
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Using photoelectron spectroscopy to observe oxygen spillover to zirconia†
Peter
Lackner
 ,
Zhiyu
Zou
,
Sabrina
Mayr
,
Ulrike
Diebold
and
Michael
Schmid
*
,
Zhiyu
Zou
,
Sabrina
Mayr
,
Ulrike
Diebold
and
Michael
Schmid
*
Institute of Applied Physics, TU Wien, 1040 Vienna, Austria. E-mail: schmid@iap.tuwien.ac.at; Fax: +43 1 58801 13499; Tel: +43 1 58801 13401
First published on 29th July 2019
Abstract
X-ray photoelectron spectroscopy (XPS) of five-monolayer-thick ZrO2 films reveals a core level binding energy difference of up to 1.8 eV between the tetragonal and monoclinic phase. This difference is explained by positively charged oxygen vacancies in the tetragonal films, which are slightly reduced. Due to the large band gap of zirconia (≈5–6 eV), these charges shift the electron levels, leading to higher binding energies of reduced tetragonal films w.r.t. fully oxidized monoclinic films. These core level shifts have the opposite direction than what is usually encountered for reduced transition metal oxides. The vacancies can be filled via oxygen spillover from a catalyst that enables O2 dissociation. This can be either a metal deposited on the film, or, if the film has holes, the metallic (in our case, Rh) substrate. Our study also confirms that tetragonal ZrO2 is stabilized via oxygen vacancies and shows that the XPS binding energy difference between O 1s and Zr 3d solely depends on the crystallographic phase.
1 Introduction
The spillover of oxygen from a metal or oxide onto zirconia (ZrO2) is of immense importance in many applications, as ZrO2 is used heavily as an oxygen-conducting electrolyte1 in solid oxide fuel cells2 and gas sensors.3 Furthermore, this process is of interest in catalysis, where zirconia is sometimes used as a catalyst, but mostly as support;4 oxygen spillover and reverse spillover would enable Mars–van-Krevelen-type reactions.5 Especially when using (yttria-stabilized) zirconia as a solid-state electrolyte, its electronic structure and band alignment are crucial for its performance.While in recent years, zirconia has been studied increasingly using surface science methods, the details of oxygen incorporation have not yet been tackled due to a lack of reliable model systems that mimic bulk properties reasonably well. The main difficulty of such studies is the lack of electronic conductivity: zirconia (pure or yttria-stabilized) has a band gap of 5–6 eV.6,7 For analysis methods involving charged particles, one has to rely on thin films to circumvent charging of the material. Only then, surface science techniques such as scanning tunneling microscopy (STM), low-energy electron diffraction (LEED), or X-ray photoelectron spectroscopy (XPS) can be used. In the last years, many studies have focussed on ultrathin zirconia films,8–10 which has helped to gain insights into molecular adsorption11,12 and metal growth.13 However, for investigating oxygen deficiency and related phenomena, thicker films are required. Inspired by work of Maurice et al.14 and Meinel et al.,15–17 but taking advantage of sputter deposition in UHV,18 we have recently demonstrated the reliable and reproducible growth of several-monolayer-thick films of bulk-like zirconia.19 Depending on the post-annealing temperature, films of both, the tetragonal or monoclinic structure can be prepared. Annealing five-monolayer-thick zirconia films at temperatures ≤730 °C yields flat, closed films; the structure of these films was identified as tetragonal by STM and LEED [(2 × 1) w.r.t. cubic ZrO2(111)].19 When annealing at T ≥ 820 °C, the films fully transform to monoclinic zirconia. In the same temperature range where the tetragonal → monoclinic transformation occurs, the films dewet the Rh(111) substrate, which becomes accessible within holes of the film. The monoclinic surface structure resembles a distorted (2 × 2) structure w.r.t. cubic ZrO2(111); this makes the two structures easily distinguishable in STM, and the monoclinic distortion leads to a clear splitting of the spots in LEED.19 The surfaces of both, the tetragonal and monoclinic films, appear essentially bulk-terminated.
Building on these results, in this work we present a thorough XPS study of these structurally well-defined 5 ML-thick zirconia films. Unexpectedly large differences of the XPS binding energies between tetragonal and monoclinic films are shown in Section 3.1. In Section 3.2, commonly-encountered reasons for such differences are discussed and excluded as main effects, and reduction of the tetragonal films is revealed as the true reason; surprisingly, the reduced tetragonal films show a higher binding energy – in contrast to what is expected from usual XPS interpretations. Reduction of tetragonal zirconia films is confirmed in Section 3.3 by a test experiment with direct observation of oxygen spillover from Rh on to the thin zirconia films, and valence-band spectra are discussed in Section 3.4. Finally, in Section 3.5, the experimental results are discussed in the light of previous theoretical findings.
2 Experimental methods
The two-chamber UHV system used in this work was described earlier.19 The Rh(111) single crystal used as a substrate (diameter 9 mm, height 2 mm, from MaTecK, Germany) was cleaned in the preparation chamber (pbase < 1 × 10−10 mbar) by repeated sputter/anneal cycles. A UHV-compatible sputter source used for Zr deposition18 was mounted in the preparation chamber. XPS measurements were conducted in the analysis chamber (pbase = 7 × 10−11 mbar) at room temperature (RT) using a Specs Phoibos 100 hemispherical analyzer (pass energy = 16 eV, 5-channel detection, energy calibrated with pure-metal Ag 3d5/2, Ag M4NN and Au 4f7/2) and near-normal emission (15° off-normal). Non-monochromatized Mg Kα radiation was used for excitation (VG XR3E2 source operated at 15 kV, 15 mA emission). The use of a flood gun was not necessary for charge compensation, as the thin zirconia films used in this work showed no charging effects, see below. The program CasaXPS was used for peak deconvolution and background subtraction. For Zr 3d, a Shirley-type background, and for O 1s, a linear background was used. The following parameters were used for the Zr 3d doublets: ΔE = 2.4 eV, ratio of areas 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.6920 (tested over several ZrO2 systems). The binding energies for Zr given in the following are the Zr 3d5/2 peak positions. The line shape was set to the Gaussian-Lorentzian product function “GL(60)” (60% Lorentzian) for all peaks except for the Zr 3d interface peaks, which behave similarly to ultrathin films9 and thus appear metallic; they are therefore fitted with an additional exponential tail “GL(60)T(1.8)”.
:0.6920 (tested over several ZrO2 systems). The binding energies for Zr given in the following are the Zr 3d5/2 peak positions. The line shape was set to the Gaussian-Lorentzian product function “GL(60)” (60% Lorentzian) for all peaks except for the Zr 3d interface peaks, which behave similarly to ultrathin films9 and thus appear metallic; they are therefore fitted with an additional exponential tail “GL(60)T(1.8)”.
The preparation of the zirconia films is described in detail elsewhere.19 In short, the UHV sputter source18 is used to deposit Zr at RT in a mixed Ar/O2 atmosphere (pO2 = 1 × 10−6 mbar, pAr = 8 × 10−6 mbar in the UHV chamber, ≈27× higher Ar pressure inside the source). Due to the good reproducibility of the deposition rate, the film thickness was simply determined by the deposition time (after initial calibration18,19); we define one monolayer (1 ML) as one O–Zr–O repeat unit of tetragonal ZrO2(101) or monoclinic ZrO2(![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 11), which corresponds to ≈9 × 1018 Zr atoms per m2 or ≈0.3 nm thickness. After deposition, the films are post-annealed at various temperatures for 10 min in pO2 = 5 × 10−7 mbar to achieve good order and to oxidize them to the extent possible at these conditions (tetragonal films stay slightly substoichiometric, see below). After annealing, the O2 pressure was kept constant until the sample temperature reached 300 °C. The thinnest film showing a bulk-terminated surface structure was found to be 5 ML for tetragonal films.19 We have therefore used films with 5 ML thickness unless noted otherwise. As mentioned above, during the tetragonal → monoclinic transformation, the film breaks and holes appear (dewetting). The material from the holes forms additional layers on the rest of the film. Here we report nominal film thicknesses (of the deposited material), thus the actual thickness of the monoclinic films is higher. For a 5 ML-thick preparation, one additional layer typically covers 35–50% of the area, depending on the size of the holes (which depends on the annealing temperature). The structure of all films was checked using STM and LEED, which are both available in the analysis chamber of the UHV system.19
11), which corresponds to ≈9 × 1018 Zr atoms per m2 or ≈0.3 nm thickness. After deposition, the films are post-annealed at various temperatures for 10 min in pO2 = 5 × 10−7 mbar to achieve good order and to oxidize them to the extent possible at these conditions (tetragonal films stay slightly substoichiometric, see below). After annealing, the O2 pressure was kept constant until the sample temperature reached 300 °C. The thinnest film showing a bulk-terminated surface structure was found to be 5 ML for tetragonal films.19 We have therefore used films with 5 ML thickness unless noted otherwise. As mentioned above, during the tetragonal → monoclinic transformation, the film breaks and holes appear (dewetting). The material from the holes forms additional layers on the rest of the film. Here we report nominal film thicknesses (of the deposited material), thus the actual thickness of the monoclinic films is higher. For a 5 ML-thick preparation, one additional layer typically covers 35–50% of the area, depending on the size of the holes (which depends on the annealing temperature). The structure of all films was checked using STM and LEED, which are both available in the analysis chamber of the UHV system.19
3 Results and discussion
3.1 XPS of tetragonal and monoclinic films
XPS measurements of both, tetragonal and monoclinic 5 ML-thick films are shown in Fig. 1. The films were annealed in O2 at 670 and 810 °C, respectively. The Zr 3d5/2 binding energies of tetragonal (182.9 eV) and monoclinic films (181.6 eV) differ by 1.3 eV. In the O 1s region, this difference is 1.1 eV (530.6 eV and 529.5 eV, respectively). A closer look at the Zr 3d signal of the tetragonal film, see Fig. 1a, reveals a shoulder at the low-binding-energy side. The spectrum can be fitted with two doublets - the larger one at 182.9 eV, the smaller at 180.9 eV. As the smaller signal decreases with increasing thickness, see the inset of Fig. 1a, we assign it to the interface layer bonded to the Rh substrate. This low-binding-energy feature appears in the same energy range as the Zr 3d signal of ultrathin zirconia films on Pt3Zr(0001) (180.7 eV).9 It was already predicted by density functional theory (DFT) that the interface layer of few-ML-thick ZrO2 films would have a distinctively different binding energy (EB) from the rest of the film, close to the value of ultrathin zirconia.9 For 5 ML films, the area of the interface doublet is between 8% and 10% of the total Zr 3d intensity, depending on the exact preparation; this is somewhat less than the expected 14% from an attenuation simulation with the SESSA code.21 The interface peak is discussed in more detail in the ESI.†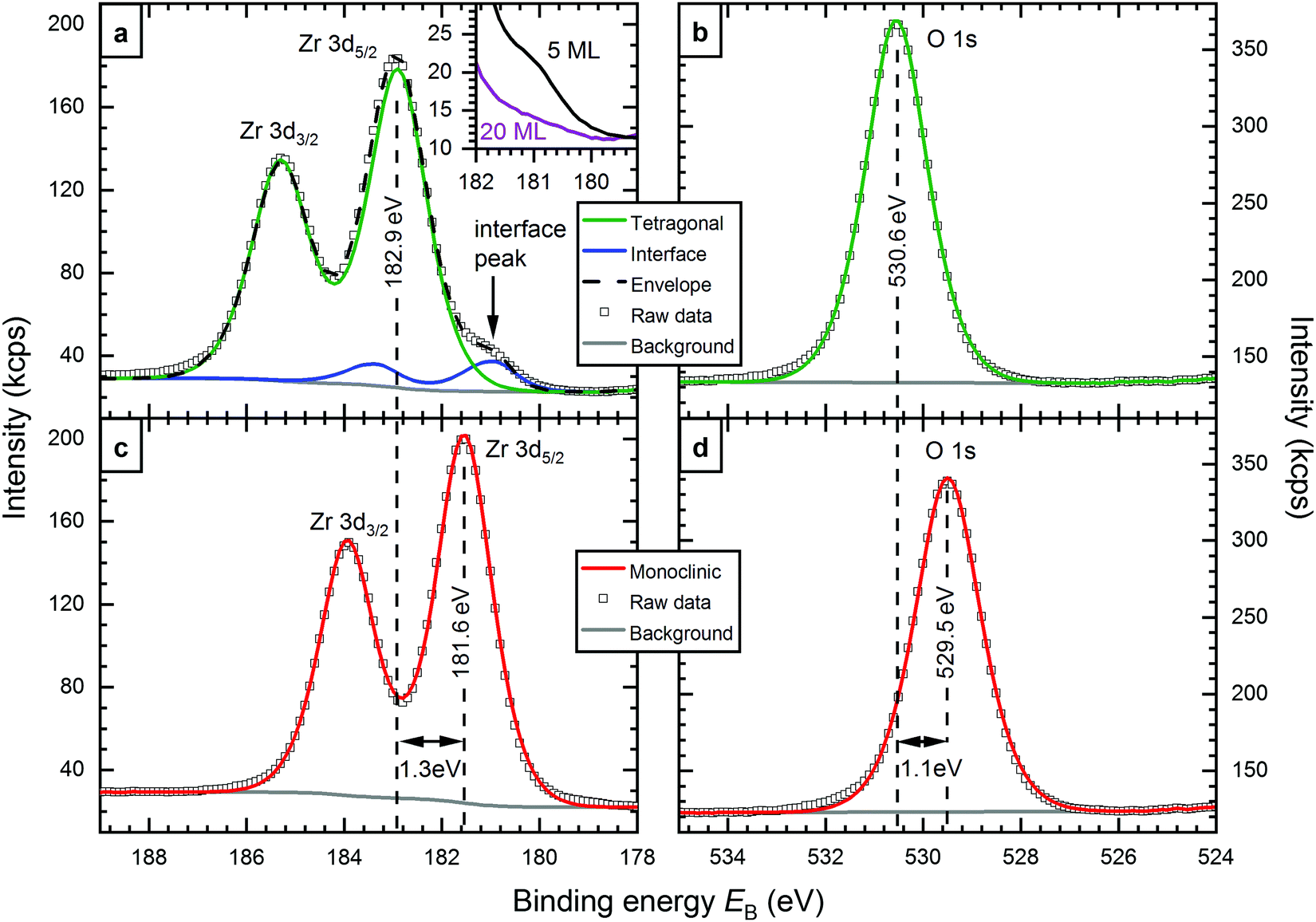 | ||
| Fig. 1 XPS spectra of (a and b) tetragonal and (c and d) monoclinic zirconia films: All spectra can be fitted with one component, except for Zr 3dtetragonal, which features a shoulder resulting from the interface layer of the film. The main signal shows a ΔEB between monoclinic and tetragonal ZrO2 of 1.3 eV in Zr 3d and 1.1 eV in O 1s. The inset of (a) shows the interface peak for a 5 ML (black) and a 20 ML-thick (violet) film. The interface peak is strongly attenuated at 20 ML thickness. | ||
The Zr 3d peak of monoclinic films shows no shoulder, and can be fitted with only one doublet. It is likely that an interface peak also exists in this case, but we cannot resolve it experimentally, as the main peak is much closer to the energy of the interface peak. For the O 1s region, where the differences between the interface and main signal are even smaller,9 one peak was sufficient for a good fit in both cases, tetragonal and monoclinic.
The Zr 3d and O 1s binding energies depend on the exact preparation parameters, i.e. O2 partial pressures during deposition and annealing, annealing temperature, and film thickness. For monoclinic films, a small spread of 0.2 eV was encountered (181.6–181.8 eV). Tetragonal films show binding energies in a larger range, between 182.6 eV and 183.4 eV; higher annealing temperatures lead to higher EB.
As mentioned above, typical monoclinic films contain holes reaching down to the substrate, in contrast to tetragonal films created with the standard preparation parameters. Using different sputter deposition parameters however,19 we can also create tetragonal films that break up and form holes when annealing to 670 °C, thus the Rh(111) substrate is exposed. These films show a lower EB than usual (182.1 eV); the EB difference with respect to monoclinic ZrO2 is only 0.5 eV for Zr 3d and 0.3 eV for O 1s (EB = 529.9 eV). Another preparation method for tetragonal films with holes starts with a monoclinic film (containing holes). By annealing at 920 °C in UHV, the film can be transformed back to the tetragonal structure; also this film does not fully cover the substrate. We attribute this transformation to reduction of the film, i.e. the formation of oxygen vacancies, which stabilize the tetragonal phase.22 After annealing this film at 610 °C in 5 × 10−7 mbar O2, the Zr 3d levels again exhibit a binding energy of 182.1 eV. In both preparations, the Rh surface in the holes is covered by a (2 × 1)-O superstructure as is usual for Rh annealed in O2 at these conditions.23,24
Additionally, elemental ratios can be extracted from XPS measurements. Here, one has to act with caution however, as truly quantitative XPS results are difficult to achieve. The main reason for this is that the transmission function (sensitivity over kinetic energy) of typical XPS analysers is not known in detail, and the attenuation of photoelectrons also depends on their kinetic energy. Therefore, comparing peak areas several hundred eV apart – as in the case of Zr 3d and O 1s – is only possible if a trustworthy reference is available. This is not the case for zirconia; pure bulk single crystals are not available, and the surface stoichiometry and composition of thick films or powders are not known. We therefore only compare tetragonal and monoclinic films. To compensate for the difference in film thickness (monoclinic films dewet the substrate and become thicker as a result) and the signal of O adsorbed on the uncovered Rh substrate (if any), we resort to XPS simulations with the program SESSA.21 These simulations are based on the morphology determined by STM. Then, the different preparations can be compared with one another. All 5 ML-thick films show similar Zr:O ratios; the variations lie within ±2%. These variations are within the error bars of our analysis.
3.2 Possible reasons for differences in binding energy
Let us focus on the large difference of ≈1.3 eV between the Zr 3d levels of the tetragonal and monoclinic films. This section first presents explanations that are commonly encountered in XPS studies, but can be excluded for the shifts encountered in this work. Finally, oxygen vacancies in the tetragonal film are presented as the favoured explanation.At first glimpse, the large ΔEB is surprising, as tetragonal and monoclinic zirconia are not vastly different; in both, Zr is present solely in the 4+ state. The same can be assumed for the majority of the Zr atoms in thin films, as the Zr:O ratio is nearly identical in both films. Thus, different Zr oxidation states can be excluded as a reason for differences of the films. However, tetragonal zirconia features eightfold-coordinated (8c) Zr and fourfold-coordinated (4c) O,25 while in the monoclinic structure, Zr is 7c and O is half 4c and half 3c; yet from such a coordination difference no large impact on the binding energy is expected.
A further possible difference is the band gap: according to ab initio calculations, the band gap of tetragonal ZrO2 is larger than that of monoclinic ZrO2 by 0.5–1.0 eV, with the more recent works favouring values at the lower end of this range.7,26 However, in an experimental comparison of monoclinic and Y-stabilized tetragonal zirconia, the monoclinic band gap was found to be larger by 0.05–0.5 eV.6 This disagreement makes a prediction for the band gap of the thin films difficult, but an upper limit of 1 eV can be used for an upper estimate of the influence of the band gap on the EB difference. Assuming the Fermi energy is in the middle of the band gap, half of the band gap difference – up to 0.5 eV – could be expected as an increase of the binding energy of XPS, see Fig. 2b. For the Fermi level at the conduction band edge (n-doped material), core level shifts equal to the band-gap differences would be conceivable. Only the largest values of the theoretically predicted band-gap differences (which we do not consider very realistic) would explain a substantial part of the EB difference.
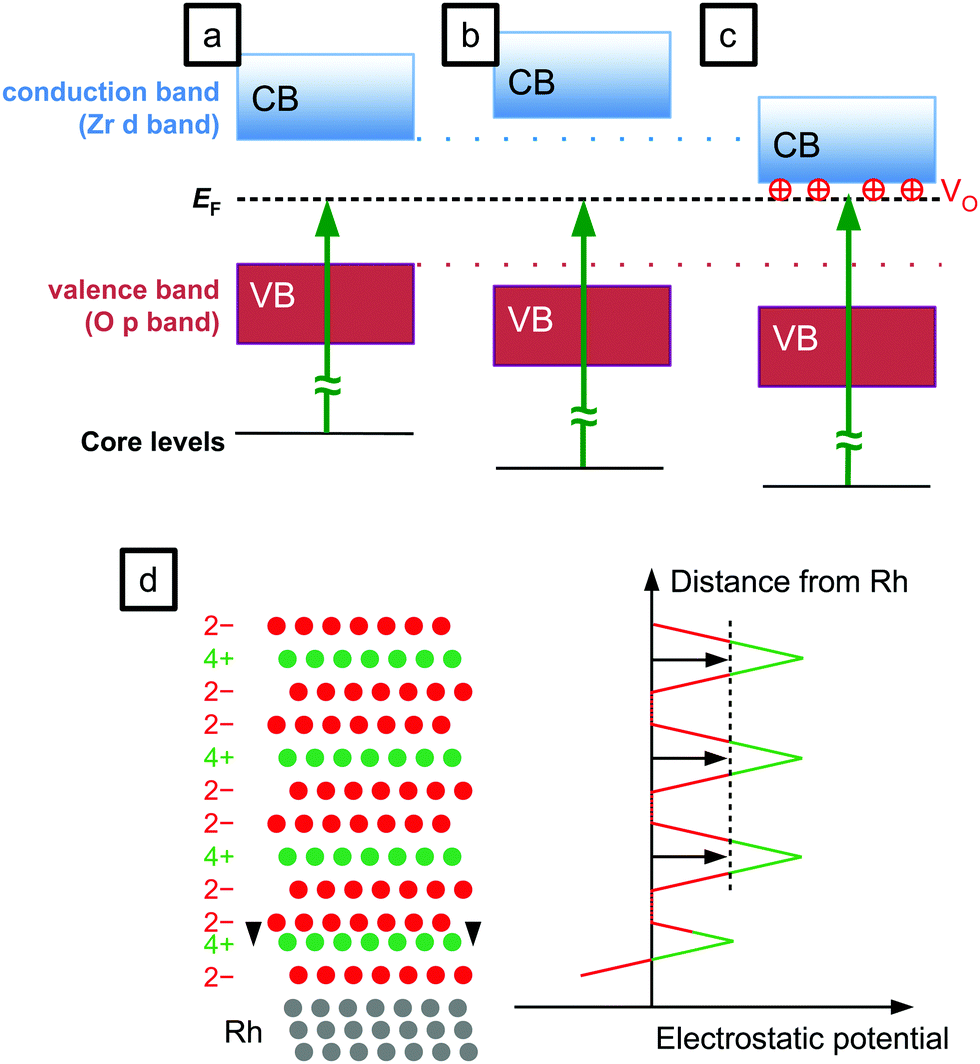 | ||
| Fig. 2 Sketch of the different possibilities for changes of the core level binding energy. (a) Reference (no change), (b) larger band gap, (c) bands shifted by positive oxygen vacancies, and (d) change of an interlayer distance. | ||
In a previous XPS study, charging was suggested as an explanation for XPS shifts of ZrO2 films.27 Although zirconia is an insulating material, the possibility of STM measurements on up to 10 ML-thick films19 excludes charging during XPS measurements: If any charging would occur, in an STM measurement it would be stronger by orders of magnitude due to the much higher current density (STM: nA nm−2, XPS: nA mm−2), thus rendering STM impossible. Charging can be excluded also for film thicknesses above 10 ML, because LEED measurements did not show any indications of charging at current densities in the μA mm−2 range. Also, changing the incident X-ray flux did not shift EB, and applying −10 V to the sample (to reduce neutralization via secondary electrons from the sample holder) only led to the expected voltage-induced shift. No time dependence of EB was found.
Further explanations for the EB difference may be based on a different structure at the interface. Measurements presented in ref. 19 indicate that the tetragonal film is stabilized by slight ion bombardment occurring during sputter deposition. This could lead to intermixing between Rh and ZrO2. Assuming that this induces a structural change with suitable changes of the interlayer distances, the resulting electrostatic potential could shift the states of the film in the higher layers to higher binding energies, see Fig. 2d. Then, the second oxygen plane would have to be ≈15 pm, or 20%, closer to the Zr plane below, to shift the potential of the layers above by 1 eV. (This calculation is based on a relative permittivity of εr = n2 = 4.8, estimated from the refractive index n, i.e. taking only the polarizability of the electrons into account, with the ion positions frozen.) Such a large change of interlayer distances is unlikely. When related to Rh–ZrO2 intermixing, this effect would depend on the intensity of ion bombardment during the deposition. We have modified the flux and energies of the ions impinging on the surface by varying the grid voltages of the sputter source18 and did not find an effect on the binding energy. Furthermore, intermixing at the interface would not explain the increased binding energy measured when annealing tetragonal films at higher temperatures in O2. The opposite behaviour would then be expected, as Rh and ZrO2 would phase separate due to the higher oxygen affinity of Zr.
The large band gap of zirconia leads to large shifts of the levels when the Fermi level gets pinned by gap states, or in the presence of electric fields caused e.g. by electronic doping, see Fig. 2c. A careful study of the cleanliness of the films grown by our sputter source revealed no contaminations that could act as dopants.18 The films can however be doped by oxygen vacancies (VOs). Strong reduction with stoichiometry changes ≳2% in the near-surface region can be excluded from XPS measurements showing roughly the same Zr:O ratio for both monoclinic and tetragonal ZrO2. However, a slight reduction would be enough to induce a shift of 1–2 eV due to the lack of screening charges in an insulator such as zirconia. A slightly reduced tetragonal film compared with a fully oxidized monoclinic film can therefore explain the differences measured with XPS. This mechanism was suggested previously for EB shifts induced by reduction of perovskites28 and ceria29 at near-ambient pressures. The suggestion of VOs in tetragonal ZrO2 is in agreement with the literature; VOs are the main candidates for explaining the stability of the tetragonal phase in powders,22 and in a previous work we have also suggested that VOs could explain the band bending effects observed in STM images of monoclinic ZrO2 films, and the lack thereof for tetragonal zirconia films.19
If the core level shift is mainly due to reduction, tetragonal films with a binding energy around 182 eV – less than usual 182.6–183.4 eV – are less reduced or not reduced at all. Both preparations with such a low EB share one similarity: these films are not continuous, but have holes reaching down to the Rh(111) substrate, which is a good catalyst for O2 dissociation.30 When annealing in O2, the accessibility of the catalyst will lead to oxidation of the film.
3.3 Oxidation of tetragonal zirconia by spillover from Rh clusters
A test experiment was designed to confirm that the (slightly) reduced state of the tetragonal films is the reason for the large EB difference between tetragonal and monoclinic core levels. The experiment is sketched in the top inset of Fig. 3b. A standard 5 ML tetragonal film was prepared (Tanneal = 630 °C, pO2 = 5 × 10−7 mbar). Using STM and LEED, we confirmed that it had no holes reaching to the substrate. XPS showed the Zr 3d5/2 level at 183.4 eV, see Fig. 3a. (Here, EB is somewhat higher than usual for tetragonal films, which we relate to a slightly lower pO2 during sputter deposition, 5 × 10−7 mbar.) Then, 1.6% of a monolayer of Rh (1.4 × 1013 cm−2) was deposited on top of the zirconia film, yielding single atoms or small clusters visible in STM (yellow circles in the lower inset of Fig. 3b). This yielded a small EB shift of −0.1 eV. After another annealing step at a lower temperature (T = 610 °C, pO2 = 5 × 10−7 mbar), the Zr 3d core levels shifted by 1.1 eV to 182.3 eV. The Zr:O ratio remained constant within the error bars of ±2%. The structure was checked by STM and LEED and a tetragonal film with only few areas transformed to the monoclinic structure was found. This is however a minority (according to STM, less than 7% of the surface) and therefore cannot explain the substantial shift of the XPS peak. Thus, oxidation by oxygen spillover from Rh reduces the EB difference between the tetragonal film and typical values for the monoclinic film from 1.8 eV to 0.7 eV. This remaining difference most likely originates from remaining non-stoichiometry, and, to a lesser degree, from the different band gap and different structure, as discussed in the previous section. The O 1s peak shifts by the same energy as Zr 3d, confirming our model of a shift of all bands with respect to the Fermi level.
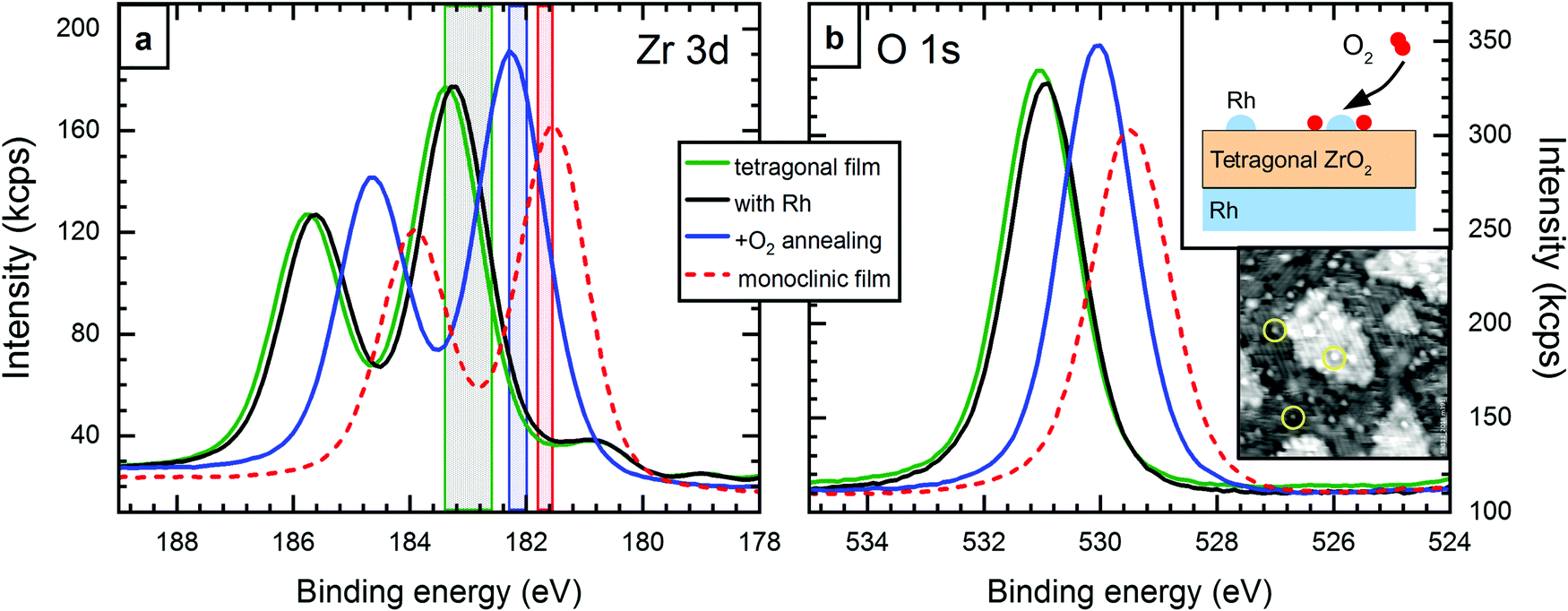 | ||
| Fig. 3 XPS results for oxidation of 5 ML-thick tetragonal films by spillover. (a) Zr 3d and (b) O 1s levels of a closed, tetragonal film before (green) and after (black) deposition of 0.016 ML Rh, and after annealing in O2 at 610 °C (blue). By annealing in the presence of Rh, the main peaks shift by 1.1 eV to 182.3 eV. The signal of a monoclinic film is shown as reference (red, dashed). The colored areas in (a) show the energy range of different preparations. The oxidized, tetragonal range (blue) includes broken tetragonal films, see Section 3.1. Top inset in (b): sketch of the setup of this experiment. Bottom inset: STM image of deposited Rh (yellow circles) on tetragonal ZrO2. 27 × 27 nm2, Vbias = +3.7 V, I = 70 pA. | ||
3.4 Valence band spectroscopy
Valence band (VB) photoelectron spectroscopy measurements further corroborate the explanation of the EB difference. For 5 ML-thick films, the XPS intensity in the valence band region is dominated by the Rh substrate, because the Rh 4d photoelectron cross section is much higher than that of oxygen, which forms the VB of ZrO2.20 Thus, the band gap of these ZrO2 films and the position of its valence band cannot be measured directly; the signal seen in Fig. 4a for a 5 ML film (red) stems mainly from Rh and, hence, reaches up to the Fermi level (EB = 0). We have therefore prepared a 50 ML-thick tetragonal film (annealed at 670 °C in pO2 = 5 × 10−7 mbar for 100 min; no difference to tanneal = 10 min). This film is substantially more oxygen-deficient than the thinner films; a comparison with an oxidized 5 ML-thick film gives a stoichiometry of ZrO1.83 for the tetragonal 50 ML-thick film. In continuation of the trend of higher EB with stronger reduction, this highly reduced film also shows the highest EB ever observed by us (Zr 3d5/2 at 183.7 eV). The large nonstoichiometry of the 50 ML film is probably due to the fact that oxidation is increasingly hindered with increasing film thickness; charge transfer through the film would be needed to activate O2.31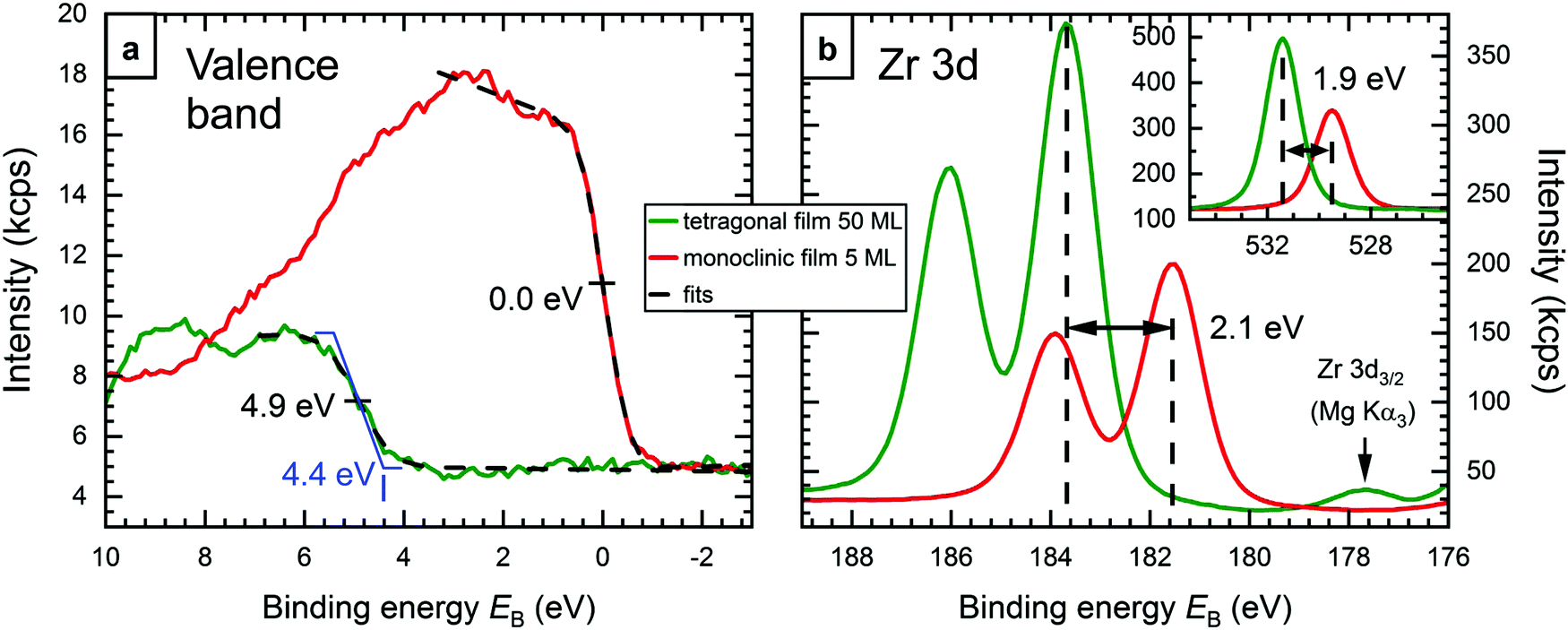 | ||
| Fig. 4 XPS measurements of a 50 ML-thick tetragonal zirconia film (green), and a 5 ML-thick monoclinic zirconia film for comparison (red). (a) Valence band region. The trapezoidal shape of the VB of the 50 ML film shown in blue results in the experimental spectrum when broadened by the instrument resolution. This indicates a VB edge at EB ≈ 4.4 eV. For the 5 ML film, the cutoff is at 0.0 eV, as expected, since the signal is dominated by the Fermi edge of the Rh substrate (see text). (b) Zr 3d region of the two films, which show a binding energy difference of 2.1 eV. (inset) The O 1s region shows a slightly smaller difference, 1.9 eV. The 50 ML data were normalized to a matching intensity at low EB values. | ||
We can clearly see the valence band of the 50 ML film in XPS (green in Fig. 4); no background from the Rh substrate is detected. The figure also shows the corresponding spectra of the 5 ML-thick monoclinic film from Fig. 1 for comparison (red). The VB edges of both preparations were fitted using an error function, which yields inflection points at EB = 4.9 eV for the thicker film, and 0.0 eV for the thinner film, as expected for a signal dominated by the Fermi edge of metallic Rh. The corresponding full width at half maximum (FWHM) values are 1.2 eV and 0.95 eV, respectively. While the FWHM value for the Rh Fermi edge is close to the expected instrument resolution, the one for the thick (tetragonal) film is higher, which indicates that its VB edge is not a sharp step. Assuming a trapezoid-like density of states (blue in Fig. 4a), which gets broadened by the instrument resolution (FWHM 0.95 eV) to the observed spectrum, the VB onset would be at EB ≈ 4.4 eV. With a total band gap of 5–6 eV6,7 (probably slightly less due to the oxygen deficiency), this means that the Fermi energy of the thick tetragonal film is not too far from the conduction band. Fig. 4b shows the Zr 3d region of both films, with the Zr 3d5/2 peaks at 183.7 eV and 181.6 eV, respectively. The inset shows the O 1s region, where the difference is only 1.9 eV (EB = 531.4 eV and 529.5 eV, respectively), 0.2 eV less than for Zr 3d. ΔEB can be used to estimate the position of the bands of the monoclinic film w.r.t. EF. The whole electronic structure – including conduction and valence band – is shifted to lower EB by 2.0 ± 0.1 eV. Assuming equal band gaps of the monoclinic and strongly oxygen-deficient films, this puts the VB onset of the monoclinic film 2.4 eV below the Fermi level; when the oxygen-deficient film has a smaller band gap than the monoclinic one, this value will be somewhat higher. In any case, with a band gap of ≈5–6 eV, the Fermi energy is slightly below the mid-gap position, in reasonable agreement to density functional theory, which predicts a position in the middle of the band gap.8,16
3.5 Discussion
The main reason for the XPS binding energy shifts in 5 ML-thick zirconia films is doping: Tetragonal zirconia films are inherently slightly reduced. In most oxides, oxygen vacancies lead to n-doping via filled gap states; this shifts all energy levels down (towards higher EB). For reducible (semiconducting) oxides, oxygen-vacancy states are close to the conduction band minimum. This is in contrast to zirconia: There, the oxygen vacancy states filled by two electrons (F-center,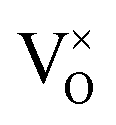 in Kröger–Vink notation) are only slightly above mid-gap.32 This is not far from the Fermi level of the metal substrate, which is also near the mid-gap position as indicated by XPS measurements of the Fermi edge (Fig. 4) and DFT calculations.8 Thus, in zirconia, a filled vacancy state would not lead to a substantial shift of the bands. It has been noted, however, that in the presence of a nearby metal, the vacancies become positively charged
in Kröger–Vink notation) are only slightly above mid-gap.32 This is not far from the Fermi level of the metal substrate, which is also near the mid-gap position as indicated by XPS measurements of the Fermi edge (Fig. 4) and DFT calculations.8 Thus, in zirconia, a filled vacancy state would not lead to a substantial shift of the bands. It has been noted, however, that in the presence of a nearby metal, the vacancies become positively charged  ; the electrons are transferred to the metal, leading to a substantial decrease of the VO formation energy from ≈6 eV to ≈3 eV.5,33 Then, the (unoccupied) vacancy state is at the bottom of the conduction band,30 and the positive charge causes downward band bending, similar to the effect of gap states in reducible oxides. In contrast to reduced zirconia films, the Fermi energy of fully oxidized (monoclinic) films is located near the middle of the band gap.8,16 This effect leads to the observed counter-intuitive shift of XPS binding energies, where (weak) reduction leads to higher EB – instead of the lower EB expected for cations in a lower oxidation state.
; the electrons are transferred to the metal, leading to a substantial decrease of the VO formation energy from ≈6 eV to ≈3 eV.5,33 Then, the (unoccupied) vacancy state is at the bottom of the conduction band,30 and the positive charge causes downward band bending, similar to the effect of gap states in reducible oxides. In contrast to reduced zirconia films, the Fermi energy of fully oxidized (monoclinic) films is located near the middle of the band gap.8,16 This effect leads to the observed counter-intuitive shift of XPS binding energies, where (weak) reduction leads to higher EB – instead of the lower EB expected for cations in a lower oxidation state.
When oxidizing the tetragonal ZrO2 films by using a catalyst, the core levels shift to lower EB, i.e. closer to the monoclinic levels. The minimum EB difference found between a (fully oxidized) monoclinic and an oxidized tetragonal film was 0.4 eV for Zr 3d (0.2 eV for O 1s), in contrast to a maximum difference of 1.8 eV for strongly reduced tetragonal 5 ML films. The large spread of EB values for the tetragonal films is nicely explained by different reduction states.
When comparing tetragonal (strongly or weakly reduced) and monoclinic films, we consistently find a difference of 0.2 eV between changes of O 1s and Zr 3d. In other words, the difference between the O 1s and Zr 3d core levels depends on whether we have the monoclinic (EO1s − EZr3d5/2 = 347.9 eV) or tetragonal (347.7 eV) phase; it must be due to the different structure and/or different band gaps of these phases and can be used to distinguish between these two phases. This difference of two binding energies may be also used to determine the ZrO2 phase for powder or bulk material, where all XPS levels can be shifted due to charging.
Full oxidation is only possible via oxygen spillover from a catalyst for O2 dissociation, such as deposited catalytic metals or the Rh substrate. The latter is accessible in case of films with holes reaching the substrate, i.e. all our monoclinic films, and specially prepared, broken tetragonal films. When annealing in O2, the molecules dissociate at the metal and spill over to the oxide, i.e. the metal clusters provide atomic oxygen.34 DFT predicts VOs to be stabilized below or near metal clusters.33,35 In other words, metallic clusters attract oxygen vacancies to the triple-phase boundary (TPB), where oxygen spillover takes place. As zirconia is a good oxygen ion conductor in the presence of oxygen vacancies,1 this attraction facilitates vacancy annihilation near the TPB.
Oxidation destabilizes tetragonal films: With Rh clusters at the surface, the transformation to monoclinic zirconia starts already at 610 °C, much lower than for (reduced) tetragonal films without a catalyst at the surface, where the transformation goes hand in hand with the formation of holes in the film19 above 730 °C. The tetragonal → monoclinic phase transformation would probably occur at even higher temperatures or not at all if the formation of holes, and, thereby, oxygen spillover could be suppressed. In any case, our study nicely confirms that the tetragonal ZrO2 phase is stabilized by oxygen vacancies, as proposed in the more recent literature,22 and in contrast to earlier studies36 suggesting the tetragonal phase stabilized by its lower surface energy.
4 Conclusions
An XPS study of zirconia films was conducted. A binding-energy difference of up to 1.8 eV between 5 ML-thick monoclinic and tetragonal films was found, which originates from the different reduction states of the two films. Tetragonal films, which exhibit core levels at higher EB, are inherently slightly reduced. The resulting positively charged oxygen vacancies bend the electronic bands, i.e. both, the O 1s and Zr 3d levels shift by equal amounts. This leads to the unusual case of a reduced oxide exhibiting higher XPS binding energies than its oxidized counterpart. These core level shifts are substantially more sensitive to the reduction state of the oxide than quantification of the XPS peak intensities. Our work also provides a way to discriminate between tetragonal and monoclinic ZrO2via the XPS binding energy difference between O 1s and Zr 3d.Tetragonal films, which cover the whole Rh substrate, cannot be fully oxidized by annealing in O2 due to the lack of a catalyst for O2 dissociation: The ZrO2 surface offers no catalytic sites for O2 from the gas phase to dissociate. The films only become more oxidized via oxygen spillover from a catalyst; this can be the substrate (if there are holes in the film) or a catalytically active metal deposited on top of the film. Monoclinic films are essentially stoichiometric ZrO2. In UHV-based preparation routes, monoclinic ZrO2 forms only in the presence of a catalyst, i.e. uncovered metal.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Austrian Science Fund (FWF) under project number F4505 (Functional Oxide Surfaces and Interfaces – FOXSI).Notes and references
- T. H. Etsell and S. N. Flengas, Electrical Properties of Solid Oxide Electrolytes, Chem. Rev., 1970, 70, 339–376 CrossRef CAS.
- S. McIntosh and R. J. Gorte, Direct Hydrocarbon Solid Oxide Fuel Cells, Chem. Rev., 2004, 104, 4845–4866 CrossRef CAS PubMed.
- Y. Liu, J. Parisi, X. Sun and Y. Lei, Solid-state gas sensors for high temperature applications - a review, J. Mater. Chem. A, 2014, 2, 9919–9943 RSC.
- T. Yamaguchi, Application of ZrO2 as a Catalyst and a Catalyst Support, Catal. Today, 1994, 20, 199–217 CrossRef CAS.
- A. R. Puigdollers and G. Pacchioni, CO Oxidation on Au Nanoparticles Supported on ZrO2: Role of Metal/Oxide Interface and Oxide Reducibility, ChemCatChem, 2017, 9, 1119–1127 CrossRef CAS.
- R. H. French, S. J. Glass, F. S. Ohuchi, Y. N. Xu and W. Y. Ching, Experimental and Theoretical Determination of the Electronic Structure and Optical Properties of Three Phases of ZrO2, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 5133–5142 CrossRef CAS PubMed.
- H. Jiang, R. I. Gomez-Abal, P. Rinke and M. Scheffler, Electronic Band Structure of Zirconia and Hafnia Polymorphs from the GW Perspective, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 085119 CrossRef.
- M. Antlanger, W. Mayr-Schmölzer, J. Pavelec, F. Mittendorfer, J. Redinger, P. Varga, U. Diebold and M. Schmid, Pt3Zr(0001): A substrate for growing well-ordered ultrathin zirconia films by oxidation, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 035451 CrossRef.
- H. Li, J.-I. J. Choi, W. Mayr-Schmölzer, C. Weilach, C. Rameshan, F. Mittendorfer, J. Redinger, M. Schmid and G. Rupprechter, Growth of an ultrathin zirconia film on Pt3Zr examined by high-resolution X-ray photoelectron spectroscopy, temperature-programmed desorption, scanning tunneling microscopy, and density functional theory, J. Phys. Chem. C, 2015, 119, 2462–2470 CAS.
- J. I. J. Choi, W. Mayr-Schmölzer, F. Mittendorfer, J. Redinger, U. Diebold and M. Schmid, The growth of ultra-thin zirconia films on Pd3Zr(0001), J. Phys.: Condens. Matter, 2014, 26, 225003 CrossRef CAS PubMed.
- P. Lackner, J. Hulva, E.-M. Köck, W. Mayr-Schmölzer, J. I. J. Choi, S. Penner, U. Diebold, F. Mittendorfer, J. Redinger, B. Klötzer, G. S. Parkinson and M. Schmid, Water adsorption at zirconia: from the ZrO2(111)/Pt3Zr(0001) model system to powder samples, J. Mater. Chem. A, 2018, 6, 17587–17601 RSC.
- H. Li, C. Rameshan, A. V. Bukhtiyarov, I. P. Prosvirin, V. I. Bukhtiyarov and G. Rupprechter, CO2 Activation on Ultrathin ZrO2 Film by H2O Co-Adsorption: In Situ NAP-XPS and IRAS Studies, Surf. Sci., 2018, 679, 139–146 CrossRef.
- J. I. J. Choi, W. Mayr-Schmölzer, I. Valenti, P. Luches, F. Mittendorfer, J. Redinger, U. Diebold and M. Schmid, Metal adatoms and clusters on ultrathin zirconia films, J. Phys. Chem. C, 2016, 120, 9920–9932 CrossRef CAS PubMed.
- V. Maurice, M. Salmeron and G. Somorjai, The epitaxial growth of zirconium oxide thin films on Pt(111) single crystal surfaces, Surf. Sci., 1990, 237, 116–126 CrossRef CAS.
- K. Meinel, K.-M. Schindler and H. Neddermeyer, Growth, Structure and Annealing Behaviour of Epitaxial ZrO2 Films on Pt(111), Surf. Sci., 2003, 532-535, 420–424 CrossRef CAS.
- K. Meinel, A. Eichler, K. M. Schindler and H. Neddermeyer, STM, LEED, and DFT characterization of epitaxial ZrO2 films on Pt(111), Surf. Sci., 2004, 562, 204–218 CrossRef CAS.
- K. Meinel, A. Eichler, S. Förster, K.-M. Schindler, H. Neddermeyer and W. Widdra, Surface and interface structures of epitaxial ZrO2 films on Pt(111): Experiment and density-functional theory calculations, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 235444 CrossRef.
- P. Lackner, J. I. J. Choi, U. Diebold and M. Schmid, Construction and evaluation of an ultrahigh-vacuum-compatible sputter deposition source, Rev. Sci. Instrum., 2017, 88, 103904 CrossRef PubMed.
- P. Lackner, Z. Zou, S. Mayr, J.-I. J. Choi, U. Diebold and M. Schmid, Surface Structures of ZrO2 Films on Rh(111): From Two Layers to Bulk Termination, Surf. Sci., 2019, 679, 180–187 CrossRef CAS.
- J. H. Scofield, Theoretical Photoionization Cross Sections from 1 to 1500 keV, California Univ., Lawrence Livermore Lab. Technical Report UCRL-51326, 1973.
- W. Smekal, W. S. M. Werner and C. J. Powell, Simulation of electron spectra for surface analysis (SESSA): a novel software tool for quantitative Auger-electron spectroscopy and X-ray photoelectron spectroscopy, Surf. Interface Anal., 2005, 37, 1059–1067 CrossRef CAS.
- S. Shukla and S. Seal, Mechanisms of Room Temperature Metastable Tetragonal Phase Stabilisation in Zirconia, Int. Mater. Rev., 2005, 50, 45–64 CrossRef CAS.
- M. V. Ganduglia-Pirovano and M. Scheffler, Structural and Electronic Properties of Chemisorbed Oxygen on Rh(111), Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 15533 CrossRef CAS.
- L. Köhler, G. Kresse, M. Schmid, E. Lundgren, J. Gustafson, A. Mikkelsen, M. Borg, J. Yuhara, J. N. Andersen, M. Marsman and P. Varga, High-Coverage Oxygen Structures on Rh(111): Adsorbate Repulsion and Site Preference Is Not Enough, Phys. Rev. Lett., 2004, 93, 266103 CrossRef PubMed.
- E. H. Kisi and C. J. Howard, Crystal Structures of Zirconia Phases and Their Inter-Relation, Key Eng. Mater., 1998, 153, 1–36 Search PubMed.
- B. Králik, E. K. Chang and S. G. Louie, Structural Properties and Quasiparticle Band Structure of Zirconia, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 7027–7036 CrossRef.
- Y. Gao, L. Zhang, Y. Pan, G. Wang, Y. Xu, W. Zhang and J. Zhu, Epitaxial Growth of Ultrathin ZrO2(111) Films on Pt(111), Chin. Sci. Bull., 2011, 56, 502–507 CrossRef CAS.
- A. Nenning, A. K. Opitz, C. Rameshan, R. Rameshan, R. Blume, M. Hävecker, A. Knop-Gericke, G. Rupprechter, B. Klötzer and J. Fleig, Ambient Pressure XPS Study of Mixed Conducting Perovskite-Type SOFC Cathode and Anode Materials under Well-Defined Electrochemical Polarization, J. Phys. Chem. C, 2016, 120, 1461–1471 CrossRef CAS PubMed.
- W. C. Chueh, A. H. McDaniel, M. E. Grass, Y. Hao, N. Jabeen, Z. Liu, S. M. Haile, K. F. McCarty, H. Bluhm and F. El Gabaly, Highly Enhanced Concentration and Stability of Reactive Ce3+ on Doped CeO2 Surface Revealed In Operando, Chem. Mater., 2012, 24, 1876–1882 CrossRef CAS.
- T. Matsushima, Dissociation of Oxygen Admolecules on Rh(111), Pt(111) and Pd(111) Surfaces at Low Temperatures, Surf. Sci., 1985, 157, 297–318 CrossRef CAS.
- M. Setvin, J. Hulva, G. S. Parkinson, M. Schmid and U. Diebold, Electron Transfer between Anatase TiO2 and an O2 Molecule Directly Observed by Atomic Force Microscopy, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E2556–E2562 CrossRef CAS PubMed.
- C. Gionco, M. C. Paganini, E. Giamello, R. Burgess, C. Di Valentin and G. Pacchioni, Paramagnetic Defects in Polycrystalline Zirconia: An EPR and DFT Study, Chem. Mater., 2013, 25, 2243–2253 CrossRef CAS.
- P. Schlexer, A. Ruiz Puigdollers and G. Pacchioni, Role of Metal/Oxide Interfaces in Enhancing the Local Oxide Reducibility, Top. Catal., 2018 DOI:10.1007/s11244-018-1056-5.
- S. Shaikhutdinov, M. Heemeier, J. Hoffmann, I. Meusel, B. Richter, M. Bäumer, H. Kuhlenbeck, J. Libuda, H. J. Freund, R. Oldman, S. D. Jackson, C. Konvicka, M. Schmid and P. Varga, Interaction of oxygen with palladium deposited on a thin alumina film, Surf. Sci., 2002, 501, 270–281 CrossRef CAS.
- A. Ruiz Puigdollers, P. Schlexer, S. Tosoni and G. Pacchioni, Increasing Oxide Reducibility: The Role of Metal/Oxide Interfaces in the Formation of Oxygen Vacancies, ACS Catal., 2017, 7, 6493–6513 CrossRef CAS.
- R. C. Garvie, Stabilization of the tetragonal structure in zirconia microcrystals, J. Phys. Chem., 1978, 82, 218–224 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cp03322j |
| This journal is © the Owner Societies 2019 |