
Low energy (1–19 eV) electron scattering from condensed thymidine (dT) I: absolute vibrational excitation cross sections†
 *a
*a
Abstract
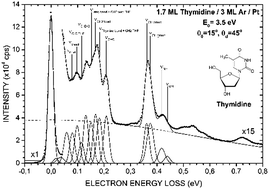
Absolute cross sections (CSs) for vibrational excitation by electrons of energy between 1–19 eV scattering from condensed thymidine (dT) were measured by means of high-resolution electron energy loss spectroscopy (HREELS). The CSs were extracted from electron energy loss spectra of dT condensed on multilayers film of Ar held at about 20 K under ultra-high vacuum (∼1 × 10−11 Torr). dT is one of the most complex molecules to be studied in condensed phase by HREELS. The magnitudes of the vibrational CSs lie within the 10−17 cm2 range. Structures observed in the energy dependence of the vibrational CSs under 3 eV and around 4 eV were compared with previous results of gas- and solid-phase studies on dT and related molecules (e.g., thymine and tetrahydrofuran). These structures were attributed to the formation of shape resonances.
- This article is part of the themed collection: 2019 PCCP HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

