
Nanocavity effects of various zeolite frameworks on n-pentane cracking to light olefins: combination studies of DFT calculations and experiments†

Abstract
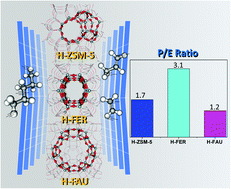
Better control of the product selectivity of light olefins (e.g., ethylene and propylene) obtained from the n-pentane catalytic cracking process has attracted considerable attention from both scientific and petrochemical industrial points of view. In this context, we report insights into the effects of the nanocavities of various zeolite frameworks, including H-FER, H-ZSM-5, and H-FAU, representing small, medium, and large cavities, on the reaction mechanism of n-pentane cracking to light olefins by using M06-2X/6-31G(d,p) density functional calculations, eventually leading to fine-tuning the product distribution of light olefins. The reaction mechanism consists of the following two main steps: (i) the protolytic cracking of n-pentane to form a pentonium intermediate; and (ii) the subsequent dissociation of the intermediate to either ethane–propylene or ethylene–propane. The key reaction pathways controlling the product distribution of light olefins relate to the dissociation of the pentonium intermediate, which can produce selectively either propylene (P) or ethylene (E), resulting in a controllable P/E ratio. The differences in the activation energies for ethylene production compared with those of propylene production over H-FER, H-ZSM-5, and H-FAU are 6.7, 5.0, and 0.5 kcal mol−1, respectively. Compared with H-ZSM-5 and H-FAU, the higher difference in the activation energy of these two pathways over H-FER implies that the preferable production of ethane–propylene compared with ethylene–propane is more pronounced. It is therefore reasonable to conclude that a smaller pore zeolite such as H-FER eventually leads to a high ratio of production of propylene to ethylene, in accordance with experimental observations.
- This article is part of the themed collection: 2019 PCCP HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

