
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Growth of well-ordered iron sulfide thin films†
Earl Matthew
Davis
 *a,
Giulia
Berti
a,
Helmut
Kuhlenbeck
a,
Vedran
Vonk
b,
Andreas
Stierle
bc and
Hans-Joachim
Freund
a
*a,
Giulia
Berti
a,
Helmut
Kuhlenbeck
a,
Vedran
Vonk
b,
Andreas
Stierle
bc and
Hans-Joachim
Freund
a
aFritz-Haber-Institut der Max-Planck-Gesellschaft, Faradayweg 4-6, 14195 Berlin, Germany. E-mail: davis@fhi-berlin.mpg.de
bDeutsches Elektronen-Synchrotron (DESY), 22607 Hamburg, Germany
cPhysics Department, University of Hamburg, Jungiusstrasse 9, 20355 Hamburg, Germany
First published on 28th August 2019
Abstract
In this paper a growth recipe for well-ordered iron sulfide films and the results of their characterisation are presented. The film was studied using X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS), low energy electron diffraction (LEED), and scanning tunneling microscopy (STM). XRD data reveal that the film has a NiAs-like structure with Fe vacancies, similar to iron sulfides such as pyrrhotite and smythite, although no indication of any ordering of these vacancies was observed. LEED and STM results show that the film exhibits a 2 × 2 surface reconstruction. XPS data provide additional evidence for a large number of Fe vacancies, and the oxidation states of the Fe and S in the film are analysed.
Introduction
The iron sulfides are a diverse group of minerals that are abundantly found in the earth's crust. They play important roles in planetary evolution and within various biogeochemical processes.1–3 Broad knowledge of the many processes involving iron sulfides is relevant for understanding the evolution of the Earth and its environment,4,5 as well as for iron sulfide-based theories of abiogenesis.6,7 Iron sulfides have also been shown to catalyse reactions such as N2 conversion to ammonia,8 and it has been demonstrated that pyrrhotite slurries are able to facilitate Fischer–Tropsch chemistry.9,10 Additionally, iron sulfide chemistry is an essential aspect in the extraction of valuable metals from ores in processes such as froth flotation, an industry worth $100 million each year,2,11 and the processing of waste from mines to prevent acid mine drainage.12 Iron sulfides also show promise for use in electrical energy storage systems.13 Despite the importance of iron sulfides, detailed knowledge of surface processes at the atomic scale has been difficult to obtain. A potentially viable pathway to increased understanding in this area is the use of simplified model systems. This approach has been shown to provide insight into fundamental phenomena at metal surfaces.14 Such a strategy, using high-quality thin films in the study of metal oxides, has already contributed a great deal to the increase in knowledge of oxide surfaces and reactivity over the past two decades.15 For this reason we set out to develop a growth recipe for well-ordered iron sulfide thin films which could then be used as a foundation for further studies using surface science techniques.A wide range of stoichiometries from FeS to FeS2 exists in nature with various structures. Among them, pyrite (FeS2) is the most abundant mineral on the earth's surface and has received the most attention in literature for its reactivity (see ref. 11 and references therein), electrical properties,16 and role in geochemistry.17,18 Mackinawite (tetragonal FeS) is a metastable iron sulfide that is a precursor for pyrite formation19 and is widespread in low-temperature aqueous environments. It has recently been shown to exhibit superconductivity,20 and is promising as a platform for the realisation of high-temperature superconductors. The formation of pyrite from mackinawite has been shown to proceed via greigite (Fe3S4),19 an inverse spinel iron sulfide which is interesting in its own right. This interest is due to structural similarities with the cubane Fe4S4 clusters found in ferrodoxins, and it shows promise for efficient conversion of CO2.21,22
Pyrrhotite (Fe1−xS (0 ≤ x ≤ 0.125)), smythite (a rare mineral whose stoichiometry is estimated to be approximately Fe9S11 or Fe13S1623–25), troilite (FeS), and high-temperature forms of hexagonal FeS all have structures derived from the NiAs structure. The various phases and superstructures associated with these iron sulfides are extremely complex and are still not well understood, particularly at lower temperatures.26–28 In troilite the superstructures arise from the clustering of metal atoms, in pyrrhotite they are caused by the ordering of Fe vacancies, and in smythite periodic stacking faults are responsible for the superstructure. In the minerals industry, understanding the differences in the reactivity of different crystallographic superstructures of pyrrhotite is valuable. The literature on the superstructures of pyrrhotite in this respect has recently been summarised by Multani and Waters.28 It has been shown that oxygen is easily incorporated into pyrrhotite and troilite surfaces,29–32 but further studies are necessary for understanding what effect this has on catalytic mechanisms. There is, additionally, a lack of theoretical literature aiding understanding in this field.33,34 Pyrrhotite is the most abundant iron sulfide in the earth and the solar system, therefore studies of it and its related compounds such as smythite and troilite are of importance for understanding a large variety of mechanisms.
In this study, we present a preparation method to grow well-ordered iron sulfide films with an Fe-deficient NiAs-like structure similar to that of pyrrhotite and the related smythite. This growth method can form the basis for further studies of the structure and surface reactivity of iron sulfides.
Experimental
Film growth was performed in a purpose-built preparation chamber, which was required in order to avoid sulfur contamination in the connected analysis chamber. The preparation chamber contained a sample stage with heating provided by a lamp, a quartz microbalance (QMB) for calibration of the Fe deposition rate, an EFM3 evaporator from Omicron fitted with a 99.99+% purity Fe rod from Goodfellow, and a sulfur source. The base pressure of the deposition chamber was in the 10−10 mbar range. The sulfur source was an electrochemical cell, characterised for the first time by Wagner,35,36 and built in-house following the design of Heegemann et al.37 The cell consists of a Ag plate as an anode, a AgI pellet, a Ag2S pellet, and a Pt mesh as a cathode. AgI is an ionic conductor, and Ag+ ions are mobile in Ag2S above ∼200 °C.38 Ag can be added to or removed from the Ag2S pellet when a potential is applied across the cell. Below a critical concentration of Ag in the Ag2S pellet, sulfur sublimes into the chamber. This sulfur cell allows precise control of the rate of sulfur deposition, as the sulfur removed from the cell is proportional to the current through the cell. Here both the current and the average chamber pressure, as measured by an ion gauge, are reported for ease of comparison with other setups. The sulfur cell was operated at 210 °C, and at this temperature it has been found that almost exclusively S2 molecules are produced.39 Sublimation of sulfur into the chamber is not immediate upon applying a potential across the cell, as first the required Ag concentration in the Ag2S pellet must be reached. Fe deposition was started once the partial pressure of sulfur in the chamber had risen to 5 × 10−8 mbar as measured by an ion gauge. We found that sulfur within the chamber reacted with standard copper sealing gaskets used in our ultra-high vacuum (UHV) system, causing corrosion, and prompting their replacement with Ag-plated gaskets. The preparation chamber was directly connected to the analysis chamber via a gate valve.The analysis chamber was an Omicron UHV chamber equipped with LEED with a MCP detector, a room temperature STM (STM.1), and an X-ray source with Al and Mg anodes and a hemispherical analyser for XPS measurements, all from Omicron. The surface sensitivity of XPS measurements could be varied by changing the angle between the surface normal of the sample and the axis of the analyser. Unless stated, measurements were made at normal emission geometry (0°), using Mg K-alpha radiation. The analyser was run in Constant Analyser Energy mode, with a pass energy of 20 eV. The binding energy was calibrated using the Fermi edge and 4f peaks of a Au(111) crystal. Also within the chamber was a quadropole mass spectrometer (QMS) for temperature programmed desorption (TPD) measurements, facilities for Ar+ bombardment, and a W filament behind the sample for annealing. The sample temperature was measured via a K-type thermocouple spot-welded to the side of the substrate.
For XRD measurements, the sample was transferred in vacuo into a mobile UHV chamber (base pressure 4 × 10−8 mbar), which was subsequently mounted on a 6-circle diffractometer. The Cu K-alpha radiation from a standard X-ray tube was focused into an approximately 300 μm diameter spot on the sample.40 Data were collected by taking rocking scans of the Bragg reflections, whereby the beam was always kept at a grazing angle with respect to the thin film surface. This geometry is beneficial for reducing the scattering from the underlying Au substrate.
Results and discussion
Thin film growth
Preliminary attempts at iron sulfide film growth were attempted using a Pt(001) substrate. This substrate was chosen because initial growth attempts were aimed at producing the (001) surface of greigite (Fe3S4), due to a recent study showing CO2 conversion by greigite nanoparticles with (001) faces.22 The surface unit cell parameter of the Pt(001) is 2.77 Å, which for a cubic metal with an fcc structure corresponds to the nearest-neighbour distance in the (001) plane. The surface unit cell of the (001) surface of greigite is 6.99 Å, giving a lattice mismatch of 0.8% for a 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio between the substrate and film. It was possible to grow hexagonal iron sulfide films on this substrate, however they were of poor crystalline order, as judged by the LEED pattern (ESI,† Fig. S1). Additionally, the films were unstable when annealed, with the Pt signal increasing in XPS (ESI,† Fig. S2). This indicated that either dewetting or loss of material from the film was occurring.
:2 ratio between the substrate and film. It was possible to grow hexagonal iron sulfide films on this substrate, however they were of poor crystalline order, as judged by the LEED pattern (ESI,† Fig. S1). Additionally, the films were unstable when annealed, with the Pt signal increasing in XPS (ESI,† Fig. S2). This indicated that either dewetting or loss of material from the film was occurring.
Because of the difficulties encountered with growing these iron sulfide films on Pt(001), a Au substrate was tried instead. We considered that using Au could potentially provide a more suitable substrate for the stability of the film because Au is less miscible than Pt with Fe. In view of the preference for hexagonal iron sulfide film growth on Pt(001), the (111) surface of Au was chosen in order to provide a hexagonal template. As the surface unit cell parameter of Au(111) is 2.86 Å, similar to that of Pt(001), the hexagonal template should promote film growth that is commensurate with a ratio of 5:2 along both surface lattice vectors. It was reasoned that this could provide a better template for growth of well-ordered iron sulfide films. The substrate was prepared via cycles of Ar+ sputtering followed by annealing at 900 K until no carbon or oxygen were visible in XPS (detection limit for C/O ≈ 1% atomic concentration) and the LEED pattern was as shown in Fig. 1a. The broad diffraction spots are due to the presence of the herringbone reconstruction on the Au(111) surface. Fe was deposited at rates of 1.65–2.10 Å min−1 (calibrated with a QMB), with S deposited simultaneously with IS = 5 mA. The average sulfur partial pressure during deposition was 5 × 10−7 mbar. The sample was held at 625 K during film deposition. This temperature was chosen to promote well-ordered growth, while remaining below the temperature where dewetting and decomposition occurs (see ESI,† Fig. S1c). We found that reducing the Fe deposition rate to <1.0 Å min−1 led to poorly ordered films with additional spots in the LEED pattern (not shown). Films were deposited with various thicknesses between 5 and 25 nm. The experimental results obtained on these films were essentially identical, therefore, unless stated, the data from the 25 nm thick film is shown. XPS spectra of the iron sulfide films showed no C or O above the detection limit (<1 atomic percent).
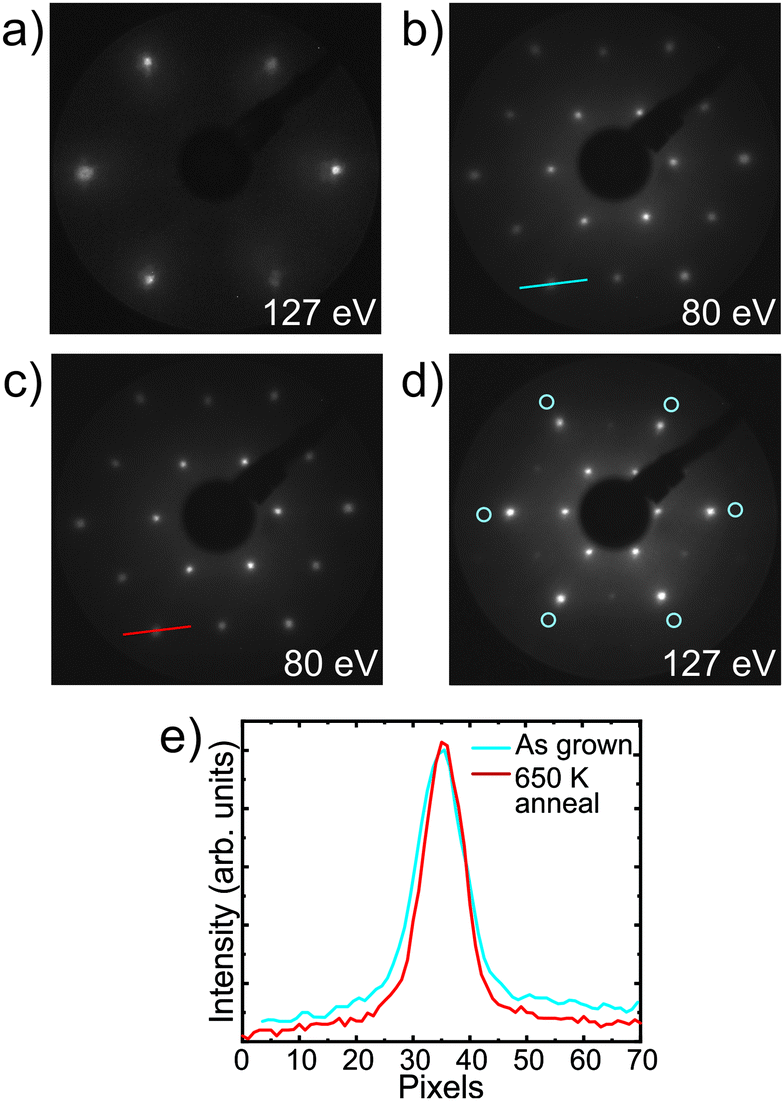 | ||
| Fig. 1 LEED images of (a) the Au(111) substrate prior to film growth, (b) the iron sulfide film immediately following growth, (c and d) following annealing at 650 K for 10 min, with blue circles in (d) indicating the positions of the substrate spots. Images have been brightened for clarity. (e) Shows a comparison of the spot profiles along the lines indicated in (b) and (c), showing the improvement in background intensity and spot sharpness. | ||
The LEED pattern shown in Fig. 1b was visible immediately following deposition. Upon further annealing at 650 K for 10 min the LEED pattern was improved, with sharper spots and reduced background intensity, as shown in Fig. 1c and the comparison spot profiles in Fig. 1e. Following this annealing step, no increase in signal from Au was observed in XPS, showing that no dewetting and no diffusion of Au to the surface had occurred. Annealing was limited to temperatures <700 K to preserve the film, as TPD experiments on preliminary films on Pt(001) showed rapid desorption of sulfur from the iron sulfide film above this temperature (see ESI,† Fig. S1c). Fig. 1d shows the LEED pattern from the iron sulfide film, with the positions of the spots from the underlying Au crystal marked. As in the case of the film grown on Pt(001), by comparing the distances of the LEED spots from the centre of the LEED pattern, the surface unit cell parameter of the iron sulfide film was estimated to be ∼7 Å.
The spot intensities observed in the LEED pattern reveal sixfold symmetry, in contrast to the symmetry of the Au(111) substrate which has P3m1 symmetry. This could be due to a film with 6-fold rotational symmetry, or due to superposition of intensities either from different rotational domains of the film or from terraces separated by steps that are half the unit cell height.
XRD measurements
To determine the structure of the film, XRD experiments were performed on a film with a thickness of 8 nm. In total, 21 reflections were observed and measured. Of these, there were 8 non-equivalent reflections. Though this data set is quite limited, it was sufficient for evaluating the unit cell parameters, the stacking of the film, and providing an estimate for the stoichiometry. Lorentz, polarisation, and intercepted beam corrections were used to deduce the observed structure factors. These can be found in Table S1 in the ESI.† The unit cell parameters for the film were calculated taking the Au lattice parameters as a reference. They were revealed to be a = b = 3.477 ± 0.005 Å, c = 5.790 ± 0.010 Å, α = β = 90.04 ± 0.05°, and γ = 120.06 ± 0.05°. The cell parameters are very similar to those found for Fe0.94S41 and Fe0.95S.42Indexing of the spots and analysing their symmetry showed that the film belongs to the hexagonal space group 186 (P63mc) with AbAc stacking. Cubic iron sulfides such as greigite or pyrite were ruled out because an ABC-type stacking sequence is not compatible with the diffraction spots observed here – (1,0,0) and (1,1,2) Bragg peaks were observed, which would be forbidden in ABC-stacked cubic materials and are direct evidence of a hexagonal structure. Although the spots observed could correspond to a wurtzite-type structure, to our knowledge no wurtzite iron sulfide has ever been observed, though it has been discussed as a hypothetical possibility.43 Moreover, the 4-fold coordination of S atoms in the wurtzite structure is not supported by our XPS results presented later, therefore this structure was ruled out.
The a and b unit cell parameters measured by XRD are half the size of the surface unit cell parameters observed in LEED. This reveals that the periodicity observed in the LEED pattern is due to a surface reconstruction. The sixfold symmetry of the bulk structure also explains the sixfold symmetry of the LEED pattern. The structure providing the best fit to the data was a NiAs-like structure with the S atoms slightly displaced along the c-axis. This structure is shown in Fig. S3 in the ESI.† The fitting was refined using an overall scale factor, Fe occupancy, and the z-position of the S atoms. The final atomic coordinates are given in Table 1. The R-value for the best fit was R = 0.157, where R = ∑|Fobs − Fcalc|/∑|Fobs|. Here, Fcalc and Fobs are the calculated and observed structure factors of each spot. Although X-ray structure refinement is not commonly used for structural determination of thin films, this has been shown to be a valid approach.44 The R-value obtained from such studies is typically worse than obtained for a single crystal sample, and the R-value we achieve here is comparable to that obtained in tests of this approach.44
| Atom | Site occupancy | Wyckoff symbol | Position | 〈u2〉 (Å2) |
|---|---|---|---|---|
| Fe | 0.74 ± 0.09 | 2a | (0, 0, 0) | 0.025 |
| S | 1 | 2b | (1/3, 2/3, 0.26 ± 0.01) | 0.025 |
Refinement was performed using the same isotropic Debye–Waller factor, B, for both Fe and S. Refining B for Fe and S independently results in too many fit parameters for a data set of only 8 unique reflections. This is because adding more fitting parameters risks having a large correlation between them, making the fitting unreliable. A value of B = 2 Å2 gave the best result, and represents a static disorder of about 0.15 Å. Although large, this is still less than some values found in literature for powders and single crystals.45 Thin films in general suffer from enlarged static disorder due to having inferior crystal quality when compared to single crystals. It could also be an indication of small but systematic deviations from the special positions imposed by the chosen space group. It is known that many different polytypes of iron sulfides with the NiAs structure exist. For example, Fe0.9857S can form a superstructure of the underlying NiAs-type building block, whereby the a and b axes are doubled and the c-axis remains identical.46 This structure, which has the same symmetry as NiAs, is characterised by Fe-vacancy ordering and small excursions from ideal positions as defined by the NiAs-type lattice. Such a crystal structure should result in additional, but very weak diffraction spots, besides the ones observed by us. Although we did not observe any of these reflections, we cannot completely exclude that the thin film sample studied here has such a crystal structure with such small deviations. The doubling of the a and b axes of this structure would be in line with the observed LEED pattern.
The refinement gives an Fe content of 74 ± 9% relative to a fully occupied NiAs structure. An error bar of the order of 10% is what is expected for XRD. The only conclusion one can draw from this data alone is that the film contains many Fe vacancies. We do not observe additional spots in the diffraction data, which suggests that either the Fe vacancies are not ordered, or that superstructure spots due to ordering were too weak to be detected. Attempts were made to modify the stoichiometry of the film by annealing to higher temperatures in UHV to remove excess sulfur. This however resulted in a large increase of the intensity of the signal from Au in XPS (not shown). Annealing the film in an S2 partial pressure of 5 × 10−7 mbar at 625 K, as an attempt to increase the S concentration of the film, did not change the properties of the film. The Fe occupancy, even at the upper limit of the error margin, puts it outside the normally given range of the pyrrhotite system (87.5–100%), but perhaps could refer to an Fe-deficient pyrrhotite. Smythite (Fe9S11 or Fe13S16) falls within the range of Fe concentration estimated from the XRD data. This material essentially has the NiAs structure with periodic stacking faults, however peaks associated with these stacking faults are not observed.
XPS
XPS spectra of the S 2p region of the film were obtained with varying surface sensitivity. From Fig. 2b it is clear that there must be a surface component to the shape of the S 2p peak shape. This is highlighted by taking the difference of the normalised spectra obtained at 0° and 70° emission geometry. A doublet with the 2p3/2 peak at 161.0 eV is revealed as the surface component. This is in line with the previously observed sulfur surface state in pyrrhotite.47 The regions at both 0° (Fig. 2a) and 70° (Fig. 2c) were fitted with doublet peaks separated by 1.19 eV with a lineshape given by a Gaussian–Lorentzian product approximation of the Voigt function (50% Gaussian). The full-width at half-maximum (FWHM) of both peaks of each doublet was constrained to be the same, and the 2p1/2 peak was given half of the area of the 2p3/2 peak. A Shirley background was subtracted from the spectra.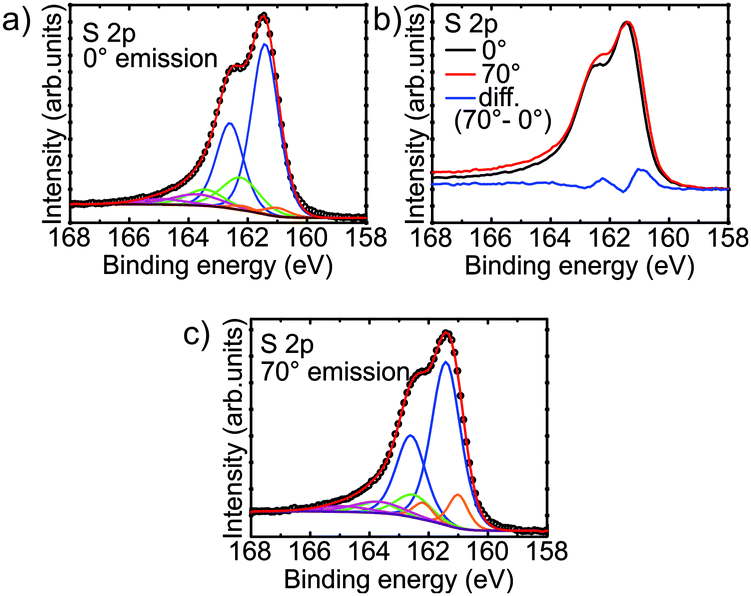 | ||
| Fig. 2 S 2p XPS spectra from the iron sulfide film obtained at (a) normal emission (0°), and (c) 70°. (b) Difference spectrum showing the presence of a surface peak at 161.0 eV. | ||
In addition to the surface doublet, the fitting revealed a large main doublet with the 2p3/2 peak at 161.4 eV, with a secondary doublet with the 2p3/2 peak at 162.5 eV. A third, broad doublet is placed with its 2p3/2 peak at ∼163.7 eV. The doublets at 161.4 eV and 162.5 eV are assigned to fivefold and sixfold coordinated sulfur respectively, in analogy to pyrrhotite which shares the basic NiAs structure.30,47 The origin of the intensity encompassed by the broad doublet at higher binding energy is not clear. Intensity in this region is observed in pyrrhotite and pyrite, and has been variously assigned to polysulfide species, inadequacy of the fitted peak shape, or energy loss features, with the latter being most likely.47–50
Discounting the contribution from the surface and the additional intensity at higher binding energy, the doublet corresponding to fivefold coordinated sulfur accounts for approximately 82% of the total S 2p peak area. This corresponds to an average coordination of the sulfur atoms of 5.18, which suggests an Fe occupancy of 86%. This is close to that of pyrrhotite Fe7S8 (87.5%). This estimate of the stoichiometry puts the Fe occupancy a little more than one standard deviation away from the value obtained from XRD measurements (74 ± 9%), but confirms that the film contains a significant amount of Fe vacancies. In fact, if the film had an Fe occupancy of 74% (and a uniform distribution of vacancies) we would not expect to see any sixfold coordinated sulfur at all, as the average coordination of the sulfur atoms would be 6 × 0.74 = 4.44. There are, however, some possible explanations for the analysis of the S 2p XPS spectra estimating larger values for the Fe occupancy. Firstly, there is a fairly large error margin for the quantification of the peak area using XPS. This is caused by factors such as photoelectron diffraction from the surrounding atoms.51–53 The photoelectron diffraction effect could have some effect in this case, as the fivefold and sixfold coordinated sulfur atoms are clearly in a different environment. Secondly, as XPS is a surface sensitive technique, it is conceivable that the Fe vacancy concentration varies as a function of depth. For example, there could be a loss of Fe from near the film/substrate interface into the Au substrate, or there may be more Fe near the surface due to a surface reconstruction.
The XPS spectrum of the Fe 2p3/2 region measured at normal emission is shown in Fig. 3. The peaks were fitted with multiplets as assigned by Pratt et al. for pyrrhotite (Fe7S8) using the calculations of Gupta and Sen.30,54,55 The large peak at 707.5 eV was assigned to Fe2+, with multiplet peaks at ±0.9 eV. The peaks at 709.1 eV, 710.2 eV, 711.3 eV, and 712.3 eV were assigned to Fe3+ multiplets. Additionally, the peak at 713.6 eV was assigned to an Fe2+ satellite peak. The presence of the multiplets indicates that the Fe within the film is in a high-spin state. Approximately 43% of the signal comes from ferric (Fe3+) iron. The ratio of ferrous (Fe2+) and ferric iron also gives information about the stoichiometry of the film. In FeS in the NiAs structure, all of the iron is in the Fe2+ state. In order to maintain charge neutrality, each Fe vacancy must lead to two other Fe ions being in the Fe3+ state. The ferric iron concentration observed here corresponds to an Fe occupancy of ∼82%, which is comparable to the Fe occupancy derived from the S 2p spectra. This is also a higher Fe occupancy than estimated from XRD, but still falls within one standard deviation of it (74 ± 9%). As mentioned above for the S 2p region, the ratios of ferrous and ferric iron also have error margins related to effects such as photoelectron diffraction. Additionally, this value could also be affected by the presence of a surface reconstruction, with potentially a lower vacancy density in the surface region. However, unlike for the S 2p region, no significant change is observed in spectra measured at different angles of photoemission. This suggests that the film has a sulfur termination. The observed ∼43% ferric iron is significantly higher than the 29% observed in pyrrhotite Fe7S8 and predicted from its stoichiometry.28,30
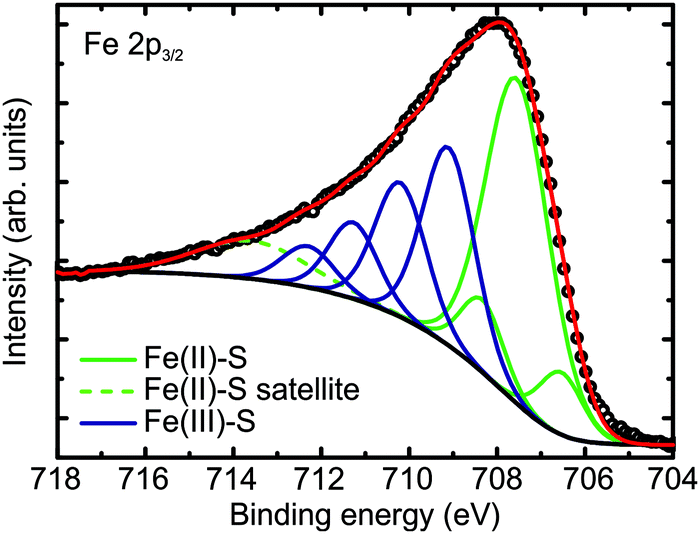 | ||
| Fig. 3 Fe 2p3/2 XPS spectrum from the iron sulfide film, fitted with multiplets corresponding to Fe2+ and Fe3+ bonded to S. | ||
STM
STM images from the iron sulfide film show terraces several tens of nanometers wide. The terraces have occasional pits, as shown in Fig. 4a, which are ∼3 Å deep. An example of terrace steps is shown in Fig. 4b. The minimum step height was measured to be ∼3 Å, corresponding to half of the unit cell height. However, steps of ∼6 Å were observed more frequently across the surface, which could be related to step formation energies. | ||
| Fig. 4 STM images from the iron sulfide film. In (b) the inset shows a line profile across the two steps along the green line. The 7 Å surface unit cell is highlighted in blue in (e), with the unit cell of the bulk (001) plane measured in XRD (black dashed line) alongside for comparison. Tunnelling conditions: (a) sample bias V = −1.61 V, current I = 0.10 nA, (b) V = −2.0 V, I = 0.19 nA, (c) V = −2.0 V, I = 1 nA, (d) V = +1.50 V, I = 2.50 nA, (e) V = +1.97 V, I = 5.04 nA. | ||
Fig. 4c–e show atomically resolved STM images of the iron sulfide film, revealing a periodicity of ∼7 Å for the unit cell, which is highlighted in Fig. 4e. This periodicity is in line with what is observed in the LEED pattern, and points to the film having a 2 × 2 reconstruction relative to the bulk structure. The surface unit cell has P3m1 symmetry. Within the unit cell four protrusions are seen, with three of them in symmetrically equivalent positions. The images look similar to those obtained by Becker et al. on pyrrhotite samples,33 however in their work the fourth, single protrusion was not visible, and the other three protrusions were found to be inequivalent when using a sample bias between 0 eV and −3 eV.
Defect structures can be observed at the surface of the film. In Fig. 4c and d, there are missing protrusions which have the appearance of vacancies. The missing protrusions can either be from the group of three symmetrically equivalent protrusions, or from the fourth protrusion in the unit cell. In addition to this, anti-phase domain boundaries are observed on the surface, as shown in Fig. 5a and b. In Fig. 5b, coloured lines are drawn along lines of protrusions in order to highlight the domains. It appears that the domain boundaries begin/end at vacancy defects. The anti-phase domain boundaries are probably constrained to the surface, with the surface reconstruction simply shifted by one bulk unit cell parameter, corresponding to half of a surface unit cell parameter.
 | ||
| Fig. 5 STM images showing anti-phase domain boundaries at the surface of the iron sulfide film. Tunnelling conditions: (a) V = −1.61 V, I = 5.0 nA, (b) V = −2.0 V, I = 5.0 nA. In (b), the black and green lines indicate the two anti-phase domains present. | ||
I/V-LEED calculations
In order to try to understand the surface structure of the film, fully dynamical LEED intensity calculations were carried out as described in previous work.56,57 Contrary to XRD, I/V-LEED is a very surface sensitive method and we hoped that it would reveal the atomic structure of the reconstructed surface. Different bulk-terminated iron sulfide model structures were used as a starting point for the I/V-LEED structural optimization: iron sulfides with a hexagonal face having a lattice parameter in the range of ∼7 Å, i.e. pyrite, greigite, and hexagonal pyrrhotite (Fe7S8), and also iron sulfides with a surface lattice parameter of ∼3.5 Å because of the possibility of a 2 × 2 reconstruction being present at the surface. The latter include smythite and high-temperature FeS in the NiAs structure. Additionally, we tried to narrow the search to include only model structures which might match the features observed in STM images. Structural optimization was performed with the differential evolution (DE) and covariance matrix adaptation evolutionary strategy (CMA-ES) optimization algorithms58,59 by minimization of the Pendry R-factor, RP.60 Initial calculations employed a LEED intensity dataset covering an energy range of 1212 eV. The details of the calculations can be found in the ESI,† however we will summarise our findings here. Our calculations using bulk-like terminations did not produce a satisfactory R-factor, with a minimum R-factor of RP = 0.272 being achieved for a sulfur-terminated greigite layer. We then attempted to modify the surfaces in various ways to simulate possible surface reconstructions that may be present, eventually achieving RP = 0.178 for a greigite-like film with a reordering of the surface layer sequence as shown in Fig. 6a. However, upon using an extended dataset of 1807 eV, the R-factor increased to 0.274 (Fig. 6b), indicating that the model did not accurately represent the surface structure of the film, although this R-factor does suggest some resemblance. Additionally, the results generally add support for a S-terminated film, as such model structures consistently provided lower R-factors than Fe-terminated ones. The high-dimensional search space involved in searching for the surface structure of an as-yet unknown termination (stoichiometry, structure) proved to be too large for the narrow convergent range of LEED-I/V, therefore there is no conclusive surface structure determination from these studies yet.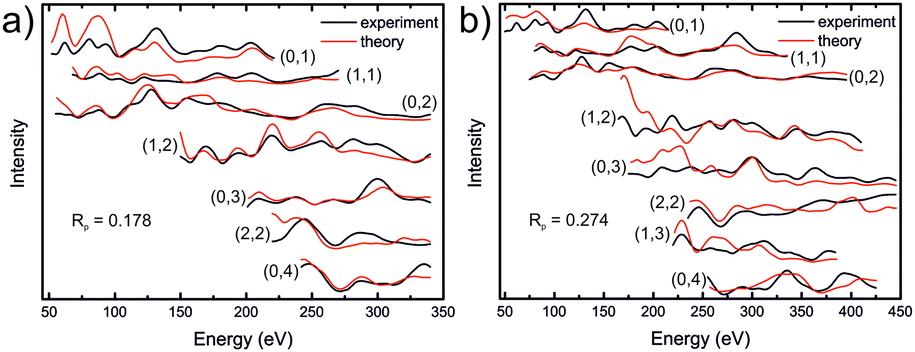 | ||
| Fig. 6 LEED-I/V curves for a modified greigite-like surface, using a total energy range of (a) 1212 eV and (b) 1807 eV. Detailed information regarding the structure model is found in the ESI.† | ||
Conclusions
We have presented a method to grow well-ordered iron sulfide thin films on a Au(111) substrate and have characterised the structure of the films. XRD measurements suggest that the film has a NiAs-like structure, similar to the pyrrhotite system. XRD and XPS data suggest that the film contains a greater concentration of Fe vacancies than pyrrhotite, and may have a stoichiometry closer to that of smythite. The vacancy concentration could potentially be due to loss of Fe from the film into the substrate, but confirmation of this would require further investigation. The surface of the film undergoes a 2 × 2 reconstruction, and large flat terraces are imaged using STM, however the exact surface structure remains unknown. The results of this study can be a foundation for further insight into characteristics of this complex material system by providing a model system on which structure and reactivity experiments can be performed.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge the contribution of the COST Action CA17120. Open Access funding provided by the Max Planck Society.References
- D. Rickard and G. W. Luther, Chem. Rev., 2007, 107, 514–562 CrossRef CAS.
- R. S. C. Smart, J. Amarantidis, W. M. Skinner, C. A. Prestidge, L. La Vanier and S. R. Grano, Solid-Liquid Interfaces, Springer, Berlin, Heidelberg, 2003, pp. 3–62 Search PubMed.
- T. Yang, M. J. Dekkers and J. Chen, J. Geophys. Res.: Solid Earth, 2018, 123, 1116–1131 CrossRef.
- A. G. Tomkins and B. Morgan, in Encyclopedia of Geochemistry, ed. W. M. White, Springer, Cham, 2017, Sulfide Minerals, pp. 1–3 Search PubMed.
- Q. Williams and R. Jeanloz, J. Geophys. Res., 1990, 95, 19299 CrossRef CAS.
- G. Wächtershäuser, Microbiol. Rev., 1988, 52, 452–484 Search PubMed.
- M. J. Russell, A. J. Hall, A. G. Cairns-Smith and P. S. Braterman, Nature, 1988, 336, 117 CrossRef.
- M. A. A. Schoonen and Y. Xu, Astrobiology, 2001, 1, 133–142 CrossRef CAS.
- G. D. Cody, N. Z. Boctor, J. A. Brandes, T. R. Filley, R. M. Hazen and H. S. Yoder, Geochim. Cosmochim. Acta, 2004, 68, 2185–2196 CrossRef CAS.
- G. D. Cody, Annu. Rev. Earth Planet. Sci., 2004, 32, 569–599 CrossRef CAS.
- R. Murphy and D. R. Strongin, Surf. Sci. Rep., 2009, 64, 1–45 CrossRef CAS.
- V. P. Evangelou, Pyrite Oxidation and Its Control, CRC Press, Boca Raton, 1995 Search PubMed.
- M.-J. Choi, J. Kim, J.-K. Yoo, S. Yim, J. Jeon and Y. S. Jung, Small, 2018, 14, 1702816 CrossRef.
- G. Ertl, Angew. Chem., Int. Ed., 2008, 47, 3524–3535 CrossRef CAS.
- H.-J. Freund, J. Am. Chem. Soc., 2016, 138, 8985–8996 CrossRef CAS.
- C. Wadia, A. P. Alivisatos and D. M. Kammen, Environ. Sci. Technol., 2009, 43, 2072–2077 CrossRef CAS.
- R. A. Berner, Am. J. Sci., 1970, 268, 1–23 CrossRef CAS.
- R. A. Berner, Geochim. Cosmochim. Acta, 1984, 48, 605–615 CrossRef CAS.
- S. Hunger and L. G. Benning, Geochem. Trans., 2007, 8, 1 CrossRef.
- X. Lai, H. Zhang, Y. Wang, X. Wang, X. Zhang, J. Lin and F. Huang, J. Am. Chem. Soc., 2015, 137, 10148–10151 CrossRef CAS.
- U.-P. Apfel and W. Weigand, Angew. Chem., Int. Ed., 2011, 50, 4262–4264 CrossRef CAS.
- A. Roldan, N. Hollingsworth, A. Roffey, H.-U. Islam, J. B. M. Goodall, C. R. A. Catlow, J. A. Darr, W. Bras, G. Sankar, K. B. Holt, G. Hogarth and N. H. de Leeuw, Chem. Commun., 2015, 51, 7501–7504 RSC.
- R. C. Erd, H. T. Evans Jr. and D. H. Richter, Am. Mineral., 1957, 42, 309–333 CAS.
- L. A. Taylor and K. L. Williams, Am. Mineral., 1972, 57, 1571–1577 CAS.
- M. E. Fleet, Phys. Chem. Miner., 1982, 8, 241–246 CrossRef CAS.
- P. Waldner and A. D. Pelton, J. Phase Equilib. Diffus., 2005, 26, 23–38 CrossRef CAS.
- H. Wang and I. Salveson, Phase Transitions, 2005, 78, 547–567 CrossRef CAS.
- R. S. Multani and K. E. Waters, Can. J. Chem. Eng., 2018, 96, 1185–1206 CrossRef CAS.
- A. N. Buckley and R. Woods, Appl. Surf. Sci., 1985, 22–23, 280–287 CAS.
- A. R. Pratt, I. J. Muir and H. W. Nesbitt, Geochim. Cosmochim. Acta, 1994, 58, 827–841 CrossRef CAS.
- Y. Mikhlin, V. Varnek, I. Asanov, Y. Tomashevich, A. Okotrub, A. Livshits, G. Selyutin and G. Pashkov, Phys. Chem. Chem. Phys., 2000, 2, 4393–4398 RSC.
- N. Belzile, Y. W. Chen, M. F. Cai and Y. Li, J. Geochem. Explor., 2004, 84, 65–76 CrossRef CAS.
- U. Becker, A. W. Munz, A. R. Lennie, G. Thornton and D. J. Vaughan, Surf. Sci., 1997, 389, 66–87 CrossRef CAS.
- U. Terranova, C. Mitchell, M. Sankar, D. Morgan and N. H. De Leeuw, J. Phys. Chem. C, 2018, 122, 12810–12818 CrossRef CAS.
- C. Wagner, Z. Elektrochem. Angew. Phys. Chem., 1934, 40, 364 CAS.
- C. Wagner, J. Chem. Phys., 1953, 21, 1819 CrossRef.
- W. Heegemann, K. Meister, E. Bechtold and K. Hayek, Surf. Sci., 1975, 49, 161–180 CrossRef CAS.
- H. Reinhold, Z. Elektrochem. Angew. Phys. Chem., 1934, 40, 361–364 CAS.
- D. Detry and J. Drowart, Z. Phys. Chem., 1967, 55, 314 CrossRef CAS.
- A. Stierle, T. F. Keller, H. Noei, V. Vonk and R. Roehlsberger, J. Large-scale Res. Facil., 2016, 2, A76 CrossRef.
- E. J. Fasiska, Phys. Status Solidi, 1972, 10, 169–173 CrossRef CAS.
- F. Li and H. F. Franzen, J. Alloys Compd., 1996, 238, 73–80 CrossRef CAS.
- A. R. Lennie and D. J. Vaughan, Mineral Spectrosc., 1996, 117–132 Search PubMed.
- P. Lee, H. Graafsma, Y. Gao, H. S. Sheu, P. Coppens, S. J. Golden and F. F. Lange, Acta Crystallogr., Sect. A: Found. Crystallogr., 1991, 47, 57–59 CrossRef.
- L.-M. Peng, G. Ren, S. L. Dudarev and M. J. Whelan, Acta Crystallogr., Sect. A: Found. Crystallogr., 1996, 52, 456–470 CrossRef.
- F. Keller-Besrest, G. Collin and R. Comès, Acta Crystallogr., Sect. B: Struct. Sci., 1983, 39, 296–303 CrossRef.
- H. W. Nesbitt, A. G. Schaufuss, M. Scaini, G. M. Bancroft and R. Szargan, Am. Mineral., 2001, 86, 318–326 CrossRef.
- M. Bronold, Y. Tomm and W. Jaegermann, Surf. Sci., 1994, 314, 0–5 CrossRef CAS.
- A. G. Schaufuß, H. W. Nesbitt, I. Kartio, K. Laajalehto, G. M. Bancroft and R. Szargan, Surf. Sci., 1998, 411, 321–328 CrossRef.
- W. M. Skinner, H. W. Nesbitt and A. R. Pratt, Geochim. Cosmochim. Acta, 2004, 68, 2259–2263 CrossRef CAS.
- O. A. Baschenko and V. I. Nefedov, J. Electron Spectrosc. Relat. Phenom., 1979, 17, 405–420 CrossRef.
- O. A. Baschenko and V. I. Nefedov, J. Electron Spectrosc. Relat. Phenom., 1980, 21, 153–169 CrossRef.
- O. A. Baschenko and V. I. Nefedov, J. Electron Spectrosc. Relat. Phenom., 1982, 27, 109–118 CrossRef CAS.
- R. P. Gupta and S. K. Sen, Phys. Rev. B, 1974, 10, 71–77 CrossRef CAS.
- R. P. Gupta and S. K. Sen, Phys. Rev. B, 1975, 12, 15–19 CrossRef CAS.
- F. E. Feiten, H. Kuhlenbeck and H.-J. Freund, J. Phys. Chem. C, 2015, 119, 22961–22969 CrossRef CAS.
- E. Primorac, H. Kuhlenbeck and H. J. Freund, Surf. Sci., 2016, 649, 90–100 CrossRef CAS.
- N. Hansen, in Towards a New Evolutionary Computation, ed. J. A. Lozano, P. Larrañaga, I. Inza and E. Bengoetxea, Springer Berlin Heidelberg, Berlin, Heidelberg, 2006, pp. 75–102 Search PubMed.
- R. Storn and K. Price, J. Global Optim., 1997, 11, 341–359 CrossRef.
- J. B. Pendry, J. Phys. C: Solid State Phys., 1980, 13, 937–944 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: (1) XRD data – observed and calculated structure factors, figure showing the NiAs-like structure derived from structure refinement. (2) Additional information about the I/V-LEED calculations performed in this study. See DOI: 10.1039/c9cp04157e |
| This journal is © the Owner Societies 2019 |