
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Resolving the chemical identity of H2SO4 derived anions on Pt(111) electrodes: they're sulfate†
Igor Ying
Zhang
 *ab,
Gregor
Zwaschka
a,
Zhenhua
Wang
c,
Martin
Wolf
a,
R. Kramer
Campen
a and
Yujin
Tong
*a
*ab,
Gregor
Zwaschka
a,
Zhenhua
Wang
c,
Martin
Wolf
a,
R. Kramer
Campen
a and
Yujin
Tong
*a
aFritz Haber Institute of the Max Planck Society, 14195 Berlin, Germany. E-mail: tong@fhi-berlin.mpg.de
bDepartment of Chemistry, Fudan University, 200433 Shanghai, China. E-mail: igor_zhangying@fudan.edu.cn
cSchool of Chemical Engineering and Environmental, Beijing Institute of Technology, 100081 Beijing, China
First published on 15th August 2019
Abstract
Understanding how electrolyte composition controls electrocatalytic reactions requires molecular-level insight into electrode/electrolyte interaction. Perhaps the most basic aspect of this interaction, the speciation of the interfacial ion, is often controversial for even relatively simple systems. For example, for Pt(111) in 0.5 M H2SO4 it has long been debated whether the adsorbed anion is SO42−, HSO4− or an H3O+⋯SO42− ion pair. Here we apply interface-specific vibrational sum frequency (VSF) spectroscopy and theory to this problem and perform an isotope exchange study: we collect VSF spectra of Pt(111) in H2SO4(H2O) and D2SO4(D2O) as a function of bias and show that at all potentials they are identical. This is the most direct spectroscopic evidence to date that SO42− is the dominant adsorbate, despite the fact that at 0.5 M H2SO4 bulk solution is dominated by HSO4−. This approach is based on the unique selection rule of the VSF spectroscopy and thus offers a new way of accessing general electrode/electrolyte interaction in electrocatalysis.
1 Introduction
Much recent work has shown that the efficiency of such important electrocatalytic reactions as CO2 reduction, hydrogen evolution and oxygen evolution or reduction, depends on the electrode electronic structure and the composition of the supporting electrolyte.1–4 While it is clear that electrolyte effects on reaction efficiency are related to the potential dependent interaction of electrode, ion and solvent, molecular level insight into how electrolyte interacts with the electrode surface is required. Gaining such insight has proven challenging: despite much recent effort5–8 very basic questions regarding the link between electrolyte, electrode and reactivity remain for even well studied systems.One such system is Pt(111) electrode in 0.5 M H2SO4. Over the last several decades this system has been extensively studied using a wide variety of approaches.1,5,9–19 While much insight has been gained, perhaps the most fundamental aspect of electrolyte/electrode interaction, i.e. the chemical identity of adsorbing H2SO4 derived anions, has remained controversial.
Because the vibrational response of sulfate (SO42−) and bisulfate (HSO4−) differ significantly in aqueous solution20 a number of prior investigators have addressed this problem using Infrared Reflection–Absorption Spectroscopy (IRRAS).10,21–23 While the data are similar in all studies, the assignment of the observed spectral features, principally the intense, narrow resonance near 1250 cm−1 that is higher in frequency than any vibrations of SO42− or HSO4− in bulk solution, has varied between adsorbed SO42−, adsorbed HSO4− and/or adsorbed SO42−⋯H3O+ ion pairs.
Recently Feliu, Lipkowski and coworkers showed that some of the challenges in assigning peaks observed in IRRAS could be overcome by conducting Subtractively Normalized Interfacial Fourier Transform Infrared Reflection Spectroscopy (SNIFTIRS) and quantitatively correcting for IR absorption in bulk solution.23 Using this approach they quantified the pH dependence of the 1250 cm−1 peak and suggested that SO42− is the dominant adsorbate based on the invariance of the spectral feature in a wide range of pH (1–5.6). However, the most direct measurement to confirm surface speciation is an isotope exchange: the spectral response of adsorbed HSO4− modes involving H should shift dramatically on deuteration while that of adsorbed SO42− should not. Unfortunately, performing this experiment in IRRAS or SNIFTIRS is extremely challenging, as the strong D2O bending absorption at 1200 cm−1 substantially complicates data interpretation.21
Vibrationally resonant sum frequency generation (VSF) spectroscopy is a coherent nonlinear optical technique in which the output of pulsed infrared and visible lasers are overlapped at an interface and the intensity of the emitted sum frequency field is detected.24,25 This SF emission is interface-specific, i.e. insensitive to ions in bulk solution, by its symmetry selection rules and emitted in a different direction than the reflected incident beams. Thus the subtraction necessary for IRRAS or SNIFTIRS is not required. Here we take advantage of these characteristics and use VSF spectroscopy to quantify the spectral response of the Pt(111)/H2SO4 in H2O and Pt(111)/D2SO4 in D2O systems from 1100–1400 cm−1 as a function of applied bias. For both systems the bias dependent spectral response is identical. This result, complemented by DFT calculations of normal mode frequencies of sulfate and bisulfate adsorbed on Pt clusters, provides the strongest experimental evidence to date that, at potentials between 0.42 and 0.9 V RHE, sulfate adsorbs at the Pt(111) surface while bisulfate is the dominant species in the adjoining bulk liquid.
2 Results and discussion
Fig. 1a shows a cyclic voltammogram (CV) of Pt(111) in 0.5 M H2SO4 aqueous solution collected with a scan rate of 50 mV s−1 in a meniscus geometry. The CV is identical to those reported previously for an atomically flat Pt(111) surface with negligible defects.1,5,9–11,13,15,16 The square wave at E < 0.3 V RHE is typically assigned to hydrogen adsorption and the current feature from 0.3 < E < 0.45 V to the adsorption of sulfuric acid derived anions. The assignment of the spike at 0.42 V has proven controversial: it has been argued to be the result of an order/disorder transition of adsorbed sulfuric acid anions and/or the deprotonation of adsorbed bisulfate.11,20,26 At potentials positive of 0.42 V the evolved current is dominated by double layer charging/discharging with tiny, irreversible peaks near 0.7 V. These small features have been argued to result from the conversion of OH coadsorption or formation,16,17 bisulfate to sulfate27 and a second order/disorder transition of the adsorbed anions.11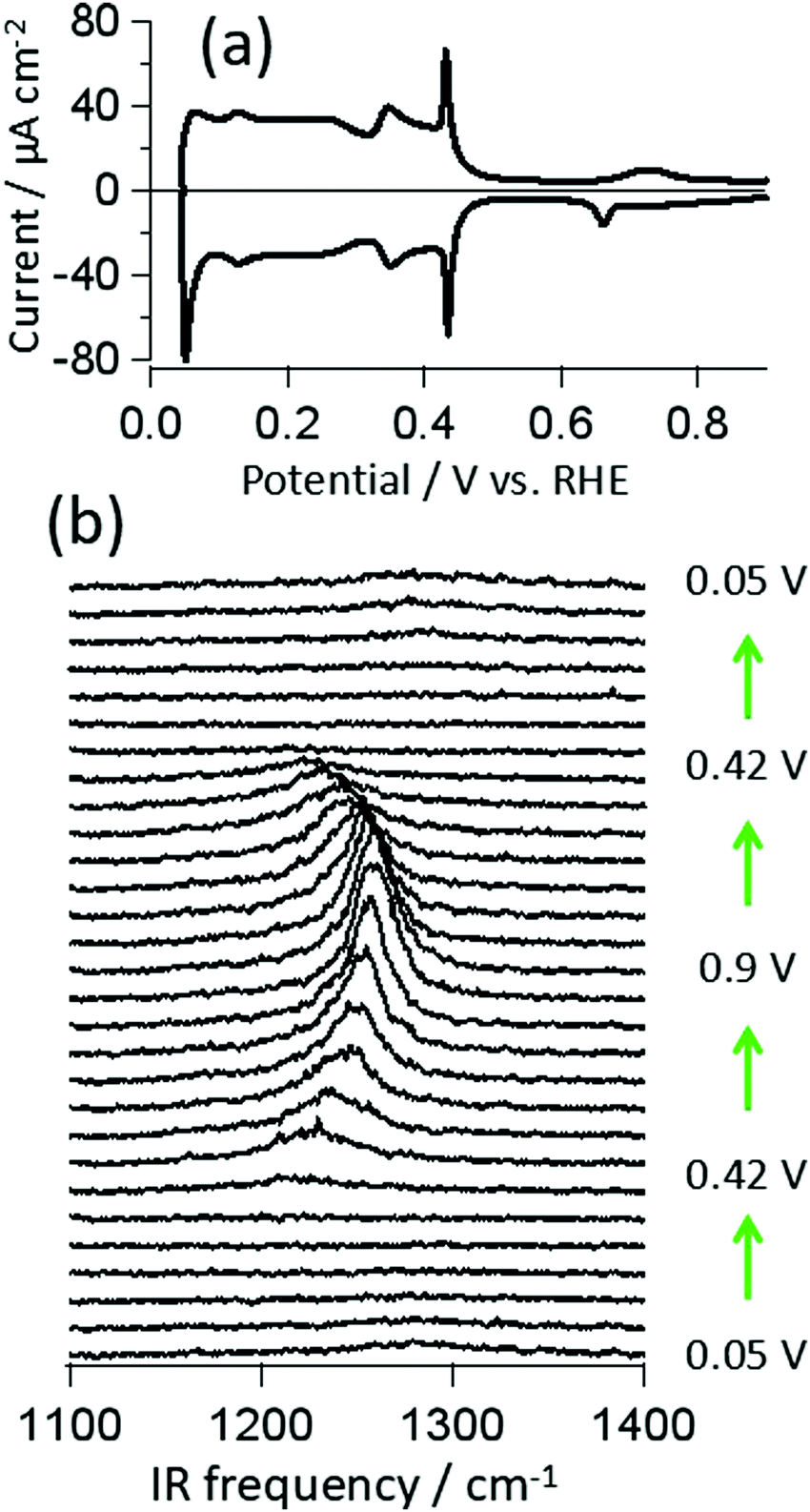 | ||
| Fig. 1 (a) Cyclic voltammogram of a Pt(111) electrode in 0.5 M H2SO4 in meniscus geometry (scanning speed 50 mV s−1), indicating a well ordered single crystal surface has been prepared; (b) potential dependent VSF spectra of the adsorbed sulfuric acid anions on a Pt(111) electrode in aqueous solution. | ||
Potential dependent VSF spectra collected at IR frequencies between 1100–1400 cm−1 for the same system are shown in Fig. 1b. A peak centered at ∼1220 cm−1 appears at 0.42 V (coincident with the current spike) and increases in intensity, blue shifts and narrows with increasing potential. As demonstrated in the figure all spectral changes are reversible. Qualitatively, similar observations have been reported previously for this system both with SNIFTIRS and VSF spectroscopy.16,23 We have recently shown that the potential dependent changes in peak amplitude, bandwidth and frequency, for E > 0.42 V, can be quantitatively understood as the result of interplay between the local field strength across the adsorbed layer and the potential dependent change in anion polarizability: the apparent changes in slope of spectral parameters with applied bias around 0.7 V can be understood without invoking a change in surface structure or chemistry.19 Here we address the assignment of this peak.
As mentioned above, SO42− and HSO4− are distinguishable in bulk solution by their vibrational response. However, in practice using the bulk spectral response of either species to assign features observed when the anions are adsorbed is challenging: adsorption may break bulk anion symmetry, and alter the vibrational response due to changes in electronic structure or the presence of interfacial fields.19,20,23,28,29 Isotope labelling avoids these issues: if an adsorbate vibrational mode includes hydrogen motion, frequency shifts should occur on deuteration. Representative VSF spectra for Pt(111) in H2SO4/H2O and D2SO4/D2O solution (red traces) at 630 mV and 870 mV are shown in Fig. 2a and b, respectively (for comparison purposes spectra of the D2SO4/D2O system have been scaled to match the maximum of the H2SO4/H2O, the complete D2SO4/D2O data set is shown in the ESI†). Both the spectra, and their potential dependent evolution, are quantitatively identical. This result clearly suggests that, for E > 0.42 V RHE, sulfate is adsorbed on Pt(111).
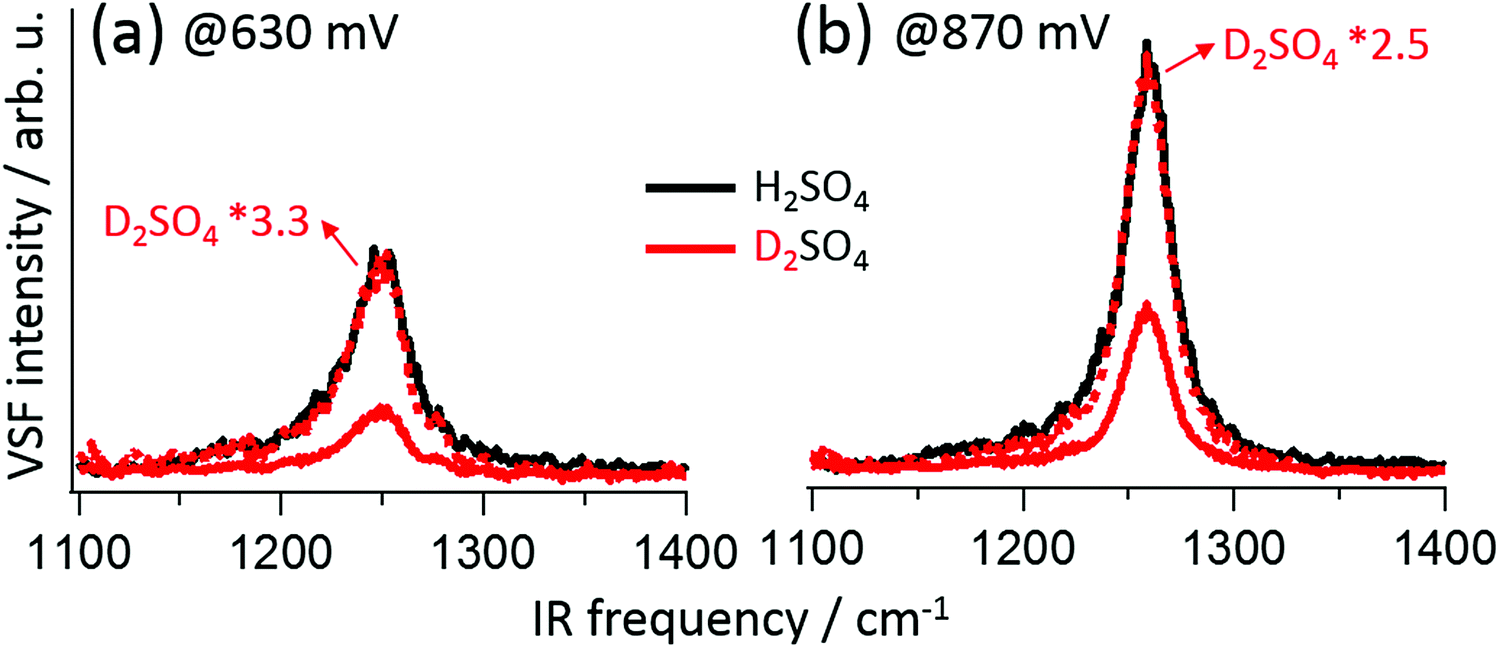 | ||
| Fig. 2 VSF spectra measured in H2SO4(H2O) solution (black traces) and D2SO4(D2O) solution (red traces) at 630 (a) and 870 mV (b); the spectra in D2SO4 solution were scaled to the same intensities as those in H2SO4 (red dotted lines). | ||
This conclusion is consistent with recent electronic structure calculations on Pt clusters,30 and extended (periodic) Pt surfaces.20 To complement these experiments we performed density functional theory calculations of a sulfate and bisulfate adsorbed on the Pt(111) surface of a cluster of 55 Pt atoms (see Fig. 3). While this system is clearly simplified – e.g. there is only a small Pt(111) surface and no water molecules – it reproduces the most important adsorbate structural features previously reported by others using both simpler and more complicated model chemistries: sulfate tends to adsorb in a tridentate geometry, bisulfate in a bidentate.31 Given the structures we next calculated the harmonic normal modes of each structure in Fig. 3. Clearly both sulfate and bisulfate have resonances in the frequency region around 1250 cm−1. For the former this frequency corresponds to the non-coordinated S–O stretching and the latter a more complicated combination of OH bending and non-coordinated S–O stretching (see ESI,† for the animation of the modes). As expected, upon isotope exchange, a significant frequency shift occurs for this combination mode. Since we did not observe any frequency shift after isotope exchange in experiment, the calculated frequencies are consistent with a scenario in which the adsorbed species at potentials from 0.42–0.9 V RHE is sulfate. It is worth noting that while calculation shows a mode around 1100 cm−1, the dynamic dipole moment of this mode is parallel to the surface and is inactive in current VSF measurement on Pt(111) surface (see orientation of the transition dipole moment of this mode in ESI†).
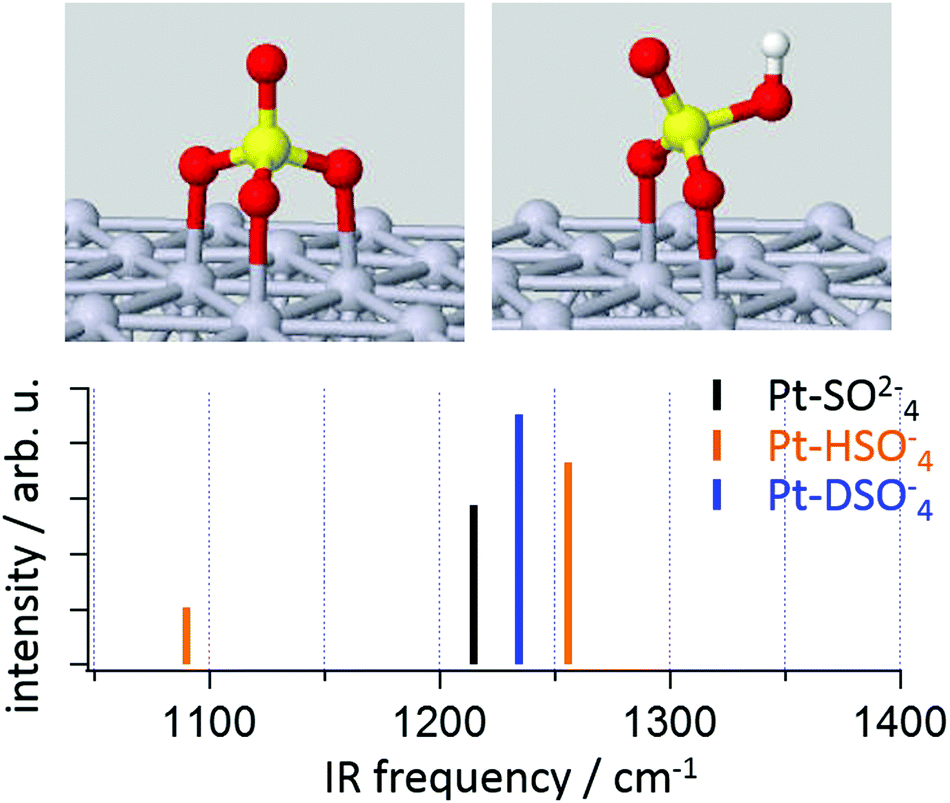 | ||
| Fig. 3 Vibrational normal modes for SO42−, HSO4− and DSO4− adsorbed on a Pt(111) surface calculated by DFT; top panel shows the optimized adsorption geometries for SO42− (top left) and HSO4− or DSO4− (top right) anions (no discernible structural difference was found between HSO4− and DSO4−). | ||
The conclusion of our current study is in agreement with that reached in thermodynamic analyses13,17 by Lipkowski, Feliu and coworkers and their SNIFTIRS study.23 However, these previous approaches are mostly indirect ways of predicting the chemical nature of the adsorbates. For instances: the thermodynamic analyses can measure the adsorption valence of the anions, which was found to be close to +2 at 0.45 V and decrease as the potential increase.13 However, as pointed out by Feliu, Lipkowski and coworkers, “Despite the values obtained (+2) the nature of the adsorbed species cannot be strictly established”.13 Infrared spectroscopic measurements with isotope exchange strategy, are in principle the most direct way to confirm the thermodynamic predictions. However, for this particular systems, implementing IRRAS or SNIFTIRS on the Pt(111) in D2O/D2SO4 system is challenged by the strong absorption of the solvent D2O bending mode at 1200 cm−1. On top of this, it is also rather difficult to quantitatively separate the spectral response of ions that are near, but only weakly interacting with the surface from those that strongly adsorb in SNIFTIRS. Because it is background free and interface-specific VSF spectroelectrochemistry avoids these correction steps.‡ Thus this study is the first to demonstrate that the bias dependent spectral response of the Pt(111)/H2SO4(H2O) and Pt(111)/D2SO4(D2O) are quantitatively identical between 0.42 and 0.9 V RHE and offers the clearest experimental evidence yet that only sulfate is present at the surface.
It is interesting to point out that the surface speciation of sulfuric acid differs markedly from the bulk. At 0.5 M H2SO4 HSO4− dominates the bulk liquid but SO42− is adsorbed: adsorption dramatically changes the equilibrium constant (Ka) of the HSO4− ⇌ H+ + SO42− reaction. Given the bulk pH close to 0 in our system, our measurement implies a pKa of HSO4− at the interface <0. In comparison the pKa of HSO4− in bulk is 2. This result points to the necessity of in situ interfacial characterization for understanding pH effects in such electrochemical systems as formic acid oxidation32–34 where reactants or products are deprotonateable. While we have observed a change in pKa of >2 for HSO4− on Pt(111) for 0.42 < E < 0.9 V RHE one might next ask whether this shift depends on bias. In this context it is interesting to note that both the current feature associated with anion adsorption (see Fig. 1a) and adsorbed sulfur (from radiolabelling studies12) appear at potentials below the appearance of the VSF signal. Electrochemical STM and operando X-ray reflectivity studies both suggest that at potentials positive of 0.42 a three fold coordinated adsorbate populates the surface (without insight into its chemical identity) and have, in conjunction with electrochemical and radiolabelling studies, been interpreted to suggest 0.42 V marks the transition from a configurationally flexible bidentate adsorbed HSO4− to a rigid, tridentate adsorbed SO42−.11,18,35 Recent electronic structure studies are consistent with this idea:20,30 from 0.35–0.41 V bidentate adsorbed bisulfate is predicted to dominate the surface, from 0.41–0.48 bidentate adsorbed HSO4− and SO42−⋯H3O+ ion pairs and above 0.48 V SO42−. Consistent with the relatively low intergrated charge transfer in the current spike, ∼8 μC cm−2, Jinnouchi et al. find that, while deprotonation occurs at 0.41 V the apparent current is dominated by a change in electrosorption valency.20
Given the adsorption of bisulfate on Pt(111) at potentials <0.42 V RHE one might ask why both previous and current VSF measurements show no resonant features at these potentials. This can be rationalized as a consequence of both the size of the transition dipole moment that perpendicular to the surface plane and the interaction strength of the anion with the substrate. Tridentate adsorbed sulfate exhibits much stronger transition dipole moment along the surface normal than that of the bidentate adsorbed bisulfate (see scheme in Fig. S5 of ESI†). In addition, the interaction of sulfate is much stronger with the electrode than that of the bisulfate, which changes significantly the polarizability as well as the permanent dipole moment of the anion as the potential increases. Such effect has been discussed in our previous work.19 It is worth noting that such effect can also explain the puzzling previous potential dependent IRRAS observations:10,21,22 while electrochemical measurements show no coverage change of anion at potential higher than 0.45 V, all reported infrared spectra show significant increase of the intensity as a function of the potential.
Our results are thus consistent with a scenario that the sulfuric acid derived anion in the potential range 0.3 < E < 0.42 is bidentate adsorbed bisulfate and in the range 0.42 < E < 0.9 tridentate adsorbed sulfate: our observation suggests the surface induces (de)protonation. While we, evidently cannot observe the bisulfate's vibrational modes, the current study undoubtedly reveals the chemical nature of the deprotonation reaction's product. Inspired by IRAS studies of bisulfate adsorption on low and high index Pt single crystals electrode,22 we currently pursue the simultaneous measurement of bi-sulfate and sulfate as a function of potential on a Pt(533) electrode.
3 Methods
Experiment
Potential dependent VSF spectra of sulfate anions adsorbed on Pt(111) surface were measured in a homebuilt thin-film spectroelectrochemical cell with the FHI VSF spectrometer. The system has been described elsewhere in detail.8,19,32 In brief, the Pt(111) working electrode (5 mm diameter, 3 mm thick, MaTeck) was flame annealed in a butane air flame, cooled in a water saturated N2 atmosphere, quenched in and covered with a droplet of Milli-Q water (after all 5 annealing steps) that was deaerated for 45 min. Quenching water and electrolyte were deaerated with N2 evaporated from liquid. Curved surface of the WE was wrapped in Teflon after flame annealing and transferred to the cell after the Teflon wrapping was rinsed thoroughly with Milli-Q water. A home-built reversible hydrogen electrode (RHE), a Pt wire in deaerated and H2(D2) saturated 0.5 M H2SO4(D2SO4), was used as a reference electrode.36 To allow sufficient IR light to reach the Pt surface we reduced the thickness of the electrolyte film to ≈1 μm (quantified by IR absorption). It is worth noting that there was some in-avoidable contamination in the spectroelectrochemical cell as shown in the CV from the in situ cell. We expect this to come from slow dissolution of the cell window: it is challenging to find relatively pure, IR transmissive materials that are also stable in strong acids for extended times. Nevertheless, as have been shown in previous studies by us and others,8,19,37 such low amount of contamination is exceedingly unlikely to affect interfacial structure sufficiently to alter the conclusion of the current study. Other information such as sources of chemicals, the cleaning procedures, the laser systems, the measurement geometry, the properties of the incidents laser beams (pulse energy, pulse durations, polarizations, foci) and the detection schemes were described in our previous paper19 and are summarized in the ESI.†Theory
First-principles electronic-structure calculations were performed using the FHI-aims package.38 The ion-electron interactions were described in an all-electron/full-potential treatment and the atomic zeroth order regular approximation (ZORA) was adopted to capture the effect of near-nuclear relativity on valence- and semicore-like electrons. The basis set employed used a set of numerical atom-centered orbitals (NAO) with the recommended “tight” setting. Density functional theory calculations were conducted in the generalized gradient approximation using the extended Perdew–Burke–Ernzerhof (xPBE) functional,39 which has been demonstrated to retain the accuracy of PBE40 for H-bond interactions but substantially improve the description of atomization energies of small molecules. The force convergence criterion in the self-consistency cycle was set to 10−5 eV Å−1. Together with the maximum residual force criterion of 10−4 eV Å−1 in the geometry relaxation procedure and a finite displacement of 0.002 Å for calculating the Hessian matrix, it ensures an accurate vibrational calculation by finite differences within FHI-aims.To model the Pt(111) surface, we extracted 55 Pt atoms from a five-layer slab with the experimental lattice constant of 3.92 Å. Except for the six platinum atoms that are closest to the adsorbed species, all the other platinum atoms were fixed in the calculations of geometry relaxation and vibration. We showed surface charging to have a small effect on calculated vibrational frequencies by comparing the results obtained from the neutral adsorption model of SO4 on the Pt(111) cluster and a model with an overall charge of −2 (see ESI†). Because differences in relative frequencies between models are minimal, and measurements of electrosorption valency find the adsorbed sulfuric acid derived anion is fully discharged, the calculated frequencies reported in the manuscript are based on neutral adsorption models.
4 Conclusions
In summary, this study demonstrates that, for one well studied system, the application of VSF spectroscopy, isotope exchange measurements, and theory provides unambiguous insight into the protonation state of surface adsorbates. Resolving the chemical nature of anions adsorbed on electrocatalysts is a prerequisite both of a mechanistic understanding of the electrolyte dependence of electrocatalytic reactions and in the operando characterization of reactants and products. However, due to the structural similarities of adsorbates and their derivatives in bulk solution, and the spectral overlap of water or deuterated water's bend mode with vibrational features of many common electrolytes (e.g. S–O and Cl–O modes) and reactants/products (e.g. carbonyl, carboxyl and ester related modes) unambiguous characterization of any such adsorbate via its vibrational response can be extremely challenging. The approach we describe should prove of general applicability in resolving electrolyte structure and chemical speciation in electrocatalytic reactions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
IYZ is grateful to the support from the 14th Recruitment Program of Young Professionals in China. We thank Professor Daniel Scherson, Professor Rolf Schuster and Professor Maki Kawai for valuable discussions. This study was supported by the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (grant agreement no 772286, to RKC). Open Access funding provided by the Max Planck Society.Notes and references
- D. Strmcnik, M. Escudero-Escribano, K. Kodama, V. R. Stamenkovic, A. Cuesta and N. M. Marković, Nat. Chem., 2010, 2, 880 CrossRef CAS PubMed.
- I. Katsounaros and M. T. Koper, in Electrochemical Science for a Sustainable Society, ed. K. Uosaki, Spring International Publishing, 2017, ch. Electrocatalysis for the Hydrogen Economy, pp. 23–50 Search PubMed.
- D. Gao, I. T. McCrum, S. Deo, Y.-W. Choi, F. Scholten, W. Wan, J. G. Chen, M. J. Janik and B. Roldan Cuenya, ACS Catal., 2018, 10012–10020 CrossRef CAS.
- T. Shinagawa, M. T.-K. Ng and K. Takanabe, ChemSusChem, 2017, 10, 4155–4162 CrossRef CAS.
- J. Drnec, D. A. Harrington and O. M. Magnussen, Curr. Opin. Electrochem., 2017, 4, 69–75 CrossRef CAS.
- M. C. Figueiredo, D. Hiltrop, R. Sundararaman, K. A. Schwarz and M. T. M. Koper, Electrochim. Acta, 2018, 281, 127–132 CrossRef CAS.
- M. Favaro, B. Jeong, P. N. Ross, J. Yano, Z. Hussain, Z. Liu and E. J. Crumlin, Nat. Commun., 2016, 7, 12695 CrossRef CAS.
- Y. Tong, F. Lapointe, M. Thaemer, M. Wolf and R. Campen, Angew. Chem., Int. Ed., 2017, 56, 4211–4214 CrossRef CAS.
- J. Clavilier, R. Faure, G. Guinet and R. Durand, J. Electroanal. Chem. Interfacial Electrochem., 1980, 107, 205–209 CrossRef CAS.
- P. Faguy, N. Marinković and R. Adžić, Langmuir, 1996, 12, 243–247 CrossRef CAS.
- A. Funtikov, U. Stimming and R. Vogel, J. Electroanal. Chem., 1997, 428, 147–153 CrossRef CAS.
- A. Kolics and A. Wieckowski, J. Phys. Chem. B, 2001, 105, 2588–2595 CrossRef CAS.
- E. Herrero, J. Mostany, J. Feliu and J. Lipkowski, J. Electroanal. Chem., 2002, 534, 79–89 CrossRef CAS.
- T. Pajkossy and D. Kolb, Electrochim. Acta, 2008, 53, 7403–7409 CrossRef CAS.
- Y. Tong, L. Lu, Y. Zhang, Y. Gao, G. Yin, M. Osawa and S. Ye, J. Phys. Chem. C, 2007, 111, 18836–18838 CrossRef CAS.
- B. Braunschweig, P. Mukherjee, D. Dlott and A. Wieckowski, J. Am. Chem. Soc., 2010, 132, 14036–14038 CrossRef CAS PubMed.
- N. Garcia-Araez, V. Climent, P. Rodriguez and J. Feliu, Langmuir, 2010, 26, 12408–12417 CrossRef CAS PubMed.
- T. Kondo, T. Masuda, N. Aoki and K. Uosaki, J. Phys. Chem. C, 2016, 120, 16118–16131 CrossRef CAS.
- G. Zwaschka, M. Wolf, R. K. Campen and Y. Tong, Surf. Sci., 2018, 678, 78–85 CrossRef CAS.
- R. Jinnouchi, T. Hatanaka, Y. Morimoto and M. Osawa, Phys. Chem. Chem. Phys., 2012, 14, 3208–3218 RSC.
- Y. Shingaya and M. Ito, J. Electroanal. Chem., 1999, 467, 299–306 CrossRef CAS.
- N. Hoshi, A. Sakurada, S. Nakamura, S. Teruya, O. Koga and Y. Hori, J. Phys. Chem. B, 2002, 106, 1985–1990 CrossRef CAS.
- Z. Su, V. Climent, J. Leitch, V. Zamlynny, J. Feliu and J. Lipkowski, Phys. Chem. Chem. Phys., 2010, 12, 15231–15239 RSC.
- Y. R. Shen, Nature, 1989, 337, 519–525 CrossRef CAS.
- L. J. Richter, T. P. Petralli-Mallow and J. C. Stephenson, Opt. Lett., 1998, 23, 1594–1596 CrossRef CAS.
- M. T. Koper and J. J. Lukkien, J. Electroanal. Chem., 2000, 485, 161–165 CrossRef CAS.
- C. Nishihara and H. Nozoye, J. Electroanal. Chem., 1994, 379, 527–530 CrossRef.
- M. Weaver and S. Wasileski, Langmuir, 2001, 17, 3039–3043 CrossRef CAS.
- A. Ge, P. E. Videla, G. L. Lee, B. Rudshteyn, J. Song, C. P. Kubiak, V. S. Batista and T. Lian, J. Phys. Chem. C, 2017, 121, 18674–18682 CrossRef CAS.
- J. A. Santana, C. R. Cabrera and Y. Ishikawa, Phys. Chem. Chem. Phys., 2010, 12, 9526–9534 RSC.
- K.-Y. Yeh, N. Restaino, M. Esopi, J. Maranas and M. Janik, Catal. Today, 2013, 202, 20–35 CrossRef CAS.
- Y. Tong, K. Cai, M. Wolf and R. Campen, Catal. Today, 2016, 260, 66–71 CrossRef CAS.
- H. Jeon, B. Jeong, J. Joo and J. Lee, Electrocatalysis, 2015, 6, 20–32 CrossRef CAS.
- J. Joo, T. Uchida, A. Cuesta, M. T. M. Koper and M. Osawa, J. Am. Chem. Soc., 2013, 135, 9991–9994 CrossRef CAS PubMed.
- O. Magnussen, Chem. Rev., 2002, 102, 679–726 CrossRef CAS PubMed.
- P.-O. Eggen, J. Chem. Educ., 2009, 86, 352 CrossRef CAS.
- P. Faguy, N. Markovic, R. Adzic, C. Fierro and E. Yeager, J. Electroanal. Chem. Interfacial Electrochem., 1990, 289, 245–262 CrossRef CAS.
- V. Blum, R. Gehrke, F. Hanke, P. Havu, V. Havu, X. Ren, K. Reuter and M. Scheffler, Comput. Phys. Commun., 2009, 180, 2175–2196 CrossRef CAS.
- X. Xu and W. A. Goddard, J. Chem. Phys., 2004, 121, 4068–4082 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details of the electrochemistry and VSF spectroscopy, full potential dependent series of VSF spectra of Pt(111) in D2SO4/D2O solution, CV collected in the measurement position of the spectroelectrochemical cell, discussion of the minimal effect of IR absorption on the measured spectra in D2O, calculated frequencies from uncharged absorption models and orientation of the transition dipole moment. See DOI: 10.1039/c9cp03397a |
| ‡ As we discuss in more detail in the ESI,† for this system bulk absorption of incident IR photons on the way to the sample changes only the intensity, and not the spectral shape, of the interfacial response. |
| This journal is © the Owner Societies 2019 |