)2(1e)4(3a1)2(2e)3 2E3/2,1/2 | (2) |
Summarising the resonance structures below the iodine 3d ionisation threshold in Fig. 7, the CHxI+ (x = 1–3) ions all show quite different behaviour in terms of line position and number of them in Table 3. However, theoretical analysis in Fig. 4 and Table 2 reveals that lines of the same origin are shifted compared relative to one another due the varying number of hydrogen atoms in the molecular ions.
| Molecular | Line label (see text, Fig. 6 and 7); all values of the chemical shift are in eV | |||||
|---|---|---|---|---|---|---|
| Reference | Target cation | A | B | C | D | E |
| IH+ | CH3I+ | 0.3 ± 0.2 | 1.0 ± 0.2 | 0.9 ± 0.2 | ||
| CH3I+ | CH2I+ | 2.1 ± 0.2 | 0.3 ± 0.2 | 1.5 ± 0.2 | 0.7 ± 0.2 | |
| CHI+ | 1.7 ± 0.2 | 0.7 ± 0.2 | 1.6 ± 0.2 | (0.7 ± 0.4) | 0.5 ± 0.2 | |
| CI+ | (1.8 ± 0.2) | 2.5 ± 0.2 | ||||
Further, the lines B and C can be resolved separately for CH2I+ and CHI+, only. Some indication for line D can be observed in CHI+, but as in IH+, it is overshadowed by the rising edge of the 3d threshold.
The changes in the resonance structure for CI+ can be explained with its different electronic ground state configuration which has the same valence electronic structure as the neutral CO molecule:80,81
| (1σ)2(2σ)2(3σ)2(4σ)2(1π)4(5σ)2 1Σ+. | (3) |
The bonding nature of the iodine in IH+ and CH3I+ can be compared to the similar isoelectronic configuration of the CH3 and H counterparts. Resonance A in the molecular cations IH+ and CH3I+ can be described by an almost pure atomic iodine 5p orbital. Weak hybridisation for the first transition in IH+ and CH3I+ compared to other molecules is evident also from Fig. 4. Peak A in CHI, CH2I and CI corresponds to the transition into the anti-bonding π orbital which is described by an iodine 5p hybridised with the 2p orbital of carbon, oriented perpendicularly to the CI bond. The latter is blocked for the CH3 constituent due to presence of three hydrogen atoms.
Resonance E in all molecules is related to the anti-bonding Σ bond between the iodine 5pz and the π orbital of the ligand directed along the C–I (or I–H) bond. The only exception is the feature marked E1 in the CHI+ spectrum in Fig. 7c which arises from the opposite-spin counterpart of the feature A shifted by exchange interaction. Resonance B in the molecules IH+ and CH3I+ is the spin–orbit counterpart of resonance A. However in CI+, CHI+, and CH2I+ resonance A is shifted to higher energies and its spin–orbit counterpart overlaps with higher-energy continuum transitions. Therefore resonance B in CI+, CHI+, and CH2I+ was assigned to the iodine 6p orbital directed perpendicularly to the C–I bond. Resonance C for IH+, CI+, and CHI+, and resonance D in CH2I+ and CH3I+ is related to the hybridised iodine 6p and 6d orbitals. The higher-lying resonances belong to the iodine 7p orbital of the np Rydberg series which is hardly observed in our spectrum and masked by the onset of the iodine 3d excitation threshold.
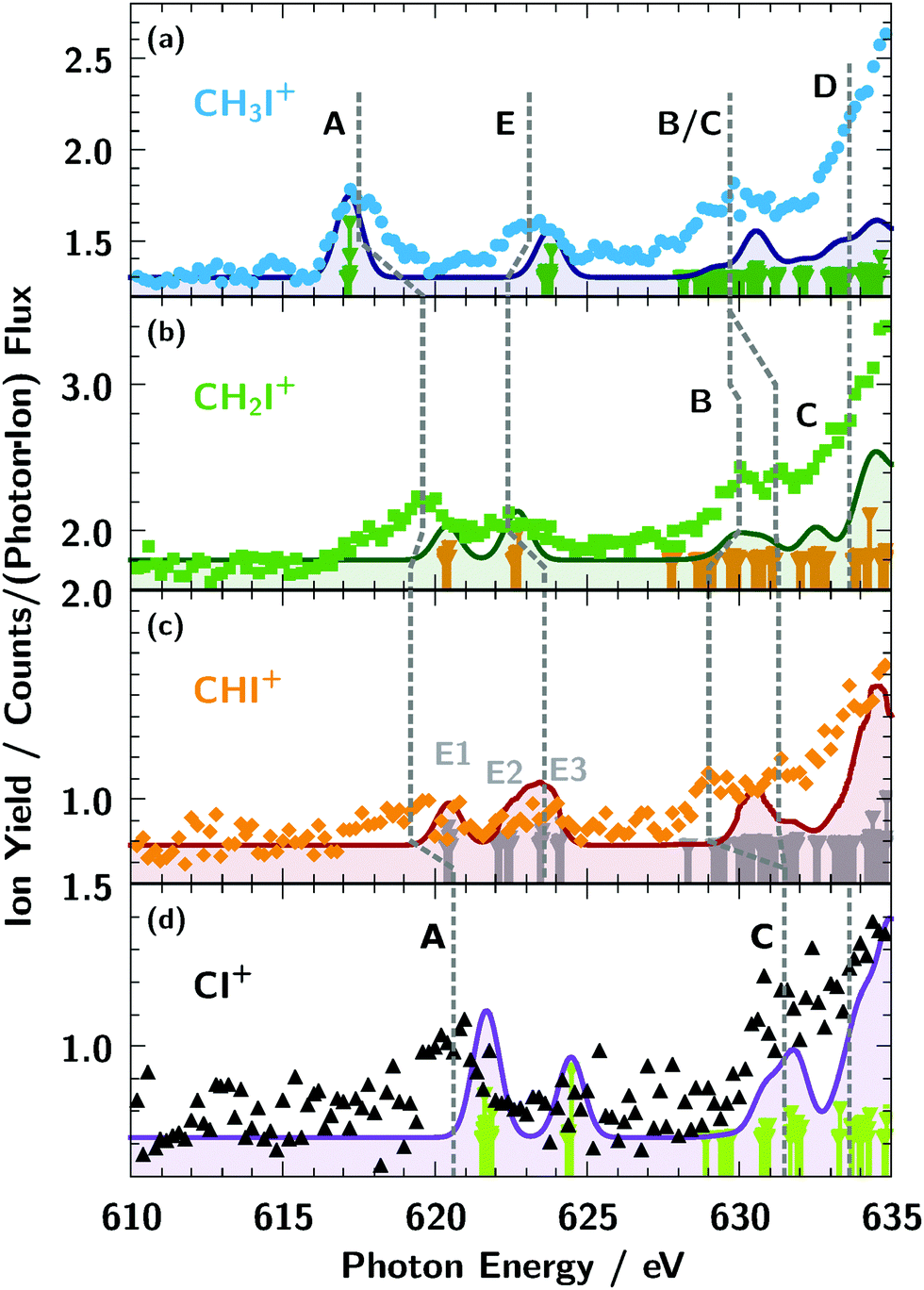 | ||
| Fig. 7 Assignment and chemical shift of the resonances of the cations CHxI+ (x = 0–3) below the iodine 3d ionisation threshold. For details see text. The dashed lines in the spectra of CHI+ and CI+ are to guide the eye to identify the respective resonances. The solid lines are the results for accompanying TDDFT calculations with a 1 eV resolution. The bars are plotted to give the line positions of the calculated resonances. | ||
Conclusions
To unravel the influence of ligands on the electronic structure of a molecule we studied the sequence of CHxI+ (x = 0–3) cations as a model system. The absorption resonances observed below the iodine 3d ionisation threshold show a strong dependence on the number of hydrogen atoms x. The observed lines for CH3I+ and CI+ were assigned to electronic states known from the literature. According to (TD)DFT calculations, the resonances observed for CH3I+ and IH+ shown in Fig. 6 have similar origins due to the similar nature of the hydrogen and CH3 ligands. The evolution of the energy position and splitting of the resonances in the pre-edge spectra in CHxI+ series is explained by a change of occupation of first molecular antibonding orbitals and value of hybridisation mainly between iodine and fragment orbitals of π-type. The two main pre-edge resonances can be well described by a molecular orbital composed of single atomic orbitals, whereas the higher lying resonances in the pre-edge are influenced by stronger hybridisation between several delocalised atomic orbitals, e.g. p and d types for iodine. Finally sharp resonances on the main edge originate from the multiplet structure of d–εf transitions.As demonstrated in this work, inner-shell spectroscopy is a powerful tool to reveal the electronic structure of molecules within a different chemical environment. In combination with an ion beam in a merged beam geometry it allows to study molecular ions not accessible by other methods and unveil their electronic structure.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was carried out at the synchrotron light source PETRA III at DESY, a member of the Helmholtz Association (HGF). We would like to thank L. Glaser, G. Hartmann, F. Scholz, J. Seltmann, and J. Viefhaus for assistance in using beamline P04. The construction of PIPE and this work were funded by the German ministry for education and research (BMBF) under contracts 05KS7RG1, 05KS7GU2, 05KS7KE1, 05KS7RF2, 05K10RG1, 05K10GUB, 05K10KEA, and 05K10RF2 within the “Verbundforschung” funding scheme. KM and MM acknowledge funding by the Deutsche Forschungsgemeinschaft (DFG) via SFB925/A3. AM acknowledges support by the Deutsche Forschungsgemeinschaft via grant Mu-1068/22. AAG acknowledges the financial support from the President's Grant of Russian Federation for young scientists MK-2730.2019.2. SB and KS are thankful for funding from the Initiative and Networking Fund of the Helmholtz Association and acknowledge financial support by the Deutsche Forschungsgemeinschaft (DFG) through Sonderforschungsbereich SFB755 Nanoscale Photonic Imaging. SK acknowledges the funding of the EUCALL project within the European Union's Horizon 2020 research and innovation programme under the grant agreement No. 654220.Notes and references
- N. Mårtensson, E. Sokolowski and S. Svensson, J. Electron Spectrosc. Relat. Phenom., 2014, 193, 27 CrossRef
.
- S. Klumpp, A. A. Guda, K. Schubert, K. Mertens, J. Hellhund, A. Müller, S. Schippers, S. Bari and M. Martins, Phys. Rev. A, 2018, 97, 033401 CrossRef CAS
- H. Kato, H. Masui, M. Hoshino, H. Cho, O. Ingólfsson, M. J. Brunger, P. Limão-Vieira and H. Tanaka, J. Chem. Phys., 2010, 133, 054304 CrossRef CAS PubMed
- C. Fotakis, M. Martin, K. Lawley and R. Donovan, Chem. Phys. Lett., 1979, 67, 1 CrossRef CAS
- G. V. Veen, T. Baller and A. D. Vries, Chem. Phys., 1985, 97, 179 CrossRef
- R. E. Continetti, B. A. Balko and Y. T. Lee, J. Chem. Phys., 1988, 89, 3383 CrossRef CAS
- A. Gilchrist, G. Hancock, R. Peverall, G. Richmond, G. A. D. Ritchie and S. Taylor, J. Phys. Chem. A, 2008, 112, 4531 CrossRef CAS PubMed
- H. Xu and S. T. Pratt, J. Chem. Phys., 2013, 139, 214310 CrossRef PubMed
- D. W. Chandler, J. W. Thoman, M. H. Janssen and D. H. Parker, Chem. Phys. Lett., 1989, 156, 151 CrossRef CAS
- R. O. Loo, H. P. Haerri, G. E. Hall and P. L. Houston, J. Chem. Phys., 1989, 90, 4222 CrossRef CAS
- A. R. Attar, A. Bhattacherjee and S. R. Leone, J. Phys. Chem. Lett., 2015, 6, 5072 CrossRef CAS PubMed
- C. Kosmidis, S. Kaziannis, P. Siozos, A. Lyras, L. Robson, K. Ledingham, P. McKenna and D. Jaroszynski, Int. J. Mass Spectrom., 2006, 248, 1 CrossRef CAS
- H. Liu, Z. Yang, Z. Gao and Z. Tang, J. Chem. Phys., 2007, 126, 044316 CrossRef PubMed
- Y. Wang, S. Zhang, Z. Wei and B. Zhang, J. Phys. Chem. A, 2008, 112, 3846 CrossRef CAS PubMed
- G. Gitzinger, M. E. Corrales, V. Loriot, R. de Nalda and L. Bañares, J. Chem. Phys., 2012, 136, 074303 CrossRef CAS PubMed
- G. Gitzinger, V. Loriot, L. Bañares and R. de Nalda, J. Mod. Opt., 2014, 61, 864 CrossRef CAS
- S. G. Walt, N. Bhargava Ram, A. von Conta, O. I. Tolstikhin, L. B. Madsen, F. Jensen and H. J. Wörner, J. Phys. Chem. A, 2015, 119, 11772 CrossRef CAS PubMed
- Y. Wang, Y. Song, W. Liu, Y. Liu, L. Duo, L. Jiang and Y. Yang, Chem. Phys. Lett., 2015, 633, 126 CrossRef CAS
- R. de Nalda, J. Durá, A. García-Vela, J. G. Izquierdo, J. González-Vázquez and L. Bañares, J. Chem. Phys., 2008, 128, 244309 CrossRef PubMed
- M. E. Corrales, G. Gitzinger, J. González-Vázquez, V. Loriot, R. de Nalda and L. Bañares, J. Phys. Chem. A, 2012, 116, 2669 CrossRef CAS PubMed
- G. Rottman, Space Sci. Rev., 2006, 125, 39 CrossRef
- S. K. Solanki, N. A. Krivova and J. D. Haigh, Annu. Rev. Astron. Astrophys., 2013, 51, 311 CrossRef CAS
- K. L. Yeo, N. A. Krivova and S. K. Solanki, Space Sci. Rev., 2014, 186, 137 CrossRef
- K. L. Yeo, W. T. Ball, N. A. Krivova, S. K. Solanki, Y. C. Unruh and J. Morrill, J. Geophys. Res.: Space Phys., 2015, 120, 6055 Search PubMed
- L. J. Carpenter, Chem. Rev., 2003, 103, 4953 CrossRef CAS PubMed
- R. von Glasow, Nature, 2008, 453, 1195 CrossRef CAS PubMed
- I. Stemmler, M. Rothe, I. Hense and H. Hepach, Biogeosciences, 2013, 10, 4211 CrossRef
- S. Solomon, R. R. Garcia and A. R. Ravishankara, J. Geophys. Res.: Atmos., 1994, 99, 20491 CrossRef
- R. Vogt, R. Sander, R. von Glasow and P. J. Crutzen, J. Atmos. Chem., 1999, 32, 375 CrossRef CAS
- R. Teiwes, J. Elm, K. Handrup, E. P. Jensen, M. Bilde and H. B. Pedersen, Phys. Chem. Chem. Phys., 2018, 20, 28606 RSC
- A. Saiz-Lopez, J. M. C. Plane, A. R. Baker, L. J. Carpenter, R. von Glasow, J. C. Gómez Martín, G. McFiggans and R. W. Saunders, Chem. Rev., 2012, 112, 1773 CrossRef CAS PubMed
- C. D. O'Dowd, J. L. Jimenez, R. Bahreini, R. C. Flagan, J. H. Seinfeld, K. Hameri, L. Pirjola, M. Kulmala, S. G. Jennings and T. Hoffmann, Nature, 2002, 417, 632 CrossRef PubMed
- T. N. Olney, G. Cooper and C. Brion, Chem. Phys., 1998, 232, 211 CrossRef CAS
- S. Eden, P. Limao-Vieira, S. Hoffmann and N. Mason, Chem. Phys., 2007, 331, 232 CrossRef CAS
- R. D. Molloy, A. Danielsson, L. Karlsson and J. H. Eland, Chem. Phys., 2007, 335, 49 CrossRef CAS
- R. Locht, B. Leyh, H. Jochims and H. Baumgärtel, Chem. Phys., 2009, 365, 109 CrossRef CAS
- R. Locht, D. Dehareng, K. Hottmann, H. W. Jochims, H. Baumgärtel and B. Leyh, J. Phys. B: At., Mol. Opt. Phys., 2010, 43, 105101 CrossRef
- D. Holland, I. Powis, G. Öhrwall, L. Karlsson and W. von Niessen, Chem. Phys., 2006, 326, 535 CrossRef CAS
- G. O'Sullivan, J. Phys. B: At. Mol. Phys., 1982, 15, L327 CrossRef
- D. W. Lindle, P. H. Kobrin, C. M. Truesdale, T. A. Ferrett, P. A. Heimann, H. G. Kerkhoff, U. Becker and D. A. Shirley, Phys. Rev. A: At., Mol., Opt. Phys., 1984, 30, 239 CrossRef CAS
- P. Morin and I. Nenner, Phys. Scr., 1987, 1987, 171 CrossRef
- J. N. Cutler, G. M. Bancroft and K. H. Tan, J. Chem. Phys., 1992, 97, 7932 CrossRef CAS
- M. Pernpointner, J. P. Zobel, E. Fasshauer and A. N. Sil, Chem. Phys., 2012, 407, 39 CrossRef CAS
- S. Hayakawa, T. Tsujinaka and A. Fujihara, J. Chem. Phys., 2012, 137, 184308 CrossRef PubMed
- K. Mertens, N. Gerken, S. Klumpp, M. Braune and M. Martins, J. Mod. Opt., 2015, 63, 383 CrossRef
- B. Erk, R. Boll, S. Trippel, D. Anielski, L. Foucar, B. Rudek, S. W. Epp, R. Coffee, S. Carron, S. Schorb, K. R. Ferguson, M. Swiggers, J. D. Bozek, M. Simon, T. Marchenko, J. Küpper, I. Schlichting, J. Ullrich, C. Bostedt, D. Rolles and A. Rudenko, Science, 2014, 345, 288 CrossRef CAS PubMed
- K. Motomura, E. Kukk, H. Fukuzawa, S. ichi Wada, K. Nagaya, S. Ohmura, S. Mondal, T. Tachibana, Y. Ito, R. Koga, T. Sakai, K. Matsunami, A. Rudenko, C. Nicolas, X.-J. Liu, C. Miron, Y. Zhang, Y. Jiang, J. Chen, M. Anand, D. E. Kim, K. Tono, M. Yabashi, M. Yao and K. Ueda, J. Phys. Chem. Lett., 2015, 6, 2944 CrossRef CAS PubMed
- R. Boll, B. Erk, R. Coffee, S. Trippel, T. Kierspel, C. Bomme, J. D. Bozek, M. Burkett, S. Carron, K. R. Ferguson, L. Foucar, J. Küpper, T. Marchenko, C. Miron, M. Patanen, T. Osipov, S. Schorb, M. Simon, M. Swiggers, S. Techert, K. Ueda, C. Bostedt, D. Rolles and A. Rudenko, Struct. Dyn., 2016, 3, 043207 CrossRef PubMed
- K. Nagaya, K. Motomura, E. Kukk, Y. Takahashi, K. Yamazaki, S. Ohmura, H. Fukuzawa, S. Wada, S. Mondal, T. Tachibana, Y. Ito, R. Koga, T. Sakai, K. Matsunami, K. Nakamura, M. Kanno, A. Rudenko, C. Nicolas, X.-J. Liu, C. Miron, Y. Zhang, Y. Jiang, J. Chen, M. Anand, D. E. Kim, K. Tono, M. Yabashi, M. Yao, H. Kono and K. Ueda, Faraday Discuss., 2016, 194, 537 RSC
- A. Rudenko, L. Inhester, K. Hanasaki, X. Li, S. J. Robatjazi, B. Erk, R. Boll, K. Toyota, Y. Hao, O. Vendrell, C. Bomme, E. Savelyev, B. Rudek, L. Foucar, S. H. Southworth, C. S. Lehmann, B. Kraessig, T. Marchenko, M. Simon, K. Ueda, K. R. Ferguson, M. Bucher, T. Gorkhover, S. Carron, R. Alonso-Mori, J. E. Koglin, J. Correa, G. J. Williams, S. Boutet, L. Young, C. Bostedt, S.-K. Son, R. Santra and D. Rolles, Nature, 2017, 546, 129 CrossRef CAS PubMed
- A. Bhattacherjee, A. R. Attar and S. R. Leone, J. Chem. Phys., 2016, 144, 124311 CrossRef PubMed
- L. Drescher, M. C. E. Galbraith, G. Reitsma, J. Dura, N. Zhavoronkov, S. Patchkovskii, M. J. J. Vrakking and J. Mikosch, J. Chem. Phys., 2016, 145, 011101 CrossRef CAS PubMed
- A. Hitchcock and C. Brion, J. Electron Spectrosc. Relat. Phenom., 1979, 17, 139 CrossRef CAS
- A. Hitchcock and C. Brion, J. Electron Spectrosc. Relat. Phenom., 1978, 13, 193 CrossRef CAS
- D. Kovaček, K. Kovačević, D. Korenić and Z. B. Maksić, THEOCHEM, 1994, 304, 163 CrossRef
- R. Forbes, A. De Fanis, C. Bomme, D. Rolles, S. T. Pratt, I. Powis, N. A. Besley, M. Simon, S. Nandi, A. R. Milosavljević, C. Nicolas, J. D. Bozek, J. G. Underwood and D. M. P. Holland, J. Chem. Phys., 2018, 149, 144302 CrossRef PubMed
- S. Schippers, S. Ricz, T. Buhr, A. B. Jr, J. Hellhund, K. Holste, K. Huber, H.-J. Schäfer, D. Schury, S. Klumpp, K. Mertens, M. Martins, R. Flesch, G. Ulrich, E. Rühl, T. Jahnke, J. Lower, D. Metz, L. P. H. Schmidt, M. Schöffler, J. B. Williams, L. Glaser, F. Scholz, J. Seltmann, J. Viefhaus, A. Dorn, A. Wolf, J. Ullrich and A. Müller, J. Phys. B: At., Mol. Opt. Phys., 2014, 47, 115602 CrossRef
- A. Müller, D. Bernhardt, A. Borovik Jr., T. Buhr, J. Hellhund, K. Holste, A. Kilcoyne, S. Klumpp, M. Martins, S. Ricz, J. Seltmann, J. Viefhaus and S. Schippers, Astrophys. J., 2017, 836, 166 CrossRef
- S. Schippers, T. Buhr, A. Borovik Jr., K. Holste, A. Perry-Sassmannshausen, K. Mertens, S. Reinwardt, M. Martins, S. Klumpp, K. Schubert, S. Bari, R. Beerwerth, S. Fritzsche, S. Ricz, J. Hellhund and A. Müller, X-Ray Spectrom., 2019 DOI:10.1002/xrs.3035
- J. Viefhaus, F. Scholz, S. Deinert, L. Glaser, M. Ilchen, J. Seltmann, P. Walter and F. Siewert, Nucl. Instrum. Methods Phys. Res., Sect. A, 2013, 710, 151 CrossRef CAS
- R. Trassl, P. Hathiramani, F. Broetz, J. B. Greenwood, R. W. McCullough, M. Schlapp and E. Salzborn, Phys. Scr., 1997, 1997, 380 CrossRef
- M. Berglund and M. Wieser, Pure Appl. Chem., 2011, 83, 397 CAS
- S. Schippers, A. L. D. Kilcoyne, R. A. Phaneuf and A. Müller, Contemp. Phys., 2016, 57, 215–229 CrossRef CAS
- H. Kjeldsen, J. Phys. B: At., Mol. Opt. Phys., 2006, 39, R325 CrossRef CAS
- K. Rinn, A. Müller, H. Eichenauer and E. Salzborn, Rev. Sci. Instrum., 1982, 53, 829 CrossRef CAS
- C. Fonseca Guerra, G. J. Snijders, G. te Velde and J. E. Baerends, Theor. Chem. Acc., 1998, 99, 391 Search PubMed
- G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. Fonseca Guerra, S. J. A. van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931 CrossRef CAS
- E. Van Lenthe and E. J. Baerends, J. Comput. Chem., 2003, 24, 1142 CrossRef CAS PubMed
- C. Van Wüllen, J. Comput. Chem., 2002, 23, 779–785 CrossRef PubMed
- S. van Gisbergen, J. Snijders and E. Baerends, Comput. Phys. Commun., 1999, 118, 119 CrossRef CAS
- S. Hirata and M. Head-Gordon, Chem. Phys. Lett., 1999, 314, 291 CrossRef CAS
- F. Vines, C. Sousa and F. Illas, Phys. Chem. Chem. Phys., 2018, 20, 8403–8410 RSC
- M. Hollstein, K. Mertens, S. Klumpp, N. Gerken, S. Palutke, I. Baev, G. Brenner, S. Dziarzhytski, M. Meyer, W. Wurth, D. Pfannkuche and M. Martins, New J. Phys., 2019, 21, 033017 CrossRef CAS
- J. A. Bearden and A. F. Burr, Rev. Mod. Phys., 1967, 39, 125–142 CrossRef CAS
- R. S. Mulliken, Phys. Rev., 1935, 47, 413 CrossRef CAS
- J. L. Ragle, I. A. Stenhouse, D. C. Frost and C. A. McDowell, J. Chem. Phys., 1970, 53, 178 CrossRef CAS
- M. Tadjeddine, G. Bouchoux, L. Malegat, J. Durup, C. Pernot and J. Weiner, Chem. Phys., 1982, 69, 229 CrossRef CAS
- M. Tadjeddine, J. Flament and C. Teichteil, Chem. Phys., 1987, 118, 45 CrossRef CAS
- K. Walter, R. Weinkauf, U. Boesl and E. W. Schlag, J. Chem. Phys., 1988, 89, 1914 CrossRef CAS
- H. Brion and C. Moser, J. Chem. Phys., 1960, 32, 1194 CrossRef CAS
- Y. A. Teterin and S. G. Gagarin, Russ. Chem. Rev., 1996, 65, 825 CrossRef
- R. K. Nesbet, J. Chem. Phys., 1965, 43, 4403 CrossRef CAS
Footnote |
| † These authors contributed equally to this work. |
| This journal is © the Owner Societies 2019 |