
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Visualizing reaction and diffusion in xanthan gum aerosol particles exposed to ozone†
Peter A.
Alpert
 *a,
Pablo
Corral Arroyo
ab,
Jing
Dou
c,
Ulrich K.
Krieger
c,
Sarah S.
Steimer
d,
Jan-David
Förster
e,
Florian
Ditas
e,
Christopher
Pöhlker
e,
Stéphanie
Rossignol
fg,
Monica
Passananti
fhi,
Sebastien
Perrier
f,
Christian
George
f,
Manabu
Shiraiwa
j,
Thomas
Berkemeier
ek,
Benjamin
Watts
l and
Markus
Ammann
*a
*a,
Pablo
Corral Arroyo
ab,
Jing
Dou
c,
Ulrich K.
Krieger
c,
Sarah S.
Steimer
d,
Jan-David
Förster
e,
Florian
Ditas
e,
Christopher
Pöhlker
e,
Stéphanie
Rossignol
fg,
Monica
Passananti
fhi,
Sebastien
Perrier
f,
Christian
George
f,
Manabu
Shiraiwa
j,
Thomas
Berkemeier
ek,
Benjamin
Watts
l and
Markus
Ammann
*a
aLaboratory of Environmental Chemistry, Paul Scherrer Institute, 5232 Villigen PSI, Switzerland. E-mail: Peter.Alpert@psi.ch; Markus.Ammann@psi.ch
bInstitute for Physical Chemistry, ETH Zürich, 8092 Zürich, Switzerland
cInstitute for Atmospheric and Climate Science, ETH Zürich, 8092 Zürich, Switzerland
dDepartment of Chemistry, University of Cambridge, Cambridge, CB2 1EW, UK
eMultiphase Chemistry Department, Max Planck Institute for Chemistry, 55128 Mainz, Germany
fUniv. Lyon, Université Claude Bernard Lyon 1, CNRS, IRCELYON, F-69626, Villeurbanne, France
gAix Marseille Université, CNRS, LCE UMR 7376, 13331 Marseille, France
hInstitute for Atmospheric and Earth System Research/Physics, Faculty of Science, University of Helsinki, 00710, Helsinki, Finland
iDipartimento di Chimica, Università di Torino, Via Giuria 5, 10125 Torino, Italy
jDepartment of Chemistry, University of California, Irvine, CA 92697-2025, USA
kSchool of Chemical & Biomolecular Engineering, Georgia Institute of Technology, Atlanta, GA 30332, USA
lLaboratory for Synchrotron Radiation-Condensed Matter, Paul Scherrer Institute, 5232 Villigen PSI, Switzerland
First published on 17th September 2019
Abstract
Atmospheric aerosol particles with a high viscosity may become inhomogeneously mixed during chemical processing. Models have predicted gradients in condensed phase reactant concentration throughout particles as the result of diffusion and chemical reaction limitations, termed chemical gradients. However, these have never been directly observed for atmospherically relevant particle diameters. We investigated the reaction between ozone and aerosol particles composed of xanthan gum and FeCl2 and observed the in situ chemical reaction that oxidized Fe2+ to Fe3+ using X-ray spectromicroscopy. Iron oxidation state of particles as small as 0.2 μm in diameter were imaged over time with a spatial resolution of tens of nanometers. We found that the loss off Fe2+ accelerated with increasing ozone concentration and relative humidity, RH. Concentric 2-D column integrated profiles of the Fe2+ fraction, α, out of the total iron were derived and demonstrated that particle surfaces became oxidized while particle cores remained unreacted at RH = 0–20%. At higher RH, chemical gradients evolved over time, extended deeper from the particle surface, and Fe2+ became more homogeneously distributed. We used the kinetic multi-layer model for aerosol surface and bulk chemistry (KM-SUB) to simulate ozone reaction constrained with our observations and inferred key parameters as a function of RH including Henry's Law constant for ozone, HO3, and diffusion coefficients for ozone and iron, DO3 and DFe, respectively. We found that HO3 is higher in our xanthan gum/FeCl2 particles than for water and increases when RH decreased from about 80% to dry conditions. This coincided with a decrease in both DO3 and DFe. In order to reproduce observed chemical gradients, our model predicted that ozone could not be present further than a few nanometers from a particle surface indicating near surface reactions were driving changes in iron oxidation state. However, the observed chemical gradients in α observed over hundreds of nanometers must have been the result of iron transport from the particle interior to the surface where ozone oxidation occurred. In the context of our results, we examine the applicability of the reacto-diffusive framework and discuss diffusion limitations for other reactive gas-aerosol systems of atmospheric importance.
1 Introduction
Organic matter in atmospheric aerosol stems from a variety of natural and anthropogenic sources and is found in significant quantities in almost every single particle.1–3 Aerosol particles can both scatter and absorb light to varying degrees depending on their morphology and chemical composition, and they significantly impact the global radiative balance.4,5 These particles also act as nuclei for liquid droplets and ice in clouds.6–8 The fate of aerosol particles residing in the atmosphere is largely dependent on physical processes such as rain out and gravitational settling, but also on chemical transformation.9 Atmospheric aerosol particles are largely composed of organic and inorganic species such as sulfate, nitrate, ammonium, sea salts and trace metals, and this mix of compounds can influence uptake and internal production of oxidants.10 In turn, reactions in the condensed aerosol phase can lead to significant chemical change over time.11–13 For example, chemical reactions can lead to more oxygenated functionalities in particles thus increasing hydrophilicity leading to more efficient cloud condensation nuclei.14 On the other hand, less hygroscopic sea spray aerosol particles can result from first condensing organic acids, then chlorine displacement and finally the formation of sodium-organic salts that take up less water than marine halides.14,15 Aerosol particles dominated by organic matter can be highly viscous with physical properties similar to glass or tar, or they can exist in a more liquid-like state depending on location, altitude, temperature and relative humidity.16,17 Reactions in highly viscous particles can be diffusion limited, i.e. when the transport of the involved parent species is slow. This results in a reduced overall rate of reaction.17,18 There is a wealth of research into the effects of this phenomenon on chemical and physical aerosol processes and includes, in part, aerosol growth after nucleation,19–21 condensed phase reactions in secondary organic aerosol,22 water uptake and ice nucleation7 and preservation and transport of biomass burning tracer compounds23 or pollutants.13,24Oxidants such as OH25–35 and O336–41 reacting with micrometer and submicrometer sized organic aerosol particles in a humidified environment have been previously observed and modeled to increase our understanding of how these small molecules may diffuse through and react within atmospheric particles. Further studies have made great strides in developing model frameworks to describe experimental data, including modeling of chemical reactions at the surface and within the bulk of aerosol particles.29,32,35,36,42–52 In Shiraiwa et al.,36 the chemical half life of amino acids in thin films of bovine serum albumin (BSA) at a relative humidity, RH, of 90% and temperature, T, of 25 °C was determined to be about 5 min and increased to over an hour when RH < 50%. These authors used the kinetic multi-layer model for aerosol surface and bulk chemistry (KM-SUB) to attribute this RH dependence on inhomogeneous mixing of reactants and products and calculated that at RH = 50%, the BSA diffusion coefficient was 10−20 cm2 s−1 with a viscosity close to that of a glass,36 typically greater than 1012 Pa s.53–55 Ozone had a diffusion coefficient of 10−9 cm2 s−1 and was predicted only to be present and reacting in the first tens of nanometers of the BSA films.36 Heine et al.41 found that the decay of squalene reacting with O3 over time was identical over the RH range of 0–60%, leading them to use a stochastic multilayer model, Kinetiscope, to reproduce their results having a 1 nm adsorption layer where the reaction with O3 was predicted to mainly occur. This was supported by the authors' previous studies40 and their calculations of the O3 diffusion coefficient during reaction observed for particles with a 1–2 nm squalene coating.56 In an earlier study by Steimer et al.,38 shikimic acid particles were chemically imaged with scanning transmission X-ray microscopy coupled to near-edge X-ray absorption fine structure spectroscopy (STXM/NEXAFS) during O3 exposure as a function of RH. The authors directly probed the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C 1s → π* electronic transition and observed that reactive decay was highly dependent on RH, where shikimic acid lifetime increased 3 orders of magnitude when RH was decreased from 82 to 12%. Later, Steimer et al.39 measured O3 reactive uptake coefficients to shikimic acid in a comprehensive data set with high time resolution over seconds to 14 h at RH = 92, 83, 68, 45, 24 and 0%, a constant O3 gas phase concentration, [O3]g = 178 ppb and additionally for various [O3]g = 79, 178, 495 and 1985 ppb at RH = 92 and 24%. This data set was a benchmark for a later modeling study by Berkemeier et al.49 using KM-SUB, who were able to determine physico-chemical parameters with high confidence such as chemical reaction kinetics of shikimic acid and reactive oxygen intermediates as well as changes in particle phase and molecular transport of O3, all of which would have otherwise been impossible under a narrow experimental range of RH, time and [O3]g.
C 1s → π* electronic transition and observed that reactive decay was highly dependent on RH, where shikimic acid lifetime increased 3 orders of magnitude when RH was decreased from 82 to 12%. Later, Steimer et al.39 measured O3 reactive uptake coefficients to shikimic acid in a comprehensive data set with high time resolution over seconds to 14 h at RH = 92, 83, 68, 45, 24 and 0%, a constant O3 gas phase concentration, [O3]g = 178 ppb and additionally for various [O3]g = 79, 178, 495 and 1985 ppb at RH = 92 and 24%. This data set was a benchmark for a later modeling study by Berkemeier et al.49 using KM-SUB, who were able to determine physico-chemical parameters with high confidence such as chemical reaction kinetics of shikimic acid and reactive oxygen intermediates as well as changes in particle phase and molecular transport of O3, all of which would have otherwise been impossible under a narrow experimental range of RH, time and [O3]g.
Observations and models mentioned above gave great insight to how molecules react and diffuse in aerosol particles, however there is a lack of direct observations of the internal chemical gradients throughout the bulk of a particle to compare against any model predictions until now. The term chemical gradient is defined here to describe a concentration gradient of a reactant inside an aerosol particle due to chemical reaction and diffusion. We use STXM/NEXAFS to image chemical gradients with 35 nm resolution inside organic aerosol particles in situ during O3 reaction at different RH and [O3]g. Steimer et al.38 were the first to attempt such a measurement. In a scenario where O3 loss is fast compared to its rate of transport, referred to as a reacto-diffusive limitation, O3 should not penetrate far into the particle interior. Instead, O3 would be limited to a characteristic length scale known as the reacto-diffusive length, which depends on its diffusion coefficient and first order loss rate.38 More precisely, it is the distance over which the O3 concentration in the condensed phase, [O3], drops by a factor of 1/e. In Steimer et al.,38 imaged chemical gradients in CC absorption signal were not discernible under dry conditions, while homogeneous degradation of CC occurred throughout the particles under humid conditions. Considering uncertainties due to image alignment, a low X-ray signal at thin particle edges and a limit on the contrast between CC absorption and total carbon, these authors hypothesized that shikimic acid loss was constrained to a too thin layer to become visible.38 Berkemeier et al.49 later calculated the extent of the chemical gradient under low RH conditions to be well below 100 nm and beyond the resolution of STXM/NEXAFS.
In this study, we exposed particles composed of a mixture of xanthan gum (XG) and FeCl2 to O3 and observed internal chemical reaction gradients over time, t. Iron in FeCl2 has an oxidation state of +2 and transitions to an oxidation state of +3 when reacted with O3, i.e. from Fe2+ to Fe3+.57–61 The use of iron in particles is a major benefit when employing STXM/NEXAFS due to the fact that X-ray absorption peaks are strong, narrow and thus can be observed with much higher contrast to detect Fe2+ and Fe3+ compared to the absorption from carbon functionalities such as CC used in previous measurements.38,40 Additionally, we built on previous work62 and developed novel experimental and analytical procedures to quickly and efficiently quantify Fe2+ and Fe3+ for thousands of individual particles with a well-constrained uncertainty. In general, Fe containing aerosol particles are important, e.g. for their role in generating reactive oxygen species and free radicals in lung fluid and potentially leading to detrimential human health effects.63–66 Iron deposition impacts ocean fertilization and stimulating the growth of phytoplankton.67 Iron oxidation state can affect iron solubility, and iron can bind with organic ligands in atmospheric or oceanic particles influencing bioavailability in aquatic ecosystems.68–75 Moffet et al.62 used STXM/NEXAFS to quantify the Fe2+ fraction, α, out of the total iron in ambient particles and calculated an average mass weighted value of α = 0.33 ± 0.08 during a pollution transport event from China to Japan, of which ∼5% of particles contained detectable iron. As a polysaccharide and biopolymer, XG is a unique model compound of marine derived organic matter in atmospheric aerosol.76 The change in XG composition by a few percent in water is enough to result in large changes in solution viscosity, a property that is highly desired for additives used in the food industry.77,78 XG hygroscopicity is of particular interest because when RH decreases, XG water content decreases and its viscosity increases.76,79 We note that XG is a reference compound for quantifying what is known as “transparent exopolymer particles” in oceans80–82 which have recently been found in ambient air at concentrations of 2 μg m−3 over the North Atlantic.83 For these reasons, the XG/Fe2+ system is an interesting proxy for understanding molecular diffusion and reaction limitations in atmospheric marine derived aerosol.
Here, we used iron as a tracer for STXM/NEXAFS to unambiguously identify chemical gradients in oxidation state as the direct result of reaction and molecular diffusion limitation. These data were used to experimentally derive 2-D projected α profiles within thousands of individual particles in situ yielding the first direct evidence of chemical gradients in viscous particles. We report on how gradients change when particles were dry (RH = 0%) or humidified at RH = 20, 40, 60 and 80%. KM-SUB was used to model molecular transport and reaction in spherical shells of aerosol particles and derived 3-D radial profiles of α using known chemical reaction rates. These 3-D α profiles were then used to calculate 2-D column integrated profiles of α for direct comparison with STXM/NEXAFS observations. Model parameters were diffusion coefficients for Fe and O3, DFe and DO3, and were described with a Vignes-type equation as a function of water mole fraction. Henry's Law constant for O3 in the XG/FeCl2 matrix, HO3, was also derived. Our STXM/NEXAFS spatio-chemical data allows a novel constraint for modeling aerosol internal chemical profiles, i.e. simultaneous reproduction of bulk Fe2+ depletion and the spatio-temporal evolution of O3 and Fe2+ reaction. In the context of our results, we discuss the applicability of the reacto-diffusive limiting case and the importance of direct observational constraints on model predictions of atmospheric aerosol chemical aging.
2 Methods
Mixed XG and FeCl2 particles were nebulized from dilute aqueous solution, dried and impacted onto silicon nitride membranes for exposure experiments and in situ STXM/NEXAFS analysis. Particle diameters on all samples were between 0.2 and 4.0 μm. Prior to impaction, silicon nitride substrates were pre-glued into removable sample exchange clips fitted to either of two microreactors named the PolLux environmental microreactor84 and the MPI-C aerosol micro-reactor.85 Both microreactors flow a stream of gas with controllable temperature, humidity and ozone content over particles remaining fixed on X-ray transparent membranes. Further details of the microreactors are provided in previous publications84,85 and specific operating conditions are provided in the ESI.† Particles were impacted onto the silicon nitride substrates and exposed to He, O2, H2O and O3 at a desired partial pressure and Tp = 20 °C, and thus RH and reactive gas exposure could be well-controlled.38,84 Helium was used as a carrier gas with a flow of 20 cm3 min−1 at standard temperature and pressure. A flow of O2 at 5 cm3 min−1 first passed through a custom built UV lamp O3 generator then mixed with the humidified He flow. The concentration of O3 over t was measured online using an absorption cell38 and is given in Fig. S1 of the ESI.† The calculated water partial pressure and Tp was used to determine the RH the particles were exposed to using the saturation vapor pressure parameterizations following Murphy and Koop.86 A total microreactor pressure of 150 mbar and flow rate of 25 cm3 min−1 was maintained. The operating principle of the MPI-C aerosol micro-reactor is similar to the PolLux microreactor. The MPI environmental cell was coupled to the same gas flow system as the PolLux microreactor with equal gas flow rates. However, the cell pressure was maintained at ∼450 mbar. This allowed for a greater concentration of O2 in the O3 generator, and thus a greater [O3]g for experiment with higher exposure. Detailed experimental methods are further outlined in the ESI.†The Fe oxidation state and carbon functionality was probed using STXM/NEXAFS at the PolLux endstation located at the Swiss Light Source (SLS).87 The transmission of X-ray photons through the particle was measured and converted to optical density, OD = −ln(I/I0), where I0 and I are the incident and transmitted photon count, respectively, and their uncertainty is 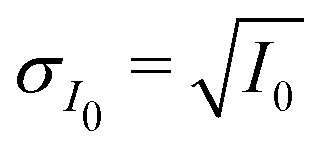 and
and 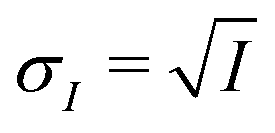 . X-ray light was focused to a 35 by 35 nm spot size while scanning a field of view (FOV) on the order of micrometers to generate an OD image at a single X-ray energy. An image was made of hundreds to thousands of individually measured pixels, where each is the OD over an area of ≃10−3 μm2. Multiple OD images over the same FOV taken over a range of X-ray energies were aligned and processed using publicly available software.88 We primarily investigated the X-ray energy range between 700–735 eV, which is the Fe L-edge absorption. When present in particles, peak absorption due to Fe2+ and Fe3+ occur at slightly different X-ray energies, allowing to differentiate between the two.89,90 The X-ray energy at Fe2+ peak absorption for FeCl2 measured here was compared with previous literature62 and an energy offset was obtained as a calibration. Peak absorptions for ferrous and ferric chloride are at 707.8 and 709.5 eV, respectively62 as seen in Fig. 1. Using the parameterization from Moffet et al.,62α was determined from measuring the peak optical density, OD, corresponding to Fe2+ and Fe3+ indicated in Fig. 1 and taking their ratio, r. A more detailed description of our experimental methods, observations and how r and α is calculated is given in the ESI.† In addition, a careful assessment of X-ray exposure used to not damage the particles while maximizing signal is also included (see Fig. S2 and S3, ESI†).
. X-ray light was focused to a 35 by 35 nm spot size while scanning a field of view (FOV) on the order of micrometers to generate an OD image at a single X-ray energy. An image was made of hundreds to thousands of individually measured pixels, where each is the OD over an area of ≃10−3 μm2. Multiple OD images over the same FOV taken over a range of X-ray energies were aligned and processed using publicly available software.88 We primarily investigated the X-ray energy range between 700–735 eV, which is the Fe L-edge absorption. When present in particles, peak absorption due to Fe2+ and Fe3+ occur at slightly different X-ray energies, allowing to differentiate between the two.89,90 The X-ray energy at Fe2+ peak absorption for FeCl2 measured here was compared with previous literature62 and an energy offset was obtained as a calibration. Peak absorptions for ferrous and ferric chloride are at 707.8 and 709.5 eV, respectively62 as seen in Fig. 1. Using the parameterization from Moffet et al.,62α was determined from measuring the peak optical density, OD, corresponding to Fe2+ and Fe3+ indicated in Fig. 1 and taking their ratio, r. A more detailed description of our experimental methods, observations and how r and α is calculated is given in the ESI.† In addition, a careful assessment of X-ray exposure used to not damage the particles while maximizing signal is also included (see Fig. S2 and S3, ESI†).
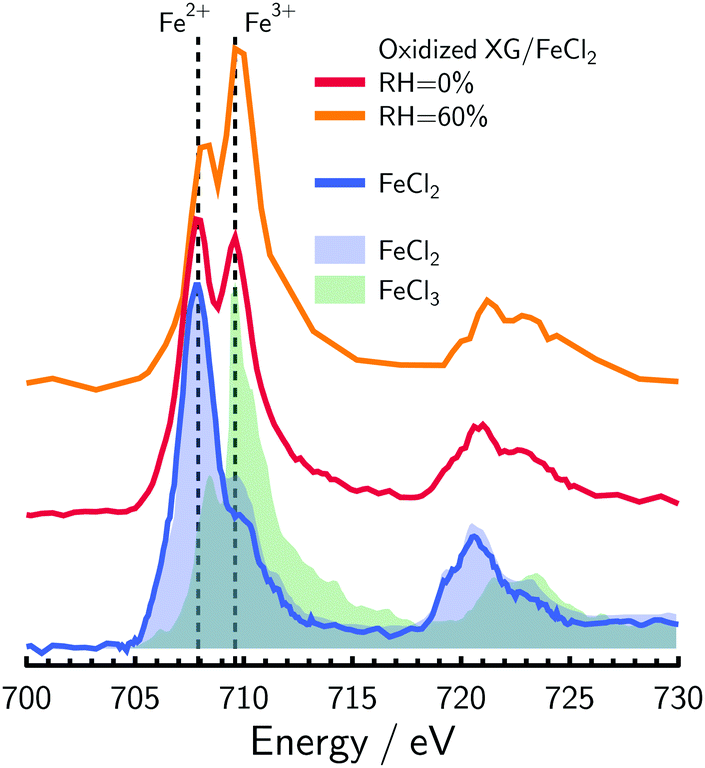 | ||
| Fig. 1 Near edge X-ray absorption fine structure (NEXAFS) spectra of oxidized XG/FeCl2 particles at RH = 0 and 60% shown as the red and orange lines, respectively, along with measured FeCl2 spectra as the blue line. Standards for ferrous and ferric chloride are shown as the blue and green shading, respectively.62 Spectra have been scaled and shifted vertically for clarity. Dashed lines indicate typical peak absorption for Fe2+ and Fe3+ at X-ray energies of 707.9 and 709.6 eV. | ||
The KM-SUB model was used to simulate chemical reaction and molecular transport inside of our XG/FeCl2 particles and infer diffusion and solubility parameters.45,49 A detailed description of KM-SUB can be found in Shiraiwa et al.45 and Berkemeier et al.49 Briefly, this model solves the system of non-linear differential equations for the time rate of change of O3 and Fe2+ in prescribed layers with defined thickness. It accounts for O3 accommodation and absorption, the O3 and Fe2+ diffusion from the surface and within bulk layers and finally, the chemical loss of O3 and Fe2+. The Knudsen number for our flow conditions was on the order of 100 and γ was on the order of 10−4–10−6 during reaction. These were used to determine the gas diffusion correction factor42 close to unity and thus was ignored. A single spherical aerosol particle geometry with dp = 0.5 μm was applied for all simulations. The model had 2 spatial regions where the first extended from the surface to a short distance into the bulk and hosted 100 to 500 finely spaced layers to resolve the O3 concentration there. The surface typically had the strongest chemical gradients because O3 molecules that entered the particle were rapidly consumed by reaction with Fe2+. Care was taken to resolve lrd for both O3 and Fe2+ with multiple layers. The remaining particle bulk had evenly spaced layers with 0.5 to 2 nm thickness, which was sufficient to resolve observed Fe2+ gradients. The total number of layers in a simulation varied between 500–1500 depending on O3 and Fe2+ gradients. α0 was approximated to be uniform through the particle. The O3 uptake coefficient, γ, was calculated over time using the O3 surface coverage, the net flux from the gas phase to the surface and the collision flux45,49 shown in Fig. S9 in the ESI.† The model was modified to include a changing gas phase O3 concentration over t.
3 Results and discussion
3.1 NEXAFS spectra
Fig. 1 shows example NEXAFS spectra of oxidized XG/FeCl2 particles at dry and humidified conditions in comparison with the reference material FeCl2 measured here and FeCl2 and FeCl3 from literature.62 Two absorption peaks at 707.8 and 709.6 eV were observed for oxidized XG/FeCl2 particles and were in agreement with Fe2+ and Fe3+ peak absorption energies for FeCl2 and FeCl3, respectively. Although nearly identical peak positions were observed, we did not investigate how humidity and the organic polysaccharide matrix influences Fe electronic excitations in detail. Important to note from Fig. 1 is that only Fe2+ and Fe3+ peaks were observed in oxidized particles meaning that the total Fe concentration, [Fetot] = [Fe2+] + [Fe3+], where [Fe2+] and [Fe3+] is the concentration of Fe2+ and Fe3+ species, respectively, and| α = [Fe2+]/[Fetot]. | (1) |
3.2 Xanthan gum/FeCl2 particles exposed to oxygen
We performed exposure experiments of XG/FeCl2 particles to O2 and observed no change in α as a function of t at any RH we investigated (see Fig. S4 in the ESI†). Therefore, any reaction taking place between O2 and Fe2+ over our time scales was considered negligible. Further discussion of reaction kinetics of O2 and O3 is discussed later in Section 4.5. The scatter in α for O2 exposure mostly fell within ±0.07, which was the standard deviation of α for individual particles with a diameter, dp, seen in Fig. S5 in the ESI.† It is apparent that α values for larger particles have a smaller error. This is due to both the greater number of pixels and the greater signal statistics, i.e. larger particles are thicker, absorb more and thus, have a better signal to noise ratio. Particles with dp < 0.2 μm (not shown) were discarded from our analysis because their error was typically larger than any physically realistic range in α. The standard deviation of each data set indicated in Fig. S5 and plotted in Fig. S4 (ESI†) was typically larger than the error propagated through quadrature. For the remainder of the manuscript, error bars on all α values are either ±0.07 or the propagated error, whichever is greater.3.3 Oxidation of xanthan gum/FeCl2 particles by ozone
Fig. 2 shows α averaged over all particles as a function of time, ozone exposure, ϕ, and RH. The most striking result is that under dry conditions where RH = 0 and 20%, very little change in α was observed. At more humid conditions of RH = 40, 60 and 80%, average α values decreased noticeably over t. Although the green circles for RH = 60% shows the greatest decrease in α, this is a consequence of using higher [O3]g (see Fig. S1 in the ESI†). At RH = 0, 20 and 60%, shown as red, pink and green circles, respectively, [O3]g was on the order of 103 ppb which was about 10 times higher than for RH = 40 and 80%. Fig. 2b shows α as a function of , where [O3]g(t) is taken from Fig. S1 (ESI†). Iron oxidation was greater at RH = 80% than at any other RH as a function of ϕ. This can be seen in particular at ϕ = 10−3 atm s, where the blue symbols reach α = 0.3 while all other data at RH < 80% have higher α. In general, as RH or [O3]g increased, O3 heterogeneous reaction rates must have also increased.
, where [O3]g(t) is taken from Fig. S1 (ESI†). Iron oxidation was greater at RH = 80% than at any other RH as a function of ϕ. This can be seen in particular at ϕ = 10−3 atm s, where the blue symbols reach α = 0.3 while all other data at RH < 80% have higher α. In general, as RH or [O3]g increased, O3 heterogeneous reaction rates must have also increased.
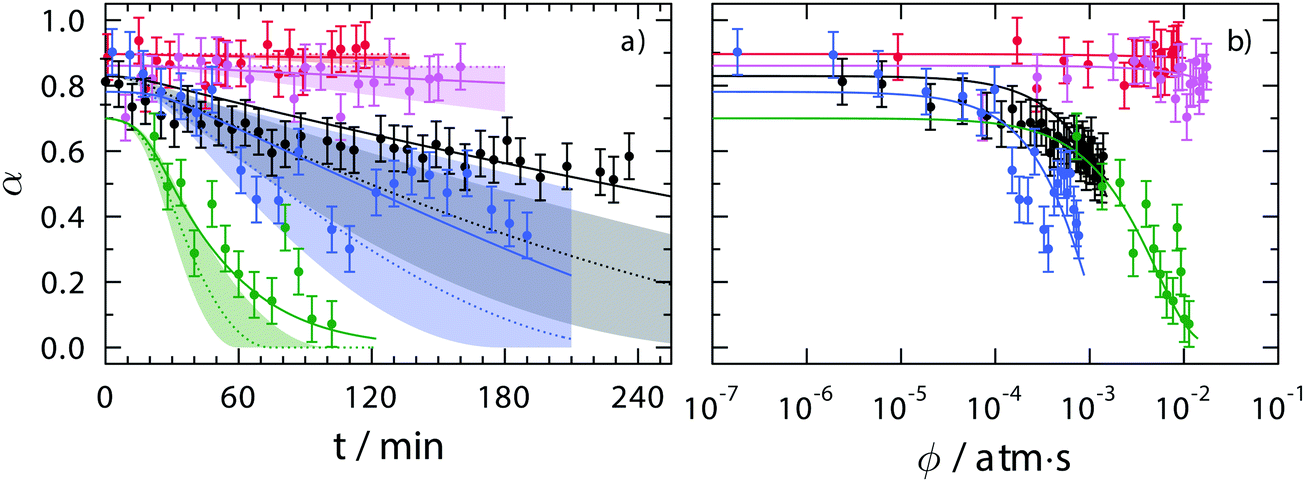 | ||
| Fig. 2 Measured and modeled depletion of the Fe2+ fraction, α, as a function of (a) time, t, and (b) ozone exposure, ϕ, for RH = 0, 22, 43, 60 and 80% as red, pink, black, green and blue circles, respectively. Each data point was determined from approximately 5–25 particles each within a field of view imaged with STXM. The error bars are either ±0.07 or the propagated error from photon counting statistics, whichever is greater. Solid lines are calculations from the KM-SUB model using parameters given in Table 1. The dotted line and shadings in panel (a) are predictions applying the reacto-diffusive framework described in more detail in the text. Shadings are not shown in panel (b) for clarity. | ||
We acquired chemical images with high spatial resolution to quantify α over the particles in two dimensions. To accomplish this, α was averaged over all pixels identified at the perimeter of particles irrespective of particle size. In other words, α was calculated from the particle perimeter to 1 pixel, or 35 nm, from the surface. Then, all adjacent concentric pixels toward the particle center (from 1 to 2 pixels from the particle surface or 35–70 nm) were identified and their corresponding α values averaged. This continued toward the center of particles and generated a 2-D concentric profile of α, which is also a column integrated profile.
Fig. 3 shows 2-D profiles of α as a function of time and the pixels (distance) from the perimeter of particles for all experiments. More oxidation occurred at the perimeter than at the center of particles, and the chemical gradients were typically shallow for all RH. For example, α increased from 0.45 to 0.65 at RH = 40% and t = 180 min (orange symbol color in Fig. 3c) over ∼0.5 μm. At RH = 80%, α increased from 0.25 to 0.35 for the same reaction time and distance. For RH = 0 and 20%, α increased only from the surface to ∼0.2 μm in depth, implying a stronger Fe2+ concentration gradient under dry conditions than for more humid conditions. Prior to exposure, initial values of α were not equivalent to 1, implying that the short time spent in contact in ambient laboratory air was enough to trigger oxidation. However, we carefully quantified initial α prior to O3 exposure to ensure a high enough value for experiments as shown by the grey symbols in Fig. 3.
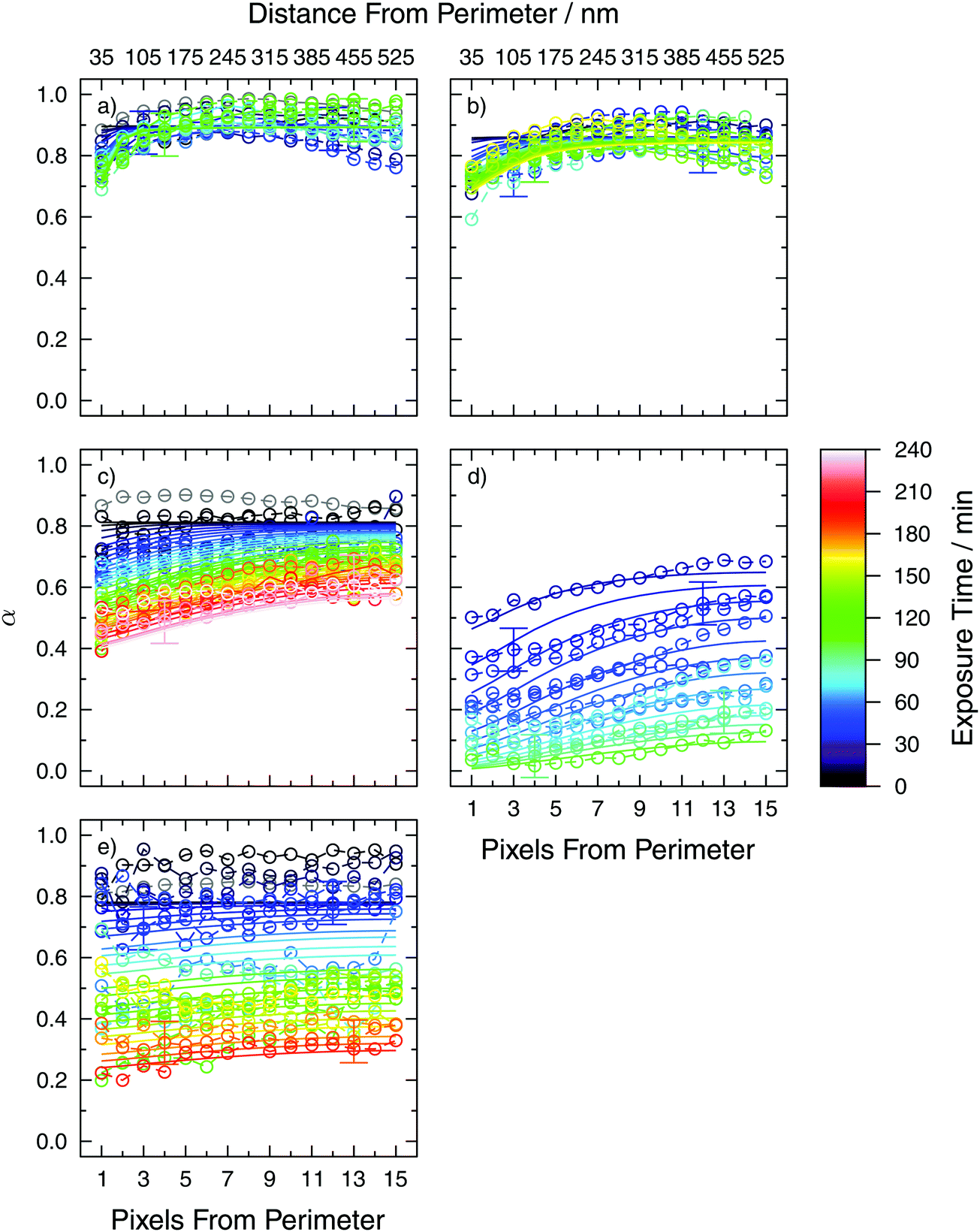 | ||
| Fig. 3 Observed and modeled 2-D profiles of the Fe2+ fraction, α, as a function of O3 exposure time as the color scale at a RH of (a) 0%, (b) 22%, (c) 43%, (d) 60% and (e) 80%. Grey symbols are initial conditions prior to O3 exposure. Example error bars are included only on some data points for clarity. The bottom and top abscissa are the pixels and distance from the particle perimeter, respectively, where each pixel has a spatial dimension of 35 by 35 nm. Solid lines are fit to this data with parameters given in Table 1. | ||
Observed 2-D profiles were likely to be influenced by 4 factors. The first is the plasticizing effect water has on viscous XG-containing particles to the extent that a greater water content likely enhanced DO3. At low RH, DO3 may have been low enough to limit penetration to the particle bulk for further reaction with Fe2+ in line with our observations that α was always less at particle surfaces than at their center. The glass transition temperature of XG was previously determined to be −16.4 and −23.3 °C for RH of 11 and 84%, respectively.79 Although our experiments were performed at 20 °C which is significantly warmer than the glass transition temperature, we still expect sufficiently low diffusion coefficients as to limit reactions due to the fact that XG is thermally stable and its viscosity does not change much with temperature.78,91 Dawson et al.76 extrapolated viscosity measurements in dilute XG aqueous solutions from Wyatt and Liberatore92 to higher concentrations and RH = 80% obtaining 1013 Pa s at 20 °C. We note that extrapolations beyond experimental conditions can be inaccurate. However, this high value is in line with our suggestion that XG remains highly viscous and possibly close to or in a glass-like state even at high humidity. As a consequence, slow molecular transport of O3 through the glassy XG matrix should contribute to our observed gradients.
The second effect on observed 2-D profiles and RH-dependent oxidation kinetics is that HO3 may increase under dry conditions in XG compared to water where the condensed phase ozone concentration, [O3] = HO3[O3]g. It was observed previously that for pure shikimic acid, HO3 in organic material was about an order of magnitude higher than for O3 in water.38,49 Solubility of O3 is known to be higher in less polar solvents, thus it is expected that higher organic content (less polar than water) leads to an increase in O3 solubility.93 Higher values of HO3 may increase condensed phase O3 concentration near the surface, increase the first order reaction rate there and cause stronger chemical gradients at low RH. A third reason is the change in the diffusion coefficient of Fe as a function of RH. As XG is a hydrogel material that takes up water in subsaturated conditions, we hypothesize that the Fe diffusion coefficient, DFe, should increase with increasing RH. A higher or lower value of DFe should result in more homogeneous or inhomogeneous α distribution, respectively, which is in line with our observations.
The fourth factor influencing 2-D profiles is dilution with water at higher RH. Water uptake will increase particle volume and cause [Fetot] to decrease and thus decrease the first order loss rate. Accounting for the water content as a function of RH between 0–80% given by Dawson et al.,76 [Fetot] varies by about 20%. This is small and expected to have little effect compared to changing Henry's Law constant or the diffusion coefficient, which is typically orders of magnitude over a relatively narrow range in RH.49,94 We argue that faster transport of Fe2+ out of the particle core and faster transport of O3 into the particle from the surface brings them together more readily allowing reactions to proceed at a faster rate at higher RH.
3.4 Prediction of molecular diffusion and solubility
We have employed the KM-SUB model49 to infer values of HO3, DO3 and DFe shown in Fig. 4 which reproduce observed 2-D profiles in α. Although KM-SUB has been previously constrained with data relating to the uptake of gas phase oxidants by particles, our data set is novel in that it is the first study to present spatially-resolved condensed-phase concentration profiles. We reiterate that KM-SUB has predicted aerosol particle internal radial composition profiles46,49,95 but up until now, there has been no direct observational evidence that they existed. For direct comparison with our observed profiles in Fig. 3, radial profiles from KM-SUB were converted to 2-D hemispherical projections of α described in more detail in the ESI† and shown in Fig. S6. We have fit modeled 2-D profiles to our observations for all RH each having 4 optimized parameters, HO3, DO3 and DFe, as well as the initial Fe2+ fraction, α0. We note that the value of α0 was constrained by our observations, i.e. it was forced to be within our measurement and uncertainty range. All other parameters used in KM-SUB were taken from previous literature49 for shikimic acid and involve surface processes with O3. Although the authors did not use iron, we assume that the iron in combination with the xanthan gum matrix behaves as shikimic acid in Berkemeier et al.49 Parameter values and their descriptions can be found in Table 1 and described in the ESI.† | ||
| Fig. 4 Fitted parameters from the KM-SUB model. (a) The diffusion coefficient for O3 (white circles), DO3, and the diffusion coefficient for Fe (solid symbols), DFe, are shown as a function of RH for mixed xanthan gum and FeCl2 particles. A Vignes type parameterization for DO3 and DFe are the dotted and solid lines, respectively. (b) Henry's Law constant for O3, HO3, as a function of RH is shown as white circles. Error bars indicate the fit sensitivity described in the text and the ESI.† The dotted and solid line is determined from a volume mixing rule and a Vignes type parameterization, respectively. | ||
| RH = 0% | RH = 22% | RH = 43% | RH = 60% | RH = 80% | ||
|---|---|---|---|---|---|---|
| k R3 | cm3 s−1 | 6.20 × 10−16 | ||||
| k slr1 | cm2 s−1 | 6.27 × 10−14 | ||||
| k slr2 | cm2 s−1 | 4.31 × 10−17 | ||||
| k bs,O3 | cm s−1 | 0.69 | 0.69 | 0.69 | 0.69 | 1.60 |
| k sb,Fe | s−1 | 9.22 × 10−5 | 3.34 × 10−4 | 6.95 × 10−4 | 8.83 × 10−4 | 4.20 × 10−4 |
| D O3 | cm2 s−1 | 2.48 × 10−16 | 1.48 × 10−15 | 1.66 × 10−10 | 3.50 × 10−10 | 4.17 × 10−8 |
| D Fe | cm2 s−1 | 5.52 × 10−18 | 2.84 × 10−15 | 3.94 × 10−14 | 1.46 × 10−13 | 2.24 × 10−13 |
| H O3 | mol cm−3 atm−1 | 2.21 × 10−2 | 4.80 × 10−3 | 1.49 × 10−3 | 5.87 × 10−4 | 3.60 × 10−4 |
| α 0 | 0.90 | 0.86 | 0.83 | 0.70 | 0.78 | |
| K bs | cm | 2.41 × 10−9 | 1.99 × 10−9 | 2.21 × 10−9 | 2.95 × 10−9 | 5.82 × 10−9 |
| τ d,O3 | s | 5.32 × 10−4 | ||||
| α s,0 | 0.50 | |||||
| σ Fe | cm2 | 2.19 × 10−16 | ||||
| [Fetot] | cm−3 | 6.15 × 1021 | 6.06 × 1021 | 5.93 × 1021 | 5.74 × 1021 | 4.99 × 1021 |
| [O3]max | cm−3 | 1.91 × 1016 | 5.71 × 1015 | 9.08 × 1013 | 7.72 × 1014 | 1.58 × 1013 |
| σ O3 | cm2 | 2.42 × 10−14 | ||||
| D g,O3 | cm2 s−1 | 0.91 |
A rate coefficient for O3 and FeCl2 was derived from previous studies57–61 following,
| Fe2+ + O3 → FeO2+ + O2, | (R1) |
| FeO2+ + Fe2+ + 2H+ → 2Fe3+ + H2O. | (R2) |
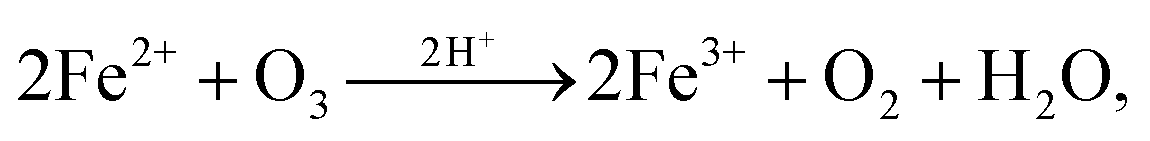 | (R3) |
Fig. 3 shows that the optimization of model-generated 2-D α profiles (solid lines) agree well with our measurements (circles), which is remarkable considering the large differences in [O3]g and the long exposure time on the order of hours. Consistently, Fe was predicted to remain more reduced in particle cores than at particle surfaces. Although not explicitly fit, the modeled α averaged over the entire particle, shown as solid lines in Fig. 2, captures the observed decay and reveals the consistency between modeled and measured α. The fitting parameters, HO3, DO3 and DFe are given in Table 1 and shown in Fig. 4. We note that there is an obvious and expected trend that as RH decreases, both DO3 and DFe values decrease and HO3 increases. A model sensitivity analysis on HO3, DO3 and DFe is detailed in the ESI† (Fig. S10 and S11). Following previous work,49 we calculated the parameter range at which the sum of the squared residual values between observed and model derived 2-D profiles in α varied by 20%, indicated as error bars in Fig. 4.
With regard to O3 transport, DO3 at RH = 0% was equal to 2.48 × 10−16 cm2 s−1 and increased about 8 orders of magnitude to 4.17 × 10−8 cm2 s−1 at RH = 80%. At this high humidity, DO3 was 3 orders of magnitude less than for water, 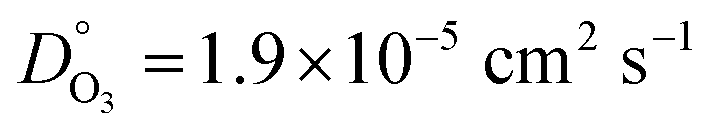 taken from previous work.49,98 Following the Stokes–Einstein relation, the molecular diffusion coefficient is inversely proportional to the viscosity of the matrix that the molecule moves through and inversely proportional to the molecular radius.99,100 In pure (dry) shikimic acid for example, DO3 = 2.90 × 10−12 cm2 s−1, which is 4 orders of magnitude faster than for dry XG/FeCl2 particles. Since DO3 for pure XG is less than for pure shikimic acid, its viscosity should also be higher. It should be noted, however, that for some organic components or when particle viscosity is in excess of 10 Pa s,94,101,102 the Stokes–Einstein relation may not necessarily be accurate. Again applying the Stokes–Einstein relation, the viscosity of an aqueous XG/FeCl2 solution at RH = 80% should be 3 orders of magnitude less than for water. However, the difference in viscosity of XG at RH = 80% (previously suggested to be glass-like at 1013 Pa s) and water viscosity (10−3 Pa s) is 16 orders of magnitude. We speculate that this discrepancy may be due to the porous nature of XG, and that pores may have swelled or expanded during water uptake103 similar to other hydrogels.104 Therefore, O3 mobility may be enhanced in hydrogels that swell compared to what is predicted from the Stokes–Einstein relation.
taken from previous work.49,98 Following the Stokes–Einstein relation, the molecular diffusion coefficient is inversely proportional to the viscosity of the matrix that the molecule moves through and inversely proportional to the molecular radius.99,100 In pure (dry) shikimic acid for example, DO3 = 2.90 × 10−12 cm2 s−1, which is 4 orders of magnitude faster than for dry XG/FeCl2 particles. Since DO3 for pure XG is less than for pure shikimic acid, its viscosity should also be higher. It should be noted, however, that for some organic components or when particle viscosity is in excess of 10 Pa s,94,101,102 the Stokes–Einstein relation may not necessarily be accurate. Again applying the Stokes–Einstein relation, the viscosity of an aqueous XG/FeCl2 solution at RH = 80% should be 3 orders of magnitude less than for water. However, the difference in viscosity of XG at RH = 80% (previously suggested to be glass-like at 1013 Pa s) and water viscosity (10−3 Pa s) is 16 orders of magnitude. We speculate that this discrepancy may be due to the porous nature of XG, and that pores may have swelled or expanded during water uptake103 similar to other hydrogels.104 Therefore, O3 mobility may be enhanced in hydrogels that swell compared to what is predicted from the Stokes–Einstein relation.
For iron transport, DFe was similar to DO3 at low RH, and a few orders of magnitude less than DO3 at higher RH. Values of DFe had a range of about 5 orders of magnitude from dry to RH = 80%. We note that we have not reported on a self-diffusion coefficient of XG in aqueous solution, but it can be found for dilute aqueous solution in previous literature,105e.g. for a 10 g L−1 solution (having a water activity > 0.99) attaining 10−8 cm2 s−1 which is about 3 orders of magnitude less than 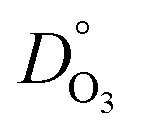 or
or 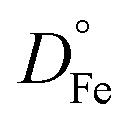 . We suggest that XG self-diffusion coefficients should be less than DO3 or DFe at all RH. A caveat of our analysis is that Fe2+ may complex with XG106,107 and therefore, DFe may be considered an apparent transport of free ions and those in complex which may have higher and lower diffusion coefficient values, respectively.
. We suggest that XG self-diffusion coefficients should be less than DO3 or DFe at all RH. A caveat of our analysis is that Fe2+ may complex with XG106,107 and therefore, DFe may be considered an apparent transport of free ions and those in complex which may have higher and lower diffusion coefficient values, respectively.
In dry XG/FeCl2 particles, HO3 = 2.21 × 10−2 mol cm−3 atm−1, which is about 3 orders of magnitude higher than for pure water, 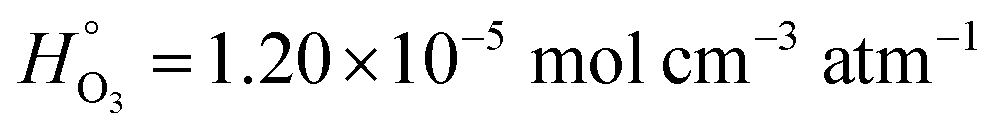 . Greater solubility of O3 in organic liquids compared to water is well known38,49,93 and can be explained by a “salting-in” effect. This is characterized by an increase in the product of ionic strength and activity coefficients of the solution (i.e. decreasing water content) and thus causes an increase in gas solubility. We note that a similar result is found for O2 solubility, which is generally higher in organic liquids than in water.108,109 The degree to which activity of XG/FeCl2 solutions changes as a function of RH is not known. Polymer chains in XG can overlap, aggregate and cross-link105 giving rise to anomalous behavior of XG and oxygen diffusion coefficients in aqueous XG solution.105,110 It may be possible that our ternary XG–FeCl2–water system is non-ideal and deviates from ideal mixing. To our knowledge, this is the first report of O3 solubility in XG. In some polymers, HO3 can be greater or less than
. Greater solubility of O3 in organic liquids compared to water is well known38,49,93 and can be explained by a “salting-in” effect. This is characterized by an increase in the product of ionic strength and activity coefficients of the solution (i.e. decreasing water content) and thus causes an increase in gas solubility. We note that a similar result is found for O2 solubility, which is generally higher in organic liquids than in water.108,109 The degree to which activity of XG/FeCl2 solutions changes as a function of RH is not known. Polymer chains in XG can overlap, aggregate and cross-link105 giving rise to anomalous behavior of XG and oxygen diffusion coefficients in aqueous XG solution.105,110 It may be possible that our ternary XG–FeCl2–water system is non-ideal and deviates from ideal mixing. To our knowledge, this is the first report of O3 solubility in XG. In some polymers, HO3 can be greater or less than 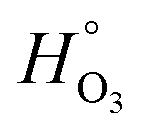 such as Hyflon AD80 and Teflon, having HO3 = 1 × 10−4 and 4 × 10−6 mol cm−3 atm−1, respectively. In XG/FeCl2 particles, the optimized HO3 parameter is higher than expected for polymers, and the reason is not yet entirely clear. It may have to do with our optimization and model sensitivity described in the ESI.† We suggest further measurements should be performed in dilute and concentrated XG solutions to understand and quantify HO3 in XG and the trends with RH and water content.
such as Hyflon AD80 and Teflon, having HO3 = 1 × 10−4 and 4 × 10−6 mol cm−3 atm−1, respectively. In XG/FeCl2 particles, the optimized HO3 parameter is higher than expected for polymers, and the reason is not yet entirely clear. It may have to do with our optimization and model sensitivity described in the ESI.† We suggest further measurements should be performed in dilute and concentrated XG solutions to understand and quantify HO3 in XG and the trends with RH and water content.
Fig. 5 shows 3-D profiles of α and [O3] normalized to its maximum concentration, [O3]max, at each RH derived from the KM-SUB model. Values of [O3]max are given in Table 1. The 3-D profiles of α in Fig. 5 were used to derive the 2-D derived profiles seen in Fig. 3 (solid lines). Gradients in α at RH = 0% (Fig. 5a) spanned a few nanometers, but extended hundreds of nanometers through the particles at RH = 80% (Fig. 5i). Despite the extent of gradients in α, O3 was found only in the first picometer to 2.7 nm at RH = 0 to 80%, respectively. We note that a length of 1 pm is much smaller than the molecular scale and the Fe2+ ionic diameter of 0.7 Å. However, constraining layer thickness to 0.3 nm as previously done49 could not resolve O3 chemical gradients in our case. We defined layer thickness in KM-SUB without a lower limit to satisfy continuum conditions. This had no consequence, however, because bulk transport follows Fick's Law treating an ensemble of molecules. In general, oxidation reactions can have only taken place where O3 was present at or near the surface of particles, and so gradients in α extending far past O3 penetration depths to nanometers and hundreds of nanometers must have been the result primarily of Fe transport.
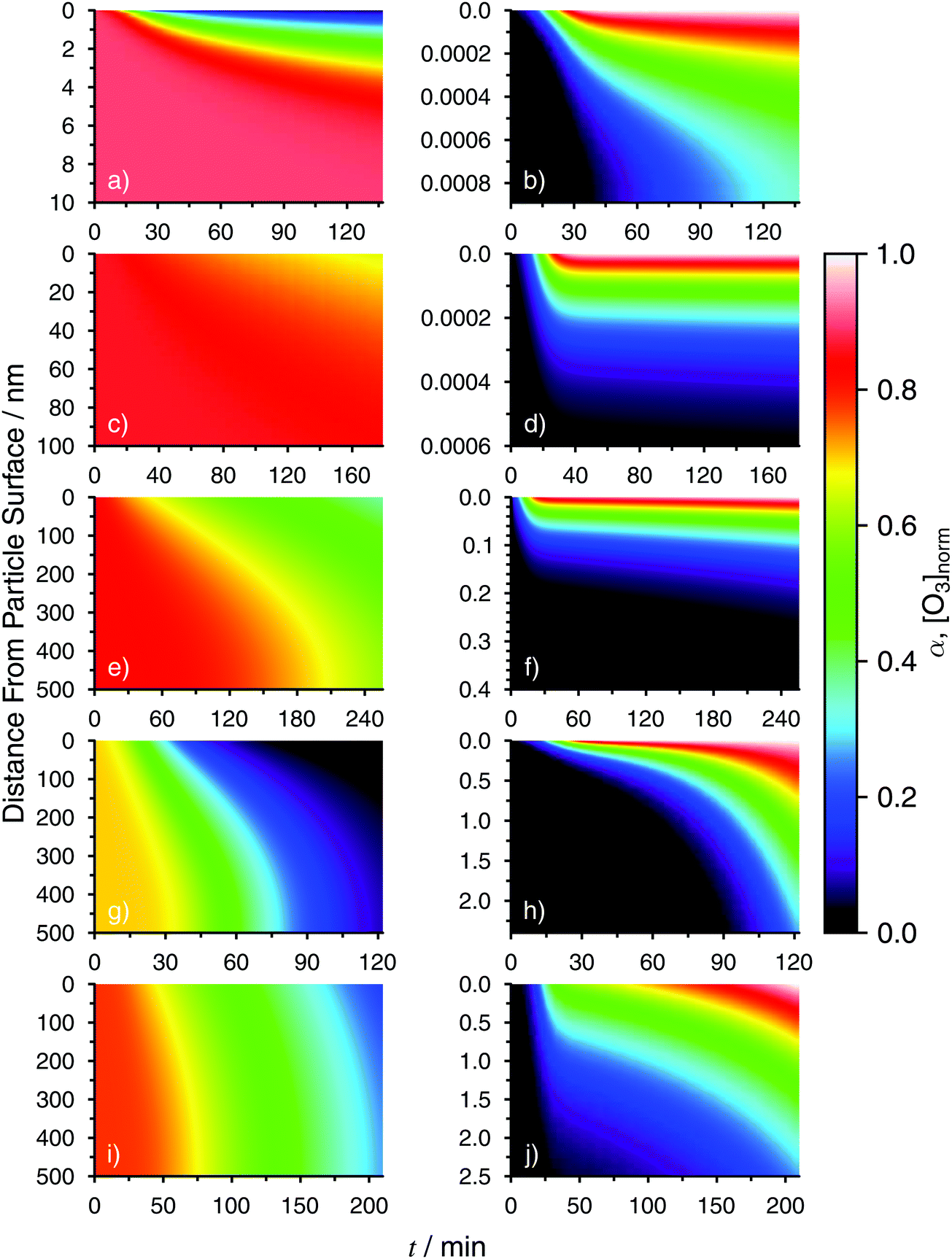 | ||
| Fig. 5 Model derived 3-D radial profiles of the Fe2+ fraction, α and normalized O3 concentration, [O3]norm = [O3]/[O3]max, where [O3]max is the maximum O3 concentration for RH = 0% (a and b), 22% (c and d), 43% (e and f), 60% (g and h) and 80% (i and j). The color scale from 0–1 is the same for both α shown in the left panels (a, c, e, g and i) and [O3]norm shown in the right panels (b, d, f, h and j). The distance from the particle surface is the ordinate. Note that the scales for all panels can be different. | ||
3.5 A case for using a reacto-diffusive framework
Our observations allowed us to test assumptions for predicting α: the most basic is that our particles were well-mixed for both Fe2+ and Fe3+ and also for O2 and O3 in equilibrium with Henry's Law. Oxidation of Fe2+ due to O2 exposure follows the reaction| Fe2+ + O2 → Fe3+ + O2˙−, | (R4) |
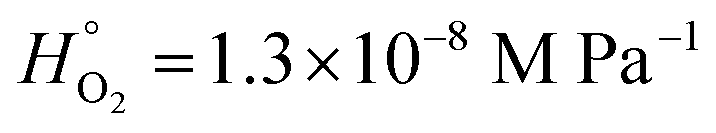 ,115 applies to our XG/FeCl2 with a O2 partial pressure of 30 mbar (for the PolLux environmental microreactor at a total pressure of 150 mbar), the equilibrium condensed phase O2 concentration was 3.9 × 10−5 M. Then, the first order loss rate for Fe2+, kI, in reaction R4 is kIR4 = 1.2 × 10−5 s−1. We compare this to kI = 1.6 × 10−2 s−1 calculated using
,115 applies to our XG/FeCl2 with a O2 partial pressure of 30 mbar (for the PolLux environmental microreactor at a total pressure of 150 mbar), the equilibrium condensed phase O2 concentration was 3.9 × 10−5 M. Then, the first order loss rate for Fe2+, kI, in reaction R4 is kIR4 = 1.2 × 10−5 s−1. We compare this to kI = 1.6 × 10−2 s−1 calculated using 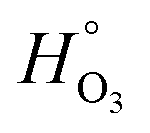 for water115 and [O3]g = 150 ppb or 0.015 Pa partial pressure. Since kIR3 is 3 orders of magnitude larger than kIR4, it comes as no surprise that to achieve a similar decay of α due to O3 observed over hours, an experiment with only O2 would need to last thousands of hours longer. This is consistent with our finding that O2 reaction in our system is negligible. Applying kIR4 = 1.2 × 10−5 s−1, we calculate the loss of α = α0
for water115 and [O3]g = 150 ppb or 0.015 Pa partial pressure. Since kIR3 is 3 orders of magnitude larger than kIR4, it comes as no surprise that to achieve a similar decay of α due to O3 observed over hours, an experiment with only O2 would need to last thousands of hours longer. This is consistent with our finding that O2 reaction in our system is negligible. Applying kIR4 = 1.2 × 10−5 s−1, we calculate the loss of α = α0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(kIR4t). After t = 2 h, α would be depleted by 8%. Using kIR3, α would be 100% depleted. Clearly, this is in disagreement with our observations. If XG acted as a ligand for Fe2+, this may change the rate of reaction and whether reaction rates are enhanced or reduced may depend on the specific system involved. A previous study114 found that rate coefficients of O2 with a Fe2+–fulvic acid complex was 100 M−1 s−1, which is over 100 times greater than kIR4. Another previous study found that iron oxidation with OH, H2O2 and HO2 was observed to be only 3 times higher with humic acid ligands from waste water than without.116 In contrast, it was found that iron in steel was complexed with XG in dilute solution and that oxidation and corrosion due to HCl was inhibited107 implying a reduction in reaction rate. Due to a lack of observations of O3 oxidation of xanthan gum ligands with Fe, we maintain our choice of kR3, and argue decreasing molecular transport of Fe and O3 leads to a significant decrease in the loss rate of Fe2+ far lower than expected when considering particles are well-mixed and in Henry's Law equilibrium.
exp(kIR4t). After t = 2 h, α would be depleted by 8%. Using kIR3, α would be 100% depleted. Clearly, this is in disagreement with our observations. If XG acted as a ligand for Fe2+, this may change the rate of reaction and whether reaction rates are enhanced or reduced may depend on the specific system involved. A previous study114 found that rate coefficients of O2 with a Fe2+–fulvic acid complex was 100 M−1 s−1, which is over 100 times greater than kIR4. Another previous study found that iron oxidation with OH, H2O2 and HO2 was observed to be only 3 times higher with humic acid ligands from waste water than without.116 In contrast, it was found that iron in steel was complexed with XG in dilute solution and that oxidation and corrosion due to HCl was inhibited107 implying a reduction in reaction rate. Due to a lack of observations of O3 oxidation of xanthan gum ligands with Fe, we maintain our choice of kR3, and argue decreasing molecular transport of Fe and O3 leads to a significant decrease in the loss rate of Fe2+ far lower than expected when considering particles are well-mixed and in Henry's Law equilibrium.
We test the applicability of a reacto-diffusive framework described in Steimer et al.38 to predict ozone transport and reaction into our particles. This is an analytical solution to the chemical loss as a function of t, which assumes that O3 reaction occurs in a thin shell or over a much smaller length scale than the particle diameter and that Fe2+ is homogeneously mixed. Following Steimer et al.38 the time rate of change of α is written as
 | (2) |
 | (3) |
 | (4) |
The left hand side of eqn (4) is entirely dependent on measurable and available quantities while the right hand side is in terms of the fitted parameters used in KM-SUB. Therefore, this provides a point of comparison to evaluate the suitability of the reacto-diffusive framework to predict Fe oxidation reaction and can be calculated for each individually probed particle. Fig. 6 shows a box plot of experimentally derived 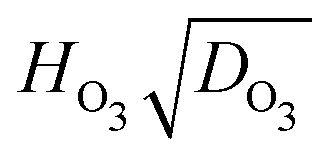 for all particles investigated as a function of RH. The median and quartile range of
for all particles investigated as a function of RH. The median and quartile range of 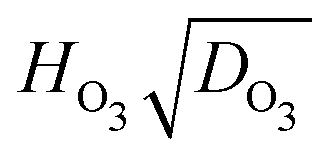 values for thousands of particles are shown. Values for individual particles are shown in Fig. S12 in the ESI.† Also included in Fig. 6 is the value of
values for thousands of particles are shown. Values for individual particles are shown in Fig. S12 in the ESI.† Also included in Fig. 6 is the value of  derived using optimized parameters from the KM-SUB model for comparison. The 25 and 75 percentiles of
derived using optimized parameters from the KM-SUB model for comparison. The 25 and 75 percentiles of  data capture the values of the fitting parameters from KM-SUB (Table 1). Median values are indicated by a horizontal solid line within the boxes. It is important to note that for some particles, mainly those investigated within minutes of the start of reaction, depletion of α is small and scatters around α0. This leads to negative values of
data capture the values of the fitting parameters from KM-SUB (Table 1). Median values are indicated by a horizontal solid line within the boxes. It is important to note that for some particles, mainly those investigated within minutes of the start of reaction, depletion of α is small and scatters around α0. This leads to negative values of  and thus negative values of
and thus negative values of 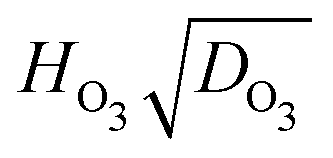 (see Fig. S12 in the ESI†), which is physically unrealistic. However, we choose to include these in Fig. S12 (ESI†) as they contribute to the scatter in our data. If a data point falls outside of three times the median absolute deviation, it is considered an outlier and shown as a symbol with a cross in Fig. S12 (ESI†).
(see Fig. S12 in the ESI†), which is physically unrealistic. However, we choose to include these in Fig. S12 (ESI†) as they contribute to the scatter in our data. If a data point falls outside of three times the median absolute deviation, it is considered an outlier and shown as a symbol with a cross in Fig. S12 (ESI†).
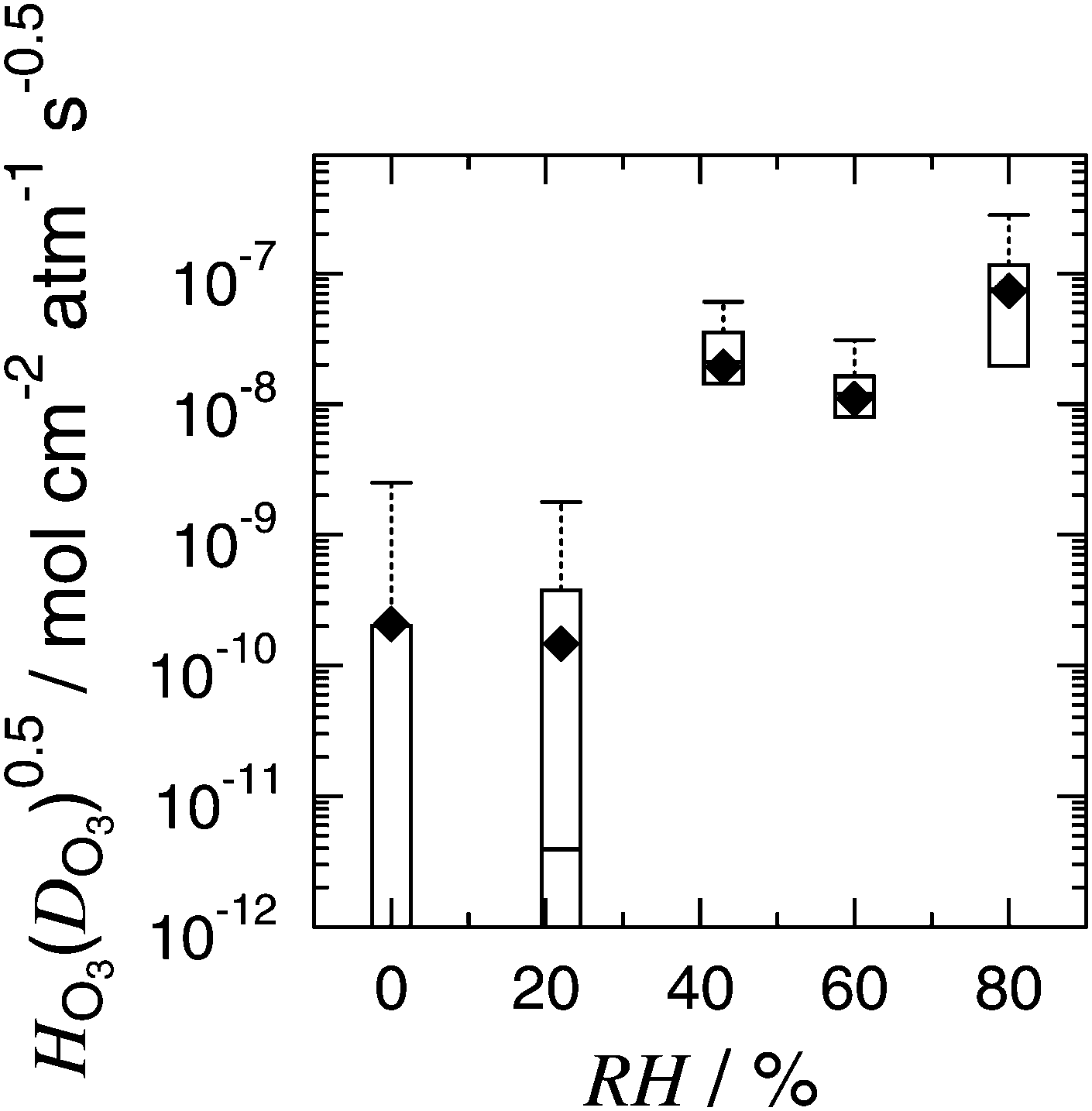 | ||
Fig. 6 A box plot of the product of Henry's Law constant for ozone, HO3, and the square root of the diffusion coefficient of ozone, DO3, or  as a function of RH. The bottom and top of the boxes represent 25 and 75 percentiles of about 1550 particles. Horizontal lines within boxes are median values. Upper error bars indicate 3× the median absolute deviation (MAD). The lower 3× MAD interval extends to negative values, and thus lower error bars are not depicted. All data for individual particles is shown in Fig. S12 (ESI†). Solid diamonds are derived from fitted parameters in Table 1. as a function of RH. The bottom and top of the boxes represent 25 and 75 percentiles of about 1550 particles. Horizontal lines within boxes are median values. Upper error bars indicate 3× the median absolute deviation (MAD). The lower 3× MAD interval extends to negative values, and thus lower error bars are not depicted. All data for individual particles is shown in Fig. S12 (ESI†). Solid diamonds are derived from fitted parameters in Table 1. | ||
Direct comparison of data and model (eqn (4)) validate the use of the reacto-diffusive framework. An important feature of eqn (2) and (3) is that DFe is not required, because reacto-diffusive framework assumes a homogeneous distribution in α. Despite that we observed chemical gradients (Fig. 3), this assumption is “good enough” within our experimental conditions due to agreement in Fig. 6. We rearrange eqn (4) to calculate α(t) following
 | (5) |
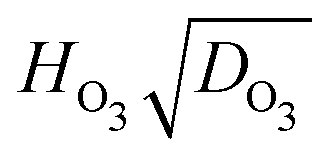 and using the geometric mean of dp respective to each experiment. We note that eqn (5) used only our observations and no fitted parameters. There is generally good agreement between our data, the reacto-diffusive framework and the KM-SUB model considering all uncertainties. At RH = 0 and 20%, there is a significant data scatter and the 25th percentile level goes below zero. It is important to note that the product
and using the geometric mean of dp respective to each experiment. We note that eqn (5) used only our observations and no fitted parameters. There is generally good agreement between our data, the reacto-diffusive framework and the KM-SUB model considering all uncertainties. At RH = 0 and 20%, there is a significant data scatter and the 25th percentile level goes below zero. It is important to note that the product  should not depend on particle surface to volume ratio (Fig. S12 in the ESI†) if the reacto-diffusive framework is valid. This was generally the case for all RH within the scatter of our data, expect for experiments at RH = 60% when [O3] was relatively high and α approached close to zero. Eqn (5) does not take into account all size dependent factors117,118 and will miss out on those induced by the diffusion limitation of Fe. This may lead to an over-prediction of the decay of α by the reacto-diffusive framework. For example, the lifetime of Fe2+ calculated using the reacto-diffusive framework is about 22 and 3 times shorter than for KM-SUB at RH = 0 and 80%, respectively, using the same parameters as in Table 1 and constant [O3]g = 50 ppb. Eqn (3) describes how, in the reacto-diffusive regime, the rate constant is inversely dependent on dp. Increasing or decreasing the modeled dp from 1 μm will tend to over or under predict α, respectively. In other words, particles that are expanding due to water uptake would appear to have a lower rate of iron oxidation. Note that the model employs a fixed dp at different RH and thus corresponds to effectively different dry diameters. The same applies to the experiment, due to the fact that different particles are probed at each time (a different field of view) under different RH. A new random population of particles is imaged at each time point. These humidity effects should not significantly affect our parameter optimization because the scatter of our data shown in Fig. S12 (ESI†) is generally larger than effects due to particle size. The vast number of particles and the high time resolution used in our observation ensures enough data is available to effectively conclude that the reacto-diffusive framework is an acceptable approximation for the KM-SUB model to describe O3 oxidation in our experiments.
should not depend on particle surface to volume ratio (Fig. S12 in the ESI†) if the reacto-diffusive framework is valid. This was generally the case for all RH within the scatter of our data, expect for experiments at RH = 60% when [O3] was relatively high and α approached close to zero. Eqn (5) does not take into account all size dependent factors117,118 and will miss out on those induced by the diffusion limitation of Fe. This may lead to an over-prediction of the decay of α by the reacto-diffusive framework. For example, the lifetime of Fe2+ calculated using the reacto-diffusive framework is about 22 and 3 times shorter than for KM-SUB at RH = 0 and 80%, respectively, using the same parameters as in Table 1 and constant [O3]g = 50 ppb. Eqn (3) describes how, in the reacto-diffusive regime, the rate constant is inversely dependent on dp. Increasing or decreasing the modeled dp from 1 μm will tend to over or under predict α, respectively. In other words, particles that are expanding due to water uptake would appear to have a lower rate of iron oxidation. Note that the model employs a fixed dp at different RH and thus corresponds to effectively different dry diameters. The same applies to the experiment, due to the fact that different particles are probed at each time (a different field of view) under different RH. A new random population of particles is imaged at each time point. These humidity effects should not significantly affect our parameter optimization because the scatter of our data shown in Fig. S12 (ESI†) is generally larger than effects due to particle size. The vast number of particles and the high time resolution used in our observation ensures enough data is available to effectively conclude that the reacto-diffusive framework is an acceptable approximation for the KM-SUB model to describe O3 oxidation in our experiments.
We argue that a reacto-diffusive limitation may apply to O2 as it does for O3. We observed no loss of Fe2+ during O2 exposure over hours. At RH = 80%, α0 = 0.78, and 2–4 hours of O2 reaction assuming homogeneous mixing of both O2 and Fe2+ would result in α = 0.71–0.65. We should have been able to observe this decay considering our uncertainty in α, especially when using a three times higher O2 particle pressure for experiments at RH = 0%, 20% and 60%. Assuming that DO2 was identical to that of DO3, we determined the reacto-diffusive length scale, 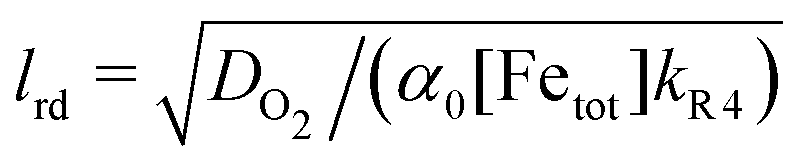 , at dry conditions and RH = 80% for O2 on the order of 0.1 nm to 1 μm. These estimates indicate that O2 reaction may have also been diffusion limited similar to O3. However, to fully evaluate this, time scales of days would be required to study O2 transport and reaction in this system.
, at dry conditions and RH = 80% for O2 on the order of 0.1 nm to 1 μm. These estimates indicate that O2 reaction may have also been diffusion limited similar to O3. However, to fully evaluate this, time scales of days would be required to study O2 transport and reaction in this system.
Our findings that O3 reaction is diffusion limited led us to speculate that other condensed phase reactions involving organic species can also be slowed due to low diffusion coefficients. There is increasing evidence that predicting secondary organic aerosol (SOA) mass must take into account condensed phase chemical reactions that include in part, oligomerization or acid catalyzed and accretion reactions that significantly increase molecular weight, reduce re-evaporation of organic compounds to the gas phase and thus add to the total mass of SOA.11,13,119,120 Examples of these are glyoxal uptake and reaction,121 oligomerization of volatile condensed phase molecules,122 aldol condensation and esterification.20,123 Isoprene epoxydiols (IEPOX) are major gas phase products of isoprene oxidation that are highly soluble and reactive in the condensed phase.11,13 When depleted in the condensed phase due to chemical reaction there, a net reactive uptake of IEPOX has been shown to lead to increasing organic and organosulfate formation contributing significantly to aqueous SOA mass in atmospheric environments.124 However, Zhang et al.125 showed in a laboratory study that when organic coatings were present on acidic aerosol particles, IEPOX reactive uptake decreased as coating thicknesses increased up to 32 nm and when RH was decreased from 50 − 15%. We speculate that irreversible uptake of IEPOX may also be diffusion limited. In general, diffusion limitations slow reactions compared to predictions considering homogeneous mixing and equilibrium with Henry's Law. For this reason, we suggest that aerosol chemical studies should be evaluated with the reacto-diffusive framework described here and in previous work38,117,118 to test its predictive capability under a range of atmospheric conditions. We call for a database of diffusion coefficients and Henry's Law constants applicable for oxidants and reactants in highly concentrated aerosol particles.94 This would preferably be for a wide range of temperature and humidity and for materials such as secondary organic aerosol and their proxies.16 Doing so would allow further evaluation of when reaction and diffusion limitations apply and elucidate how pervasive they are to atmospheric multi-phase chemistry.
4 Conclusions
We have measured chemical gradients within individual aerosol particles containing xanthan gum and FeCl2 due to heterogeneous reaction with O3. Oxidation from Fe2+ to Fe3+ was imaged in 2-D using STXM/NEXAFS prior to and during in situ O3 exposure for hours. Using a novel 2-D concentric perimeter analysis and careful error propagation, we were able to derive 2-D reaction profiles with well-defined uncertainties. Thousands of particles were probed, each with hundreds of pixels that provided quantitative spatial and chemical information about the reaction with O3. We found that during O3 exposure, increased oxidation occurred with increasing RH and [O3]g. At all reaction time and at particle perimeters, i.e. the outer most pixels of all particles, α was consistently less than concentric pixels toward the particle center. Therefore, we conclude that O3 oxidized particle surfaces more than interiors. Observed 2-D gradients in α from perimeter to center were shallow and ranged roughly by about 0.2 over the first 200 nm at RH = 0 and 22%. At RH = 43, 60 and 80%, a difference of 0.2 in α occurred for ≥500 nm. We conclude that changes in O3 and Fe transport must have occurred and were driven by the water content in xanthan gum, which is known to increase with increasing RH. To test this conclusion, we used the kinetic multi-layer model for aerosol surface and bulk chemistry, KM-SUB. Using previous literature values where possible and fitting parameters for diffusion coefficients of O3 and Fe2+, as well as Henry's Law constants for O3 as a function of RH, we were able to predict 2-D concentration profiles. This is the first time that a direct comparison between observed and predicted internal aerosol spatio-chemical changes has been achieved. Resulting diffusion coefficients increased exponentially as a function of RH and was parameterized using a Vignes-type equation. HO3 decreased exponentially as a function of RH and could also be described using a Vignes-type equation. A volume mixing rule over-predicted HO3 and is advised not to be used for this system. Our findings may apply for ozone in marine derived organic aerosol due to XG being a proxy of polysaccharide and exopolymer particles found to be aerosolized from oceans.We have used a limiting case in heterogeneous aerosol chemistry referred to as a reacto-diffusive kinetic regime to describe our results following a square root dependent loss rate of α. The corresponding framework described oxidation by O3 to occur in a thin layer at the aerosol surface with uniformly distributed (or well-mixed) condensed phase Fe2+ concentration in the bulk. However, this was not supported because shallow gradients in Fe2+ were, in fact, observed. Despite this, we tested the appropriateness of this approximation using only observed values to derive the product of the Henry's Law constant and the square root of the diffusion coefficient for O3, or  , at all experimental conditions. This product could be directly compared with those derived from KM-SUB. It was found that within experimental uncertainties, total depletion of Fe2+ could be reproduced for all experiments, and we conclude that the reacto-diffusive framework was applicable to describe the chemical gradients observed here. This approximation makes predicting the loss rate of aerosol components significantly easier with no computational expense compared to the KM-SUB or any other multilayer reaction and diffusion models. Furthermore, important chemical and physical parameters in the reacto-diffusive framework such as diffusion coefficients, Henry's Law constants, and second order bulk reaction rates are currently capable of being derived in a laboratory setting or a controlled setting in the ambient atmosphere. We have derived these parameters for O3 and Fe using KM-SUB in xanthan gum, a proxy for a natural biogenic source of organic matter emitted directly into the marine atmosphere. Their use should give better representation of condensed phase loss and production rates of reactants and products in marine aerosol with O3. Furthermore, we also suggest that the reacto-diffusive framework should be evaluated for various oxidants and reactants under a wide range of temperature and humidity to further validate its use in atmospheric heterogeneous chemical reactions.
, at all experimental conditions. This product could be directly compared with those derived from KM-SUB. It was found that within experimental uncertainties, total depletion of Fe2+ could be reproduced for all experiments, and we conclude that the reacto-diffusive framework was applicable to describe the chemical gradients observed here. This approximation makes predicting the loss rate of aerosol components significantly easier with no computational expense compared to the KM-SUB or any other multilayer reaction and diffusion models. Furthermore, important chemical and physical parameters in the reacto-diffusive framework such as diffusion coefficients, Henry's Law constants, and second order bulk reaction rates are currently capable of being derived in a laboratory setting or a controlled setting in the ambient atmosphere. We have derived these parameters for O3 and Fe using KM-SUB in xanthan gum, a proxy for a natural biogenic source of organic matter emitted directly into the marine atmosphere. Their use should give better representation of condensed phase loss and production rates of reactants and products in marine aerosol with O3. Furthermore, we also suggest that the reacto-diffusive framework should be evaluated for various oxidants and reactants under a wide range of temperature and humidity to further validate its use in atmospheric heterogeneous chemical reactions.
Author contributions
P. A. A. envisioned this project, conducted experiments, performed data analysis, modeled results and wrote the paper. P. A. A. and M. A. oversaw the entire project. P. C. A., J. D., S. S. S., J.-D. F., F. D., S. R., M. P. and S. P. acquired data and performed data analysis. M. S. and T. B. wrote the KM-SUB model and evaluated model results. T. B. supervised the modeling process. B. W. evaluated data analysis and maintained operation of experiment. U. K. K., C. P., C. G., M. S., B. W. and M. A. supervised the project. All authors contributed to the writing of the manuscript.Conflicts of interest
We declare no conflict of interest.Acknowledgements
This work was funded by the Swiss National Science Foundation (Grant 163074) and by the European Union's Horizon 2020 research and innovation program under the Marie Skodowska-Curie grant agreement (No 701647). J.-D. F., F. D. and C. P. acknowledge support by the Max Planck Society. P. A. A., S. R., M. P., S. P. and C. G. received support from the European Research Council under the European Union's Seventh Framework Program (FP/2007-2013)/ERC Grant Agreement (290852-AIRSEA). M. S. acknowledges funding from the National Science Foundation (AGS-1654104). T. B. acknowledges support by the Eckert Postdoctoral Fellowship from Georgia Tech School of Chemical and Biomolecular Engineering. The PolLux end station was financed by the German Ministerium für Bildung und Forschung (BMBF) through contracts 05KS4WE1/6 and 05KS7WE1.References
- Q. Zhang, J. L. Jimenez, M. R. Canagaratna, J. D. Allan, H. Coe, I. Ulbrich, M. R. Alfarra, A. Takami, A. M. Middlebrook, Y. L. Sun, K. Dzepina, E. Dunlea, K. Docherty, P. F. DeCarlo, D. Salcedo, T. Onasch, J. T. Jayne, T. Miyoshi, A. Shimono, S. Hatakeyama, N. Takegawa, Y. Kondo, J. Schneider, F. Drewnick, S. Borrmann, S. Weimer, K. Demerjian, P. Williams, K. Bower, R. Bahreini, L. Cottrell, R. J. Griffin, J. Rautiainen, J. Y. Sun, Y. M. Zhang and D. R. Worsnop, Geophys. Res. Lett., 2007, 34, L13801 Search PubMed
.
- L. M. Russell, L. N. Hawkins, A. A. Frossard, P. K. Quinn and T. S. Bates, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 6652–6657 CrossRef CAS
- A. Laskin, M. K. Gilles, D. A. Knopf, B. Wang and S. China, Annu. Rev. Anal. Chem., 2016, 9, 117–143 CrossRef PubMed
- J. Haywood and O. Boucher, Rev. Geophys., 2000, 38, 513–543 CrossRef CAS
-
O. Boucher, D. Randall, P. Artaxo, C. Bretherton, G. Feingold, P. Forster, V.-M. Kerminen, Y. Kondo, H. Liao, U. Lohmann, P. Rasch, S. K. Satheesh, S. Sherwood, S. B. and Z. X.-Y., in Climate Change 2013: The Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change, ed. S. Fuzzi, J. Penner, V. Ramaswamy and C. Stubenrauch, Cambridge University Press, Cambridge, United Kingdom and New York, NY, USA, 2013, ch. 8, Clouds and Aerosols, pp. 571–657 Search PubMed
- T. Storelvmo, Annu. Rev. Earth Planet. Sci., 2017, 45, 199–222 CrossRef CAS
- D. A. Knopf, P. A. Alpert and B. Wang, ACS Earth Space Chem., 2018, 2, 168–202 CrossRef CAS
- Z. A. Kanji, L. A. Ladino, H. Wex, Y. Boose, M. Burkert-Kohn, D. J. Cziczo and M. Krämer, Meteor. Mon., 2017, 58, 1.1–1.33 CrossRef
-
J. H. Seinfeld and S. N. Pandis, Atmospheric Chemistry and Physics - From Air Pollution to Climate Change, John Wiley & Sons., Hoboken, NJ, 2nd edn, 2006 Search PubMed
- A. Tilgner, P. Bräuer, R. Wolke and H. Herrmann, J. Atmos. Chem., 2013, 70, 221–256 CrossRef CAS
- H. Herrmann, T. Schaefer, A. Tilgner, S. A. Styler, C. Weller, M. Teich and T. Otto, Chem. Rev., 2015, 115, 4259–4334 CrossRef CAS
-
B. Ervens, in Multiphase Environmental Chemistry in the Atmosphere, ed. S. W. Hunt, A. Laskin and S. A. Nizkorodov, American Chemical Society, Washington, DC, 2018, ch. Progress and problems in modeling chemical processing in cloud droplets and wet aerosol particles, pp. 327–345 Search PubMed
- M. Shrivastava, S. Lou, A. Zelenyuk, R. C. Easter, R. A. Corley, B. D. Thrall, P. J. Rasch, J. D. Fast, S. L. Massey Simonich, H. Shen and S. Tao, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 1246–1251 CrossRef CAS
- D. K. Farmer, C. D. Cappa and S. M. Kreidenweis, Chem. Rev., 2015, 115, 4199–4217 CrossRef CAS
- A. Laskin, R. C. Moffet, M. K. Gilles, J. D. Fast, R. A. Zaveri, B. Wang, P. Nigge and J. Shutthanandan, J. Geophys. Res., 2012, 117, D15302 CrossRef
- M. Shiraiwa, Y. Li, A. P. Tsimpidi, V. A. Karydis, T. Berkemeier, S. N. Pandis, J. Lelieveld, T. Koop and U. Pöschl, Nat. Commun., 2017, 8, 15002 CrossRef
- J. P. Reid, A. K. Bertram, D. O. Topping, A. Laskin, S. T. Martin, M. D. Petters, F. D. Pope and G. Rovelli, Nat. Commun., 2018, 9, 15002 Search PubMed
- B. R. Bzdek and J. P. Reid, J. Chem. Phys., 2017, 147, 220901 CrossRef
- V. Perraud, E. A. Bruns, M. J. Ezell, S. N. Johnson, Y. Yu, M. L. Alexander, A. Zelenyuk, D. Imre, W. L. Chang, D. Dabdub, J. F. Pankow and B. J. Finlayson-Pitts, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 2836–2841 CrossRef CAS
- M. Shiraiwa, L. D. Yee, K. A. Schilling, C. L. Loza, J. S. Craven, A. Zuend, P. J. Ziemann and J. H. Seinfeld, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 11746–11750 CrossRef CAS
- L. Renbaum-Wolff, J. W. Grayson, A. P. Bateman, M. Kuwata, M. Sellier, B. J. Murray, J. E. Shilling, S. T. Martin and A. K. Bertram, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 8014–8019 CrossRef CAS PubMed
-
M. Kuwata and S. T. Martin, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 17354–17359
- J. H. Slade and D. A. Knopf, Geophys. Res. Lett., 2014, 41, 5297–5306 CrossRef CAS
- J. Socorro, P. S. J. Lakey, L. Han, T. Berkemeier, G. Lammel, C. Zetzsch, U. Pöschl and M. Shiraiwa, Environ. Sci. Technol., 2017, 51, 13749–13754 CrossRef CAS
- J. D. Smith, J. H. Kroll, C. D. Cappa, D. L. Che, C. L. Liu, M. Ahmed, S. R. Leone, D. R. Worsnop and K. R. Wilson, Atmos. Chem. Phys., 2009, 9, 3209–3222 CrossRef CAS
- M. N. Chan, H. Zhang, A. H. Goldstein and K. R. Wilson, J. Phys. Chem. C, 2014, 118, 28978–28992 CrossRef CAS
- A. M. Arangio, J. H. Slade, T. Berkemeier, U. Pöschl, D. A. Knopf and M. Shiraiwa, J. Phys. Chem. A, 2015, 119, 4533–4544 CrossRef CAS
- J. F. Davies and K. R. Wilson, Chem. Sci., 2015, 6, 7020–7027 RSC
- H. Fan, M. R. Tinsley and F. Goulay, J. Phys. Chem. A, 2015, 119, 11182–11190 CrossRef CAS PubMed
- M. M. Chim, C. T. Cheng, J. F. Davies, T. Berkemeier, M. Shiraiwa, A. Zuend and M. N. Chan, Atmos. Chem. Phys., 2017, 17, 14415–14431 CrossRef CAS
- M. M. Chim, C. Y. Chow, J. F. Davies and M. N. Chan, J. Phys. Chem. A, 2017, 121, 1666–1674 CrossRef CAS PubMed
- M. J. Liu, A. A. Wiegel, K. R. Wilson and F. A. Houle, J. Phys. Chem. A, 2017, 121, 5856–5870 CrossRef CAS
- K. C. Kwong, M. M. Chim, E. H. Hoffmann, A. Tilgner, H. Herrmann, J. F. Davies, K. R. Wilson and M. N. Chan, ACS Earth Space Chem., 2018, 2, 895–903 CrossRef CAS
- M. M. Chim, C. Y. Lim, J. H. Kroll and M. N. Chan, ACS Earth Space Chem., 2018, 2, 1323–1329 CrossRef CAS
- Z. Li, K. A. Smith and C. D. Cappa, Atmos. Chem. Phys., 2018, 18, 14585–14608 CrossRef CAS
- M. Shiraiwa, M. Ammann, T. Koop and U. Pöschl, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 11003–11008 CrossRef CAS
- S. Zhou, M. Shiraiwa, R. D. McWhinney, U. Pöschl and J. P. D. Abbatt, Faraday Discuss., 2013, 165, 391–406 RSC
- S. S. Steimer, M. Lampimäki, E. Coz, G. Grzinic and M. Ammann, Atmos. Chem. Phys., 2014, 14, 10761–10772 CrossRef
- S. S. Steimer, T. Berkemeier, A. Gilgen, U. K. Krieger, T. Peter, M. Shiraiwa and M. Ammann, Phys. Chem. Chem. Phys., 2015, 17, 31101–31109 RSC
- M. I. Jacobs, B. Xu, O. Kostko, N. Heine, M. Ahmed and K. R. Wilson, J. Phys. Chem. A, 2016, 120, 8645–8656 CrossRef CAS PubMed
- N. Heine, F. A. Houle and K. R. Wilson, Environ. Sci. Technol., 2017, 51, 13740–13748 CrossRef CAS PubMed
- U. Pöschl, Y. Rudich and M. Ammann, Atmos. Chem. Phys., 2007, 7, 5989–6023 CrossRef
- M. Ammann and U. Pöschl, Atmos. Chem. Phys., 2007, 7, 6025–6045 CrossRef CAS
- C. Pfrang, M. Shiraiwa and U. Pöschl, Atmos. Chem. Phys., 2010, 10, 4537–4557 CrossRef CAS
- M. Shiraiwa, C. Pfrang and U. Pöschl, Atmos. Chem. Phys., 2010, 10, 3673–3691 CrossRef CAS
- M. Shiraiwa, C. Pfrang, T. Koop and U. Pöschl, Atmos. Chem. Phys., 2012, 12, 2777–2794 CrossRef CAS
- T. Berkemeier, A. J. Huisman, M. Ammann, M. Shiraiwa, T. Koop and U. Pöschl, Atmos. Chem. Phys., 2013, 13, 6663–6686 CrossRef
- F. A. Houle, W. D. Hinsberg and K. R. Wilson, Phys. Chem. Chem. Phys., 2015, 17, 4412–4423 RSC
- T. Berkemeier, S. S. Steimer, U. K. Krieger, T. Peter, U. Pöschl, M. Ammann and M. Shiraiwa, Phys. Chem. Chem. Phys., 2016, 18, 12662–12674 RSC
- A. A. Wiegel, M. J. Liu, W. D. Hinsberg, K. R. Wilson and F. A. Houle, Phys. Chem. Chem. Phys., 2017, 19, 6814–6830 RSC
- F. A. Houle, A. A. Wiegel and K. R. Wilson, Environ. Sci. Technol., 2018, 52, 13774–13781 CrossRef CAS
- F. A. Houle, A. A. Wiegel and K. R. Wilson, J. Phys. Chem. Lett., 2018, 9, 1053–1057 CrossRef CAS PubMed
- C. A. Angell, Science, 1995, 267, 1924–1935 CrossRef CAS PubMed
- P. G. Debenedetti and F. H. Stillinger, Nature, 2001, 410, 259–267 CrossRef CAS
-
T. Koop, J. Bookhold, M. Shiraiwa and U. Pöschl, Phys. Chem. Chem. Phys., 2011, 13, 19238–19255 Search PubMed
- L. Lee and K. Wilson, J. Phys. Chem. A, 2016, 120, 6800–6812 CrossRef CAS PubMed
- T. J. Conocchioli, E. J. Hamilton and N. Sutin, J. Am. Chem. Soc., 1965, 87, 926–927 CrossRef CAS
- J. Hoigné, H. Bader, W. Haag and J. Staehelin, Water Res., 1985, 19, 993–1004 CrossRef
- T. Løegager, J. Holcman, K. Sehested and T. Pedersen, Inorg. Chem., 1992, 31, 3523–3529 CrossRef
- O. Pestovsky and A. Bakac, Inorg. Chem., 2006, 45, 814–820 CrossRef CAS PubMed
- S. Enami, Y. Sakamoto and A. J. Colussi, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 623–628 CrossRef CAS PubMed
- R. C. Moffet, H. Furutani, T. C. Rödel, T. R. Henn, P. O. Sprau, A. Laskin, M. Uematsu and M. K. Gilles, J. Geophys. Res., 2012, 117, D07204 CrossRef
- S. Stohs and D. Bagchi, Free Radical Biol. Med., 1995, 18, 321–336 CrossRef CAS PubMed
- N. Li, C. Sioutas, A. Cho, D. Schmitz, C. Misra, J. Sempf, M. Wang, T. Oberley, J. Froines and A. Nel, Environ. Health Persp., 2003, 111, 455–460 CrossRef CAS PubMed
- M. Shiraiwa, K. Selzle and U. Pöschl, Free Radical Res., 2012, 46, 927–939 CrossRef CAS
- H. Tong, P. S. J. Lakey, A. M. Arangio, J. Socorro, C. J. Kampf, T. Berkemeier, W. H. Brune, U. Pöschl and M. Shiraiwa, Faraday Discuss., 2017, 200, 251–270 RSC
- T. D. Jickells, Z. S. An, K. K. Andersen, A. R. Baker, G. Bergametti, N. Brooks, J. J. Cao, P. W. Boyd, R. A. Duce, K. A. Hunter, H. Kawahata, N. Kubilay, J. laRoche, P. S. Liss, N. Mahowald, J. M. Prospero, A. J. Ridgwell, I. Tegen and R. Torres, Science, 2005, 308, 67–71 CrossRef CAS PubMed
- M. T. Maldonado and N. M. Price, Deep-Sea Res. Pt. II, 1999, 46, 2447–2473 CrossRef CAS
- A. Baker and P. Croot, Mar. Chem., 2010, 120, 4–13 CrossRef CAS
- Z. Shi, M. D. Krom, T. D. Jickells, S. Bonneville, K. S. Carslaw, N. Mihalopoulos, A. R. Baker and L. G. Benning, Aeolian Res., 2012, 5, 21–42 CrossRef
- H. Chen and V. H. Grassian, Environ. Sci. Technol., 2013, 47, 10312–10321 CAS
- R. Paris and K. V. Desboeufs, Atmos. Chem. Phys., 2013, 13, 4895–4905 CrossRef
- A. Ito and Z. Shi, Atmos. Chem. Phys., 2016, 16, 85–99 CrossRef CAS
- N. M. Mahowald, R. Scanza, J. Brahney, C. L. Goodale, P. G. Hess, J. K. Moore and J. Neff, Curr. Clim. Change Rep., 2017, 3, 16–31 CrossRef
- M. Kanakidou, S. Myriokefalitakis and K. Tsigaridis, Environ. Res. Lett., 2018, 13, 063004 CrossRef
- K. W. Dawson, M. D. Petters, N. Meskhidze, S. S. Petters and S. M. Kreidenweis, J. Geophys. Res., 2016, 121, 11803–11818 CAS
-
I. A. Challen, in Food Hydrocolloids: Structures, Properties, and Functions, ed. K. Nishinari and E. Doi, Plenum Press, New York, NY, USA, 1993, ch. Xanthan Gum: A Multifunctional Stabiliser for Food Products, pp. 135–140 Search PubMed
- B. Katzbauer, Polym. Degrad. Stab., 1998, 59, 81–84 CrossRef CAS
- S. Basu, U. S. Shivhare and A. S. Mujumdar, Dry. Technol., 2007, 25, 1581–1586 CrossRef CAS
- U. Passow and A. L. Alldredge, Limnol. Oceanogr., 1995, 40, 1326–1335 CrossRef CAS
- U. Passow, Prog. Oceanogr., 2002, 55, 287–333 CrossRef
-
A. Engel, in Practical Guidelines for the Analysis of Seawater, ed. O. Wurl, CRC Press, Boca Raton, FL, USA, 2009, ch. 7. Determination of Marine Gel Particles, pp. 125–142 Search PubMed
- J. Y. Aller, J. C. Radway, W. P. Kilthau, D. W. Bothe, T. W. Wilson, R. D. Vaillancourt, P. K. Quinn, D. J. Coffman, B. J. Murray and D. A. Knopf, Atmos. Environ., 2017, 154, 331–347 CrossRef CAS
- T. Huthwelker, V. Zelenay, M. Birrer, A. Krepelova, J. Raabe, G. Tzvetkov, M. G. C. Vernooij and M. Ammann, Rev. Sci. Instrum., 2010, 81, 113706 CrossRef CAS PubMed
- J.-D. Förster, C. Pöhlker, C. Gurk, M. Lamneck, M. Ammann, U. Pöschl and M. O. Andreae, Atmos. Mech. Tech., 2019 Search PubMed
- D. M. Murphy and T. Koop, Q. J. R. Meteorol. Soc., 2005, 131, 1539–1565 CrossRef
- J. Raabe, G. Tzvetkov, U. Flechsig, M. Böge, A. Jaggi, B. Sarafimov, M. G. C. Vernooij, T. Huthwelker, H. Ade, D. Kilcoyne, T. Tyliszczak, R. H. Fink and C. Quitmann, Rev. Sci. Instrum., 2008, 79, 113704 CrossRef CAS PubMed
- R. C. Moffet, T. Henn, A. Laskin and M. K. Gilles, Anal. Chem., 2010, 82, 7906–7914 CrossRef CAS
- G. Cressey, C. M. B. Henderson and G. van der Laan, Phys. Chem. of Miner., 1993, 20, 111–119 CrossRef CAS
- L. A. J. Garvie, A. J. Craven and R. Brydson, Am. Mineral., 1994, 79, 411–425 CAS
- R. A. Speers and M. A. Tung, J. Food Sci., 1986, 51, 96–98 CrossRef CAS
- N. B. Wyatt and M. W. Liberatore, J. Appl. Polym. Sci., 2009, 114, 4076–4084 CrossRef CAS
- A. K. Biń, Ozone-Sci. Eng., 2006, 28, 67–75 CrossRef
- F. H. Marshall, R. E. H. Miles, Y.-C. Song, P. B. Ohm, R. M. Power, J. P. Reid and C. S. Dutcher, Chem. Sci., 2016, 7, 1298–1308 RSC
- M. Shiraiwa, U. Pöschl and D. A. Knopf, Environ. Sci. Technol., 2012, 46, 6630–6636 CrossRef CAS
- F. Jacobsen, J. Holcman and K. Sehested, Int. J. Chem. Kinet., 1998, 30, 215–221 CrossRef CAS
- D. T. Sawyer, H. Sugimoto and T. S. Calderwood, Proc. Natl. Acad. Sci. U. S. A., 1984, 81, 8025–8027 CrossRef CAS PubMed
- R. S. Smith and B. D. Kay, Nature, 1999, 398, 788–791 CrossRef CAS
- A. Einstein, Ann. Phys., 1905, 17, 549–560 CrossRef CAS
- W. Sutherland, Philos. Mag., 1905, 9, 781–785 CAS
- D. M. Lienhard, A. J. Huisman, U. K. Krieger, Y. Rudich, C. Marcolli, B. P. Luo, D. L. Bones, J. P. Reid, A. T. Lambe, M. R. Canagaratna, P. Davidovits, T. B. Onasch, D. R. Worsnop, S. S. Steimer, T. Koop and T. Peter, Atmos. Chem. Phys., 2015, 15, 13599–13613 CrossRef CAS
- R. M. Power and J. P. Reid, Rep. Prog. Phys., 2014, 77, 074601 CrossRef PubMed
- J. Sujja-areevath, D. Munday, P. Cox and K. Khan, Eur. J. Pharm. Sci., 1998, 6, 207–217 CrossRef CAS PubMed
-
R. A. Gemeinhart and C. Guo, in Reflexive Polymers and Hydrogels: Understanding and Designing Fast Responsive PolymericSystems, ed. N. Yui, R. J. Mrsny and K. Park, CRC Press, Boca Raton, Fl, 2004, ch. 13: Fast Swelling Hydrogel Systems, pp. 245–257 Search PubMed
- P. D. Oliveira, R. C. Michel, A. J. A. McBride, A. S. Moreira, R. F. T. Lomba and C. T. Vendruscolo, PLoS One, 2013, 8, 1–7 Search PubMed
- D. Bergmann, G. Furth and C. Mayer, Int. J. Biol. Macromol., 2008, 43, 245–251 CrossRef CAS PubMed
- M. Mobin and M. Rizvi, Carbohydr. Polym., 2016, 136, 384–393 CrossRef CAS PubMed
- R. Battino, T. R. Rettich and T. Tominaga, J. Phys. Chem. Ref. Data, 1983, 12, 163–178 CrossRef CAS
- P. Corral Arroyo, T. Bartels-Rausch, P. A. Alpert, S. Dumas, S. Perrier, C. George and M. Ammann, Environ. Sci. Technol., 2018, 52, 7680–7688 CrossRef CAS PubMed
- L.-K. Ju and C. S. Ho, Can. J. Chem. Eng., 1989, 67, 471–477 CrossRef CAS
- F. J. Millero, S. Sotolongo and M. Izaguirre, Geochim. Cosmochim. Acta, 1987, 51, 793–801 CrossRef CAS
- J. W. Moffett and R. G. Zika, Environ. Sci. Technol., 1987, 21, 804–810 CrossRef CAS PubMed
- D. W. King, H. A. Lounsbury and F. J. Millero, Environ. Sci. Technol., 1995, 29, 818–824 CrossRef CAS
- A. L. Rose and T. D. Waite, Environ. Sci. Technol., 2002, 36, 433–444 CrossRef CAS PubMed
- R. Sander, Atmos. Chem. Phys., 2015, 15, 4399–4981 CrossRef CAS
- Y. P. Lee, M. Fujii, K. Terao, T. Kikuchi and C. Yoshimura, Water Res., 2016, 103, 160–169 CrossRef CAS PubMed
- D. R. Hanson and E. R. Lovejoy, Science, 1995, 267, 1326–1328 CrossRef CAS PubMed
- D. R. Worsnop, J. W. Morris, Q. Shi, P. Davidovits and C. E. Kolb, Geophys. Res. Lett., 2002, 29, GL015542 CrossRef
- J. H. Kroll and J. H. Seinfeld, Atmos. Environ., 2008, 42, 3593–3624 CrossRef CAS
- M. Hallquist, J. C. Wenger, U. Baltensperger, Y. Rudich, D. Simpson, M. Claeys, J. Dommen, N. M. Donahue, C. George, A. H. Goldstein, J. F. Hamilton, H. Herrmann, T. Hoffmann, Y. Iinuma, M. Jang, M. E. Jenkin, J. L. Jimenez, A. Kiendler-Scharr, W. Maenhaut, G. McFiggans, T. F. Mentel, A. Monod, A. S. H. Prévôt, J. H. Seinfeld, J. D. Surratt, R. Szmigielski and J. Wildt, Atmos. Chem. Phys., 2009, 9, 5155–5236 CrossRef CAS
- R. Volkamer, F. San Martini, L. T. Molina, D. Salcedo, J. L. Jimenez and M. J. Molina, Geophys. Res. Lett., 2007, 34, L19807 CrossRef
- X. Zhang, A. T. Lambe, M. A. Upshur, W. A. Brooks, A. Gray Bé, R. J. Thomson, F. M. Geiger, J. D. Surratt, Z. Zhang, A. Gold, S. Graf, M. J. Cubison, M. Groessl, J. T. Jayne, D. R. Worsnop and M. R. Canagaratna, Environ. Sci. Technol., 2017, 51, 5932–5940 CrossRef CAS PubMed
- T. P. Riedel, Y.-H. Lin, Z. Zhang, K. Chu, J. A. Thornton, W. Vizuete, A. Gold and J. D. Surratt, Atmos. Chem. Phys., 2016, 16, 1245–1254 CrossRef CAS
- V. F. McNeill, J. L. Woo, D. D. Kim, A. N. Schwier, N. J. Wannell, A. J. Sumner and J. M. Barakat, Environ. Sci. Technol., 2012, 46, 8075–8081 CrossRef CAS PubMed
- Y. Zhang, Y. Chen, A. T. Lambe, N. E. Olson, Z. Lei, R. L. Craig, Z. Zhang, A. Gold, T. B. Onasch, J. T. Jayne, D. R. Worsnop, C. J. Gaston, J. A. Thornton, W. Vizuete, A. P. Ault and J. D. Surratt, Environ. Sci. Technol. Lett., 2018, 5, 167–174 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cp03731d |
| This journal is © the Owner Societies 2019 |