

Abstract
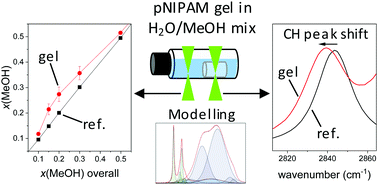
Crosslinked poly-N-isopropylacrylamide (pNIPAM) gels adapt to their environment by a unique transition from a flexible, swollen macromolecular network to a collapsed particle. pNIPAM gels are swollen in both, pure water and pure methanol (MeOH). However, a drastic volume loss is observed in mixtures of water and methanol over a wide composition range. This effect is referred to as cononsolvency. Cononsolvency couples the volume phase transition to the transport of the cosolvent into the polymeric network. So far, the mechanisms underlying cononsolvency have not been fully elucidated. To obtain insights on cononsolvency, Raman microspectroscopy was applied to capture spatially resolved spectra distinguishing between the surroundings and the inside of the gel. Here, we used Indirect Hard Modelling (IHM) for the spectral analysis. Mass balancing allowed the calculation of the solvent composition inside the pNIPAM gel. The results show an increased methanol fraction inside the collapsed gel as compared to its surroundings. Furthermore, the sensitivity of the vibrational bands of methanol to its local hydrogen bonding environment allow to derive information about the molecular interactions. The methanol peak shifts measured inside the gel point towards donor-type hydrogen bonds between methanol and the peptide group of pNIPAM in the cononsolvency-induced collapse. The presented data should enhance our understanding of cononsolvency.
- This article is part of the themed collections: PCCP Editor’s Choice, 2020 and 2019 PCCP HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

