
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDefects on a pyrite(100) surface produce chemical evolution of glycine under inert conditions: experimental and theoretical approaches
Santos
Galvez-Martinez
a,
Elizabeth
Escamilla-Roa
b,
Maria-Paz
Zorzano
ab and
Eva
Mateo-Marti
 *a
*a
aCentro de Astrobiología (CSIC-INTA), Ctra. Ajalvir, Km. 4, 28850 Torrejón de Ardoz, Madrid, Spain. E-mail: mateome@cab.inta-csic.es
bDepartment of Computer Science, Electrical and Space Engineering, Luleå Universit of Technology, 97187 Luleå, Sweden
First published on 10th October 2019
Abstract
The presence of non-stoichiometric sites on the pyrite(100) surface makes it a suitable substrate for driving the chemical evolution of the amino acid glycine over time, even under inert conditions. Spectroscopic molecular fingerprints prove a transition process from a zwitterionic species to an anionic species over time on the monosulfide enriched surface. By combining experimental and theoretical approaches, we propose a surface mechanism where the interaction between the amino acid species and the surface will be driven by the quenching of the surface states at Fe sites and favoured by sulfur vacancies. This study demonstrates the potential capability of pyrite to act as a surface catalyst.
Introduction
Understanding the mechanism of adsorption of bio-molecules, such as amino acids, peptides and proteins, on mineral surfaces is a topic of great interest in several fields such as materials science, surface chemistry, nanotechnology, chemical energy storage, prebiotic chemistry and catalysis.1–6 Iron sulfides are central to our understanding of the evolution of the Earth. They have been described as catalysts in a number of key biochemical reactions associated with the “iron–sulfur world” hypothesis for the development of life.7 In addition to the widely accepted prebiotic soup theory, Wachterhauser proposed the “iron–sulfur world” theory, which states that the first reactions that enabled the formation of amino acids occurred on the surfaces of sulfide minerals, such as pyrite, because they can catalyze chemical reactions between organic molecules.8,9 In fact, researchers have recently focused much attention on studying the interactions and adsorption of organic molecules from a solution, or from the gas phase, on mineral surfaces.10,11 Of the possible minerals, pyrite (FeS2) has the highest potential to act as a catalyst in chemical reactions because it has a highly reactive surface that could drive molecular adsorption.Our recent experiments suggest that amino acids can adsorb on the pyrite surface, depending on its structure, the latter being affected by the surroundings or surface pre-treatment conditions.12,13 These studies provided some new insights into the adsorption process on pyrite surfaces; however, the understanding of amino acid–surface interactions at the atomic level remains limited. Unfortunately, only a few spectroscopic studies on the adsorption of amino acids from the gas phase onto mineral surfaces have been reported. Moreover, the interactions of amino acids with pyrite are key parameters to understanding the capability of this mineral to adsorb and pattern biomolecules of increasing complexity. In this context of surface chemistry, the interaction mechanism and molecular reactivity of amino acids such as glycine on a pyrite surface were studied by X-ray photoelectron spectroscopy (XPS). A combination of experimental and theoretical techniques was used to characterize how the structure defects at the pyrite(100) surface can bind amino acids. These defects on the fracture surface are sulfur vacancies and uncoordinated iron atoms. The purpose of this study is to investigate the possible role of sulfur vacancies in stabilizing amino acids at pyrite surfaces and the role played by the generated iron dangling bonds in the molecular adsorption process. To elucidate the adsorption mechanisms of simple molecules on pyrite surfaces, it is essential to investigate the adsorption behavior at these interfaces on a molecular scale.
We focused our efforts on the spectroscopic characterization of the simplest amino acid glycine, which is thought to have been present on primordial earth.14,15 We investigated its adsorption on pyrite surfaces, and the changes in their chemical states in the adsorbed phase as a function of time. In all the experiments, the molecules were successfully adsorbed on the surface, showing interesting changes as a function of time and demonstrating that the nature of the surface is critical in driving the molecular evolution in the chemical form adsorbed over time. In the present work, by combining the adsorption of amino acid, XPS spectroscopic characterization and theory, we identify spectroscopic molecular signatures of the evolution process, which suggest the possible co-existence and evolution of various chemical forms of the amino acids over time. Therefore, this experimental study aims to investigate the adsorption affinities of a simple amino acid on pyrite and to experimentally expose the essential role played by the pyrite surface on the molecular bonding. Complementary theoretical studies based on quantum mechanical calculations, that rely on Density Functional Theory (DFT), help us to propose the surface mechanisms of interaction between the amino acid species and the surface and will characterize the role of the iron dangling bonds due to the presence of sulfur vacancies.
Experimental
Experimental procedure
A pyrite(100) single crystal was provided by the Surface Preparation Laboratory and mounted in a multi-technique (XPS, UPS and LEED) ultra-high vacuum (UHV) chamber with a base pressure of 3 × 10−10 mbar. The pyrite(100) sample was cleaned by repeated cycles of Ar+ sputtering at 800 keV and annealing at 593 K under ultra-high vacuum (UHV) conditions. The surface order was confirmed by means of low-energy electron diffraction (LEED) and cleanliness was monitored using the XPS technique. The pyrite sample was not heated to temperatures above 600 K to avoid thermal decomposition.Materials and reactants
Glycine (purity ≥ 98%) powder was purchased from Sigma-Aldrich and used without further purification. A home-built molecular doser was used to sublimate the amino acid glycine (NH2–CH2–COOH), under UHV conditions. The chemical sample material was placed in a tantalum envelope in the doser with a thermocouple facing it. The tantalum envelope was heated by direct current. A K-type thermocouple was attached to the outside of the bag close to the chemical to measure its temperature. Before sublimation, glycine molecules were outgassed and then heated to 380–400 K, and the pyrite crystal was subsequently exposed to it. During sublimation, the main chamber pressure typically increased to 1 × 10−9 mbar.Analysis techniques
X-ray photoelectron spectroscopic analysis of the samples was performed in an ultra-high vacuum chamber equipped with a hemispherical electron analyser using an Al Kα X-ray source (1486.6 eV) with an aperture of 7 mm × 20 mm. The base pressure in the chamber was 1 × 10−9 mbar, and the experiments were performed at room temperature. After subtraction of the background spectra, the XPS peaks were deconvoluted into different components by fitting them with a convolution of Lorentzian and Gaussian curves using the XPS Fitt software.Computational approach: methodology and models
The atomic coordinates of the crystal cubic structure of pyrite with face-centered Fe were used to generate the (100) surface models. These values were taken from the American Mineralogist Crystal Structure Database.16 The surface was modelled as a 2 × 2 × 1 supercell with 96 atoms. The theoretical calculations are based on Density Functional Theory. The optimization processes of these models were developed with the CASTEP code of the Biovia package, and periodical boundary conditions were used. The electronic calculations were made using the Generalized Gradient Approximation (GGA) with the PBE exchange correlation functional17 and a mesh cut-off energy of 340 eV was used along with ultrasoft pseudopotentials (OTFG) with the relativistic treatment of Koelling–Harmon. With respect to the Fe atoms, these were maintained as diamagnetic states, this treatment is in good agreement with previous theoretical and experimental models using this mineral.18–20On the other hand, for the weak interactions, we applied the semiempirical Grimme dispersion correction21 by fully optimizing the adsorption complex and the reactant with the GGA/PBE/G06 method.22
In addition to manually exploring the best possible adsorption sites of the adsorbates on the pyrite surface, we used the “adsorption locator” program which is based on Monte Carlo dynamics simulations. This algorithm involves simulating the annealing method where the energy of the system is calculated with empirical interatomic potentials based on the Universal force field. This program was implemented in the Biovia package.16
To simulate the adsorption process, the chemical species were adsorbed on a 2 × 2 × 1 unit cell of the (100) surface of pyrite. The optimization of these complexes was similar to that of the pristine surface i.e., in the slab, two upper Fe planes were optimized while the rest of the planes remained fixed.
To determine the adsorption energy, we used the following equation:
| Eads = (Ex/surf + Esurf + nEadsorbates) | (1) |
Results and discussion
XPS study on glycine adsorption on the pyrite(100) surface
Fig. 1 shows the XPS spectra for glycine adsorption on the pyrite surface from the gas phase under ultra-high vacuum conditions. These XPS spectra, which were obtained immediately after glycine adsorbed on the surface, show the integration of 20 scans for each region. This work aims to monitor the chemical transition due to glycine chemical evolution on the pyrite surface over time, which explains the slight noise in the spectra, especially the N 1s spectrum. Nevertheless, the spectra can be decomposed with no ambiguities in all cases.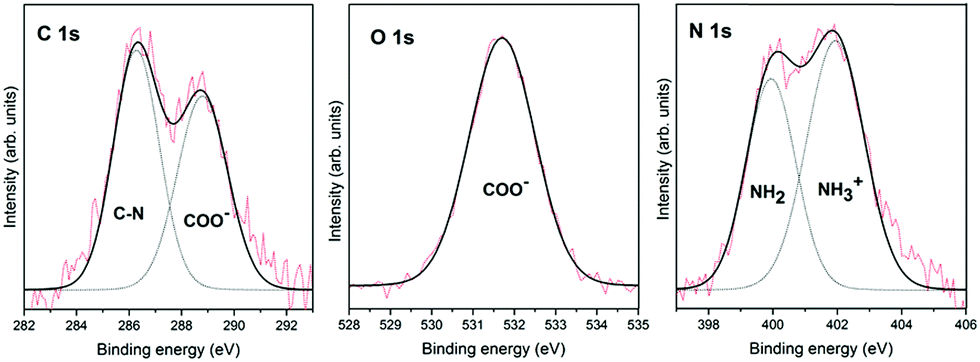 | ||
| Fig. 1 XPS photoemission spectra of the C 1s, O 1s and N 1s core-level peaks of glycine adsorbed on the pyrite(100) surface from the gas phase under ultra-high vacuum conditions. Experimental core-level spectra (red) and fitting results for all the components (black) and individual components (dotted lines). | ||
Glycine adsorption on the pyrite(100) surface was analysed by XPS, which is well suited to characterize the chemical states of adsorbed entities as well as their interactions with surfaces, thanks to its high sensitivity and non-destructive nature. The in situ XPS analysis of glycine adsorption on the pyrite surface reveals the presence of carbon, oxygen and nitrogen on the pyrite(100) surface under UHV conditions. The best-fit curve for the C 1s peak was achieved using two components for glycine (see Table 1). For glycine, the first carbon component has a binding energy (B.E.) of 286.3 eV and is attributed to the C–N group,23,24 whereas the second component is observed at 288.8 eV and is assigned to the COOH or COO− groups.25 The intensity ratio of the two components is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (CN:COO− or COOH), in close agreement with the chemical formula of the molecule. For the glycine molecule, the O 1s peak is observed at 531.7 eV and is attributed to one component, the oxygen in the COO− groups.26–29 The best-fit curve of the N 1s peak consists of two components centred at binding energies of 399.9 and 401.9 eV, which are assigned to the NH2 and NH3+ species, respectively,23,28–30 (see Fig. 1 for the glycine molecule). The presence of the NH3+ species is only observed when the molecular adsorption occurs under ultra-high vacuum conditions, which favours molecular diversity, as previously reported by our group.11 Therefore, the ratio between the N 1s components for glycine, shows that the two nitrogen components are 58% for the higher B.E. (NH3+), meaning zwitterionic state, and 42% for the lower B.E. (NH2), meaning the anionic state.
:1 (CN:COO− or COOH), in close agreement with the chemical formula of the molecule. For the glycine molecule, the O 1s peak is observed at 531.7 eV and is attributed to one component, the oxygen in the COO− groups.26–29 The best-fit curve of the N 1s peak consists of two components centred at binding energies of 399.9 and 401.9 eV, which are assigned to the NH2 and NH3+ species, respectively,23,28–30 (see Fig. 1 for the glycine molecule). The presence of the NH3+ species is only observed when the molecular adsorption occurs under ultra-high vacuum conditions, which favours molecular diversity, as previously reported by our group.11 Therefore, the ratio between the N 1s components for glycine, shows that the two nitrogen components are 58% for the higher B.E. (NH3+), meaning zwitterionic state, and 42% for the lower B.E. (NH2), meaning the anionic state.
| Element | Carbon | Oxygen | Nitrogen | ||
|---|---|---|---|---|---|
| Initial state BE (eV) | 286.3 | 288.8 | 531.7 | 399.9 | 401.9 |
| Initial (%) | 53 | 47 | 100 | 42 | 58 |
| Final state BE (eV) | 286.1 | 288.6 | 531.7 | 400.1 | 402.1 |
| Final (%) | 55 | 45 | 100 | 65 | 35 |
| Assignment | C–N | COO− | COO− | NH2 | NH3+ |
Spectroscopy evidenced chemical evolution of glycine on pyrite(100) surfaces
The XPS spectra obtained after ageing overnight unexpectedly reveals that glycine evolves on the pyrite(100) surface. During the initial step of the adsorption process (t = 1 h), NH3+ is present in higher amounts than NH2 (58% vs. 42%). Over time, evolution of the chemical forms does occur on the surface, the NH3+ intensity decreasing to 45% after 4 h, and 35% after 15 h. No significant changes in the C 1s and O 1s peaks were detected (see Table 1). The XPS spectra recorded at times longer than 15 hours do not vary, indicating that an equilibrium has been attained, and the ratio between the two chemical forms (NH2/NH3+) remains constant at 65/35 for glycine which is significantly different from the initial 42/58 ratio.Unexpectedly, the molecule/pyrite system evolves chemically over time. This specific molecular evolution phenomenon on the pyrite surface is reported here for the first time for these type of molecules (amino acids). The XPS spectra of the glycine/pyrite system recorded at different times show that the intensity of the NH3+ peak decreases with increasing time (from 58% to 35%). The change in the chemical functionality of amino acid molecules adsorbed on sputtered surfaces with time has not been previously reported for any other surface.31,32 During the chemical evolution process, the N 1s peak varies remarkably; the relative intensities of the two components are reversed (Table 1). A summary of the XPS results, that is, the C 1s, N 1s and O 1s core-level peaks, ratios and component assignments, for the glycine/pyrite (monosulfide-enriched) system obtained at different evolution times under UHV is given in Table 1.
Fig. 2a shows the XPS photoemission spectra of the N 1s core-level peak of glycine on the pyrite(100) surface over time, from the initial step (at 1 hour) to the final step (at 15 hours). Significant changes in the N 1s core-level peak intensities are observed for the glycine molecule on the pyrite surface over time (at evolution times of 1, 2 and 15 hours) (Fig. 2a). The glycine molecule undergoes molecular evolution (from NH3+ to NH2), therefore, it is proposed that the pyrite surface structure is responsible for this proton abstraction within a fraction of the adsorbed molecules.
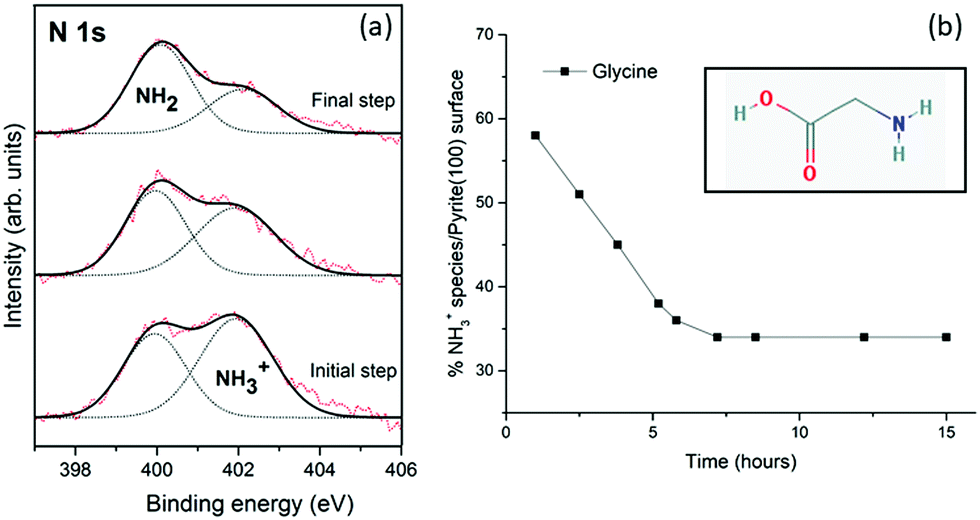 | ||
| Fig. 2 (a) XPS photoemission spectra of the N 1s core-level peak of glycine adsorbed on the pyrite(100) surface from the gas phase obtained at different times (from bottom to top at 1, 2 and 15 hours respectively). Experimental core-level spectra (red) and fitting results for all the components (black) and individual components (dotted lines). (b) Evolution of the percentage of the NH3+ species on the pyrite surface over time (1–15 hours) for glycine molecules. | ||
We will next analyse the possible influence of the pyrite surface upon its chemical reactivity. The sputtering conditions used to clean the sample lead to the preferential desorption of sulfur atoms from the pyrite surface, thus creating sulfur vacancy sites.33,34 The sputtering process preferentially removes lighter elements from the surface, in this case sulfur atoms would be favourably detached. After creating non-stoichiometric sites on pyrite and characterizing them by XPS,12 the adsorption and unique evolution behaviour of the amino acid was identified and carefully studied by XPS. The presence of sulfur vacancy sites on the pyrite surface drives the chemical evolution of the molecularly adsorbed amino acids. The presence of sulfur vacancies induces changes in the electronic structure of pyrite, and modifies its reactivity.
As previously reported by our group,12 in the case of the cystine amino acid on the (100) pyrite surface, the presence of an ordered structure on the surface (disulfide enrichment surface), as indicated by the LEED pattern, favours the cystine NH3+ chemical form, whereas the absence of surface ordering promotes cystine NH2 adsorption due to the sulfur-deficient surface (monosulfide enrichment surface). The breaking of the S–S bonds leads to an increase in the binding energy of the Fe 2p3/2 component, while the contribution of the component at 161.3 eV, which can be attributed to the sulfur monomers species (![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) S2−), to the S 2p3/2 peak increases. Thus, XPS measurements, after ion sputtering of the surface, which breaks the S–S dimer bonds, reveal the presence of iron dangling bonds.35,36 It is known that the rupture of S–S bonds generates highly unstable S− species.37–39 To compensate for the charge disequilibrium, an electron is donated to the nearest nitrogen atom. In the present work, after the molecules are adsorbed on the pyrite surface, a decrease in the NH3+ component is observed (see Fig. 2), which might be attributed to the sulfur vacancies at the surface.
S2−), to the S 2p3/2 peak increases. Thus, XPS measurements, after ion sputtering of the surface, which breaks the S–S dimer bonds, reveal the presence of iron dangling bonds.35,36 It is known that the rupture of S–S bonds generates highly unstable S− species.37–39 To compensate for the charge disequilibrium, an electron is donated to the nearest nitrogen atom. In the present work, after the molecules are adsorbed on the pyrite surface, a decrease in the NH3+ component is observed (see Fig. 2), which might be attributed to the sulfur vacancies at the surface.
Additionally, initially, most of the molecules are zwitterionic, and a second anionic species is also observed. Therefore, the pyrite surface initially favours the zwitterionic adsorption species, but these species evolve into anionic species over time, due to the non-stoichiometric pyrite (FeS2−x) surface structure. Therefore, a very significant conclusion is that amine groups (NH3+) in the structures of amino acids and peptides are unstable on non-stoichiometric pyrite (FeS2−x) surfaces, and some of them undergo a rapid transformation to the NH2 form, which should result in considerable changes in the molecular functionality.
In order to support the experimental data and to propose a possible mechanism of the chemical evolution process, energy calculations have been done for pristine and vacancy pyrite(100) surfaces. The effect of the vacancies in the adsorption process of the neutral and zwitterion glycine on the (100) pyrite surface have been studied in detail.
Computational approach
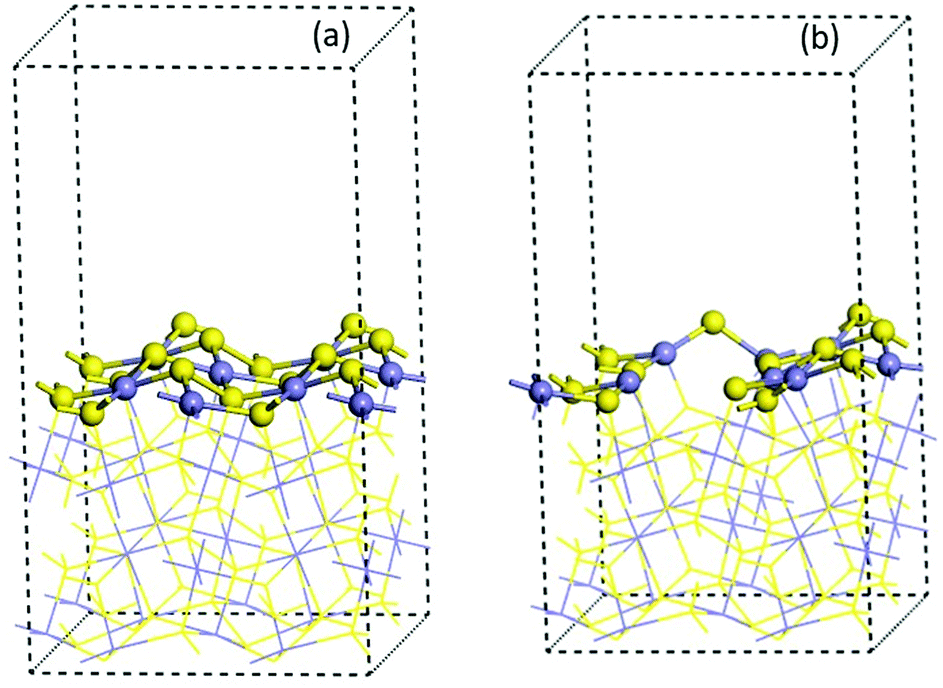 | ||
| Fig. 3 Optimized 2 × 2 × 1 cell of (100) pyrite surfaces (a) pristine model and (b) with four vacancies. Blue and yellow ball colours represent the Fe and S atoms respectively. | ||
The effect of the vacancies in the adsorption process of the neutral and zwitterion glycine on the (100) pyrite surface is studied. In order to explore the adsorption energy of the neutral glycine (Fig. 4a) and the zwitterion molecules onto pristine and vacancies surfaces, we used the “adsorption locator” program to determine the best adsorption site and the principal glycine configuration in the interaction with the mineral surface. After the adsorption test, the whole adsorption complexes were optimized. The adsorption energy indicates that a pyrite surface with four vacancies on the surface presents a significantly higher adsorption energy than the pristine surface (ΔEads = 1.16 eV). On the other hand, the glycine configuration in which the COO− moiety is adsorbed on the surface's Fe atoms is the most favourable one from the point of view of energy. After the adsorption process test of the neutral glycine onto both pristine and vacancies surfaces, the adsorption energy indicates that four vacancies can be a good model to study the adsorption processes on the pyrite defect surfaces. The zwitterion form was optimized as in the model above; it is adsorbed through the carboxylic moiety in the Fe atoms on the surface (Fig. 4b). This site is close to the defect point, which corresponds to S vacancy formation, in which the lone pair electron produced by the breaking of an S–S bond increases the interaction with the adsorbate of the surface, this behaviour was described in previous theoretical results.20 Also, the adsorption site in this surface corresponds to the under-coordinated Fe atoms (3f). The low coordination increases the reactivity of the surface, this has been observed too in studies of glycine adsorbed on forsterite.46,47 The adsorption site has an interaction distance of d(COO⋯Fe) = 1.90 Å, which, as we will see later, indicates that this one is one of the most adsorbed complexes.
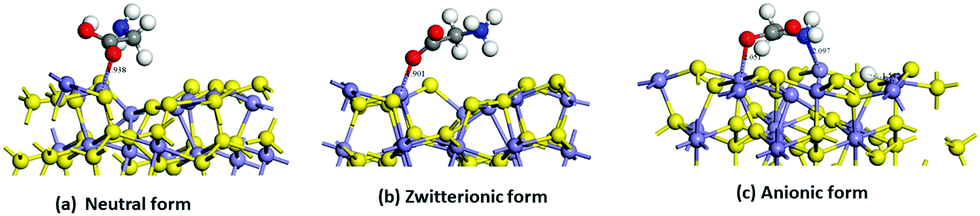 | ||
| Fig. 4 Optimized structures of the adsorption of (a) neutral glycine, (b) zwitterion, and (c) anionic forms of glycine onto 2 × 2 × 1 vacancies surface pyrite. Interaction distances are indicated in angstrom. | ||
Our theoretical models are based on the experimental evidence i.e., the proton formed with the anionic species could be removed in the vacuum step in the vapour gas phase. The second possible pathway reaction is when the proton could be transferred onto the pyrite surface to form the anionic form (COO−/H+), in this case H+ interacts with the active sites on the surface. Both models were explored. However, the vacuum model was an endothermic process. Only the stable species (exothermic) were considered i.e., when the proton of NH3 in the anionic form (COO−) was transferred to the surface.
An alternative proposed mechanism could be zwitterion/neutral isomerization, which seems plausible from a chemical point of view, nevertheless XPS measurements on the pyrite surface show a component of the carbon peak at 288.6–288.8 eV which could be assigned to the COOH or COO− groups,48 and the O 1s peak was fitted only by one component at 531.7 eV which has been unambiguously assigned to the two equivalent oxygen atoms belonging to the so-called resonant state of the deprotonated carboxylic group (carboxylate group).49–51 In addition, the absence of an oxygen component at 533.0 eV (B.E.) confirms the non-presence of the carboxyl group (COOH) on the pyrite surface, meaning the absence of possible neutral species on the surface.
We simulated the adsorption of the anionic form of glycine onto pyrite with four vacancies. We assume that this form is previously generated in the experiment and that the proton was transferred to the surface (Fig. 4c).
We have used the “adsorption locator” tool to find the most reactive site to the adsorption. The adsorption energy of the optimized anionic structure indicates that it is an exothermic process (Table 2). In the same way, glycine was adsorbed through the COO− and NH2 moiety to the under coordinated Fe atoms (3f and 4f) and the proton is adsorbed onto the 4f site of the Fe atom (Fig. 4c). These iron atoms are neighbours to the sulfur vacancies. The adsorption site has interaction distances of d(COO⋯Fe) = 2.05 Å, d(H2N⋯Fe) = 2.09 Å and d(H⋯S) = 1.5 Å. This structure has greater stability due to the ring-bond that was formed between the amino and carboxylic group and the top Fe atoms on the surface. This behaviour of the role of geometry on the interaction bond distances was observed previously in a theoretical model of pyrite with defects.20
The above calculations showed that the adsorption energy on defect pyrite, of both the glycine zwitterion and the anionic form, is greater than that on the pristine surface. This supports the experimental evidences that indicate that the adsorption energy of glycine molecules increases when the pyrite surface has vacancies. On the other hand, the theoretical calculations also support the conversion over time of the zwitterionic form (NH3+) to the anionic form (NH2) as their adsorption energy is similar, this explains why in equilibrium, the two chemical forms are present. Indeed, when the zwitterionic form was adsorbed, this molecule was found in the XPS spectra in a greater amount than the one in the (NH2) peak (58% vs. 42%) and then the NH3+ (zwitterionic form) intensity decreases to 35% over time, and it is partially converted to the (NH2) form of the anionic model. Finally, the theoretical calculations suggest that in the anionic mode, the proton was transferred to the surface in the site in which S vacancies were formed. However, this cannot be resolved by this experimental setup, which stabilizes the molecule–surface interaction, making it energetically favourable.
To summarize the molecule/surface processes and understand the mechanism of the molecular chemical conversion, Scheme 1 presents the details of the different stages and the XPS spectra that support the novel proposed mechanism. Scheme 1 shows that in stage 1, cleaning the disulfide surface (FeS2) by Ar+ sputtering leads to the preferential desorption of sulfur atoms, creating sulfur vacancies. We followed this process spectroscopically using the identified XPS sulfur peak as the spectroscopic signature, confirming the presence of the disulfide surface (FeS2) with the appearance of the main component at 162.2 eV (B.E.) (see black spectra). Then, after cleaning the disulfide surface by Ar+ sputtering which led to the preferential desorption of sulfur atoms, creating sulfur vacancies, the sulfur peak showed a main component at 161.3 eV which is assigned to FeS (see red spectra). After the sputtering process, for the sulfur species, the 162.6 eV peak assigned to the FeS2 (black line) partially disappears in favor of the main component at 161.3 eV; this component is assigned to the FeS species (red line). This process, results in the appearance of an increase of monosulfide surface (FeS2−x), the main component of the S 2p spectra shows a shift to lower binding energies that is related to the presence of sulfur vacancies.
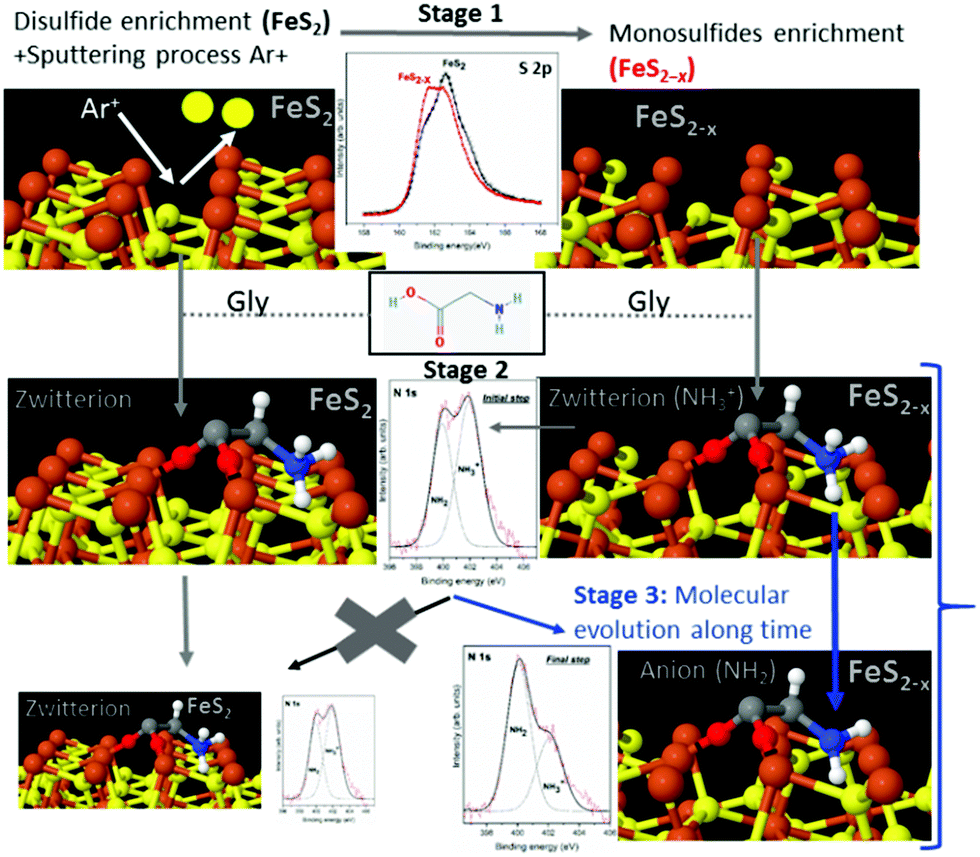 | ||
| Scheme 1 Different stages of the molecule/surface phenomenon: stage 1, disulfide (FeS2) (black sulfur spectra) conversion to monosulfides (red sulfur spectra, FeS2−x generation of sulfur vacancies); stage 2, molecular adsorption (glycine) on both surfaces (FeS2 and FeS2−x); and stage 3, chemical molecular evolution from a zwitterionic species to an anionic species over time on the monosulfide surface (with sulfur vacancies). Disulfide enrichment surfaces (without sulfur vacancies) do not show a molecular evolution phenomenon, stage 3 does not take place on this surface FeS2. | ||
Immediately after dosing pyrite with the molecules (stage 2), they adsorb on both surfaces, FeS2 and FeS2−x, mainly as a zwitterionic species (NH3+ and COO−), and no remarkable changes are observed for the S 2p spectra after adsorption of the molecules on the surface. Finally, as the molecule/surface system ages in stage 3, the molecules evolve from mainly zwitterionic to anionic on the monosulfide-enriched surface (FeS2−x). In contrast, this process does not occur on the pristine pyrite (FeS2 disulfide-enriched surface) which does not display sulfur vacancies. The sputtering process creates surface defects on pyrite, which are necessary for the evolution of the amino acid over time from zwitterion to anion species.
The in situ XPS spectroscopy experiments support the proposed mechanism for the chemical evolution of amino acids adsorbed on non-stoichiometric pyrite (FeS2−x) over time due to the presence of sulfur vacancies on the surface (see Scheme 1). A sulfur vacancy on the FeS2(100) surface has also been measured to be quite reactive and the presence of two 4-coordinated iron ions significantly increases the reactivity of the site.20 Therefore, we could suggest a mechanism where the chemical evolution of amino acids on the pyrite surface is due to the compensated sulfur vacancies from monosulfide surfaces, and complementary theoretical studies confirm that molecules compensate surface instabilities the molecule–surface interaction being more energetically favourable on the monosulfide than on the disulfide enriched pyrite surface. Furthermore, theoretical calculations reveal that iron sites close to the S defect are driving the chemisorption processes, due to the vacancies, the surface is more reactive than the pristine surface, this behaviour was previously observed in pyrite studies.20Ab initio simulations of desorption and reactivity of glycine at a water–pyrite interface have been studied,20,52–54 proving that surface point defects (sulfur vacancies) can greatly stabilize adsorbed amino acids providing strong bonding sites.20 A high concentration of amino acids on its surface, would be a necessary ingredient for any sustained surface chemical reaction on minerals. Additionally, amino acid adsorption modes play a key role in the detachment process and chemical surface reactivity.54
The fact that the pyrite surface electronic structure is dominated by iron dangling bonds means that at least the preliminary interactions between the amino acid species and the surface will be driven by the quenching of high energy surface states at Fe sites. This interaction process is expected to lead to new bonds between the molecules and surface iron sites, implying that the chemical structure and reactivity of the surface would be fundamentally different.
Understanding the interactions of a single amino acid with a surface is important for increasing our knowledge of molecule adsorption on mineral surfaces and of the general properties of pyrite. On pyrite, which displays a chemically complex surface (Fe and S2 functional groups), ion bombardment is selective to lighter atoms; unstable surface structures are created, which thus react with adsorbed species; this may have remarkable implications for pyrite used as a catalyst. Therefore, the interactions of amino acids with pyrite must be studied to assess the capability of this mineral to adsorb, react with (as a catalyst) and pattern biomolecules of increasing complexity. An anoxic early Earth atmosphere would help molecular variety, because under those conditions, the pyrite surface could provide a medium for obtaining a distribution of both the anionic and zwitterionic species and let them evolve over time. Furthermore, these species would be driven to their most stable form to compensate for and stabilize the surrounding iron and sulfur atoms. In particular, the existence of various adsorption modes for amino acids could help diverse interactions with organic molecules, leading to a variety of more complex biomolecules.
Conclusions
Our findings contribute to the understanding of chemical reactivity and the role that minerals might have played in prebiotic and surface chemistry. They also add to the knowledge of the interfacial processes of amino acids in prebiotic chemistry surface reactions, iron–sulfur scenarios and biomolecule/surface interactions, which is fundamental for assessing their use in biotechnological applications as well. Understanding the common guidelines that govern molecular adsorption on surfaces, particularly preferential adsorption and reactive sites, has significant implications in many fields, including materials science, catalysis, nanotechnology, and prebiotic chemistry, among others.Our studies evidence the importance of sulfur surface vacancies, as we have shown that the formation of such point defects has a significant impact on the reactivity of this surface and the chemical evolution of adsorbate species. Therefore, surface defects have a high impact on the chemistry of glycine adsorption on pyrite surfaces.
In summary, the pyrite surface is shown to be a suitable substrate for facilitating the chemical evolution of the amino acid glycine, even under inert conditions, due to unstable surface charges. This will likely have implications for early Earth studies and the origin of the iron–sulfur world. This work supports the fact that molecules can be adsorbed and evolve on pyrite surfaces which has implications for the sulfur–iron world theory.
Systematic step by step XPS measurements provide evidence to uncover and fully characterise the surface chemistry composition (FeS2−x generation of sulfur vacancies) and to observe how molecules evolve chemically over time. The nitrogen XPS signal is the main spectroscopic signature for monitoring molecular evolution processes on the surface. Furthermore, our calculations support the experimental results, confirming that sulfur vacancies play a significant role in the adsorption process of the amino acid glycine on a pyrite(100) surface, and thus we anticipate that they will play an important role in more complex molecular problems. Furthermore we conclude that the breaking-up of bonds in the sulfur–sulfur dimers due to the sputtering process, the variations of the coordination number of iron atoms, and the creation of defects which drives molecular adsorption and chemical evolution, should be considered in the future.
Finally, this experiment constitutes a unique observational tool for molecular surface processes that take place over a number of hours. Our future studies will also investigate other amino acids with different binding energies as well as the role of temperature in the molecular population distribution and its evolution over time.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been supported by the MINECO project ESP2017-89053. The Instituto Nacional de Técnica Aeroespacial supported the work performed at CAB. EER is thankful to Javier Martin-Torres, Alfonso Hernández-Laguna and C. M. Pradier for their support and suggestions. This Project has been partially funded by the Spanish State Research Agency (AEI) Project No. MDM-2017-0737 Unidad de Excelencia “María de Maeztu”-Centro de Astrobiología (CSIC-INTA).Notes and references
- C. Joachim, J. K. Gimzewski and A. Aviram, Nature, 2000, 408, 541–548 CrossRef CAS.
- Nanobiotechnology: Concepts, Applications and Perspectives, ed. C. M. Niemeyer and C. A. Mirkin, Wiley, 1st edn, 2004 Search PubMed.
- E. Mateo-Martí, C. Briones, C. M. Pradier and J. A. Martín-Gago, Biosens. Bioelectron., 2007, 22, 1926–1932 CrossRef PubMed.
- L. Li, M. Cabán-Acevedo, S. N. Girard and S. Jin, Nanoscale, 2014, 6, 2112–2118 RSC.
- A. Douglas, R. Carter, L. Oakes, K. Share, A. P. Cohn and C. L. Pint, ACS Nano, 2015, 9, 11156–11165 CrossRef CAS.
- J. Liu, Y. Wen, Y. Wang, P. A. van Aken, J. Maier and Y. Yu, Adv. Mater., 2014, 26, 6025–6030 CrossRef CAS PubMed.
- D. Rickard and G. W. Luther, Chem. Rev., 2007, 107, 514–562 CrossRef CAS PubMed.
- G. Wachtershauser, Proc. Natl. Acad. Sci. U. S. A., 1990, 87, 200–204 CrossRef CAS PubMed.
- C. Huber, Science, 1998, 281, 670–672 CrossRef CAS PubMed.
- J.-F. Lambert, Origins Life Evol. Biospheres, 2008, 38, 211–242 CrossRef CAS PubMed.
- M. Sanchez-Arenillas and E. Mateo-Marti, Chem. Phys., 2015, 458, 92–98 CrossRef CAS.
- M. Sanchez-Arenillas and E. Mateo-Marti, Phys. Chem. Chem. Phys., 2016, 18, 27219–27225 RSC.
- M. Sanchez-Arenillas, S. Galvez-Martinez and E. Mateo-Marti, Appl. Surf. Sci., 2017, 414, 303–312 CrossRef CAS.
- V. Moy, E. Florin and H. Gaub, Science, 1994, 266, 257–259 CrossRef CAS PubMed.
- E. Evans, Annu. Rev. Biophys. Biomol. Struct., 2001, 30, 105–128 CrossRef CAS.
- P. Bayliss, Am. Mineral., 1977, 62, 1168–1172 CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- N. Y. Dzade, A. Roldan and N. H. de Leeuw, Phys. Chem. Chem. Phys., 2014, 16, 15444–15456 RSC.
- A. Hung, J. Muscat, I. Yarovsky and S. P. Russo, Surf. Sci., 2002, 513, 511–524 CrossRef CAS.
- N. N. Nair, E. Schreiner and D. Marx, J. Am. Chem. Soc., 2006, 128, 13815–13826 CrossRef CAS PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- E. R. McNellis, J. Meyer and K. Reuter, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 205414 CrossRef.
- O. Cavalleri, G. Gonella, S. Terreni, M. Vignolo, L. Floreano, A. Morgante, M. Canepa and R. Rolandi, Phys. Chem. Chem. Phys., 2004, 6, 4042 RSC.
- G. Gonella, S. Terreni, D. Cvetko, A. Cossaro, L. Mattera, O. Cavalleri, R. Rolandi, A. Morgante, L. Floreano and M. Canepa, J. Phys. Chem. B, 2005, 109, 18003–18009 CrossRef CAS PubMed.
- S. Fischer, A. C. Papageorgiou, M. Marschall, J. Reichert, K. Diller, F. Klappenberger, F. Allegretti, A. Nefedov, C. Wöll and J. V. Barth, J. Phys. Chem. C, 2012, 116, 20356–20362 CrossRef CAS.
- D. T. Clark, J. Peeling and L. Colling, Biochim. Biophys. Acta, Protein Struct., 1976, 453, 533–545 CrossRef CAS.
- K. Uvdal, P. Bodö, A. Ihs, B. Liedberg and W. Salaneck, J. Colloid Interface Sci., 1990, 140, 207–216 CrossRef CAS.
- K. Uvdal, P. Bodö and B. Liedberg, J. Colloid Interface Sci., 1992, 149, 162–173 CrossRef CAS.
- Handbook of X-ray photoelectron spectroscopy: a reference book of standard spectra for identification and interpretation of XPS data, ed. J. F. Moulder and J. Chastain, PerkinElmer Corporation, Eden Prairie, Minn, Update, 1992 Search PubMed.
- E. Mateo-Martí, C. Briones, E. Román, E. Briand, C. M. Pradier and J. A. Martín-Gago, Langmuir, 2005, 21, 9510–9517 CrossRef PubMed.
- S. M. Barlow and R. Raval, Surf. Sci. Rep., 2003, 50, 201–341 CrossRef CAS.
- C. Méthivier, H. Cruguel, C.-M. Pradier and V. Humblot, Faraday Discuss., 2017, 204, 69–81 RSC.
- K. Andersson, M. Nyberg, H. Ogasawara, D. Nordlund, T. Kendelewicz, C. S. Doyle, G. E. Brown, L. G. M. Pettersson and A. Nilsson, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 195404 CrossRef.
- S. Chaturvedi, R. Katz, J. Guevremont, M. A. A. Schoonen and D. R. Strongin, Am. Mineral., 1996, 81, 261–264 CAS.
- G. Ruano, F. Pomiro and J. Ferrón, Surf. Sci., 2018, 667, 138–147 CrossRef CAS.
- C. Gil-Lozano, A. F. Davila, E. Losa-Adams, A. G. Fairén and L. Gago-Duport, Sci. Rep., 2017, 7, 43703 CrossRef CAS PubMed.
- H. W. Nesbitt, G. M. Bancroft, A. R. Pratt and M. J. Scaini, Am. Mineral., 1998, 83, 1067–1076 CrossRef CAS.
- A. G. Schaufuß, H. W. Nesbitt, I. Kartio, K. Laajalehto, G. M. Bancroft and R. Szargan, Surf. Sci., 1998, 411, 321–328 CrossRef.
- N. N. Nair, E. Schreiner and D. Marx, J. Am. Chem. Soc., 2006, 128, 13815–13826 CrossRef CAS PubMed.
- H. E. King, M. Stimpfl, P. Deymier, M. J. Drake, C. R. A. Catlow, A. Putnis and N. H. de Leeuw, Earth Planet. Sci. Lett., 2010, 300, 11–18 CrossRef CAS.
- E. Escamilla-Roa and C. I. Sainz-Díaz, J. Phys. Chem. C, 2014, 118, 3554–3563 CrossRef CAS.
- E. Escamilla-Roa, J. Martin-Torres and C. I. Sainz-Díaz, Planet. Space Sci., 2018, 153, 163–171 CrossRef CAS.
- E. Escamilla-Roa, F. Moreno, J. J. López-Moreno and C. I. Sainz-Díaz, Planet. Space Sci., 2017, 135, 17–26 CrossRef CAS.
- L. Zhu, B. Richardson, J. Tanumihardja and Q. Yu, CrystEngComm, 2012, 14, 4188 RSC.
- M. Birkholz, S. Fiechter, A. Hartmann and H. Tributsch, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 43, 11926–11936 CrossRef CAS.
- E. Escamilla-Roa and F. Moreno, Planet. Space Sci., 2012, 70, 1–9 CrossRef CAS.
- E. Escamilla-Roa and C. I. Sainz-Díaz, J. Phys. Chem. C, 2014, 118, 26080–26090 CrossRef CAS.
- S. Fischer, et al. , J. Phys. Chem. C, 2012, 116, 20356–20362 CrossRef CAS.
- D. T. Clark, J. Peeling and L. Colling, Biochim. Biophys. Acta, 1976, 453, 533–545 CrossRef CAS.
- K. Uvdal, P. Bodo, A. His, B. Lieberg and W. R. Salaneck, J. Colloid Interface Sci., 1990, 140, 207–216 CrossRef CAS.
- E. Mateo-Marti, et al. , Langmuir, 2004, 20, 10223–10230 CrossRef CAS PubMed.
- N. N. Nair, E. Schreiner and D. Marx, J. Am. Chem. Soc., 2008, 130, 14148–14160 CrossRef CAS PubMed.
- C. Boehme and D. Marx, J. Am. Chem. Soc., 2003, 125, 13362–13363 CrossRef CAS PubMed.
- R. Pollet, C. Boehme and D. Marx, Origins Life Evol. Biospheres, 2006, 36, 363–379 CrossRef CAS PubMed.
| This journal is © the Owner Societies 2019 |