
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bilayer thickness determines the alignment of model polyproline helices in lipid membranes†
Vladimir
Kubyshkin
 *ab,
Stephan L.
Grage
c,
Anne S.
Ulrich
cd and
Nediljko
Budisa
ab
*ab,
Stephan L.
Grage
c,
Anne S.
Ulrich
cd and
Nediljko
Budisa
ab
aInstitute of Chemistry, Technical University of Berlin, Müller-Breslau-Str. 10, Berlin 10623, Germany
bDepartment of Chemistry, University of Manitoba, Dysart Rd. 144, Winnipeg MB R3T 2N2, Canada. E-mail: vladimir.kubyshkin@umanitoba.ca
cInstitute of Biological Interfaces (IBG-2), Karlsruhe Institute of Technology (KIT), P.O.B. 3640, Karlsruhe 76021, Germany
dInstitute of Organic Chemistry, KIT, Fritz-Haber-Weg 6, Karlsruhe 76131, Germany
First published on 18th September 2019
Abstract
Our understanding of protein folds relies fundamentally on the set of secondary structures found in the proteomes. Yet, there also exist intriguing structures and motifs that are underrepresented in natural biopolymeric systems. One example is the polyproline II helix, which is usually considered to have a polar character and therefore does not form membrane spanning sections of membrane proteins. In our work, we have introduced specially designed polyproline II helices into the hydrophobic membrane milieu and used 19F NMR to monitor the helix alignment in oriented lipid bilayers. Our results show that these artificial hydrophobic peptides can adopt several different alignment states. If the helix is shorter than the thickness of the hydrophobic core of the membrane, it is submerged into the bilayer with its long axis parallel to the membrane plane. The polyproline helix adopts a transmembrane alignment when its length exceeds the bilayer thickness. If the peptide length roughly matches the lipid thickness, a coexistence of both states is observed. We thus show that the lipid thickness plays a determining role in the occurrence of a transmembrane polyproline II helix. We also found that the adaptation of polyproline II helices to hydrophobic mismatch is in some notable aspects different from α-helices. Finally, our results prove that the polyproline II helix is a competent structure for the construction of transmembrane peptide segments, despite the fact that no such motif has ever been reported in natural systems.
Introduction
Living nature operates with a few types of biopolymeric scaffolds involved in major biochemical processes. The main two are nucleic acid and polypeptide types, ubiquitous for all living systems. The ability to interact with lipid membranes is one of the key chemical differences between these two biopolymeric “worlds”. Both a lack of hydrophobic elements and high negative charge render nucleic acids unable to transverse membranes.1 Conversely, proteins possess all necessary features to insert into lipid bilayers, thereby forming functional membrane protein scaffolds. The introduction of proteins and their anchoring within lipid bilayers were a critical step in the origin of life, which is based on the utilization of compartments and gradients. The interactions of proteins with the membrane are very diverse, being associated with the presence of hydrophobic (Leu, Ile, Val),2,3 anchoring (Trp, Lys),4 and other amino acid residues that are, e.g., in contact with the polar head group region (Arg, Lys),5,6 or forming intraprotein interfaces (Gly, Ala, Ser, etc.).7 Virtually every encoded amino acid can be utilized for the construction of membrane proteins, to either interact with the lipid medium or form the interior of an integral membrane protein with desired function and specificity.8However, when considering the secondary structure elements that carry the side-chain functions, this natural versatility vanishes. Typically, when traversing the membrane with hydrophobic transmembrane (TM) elements, nature becomes extremely conservative, utilizing only two standard secondary structures to accomplish this task. The vast majority of integral TM proteins are constructed from α-helices that are often regarded simply as “TM helices”; another class of membrane proteins consists of β-barrel structures (only occur in special types of membranes).9 In the aqueous medium, nature operates with a much larger repertoire of secondary structures, but only a few are present in membrane embedded proteins. Among these structures excluded from the membrane, the most widespread is the polyproline-II (PII) helix, also regarded as a “semi-extended” helix (Fig. 1A).10,11 Interestingly, in the history of life evolution, the PII helix was present at the very beginning of protein biosynthesis, before the α-helix became the dominant protein fold.12 It is therefore quite surprising that this structural element was not utilized to form membrane spanning segments. One potential explanation for this fact could be that the structure itself is too hydrophilic, thus incompatible with the hydrophobic environment of a lipid bilayer. Indeed, in biochemical settings, the PII helix is present in polar environments: in globular proteins,13 in solvated exposed peptide stretches,14 as well as in intrinsically disordered proteins and protein fragments lacking persistent structure.15,16 If the PII helix acts as an intrinsically polar structure in nature, does this mean that it can only be polar?
 | ||
Fig. 1 (A) The polyproline-II (PII) helix, compared to an α-helix. (B) Experimental log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P scale determined for methyl esters of N-acetyl amino acids.18 Proline and its derivatives are highlighted in red. P scale determined for methyl esters of N-acetyl amino acids.18 Proline and its derivatives are highlighted in red. | ||
The answer is ‘no’, and the apparent hydrophilicity of PII helices can actually be overcome by means of peptide chemistry, when utilizing a specific amino acid composition. For example, it is possible to substantially reduce the polarity of the PII helix, simply by replacing the backbone with a tertiary amide, thereby eliminating the polar NH-bonds. In this context, oligoproline peptides are especially convenient scaffolds, because proline naturally forms a tertiary amide bond. However, the direct use of oligoproline in a nonpolar environment is precluded due to an insufficient hydrophobicity of the prolyl side-chains (Fig. 1B)17,18 as well as the weak relative stability of the PII helix.19,20
We have taken all these considerations into account and designed an oligoproline scaffold based on the proline analogue (2S,3aS,7aS)-octahydroindole-2-carboxylic acid (Oic). This amino acid is hydrophobic (Fig. 1B), and at the same time it features stronger backbone-stabilizing contacts than the parent proline structure.21 The PII helix formed by oligo-Oic sequences turned out to be both stable and highly lipophilic, and it maintains the desired fold when dissolved in nonpolar solvents such as alkanes.18,22 Based on this peptide scaffold, we have recently reported an oligo-Oic peptide with a TM alignment in model lipid bilayers.23 We have thus proven that a TM PII helix is a realistic option. Therefore, it might be quite astonishing that such a motif has never been discovered in natural proteomes. This very fact has numerous implications for membrane biophysics, protein engineering, and understanding the evolution of life. Indeed, if nature operates transmembrane peptides and PII helices, why does it not operate transmembrane PII helices?
To answer this rather general question, we need to collect much better understanding on the actual behaviour of the helix in the membrane. For example, our pioneering observation of a TM PII helix still does not explain which factors in the peptide architecture and/or the lipid membrane determine the transmembrane alignment. To further investigate this first TM PII helix, we have designed here an experimental study with the aim to test for the occurrence of the TM state under various peptide/lipid conditions. In this way, we intend to explain which elements of the peptide structure and of the lipid bilayer result in a TM alignment, and which do not. We envisage that these explanations will help in utilizing the TM PII motif in further biochemical settings. At the same time, we expect that the trends observed here with the model PII helix may provide a unique insight into the general aspects of the interaction of a folded peptide body with the lipid environment.
The PII helix is a rare example of a natural peptide structure, which is not at all based on hydrogen bonding. One of the notable features of this new TM PII helix is its simple geometric arrangement, with 3.0 residues per turn and 9 Å per turn (Fig. 1A).21,24,25 Thereby, an extended TM PII helix requires only half as many residues to traverse the membrane as compared to a “classical” TM α-helix. At the same time, it places the residues directly above one other with a more compact 9 Å periodicity (i ↔ i + 3), compared to the more extended 10.5 Å stacking of residues in an α-helix (i ↔ i + 7, Fig. 1A). Thus, a TM PII helix will offer a versatile and useful tool for peptide/protein engineering, when its membrane behaviour is properly understood.
Experimental section
Preparation of the peptides
The starting nonameric peptide 1 with a CF3-label in the middle position and C6-based positive charge linkers was prepared as described.23 Next, we synthesized a series of peptides with variations in each component of the peptide structure: we varied the position of the 19F NMR label (peptides 2, 3), the presence of the terminal charges (peptides 4–6), the length of the charge linkers (peptides 7, 8), and the overall length of the peptide (peptides 9–13). The sequences are summarized in Table 1.| # | PII length, residues | Terminal charge linkers | Peptide sequence |
|---|---|---|---|
| 1 | 9 | C6 | H3N+(CH2)5CO-(Oic4-TfmPro-Oic4)-NH(CH2)6N+H3, 2Cl− |
| 2 | 9 | C6 | H3N+(CH2)5CO-(Oic3-TfmPro-Oic5)-NH(CH2)6N+H3, 2Cl− |
| 3 | 9 | C6 | H3N+(CH2)5CO-(Oic5-TfmPro-Oic3)-NH(CH2)6N+H3, 2Cl− |
| 4 | 9 | none | H3CCO-(Oic4-TfmPro-Oic4)-OCH3 |
| 5 | 9 | C6; none | H3N+(CH2)5CO-(Oic4-TfmPro-Oic4)-OCH3, Cl− |
| 6 | 9 | none; C6 | H3CCO-(Oic4-TfmPro-Oic4)-NH(CH2)6N+H3, Cl− |
| 7 | 9 | C3 | H3N+(CH2)2CO-(Oic4-TfmPro-Oic4)-NH(CH2)3N+H3, 2Cl− |
| 8 | 9 | C8 | H3N+(CH2)7CO-(Oic4-TfmPro-Oic4)-NH(CH2)8N+H3, 2Cl− |
| 9 | 10 | C6 | H3N+(CH2)5CO-(Oic5-TfmPro-Oic4)-NH(CH2)6N+H3, 2Cl− |
| 10 | 10 | C6 | H3N+(CH2)5CO-(Oic4-TfmPro-Oic5)-NH(CH2)6N+H3, 2Cl− |
| 11 | 11 | C6 | H3N+(CH2)5CO-(Oic5-TfmPro-Oic5)-NH(CH2)6N+H3, 2Cl− |
| 12 | 12 | C6 | H3N+(CH2)5CO-(Oic7-TfmPro-Oic4)-NH(CH2)6N+H3, 2Cl− |
| 13 | 12 | C6 | H3N+(CH2)5CO-(Oic4-TfmPro-Oic7)-NH(CH2)6N+H3, 2Cl− |
The synthesis of oligomeric-Oic peptides has specific peculiarities. We found previously that oligo-Oic sequences longer than 5 amino acid residues complicate the solid-phase peptide synthesis due to their limited solubility in DMF.18 Therefore, we applied the following two strategies for the peptide synthesis. In the first approach (Scheme 1A), we synthesized peptide fragments not longer than five amino acids on resin with normal resin loading and using N,N-dimethylformamide (DMF) as a solvent. The peptides with Boc-groups at the N-termini were cleaved from the resin in a hexafluoro-2-propanol/dichloromethane cocktail, which keeps the Boc-group intact.26 The fragment meant to be at the C-terminus was either coupled with the Cbz-protected hexamethylenediamine (for C6 charge linkers) or esterified with 2,2-difluorodiazoethane (for other size linkers).27,28
 | ||
| Scheme 1 Synthesis of the target peptides 1–13. | ||
Then, we removed the Boc-group from the C-terminal fragment peptides, and these were coupled with the corresponding N-terminal fragment peptides, yielding full-length peptides. The N-terminal Boc- and C-terminal Cbz-protection groups were removed simultaneously to give target peptides 2, 3, and 11 (Scheme 1C). For the synthesis of 7 and 8, the peptides with 2,2-difluoroethyl esters at the C-terminus were mixed with liquid diamines, trimethylenediamine (at room temperature) and octamethylenediamine (at elevated temperature), respectively. The final peptides 7 and 8 were obtained after the N-terminal Boc-group was removed.
In another strategy, the full-length peptides were prepared on-resin with low-loading. A DMF-dichloromethane mixture was used as the solvent for coupling all amino acids beyond the 10th residue (Scheme 1B). The peptides were cleaved off the resin, the C-terminal charge linker was installed in solution, and the simultaneous removal of the protection groups yielded target peptides 9–13 (Scheme 1C). The peptides with removed charges (4–6) were synthesized analogously, as shown in Scheme 1D.
Identity and purity of final peptides were confirmed by mass-spectra (ESI-Orbitrap), liquid chromatography (UV detection at 220 nm), and 1H NMR (CD3OD, 600 MHz) spectra (see ESI†).
Solid state NMR
To assess the quality of lipid alignment for each sample, 31P{1H} HMR spectra were acquired at 202.5 MHz using a Hahn echo pulse sequence with 1H decoupling during acquisition (spinal64, 17 kHz). Parameters were as follows: 90 degree pulse of 5 μs at 95 W, echo delay of 20 μs, dwell time of 1 μs, time domain of 16384, recycling delay of 3 s, 256 scans, and the transmitter frequency was set around 0 ppm. The spectra were processed using exponential line broadening with a Lorentzian parameter of 100 Hz. A 5th degree polynomial function was applied for baseline correction of the frequency domain spectra. Unless stated otherwise, the spectra demonstrated 80% or higher alignment of the lipid according to integration over the 31P signal.
The 19F{1H} NMR spectra were acquired at 471 MHz, using a single 90-degree pulse experiment with 1H decoupling during acquisition (spinal64, 12.5 kHz). Parameters were as follows: 90 degree pulse of 2.5 μs at 180 W, dwell time of 1 μs, time domain of 4096, recycling delay of 2 s, 2048 or more scans, and the transmitter frequency was set around −75 ppm. For processing, 500 Hz Lorentzian line broadening and 5th degree polynomial baseline correction were applied to the spectra.
Results
Design of the peptides
The peptides were designed according to the following considerations. First, the peptide sequence was based on an oligomeric Oic structure to enable the desired selectivity for a hydrophobic environment (Fig. 2A). Sufficient hydrophobicity of the core peptides can be inferred from the fact that oligo-Oic peptides with 6 or more residues are not soluble in water, but they are soluble in hexane as well as other organic solvents, such as dichloromethane or chloroform, which are typical solvents for lipids.18 Second, the length of the sequence should be close to the thickness of the hydrophobic core of the model lipid bilayers. For example, a nonameric peptide Oic9 makes 3 turns of a PII helix, resulting in approx. 9 × 3 Å = 27 Å effective length, which should perfectly match the hydrophobic thickness reported for 14:0/14:0 PC (26 Å at 30 °C, Fig. 2B).31 Third, we included terminal positive charges, with the aim to anchor the peptide ends in the head group section of the lipid bilayer. We introduced these charges using chemical rather than biochemical building blocks; this means that the positive charges were not entire amino acid residues (such as Lys or Arg), but they were simple ammonium groups on minimalistic C6 linkers, attached via amide bonds at each of the two termini (Fig. 2C). The purpose of the C6 linkers was to avoid any deleterious impact of the terminal charge on the stability of the PII conformation.34,35 Finally, the fourth construction element is an orientation-dependent NMR label. One of the Oic residues was replaced by (2S,4S)-trifluoromethylproline (TfmPro),36 a hydrophobic proline analogue (Fig. 1B). This amino acid bears a CF3-group, which is a highly sensitive 19F NMR reporter group that reflects the backbone alignment in an anisotropic environment (Fig. 2D).37,38 | ||
| Fig. 2 Design of the study. The TM PII peptide should be based on a hydrophobic oligoproline scaffold (A) with a sufficient length to span the membrane (B), with terminal charge linkers to anchor on the polar sides of the membrane (C), and with a CF3-label as an orientation probe for a 19F NMR based method (D). | ||
We then carried out the orientational analyses on the peptides embedded in lipid bilayers, in samples that were macroscopically oriented on glass plates. In these fully hydrated samples, the membranes spontaneously form a lamellar phase, with all bilayers arranged parallel to the surface of the glass plates. They are placed in the NMR probe such that the bilayer normal is aligned parallel to the external magnetic field B0.39 In this experimental setting, any defined peptide orientation in the lamellar lipid bilayer should result in a characteristic 19F NMR signal. The anisotropic signature of the CF3-group is a triplet, due to the dipolar coupling between the three equivalent fluorine nuclei. Both the shift of the triplet position from the isotropic δiso position (chemical shift anisotropy, δCSA) and the observed distance between the triplet components (splitting value, Δ) have the same dependence on the CF3-group orientation with respect to B0, angle θ, as expressed in eqn (1) and (2).40
The splitting value is considered to be the most sensitive and reliable quantitative parameter, because it does not require referencing of any kind (external or internal), while δCSA (position of the resonance) unambiguously reveals the sign of the splitting.
 | (1) |
 | (2) |
The peptide states
For peptide 1, with an assumed hydrophobic length of about 27 Å, we have previously observed a single triplet signal of the CF3-group in short lipid bilayers (12:0/12:0 PC, ∼22 Å), which defines a unique helix alignment, while two peptide populations were observed in longer chain lipid bilayers (16:0/18:1 PC, ∼29 Å).23 Because the former signal is consistent with an upright TM orientation of the helix, while the latter two signals are not, we concluded that the TM orientation is adopted only in the short-chain lipid. We thus speculated that in the long-chain lipid, the helix would be submerged within the bilayer such that both charged termini reside on the same face of the membrane. As the helix axis lies essentially parallel to the bilayer plane in this state (which we had described as a hammock-like state), we will now refer to it as a “submerged state” (SM-state). An intermediate situation was found in 14:0/14:0 PC (∼26 Å), where we had observed the simultaneous occurrence of all three sets of splittings in the same spectrum.Clearly, a single anisotropic label in 1 is not enough to resolve the full 3D orientation of the peptide in the membrane, nor can any alternative orientations be ruled out that are compatible with the single data point. Thus, our initial conclusions regarding the peptide states need to be confirmed with a more comprehensive set of data. For this reason, here, we prepared isomeric peptides 2 and 3 with TfmPro shifted one position down or up in the helix, respectively. As a result, the relative orientations of the CF3-groups with respect to the helical axis differ by about 120° between the isomeric peptides.
Our experiments demonstrate that in the short-chain lipid, the splitting remains nearly the same for all three peptides, −5.8, −6.8 and −6.7 kHz for 2, 1 and 3, respectively (Fig. 3). This very straightforward observation already gives a clear indication that in all cases, the helix axis is aligned parallel with respect to B0, indicating a TM state of these peptides. In contrast, in the longer chain lipid, the major splitting showed alternating values of +4.0, −3.8 and +6.8 kHz, for 2, 1 and 3, respectively. These data are fully consistent with the helix axis being orthogonal to B0, thus the peptide being aligned in the SM state. We drew out the SM state based on the assumption that the helix remains in the hydrophobic core, and does not come out into polar sections of the lipid bilayer. The spectra in 14:0/14:0 PC demonstrate a co-existence of states for all three peptides, indicating that the TM ↔ SM transition is slow on the NMR time scale.
 | ||
| Fig. 3 Examination of the membrane alignment of isomeric nonanpeptides carrying a 19F NMR label in different positions. | ||
Next, we analyzed the obtained data in terms of peptide orientation, as defined by two angles τ and ρ, plus the molecular order parameter Smol that quantifies mobility. In this description, the tilt angle (τ) gives the tilt of the helical axis with respect to bilayer normal. We define the azimuthal rotation angle (ρ) as the rotational position around the helix of the Cα-carbon atom in the 5th residue (Fig. 4A).‡ In our analysis, we combined the orientations of the three CF3-labels from separate experiments to calculate the orientation of the helix based on the following assumptions: (i) the helix alignment is not influenced by the position of the label (TfmPro); (ii) the sterically bulky CF3-group in the TfmPro residues adopts an equatorial conformation;36 (iii) the maximal dipolar splitting value Δmax is the same as for other CF3-labeled amino acids (16 kHz).36,40 To obtain the helix orientation that best agrees with the experimental data, we systematically calculated the expected dipolar splitting for all possible combinations of τ, ρ and Smol. The agreement between the calculated splitting (Δcalc,i) and measured splitting (Δexp,i) was quantified by a root of mean square deviation (rmsd) according to eqn (3):
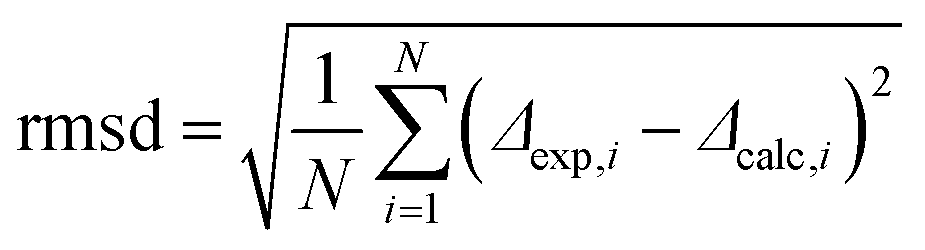 | (3) |
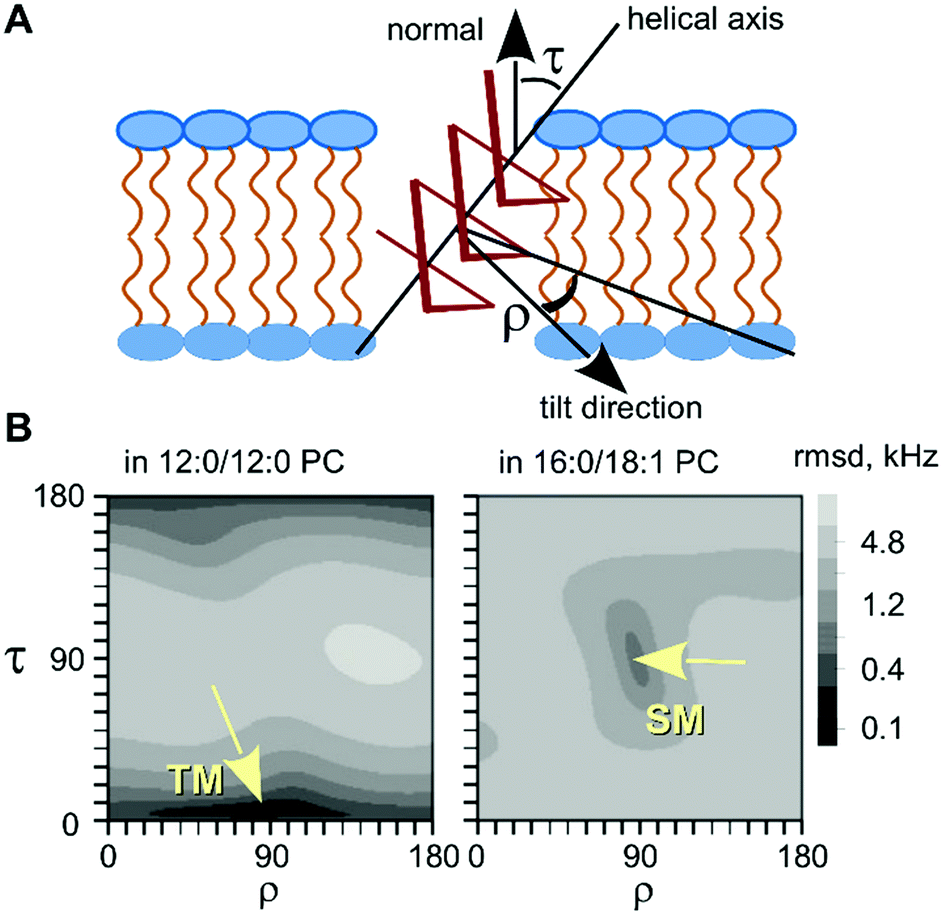 | ||
| Fig. 4 Tilt-rotation presentation of the helix alignment in lipid bilayers. (A) Definitions of the coordinate angles. (B) Results obtained from the combined data on peptides 1, 2 and 3. | ||
To find the best-fit solution, τ and ρ were varied from 0° to 180° in steps of 1°, and Smol was varied from 0 to 1.0 in steps of 0.1. The results presented in Fig. 4B clearly illustrate a TM alignment in the short-chain lipid with a small tilt angle of τ = 6° (Table 2). Conversely, in the long-chain lipid, the solution was a nearly perpendicular tilt angle with τ = 87°, which is fully consistent with our suggestion of a SM state. We thus confirmed that the peptide exists in an equilibrium between the proposed states, where the bilayer thickness has a decisive influence on the alignment of the peptide.
| Lipid | τ | ρ | S mol | rmsd, kHz |
|---|---|---|---|---|
| 12:0/12:0 PC | 6 | 84 | 0.85 | 0.13 |
| 16:0/18:1 PC | 87 | 88 | 0.75 | 1.09 |
The terminal groups
We then decided to examine the influence of the terminal charges on the helix alignment. We first compared the spectra of the peptides with two (1) and without any (4) charges, and with the peptides bearing a single charge on either terminus (5 and 6). The spectra of the short-chain lipid demonstrate that all peptides adopt the TM alignment (Fig. 5). However, the TM alignment was complete only for the peptide with two terminal anchoring charges (1), while another additional state was present for the other peptides of this series (4–6). In all cases, the TM alignment was lost in the long-chain lipid, as expected. Interestingly, in the long-chain lipid, the peptide without C-terminal charge (5) exhibited only one triplet (+2.3 kHz), whereas sets of splittings were observed for both peptides with a C-terminal charge (1 and 6). These results suggest that the rotational state of the peptide in the SM state relies on the orientation of the charge linkers relative to the peptide body. Furthermore, the co-existence of two triplets in the expected SM state may originate from rotational hopping of the terminal amide, which is a slow process on the NMR time scale (Scheme 2). We previously identified that the terminal amide rotation is the major source of the alternative conformations in oligo-Oic peptides.21 Both amides with the C- and N-terminus may lead to multiple conformations, which will necessarily result in different local orientations of the charge linkers with respect to each other, thereby generating different orientations of the peptide backbone in the lipid bilayer. Notably, different orientations of the charge linkers only generated visible differences in the SM, but not TM state where only one triplet was observed (−6.8 kHz). Indeed, in the TM state, the helix stands upright, hence mutual orientation of the linkers becomes unimportant for peptide alignment. | ||
| Fig. 5 Dependence of the nonameric peptide alignment on the presence and position of the terminal charges. | ||
 | ||
| Scheme 2 Terminal rotation of the amide moieties generates different alignments of the charge linkers with respect to each other. | ||
As a next step, we examined the importance of the length of the charge linkers. At this point, we hypothesized that the size of the charge linkers may influence the depth of the hydrophobic peptide placement in the lipid hydrophobic core, when the peptide adopts the SM state. Following this assumption, the charge linker size may influence the stability of the SM state, thereby affecting the TM ↔ SM equilibrium.
To this end, we measured peptides with C3 (7) and C8 (8) linkers at both termini of the nonameric peptide. According to our expectations, shorter charge linkers would disfavour the SM state, because they would pull the peptide out from the hydrophobic core (the pulling-out effect). The experimental 19F NMR spectra confirmed our expectations (Fig. 6). We found that in 14:0/14:0 PC, where the SM and TM states co-exist for the C6 and C8 linkers, shortening of the linker to C3 eliminated the SM state almost entirely. Nonetheless, a shorter size of the linker did not eliminate the SM state completely: in the longer chain lipid, 16:0/18:1 PC, the spectra were still fully dominated by the SM state resonances for all three peptides. Overall, we conclude that the charge linker size is only of secondary importance, while the TM ↔ SM state equilibrium depends primarily on the lipid thickness. In addition, the pulling-out effect of the terminal charges provides indirect proof for the localization of the peptide in the hydrophobic core, when the helix adopts the SM state.
 | ||
| Fig. 6 Influence of the terminal groups on the alignment of the nonameric peptide in membranes. | ||
The length of the helix
To explore the influence of the length of the helix on its behaviour in the membrane, we extended the length of the helix by adding an Oic residue to peptide 1 at the N-terminus (9), the C-terminus (10), or at both ends (11), or otherwise by adding one helical turn at the N- (12) or C-terminus (13). According to the basic architecture of the PII helix, each additional Oic residue adds 3 Å to the hydrophobic length of the peptide. As a result, we expected that the TM ↔ SM peptide re-alignment will shift to longer chain lipids.We first examined the undecameric peptide 11 in a series of lipids (Fig. 7). In the short-chain lipids, the signal appeared as a triplet with a splitting of −6.8 kHz, which is identical to the value observed in the TM alignment of the nonameric peptide.§ In the long-chain lipids, the signal changed to a triplet with a distinctly different splitting of +8.1 kHz. Hence, a re-alignment of the peptide occurred in the lipids 16:0/18:1 PC and 18:1/18:1 PC. Thus, we confirmed our original assumption that for longer peptides, the TM ↔ SM peptide transition is shifted to longer chain lipids.
 | ||
| Fig. 7 Re-alignment of the undecameric peptide (11) in a series of lipids. | ||
Interestingly, the value of the dipolar splitting in the TM state was the same in all lipids where this state was observed (≈−6.8 kHz). This finding suggests that the tilt of the PII helix does not change from the essentially upright orientation, despite the distinctly different hydrophobic thicknesses of the corresponding lipids, ranging from 22 to about 29 Å. Since the length of the hydrophobic PII helix in 11 is about 33 Å, the TM alignment of this peptide in the shortest lipids should be accompanied by a drastic mismatch between the long hydrophobic peptide and thin hydrophobic membrane core, known as “positive mismatch”. Indeed, we found indications that the lipid environment undergoes some adaptation to compensate for this mismatch. Namely, both of the shortest lipids in the series, 12:0/12:0 PC and 14:0/14:0 PC, showed a sizable fraction of non-oriented lipids in their 31P NMR spectra, indicating a notable loss of lipid orientation in the presence of peptide 11 (Fig. 7, right panel). The 19F NMR spectra of these samples also exhibited unusually broadened resonances (Fig. 7, left panel), thereby indicating that the sample orientation is severely perturbed by the presence of the peptide.
We further analysed the lineshapes of both 19F and 31P NMR spectra of the sample containing peptide 11 by fitting them with a distribution of orientations (see ESI†). No further orientation states besides the TM and SM states were required to explain the 19F NMR spectra. The broadening effects observed in the 31P and 19F NMR spectra in short-chain lipids were both caused by a similar amount of a badly/non-oriented fraction, visible in the 19F spectra as a shoulder near −70 ppm, and in the 31P spectra as a powder lineshape with a maximum near −15 ppm. This behaviour was concentration dependent. When the peptide was diluted from a peptide-to-lipid ratio of 1/40 down to 1/200, all these special features were gone, and both the 31P and 19F NMR spectra appeared sharp like the other samples (bottom spectra in Fig. 7). We can thus conclude that the adaptation to positive hydrophobic mismatch does not include any tilting of the TM PII helix in contrast to what has been reported for α-helices.41–44 We thus conclude that positive mismatch shows up as a perturbance of the lamellar lipid bilayers. We note at this point that we did not observe additional 31P NMR signals near 0 ppm or to the right of the signal at ∼30 ppm, which would be indicative of non-lamellar phases such as inverted micelles or hexagonal phases. We can hence exclude that the peptide induces severe structural changes of the lipid bilayer.
These conclusions obtained from the examination of the undecameric peptide 11 agree well with the results from the remaining series of PII peptides. For the decameric peptides (9 and 10), we observed a transition between the TM and SM state between 14:0/14:0 PC (prevalence of TM) and 16:0/18:1 PC (prevalence of SM, Fig. 8). For dodecapeptides 12 and 13, the re-alignment was shifted to longer chain lengths: a co-existence of TM and SM was observed in 20:1/20:1 PC (Fig. 9).¶ The phenomena associated with positive mismatch (resonance broadening, loss of the oriented part) were observed in short-chain lipids, and the splitting of the TM alignment was the same as for the other peptides in this series, 9–11. This fact again confirms our previous conclusion that the peptide does not tilt under positive mismatch, instead it seems that the lipids that adapt to the upright-standing TM helix.
 | ||
| Fig. 8 Re-alignment of the decameric peptides (9 and 10) in a series of lipids. | ||
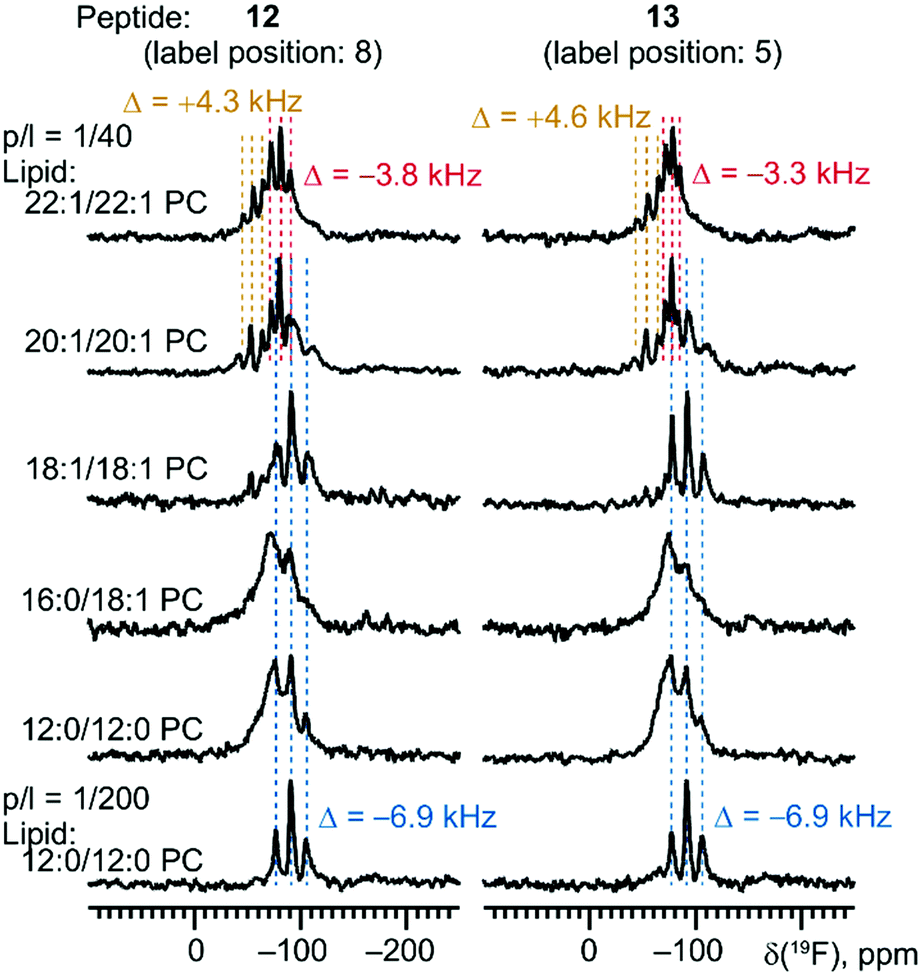 | ||
| Fig. 9 Re-alignment of the dodecameric peptides (12 and 13) in a series of lipids. | ||
Presence of lyso-lipids
Finally, we examined the influence of lyso-lipids on the TM ↔ SM equilibrium. It is well known that addition of lyso lipids reduces the lateral pressure in the hydrophobic core of the lamellar bilayer. For amphipathic α-helices and β-barrel proteins, it has been demonstrated that lyso-lipids can significantly promote the TM orientation by modulating the membrane curvature, compared to their surface-bound states.45,46 Could lyso-lipids promote the TM alignment of the PII helical system as well?To address this question, we took peptide 11 and examined its 19F NMR spectra in membranes upon the addition of lyso-lipids, judging the TM ↔ SM equilibrium by integration of the respective signals of the TM and SM states (Fig. 10). Our results demonstrate that a lyso-lipid content of up to 40 molar% does not change the ratio between the two states. These results fully agree with our earlier observations of peptide 1 in lipid bilayers, where the addition of about 1/3 molar lyso-lipid failed to promote the TM state.23 The absence of the influence from the lyso-lipid content can be explained by the fact that our PII peptides reside in the hydrophobic lipid core in both states, TM and SM. Thus, lowering of the pressure in the hydrophobic core relative to the lipid head group region does not show an effect on the equilibrium between these states.
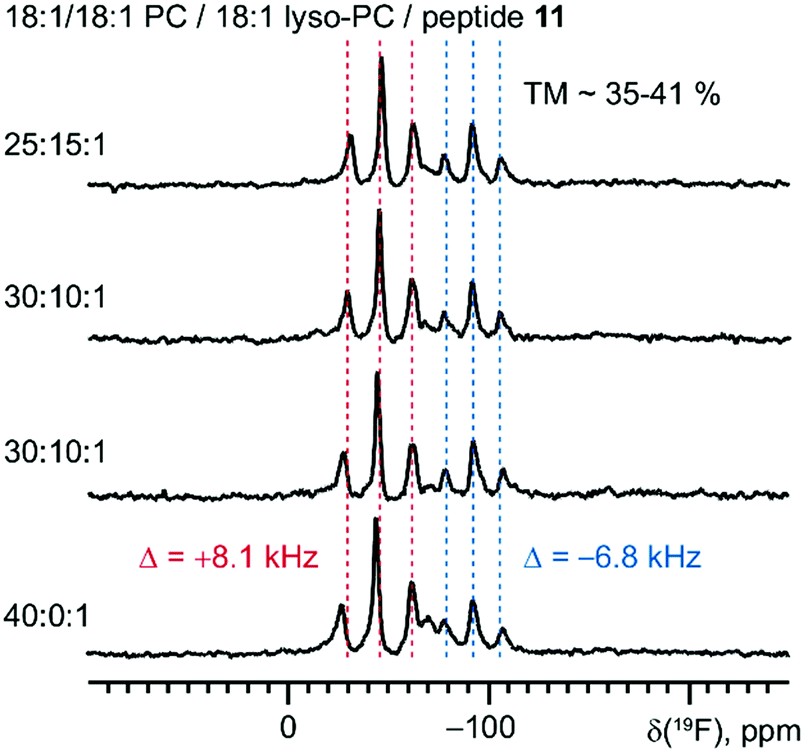 | ||
| Fig. 10 Spectra of the undecameric peptide 11 in 18:1/18:1 PC show absence of systematic dependence from the presence of 18:1 lyso-PC. | ||
Discussion
Lipid bilayers have a distinct lateral pressure profile, depending on the spontaneous curvature of the constituent lipids.47 The relative lateral pressure in the head group region and the hydrophobic core exerts a significant effect on the molecules immersed in it. For example, in well-balanced lamellar bilayers like the ones we used here (spontaneous curvature close to zero), a rigid cylindrical body would have a tendency to orient parallel to the bilayer normal, as can be exemplified by cholesterol molecules (Fig. 11).48–51 Hydrophobic helical peptides usually adopt the same transmembrane alignment, with α-helical oligo-Leu52 or oligo-AlaLeu (mainly, WALP and KALP peptides)53 as typical models.54 Other interesting but more complex examples include transmembrane assemblies such as the oligomeric alamethicin pore55–57 as well as the dimeric gramicidin A pore.58–60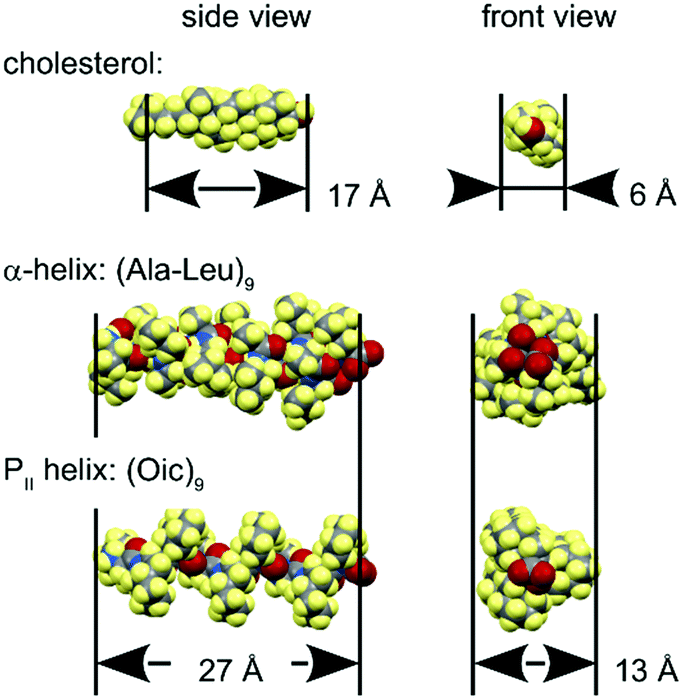 | ||
| Fig. 11 Dimensions of some relevant hydrophobic rigid bodies. | ||
In our study, we examined the alignment behaviour of a hydrophobic PII helical structure. Considering an oligo-Oic core peptide as a rigid body with similar geometric requirements to other common hydrophobic objects (Fig. 11), we would expect that this model peptide should indeed adopt a transmembrane alignment in lipid bilayers.
To prove this hypothesis, we first constructed a nonameric peptide with an estimated effective length of about 27 Å, and examined its alignment in lipid bilayers. The incorporation of a single NMR label (CF3-group) at three different positions on the helix allowed us to probe all three faces of the helix (peptides 1, 2, 3). Our results unambiguously prove that the peptide aligns parallel to the membrane normal in short-chain lipids (22 Å hydrophobic thickness). In contrast, in long-chain lipids (29 Å), it aligns preferably perpendicular to the bilayer normal, and in the matching lipid (26 Å), we observed a co-existence of states in our NMR spectra (Fig. 3, 4 and Table 2). These observations are consistent with our proposed interpretation that the peptide can adopt both an upright transmembrane (TM) alignment and a state in which the helix is submerged (SM) horizontally beneath the head group region of the lipid.
The finding that a negative mismatch (bilayer thickness greater than the peptide length) results in a SM state was rather unexpected. In the case of amphipathic membrane-associated peptides, the helix is usually oriented perpendicular to the bilayer normal (often called the ‘surface’ state).61 However, in the case of hydrophobic α-helices, there exist only a very few examples where an alignment parallel to the bilayer surface was suggested under conditions of negative mismatch.62–65 Generally, for hydrophobic α-helical peptides, a TM orientation can usually be concluded already from the simple observation that the peptide resides in the lipid hydrophobic core (for example, using Trp fluorescence assay).66
With our PII helical model, we show that the placement of a sufficiently hydrophobic helix into the hydrophobic core does not necessarily imply a TM state, but could also be compatible with a SM state. Interestingly, a similar two-state coexistence is known for bolalipids, where the corresponding states are referred to as a spanning (analogous to TM) and looping (analogous to SM) state.67 Transmembrane peptides with anchoring charges are similar to bolalipids, with the difference that the hydrophobic part is not really flexible in peptides.
We consider two possible explanations for the unexpected occurrence of the SM state in our PII model peptide. The first explanation is that the peptide has a lower density compared to an α-helix (see Fig. 11), therefore it is less influenced by the membrane lateral pressure when oriented perpendicular to the lipid chains in the SM state. Another explanation is that the anchoring linkers are much thinner and more flexible than a contiguous α-helix. Indeed, α-helical TM segments are usually decorated with anchoring residues, which tend to have a direct influence on the peptide orientational behaviour, dynamics and membrane interactions. For α-helical model peptides, the terminal anchoring regions are rather complex, involving several amino acid residues that typically include Lys, Trp, or other aromatic residues (WALP, KALP and others).53,54,62,68,69 Conversely, in our model peptides, the anchoring linkers are rather minimalistic, with a single ammonium group attached flexibly to each end of the helix. This is why results on α-helical and PII TM peptides are not directly comparable. Future studies should reveal whether more complex terminal anchors will substantially favour the TM alignment of the PII helical construct.
In our current examination of peptides with different lengths (1, 9–13), we have confirmed the tendencies observed for the first nonameric peptide, and the summary on peptide alignment is shown in Fig. 12. Considering the PII architecture with 3 Å length increment per residue, our peptide series was designed to probe lengths between 27 and 36 Å. In all instances, we observed the TM state in short-chain lipid(s), while an unusual submerged state (SM) prevailed in long chain lipids (Fig. 7–9).
 | ||
| Fig. 12 Summary of the peptide alignment dependence from the lipid thickness. | ||
These results show that the SM ↔ TM transition is a general phenomenon. The co-existence of states occurred in certain lipids with intermediate thickness. This transition was shifted towards longer lipids for longer peptides: from 14:0/14:0 PC (26 Å) for nonameric peptide (1, 27 Å), to 20:1/20:1 PC (32 Å) for dodecameric peptides (12 and 13, 36 Å).
Furthermore, we found that the orientation of the CF3-label was the same for all peptides in the TM state, even when there was significant positive mismatch between a long peptide embedded in a thin membrane. This is an indication that the peptide remains upright in the TM state, and does not tilt to adapt to the hydrophobic mismatch, in contrast to our preliminary suggestion earlier.23 This finding contrasts the typical behaviour of α-helical peptides, for which changes in peptide tilt and dynamics are usually observed in the case of a positive mismatch.41–44,53,54 Furthermore, the loss of the peptide orientation in concentrated samples (as observed in both the 31P and 19F NMR spectra) indicates adaptation of the lipid to the essentially upright standing helix (Fig. 12, right panel).62 Nonetheless, in this study, we did not collect enough data to elucidate the lipid adaptation mechanism with full confidence.
Conclusions
With this comprehensive set of data, we fully confirmed our preliminary report on the existence of a TM PII helix.23 We found that this TM state of the helix exists in equilibrium with a submerged state of the peptide. The ratio between the helix length and the hydrophobic membrane thickness plays a pivotal role in the peptide alignment.The novel PII TM helix is an artificial peptide motif, and it is the first reported transmembrane peptide where the stability is not based on hydrogen bonding. Being more hydrophobic than natural proline, the chemically modified Oic analogue allowed us to construct such a new motif de novo, which has never been found in the proteome.
We believe that this design route will help to rationalize the chemical limitations of natural protein architectures. Furthermore, these novel peptide frameworks could contribute to a bottom-up design of artificial biological diversity, such as synthetic life, with an alternative chemistry. We recently speculated that from a strategical perspective, a proteome-wide exchange of the underlying secondary structure can give rise to a completely new biological World.12b After reporting a hydrophobic PII helix, our recent study on collagen mimicking peptides suggests a possibility for oligomeric PII assemblies in hydrophobic media.70 Therefore, our discovery of the TM PII helix opens up an avenue for engineering entirely new types of de novo membrane-associated peptide structures, not present in nature but deliberately constructed under laboratory conditions.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work of VK was supported by the DFG-funded research group FOR 1805. SG and AU gratefully acknowledge support of the NMR facility by an instrument grant (INST 121384/58-1) from DFG. VK thanks Dr Oleg Babii (KIT) for his help in the laboratory.Notes and references
- A. Vlassov, Orig. Life Evol. Biosph., 2005, 35, 135–149, DOI:10.1007/s11084-005-8901-9.
- S.-C. Li and C. M. Deber, Nat. Struct. Mol. Biol., 1994, 1, 368–373, DOI:10.1038/nsb0694-368.
- C. M. Deber and T. A. Stone, Pept. Sci., 2019, 111, e24075, DOI:10.1002/pep2.24075.
- M. R. R. De Planque, J. A. W. Kruijtzer, R. M. J. Liskamp, D. Marsh, D. V. Greathouse, R. E. Koeppe, II, B. de Kruijff and J. A. Killian, J. Biol. Chem., 1999, 274, 20839–20846, DOI:10.1074/jbc.274.30.20839.
- L. Li, I. Vorobyov and T. W. Allen, J. Phys. Chem. B, 2013, 117, 11906–11920, DOI:10.1021/jp405418y.
- E. Wexselblatt, J. D. Esko and Y. Tor, J. Org. Chem., 2014, 79, 6766–6774, DOI:10.1021/jo501101s.
- M. Eilers, S. C. Shekar, T. Shieh, S. O. Smith and P. J. Fleming, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 5796–5801, DOI:10.1073/pnas.97.11.5796.
- M. B. Ulmschneider and M. S. P. Sansom, Biochim. Biophys. Acta, Biomembr., 2001, 1512, 1–14, DOI:10.1016/S0005-2736(01)00299-1.
- J. W. Fairman, N. Noinaj and S. K. Buchanan, Curr. Opin. Struct. Biol., 2011, 21, 523–531, DOI:10.1016/j.sbi.2011.05.005.
- M. Crisma, F. Formaggio, A. Moretto and C. Toniolo, Pept. Sci., 2006, 84, 3–12, DOI:10.1002/bip.20357.
- A. A. Adzhubei, M. J. E. Sternberg and A. A. Makarov, J. Mol. Biol., 2013, 425, 2100–2132, DOI:10.1016/j.jmb.2013.03.018.
- (a) V. Kubyshkin and N. Budisa, Biotechnol. J., 2017, 12, 1600097, DOI:10.1002/biot.201600097; (b) V. Kubyshkin and N. Budisa, Curr. Opin. Biotechnol., 2019, 60, 242–249, DOI:10.1016/j.copbio.2019.05.006.
- A. A. Adzhubei and M. J. E. Sternberg, J. Mol. Biol., 1993, 229, 472–493, DOI:10.1006/jmbi.1993.1047.
- B. K. Kay, M. P. Williamson and M. Sudol, FASEB J., 2000, 14, 231–241, DOI:10.1096/fasebj.14.2.231.
- Z. Shi, C. A. Olson, G. D. Rose, R. L. Baldwin and N. R. Kallenbach, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 9190–9195, DOI:10.1073/pnas.112193999.
- J. Makowska, S. Rodziewicz-Motowidło, K. Bagińska, J. A. Vila, A. Liwo, L. Chmurzyński and H. A. Scheraga, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 1744–1749, DOI:10.1073/pnas.0510549103.
- F.-X. Theillet, L. Kalmar, P. Tompa, K.-H. Han, P. Selenko, A. K. Dunker, G. W. Daughdrill and V. N. Uversky, Intrinsically Disord. Proteins, 2013, 1, e24360, DOI:10.4161/idp.24360.
- V. Kubyshkin and N. Budisa, J. Pept. Sci., 2018, 24, e3076, DOI:10.1002/psc.3076.
- T. J. El-Baba, D. R. Fuller, D. A. Hales, D. H. Russell and D. E. Clemmer, J. Am. Soc. Mass Spectrom., 2019, 30, 77–84, DOI:10.1007/s13361-018-2034-7.
- V. Kubyshkin, P. Durkin and N. Budisa, New J. Chem., 2016, 40, 5209–5220, 10.1039/C5NJ03611A.
- V. Kubyshkin and N. Budisa, Org. Biomol. Chem., 2017, 15, 619–627, 10.1039/C6OB02306A.
- Alternative hydrophobic scaffold, oligosilaproline, has been reported in: (a) C. Martin, A. Lebrun, J. Martinez and F. Cavelier, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 3103–3109, DOI:10.1002/pola.26705; (b) C. Martin, B. Legrand, A. Lebrun, D. Berthomieu, J. Martinez and F. Cavelier, Chem. – Eur. J., 2014, 20, 14240–14244, DOI:10.1002/chem.201404820.
- V. Kubyshkin, S. L. Grage, J. Bürck, A. S. Ulrich and N. Budisa, J. Phys. Chem. Lett., 2018, 9, 2170–2174, DOI:10.1021/acs.jpclett.8b00829.
- P. Wilhelm, B. Lewandowski, N. Trapp and H. Wennemers, J. Am. Chem. Soc., 2014, 136, 15829–15832, DOI:10.1021/ja507405j.
- G. Berger, M. Vilchis-Reyes and S. Hanessian, Angew. Chem., Int. Ed., 2015, 54, 13268–13272, DOI:10.1002/anie.201506208.
- R. Bollhagen, M. Schmiedberger, K. Barlos and E. Grell, Chem. Commun., 1994, 2559–2560, 10.1039/C39940002559.
- P. K. Mykhailiuk, I. Kishko, V. Kubyshkin, N. Budisa and J. Cossy, Chem. – Eur. J., 2017, 23, 13279–13283, DOI:10.1002/chem.201703446.
- S.-Q. Peng, X.-W. Zhang, L. Zhang and X.-G. Hu, Org. Lett., 2017, 19, 5689–5692, DOI:10.1021/acs.orglett.7b02866.
- N. Kučerka, D. Uhríková, J. Teixeira and P. Balgavý, Phys. B, 2004, 350, e639–e642, DOI:10.1016/j.physb.2004.03.171.
- N. Kučerka, S. Tristram-Nagle and J. F. Nagle, J. Membr. Biol., 2006, 208, 193–202, DOI:10.1007/s00232-005-7006-8.
- N. Kučerka, M.-P. Nieh and J. Katsaras, Biochim. Biophys. Acta, Biomembr., 2011, 1808, 2761–2771, DOI:10.1016/j.bbamem.2011.07.022.
- R. Koynova and M. Caffrey, Biochem. Biophys. Acta, Rev. Biomembr., 1998, 1376, 91–145, DOI:10.1016/S0304-4157(98)00006-9.
- P. L. Gor'kov, E. Y. Chekmenev, C. Li, M. Cotten, J. J. Buffy, N. J. Traaseth, G. Veglia and W. W. Brey, J. Magn. Reson., 2007, 185, 77–93, DOI:10.1016/j.jmr.2006.11.008.
- L. He, A. E. Navarro, Z. Shi and N. Kallenbach, J. Am. Chem. Soc., 2012, 134, 1571–1576, DOI:10.1021/ja2070363.
- K.-Y. Huang and J.-C. Horng, J. Phys. Chem. B, 2019, 123, 138–147, DOI:10.1021/acs.jpcb.8b10864.
- V. Kubyshkin, S. Afonin, S. Kara, N. Budisa, P. K. Mykhailiuk and A. S. Ulrich, Org. Biomol. Chem., 2015, 13, 3171–3181, 10.1039/C5OB00034C.
- V. S. Kubyshkin, I. V. Komarov, S. Afonin, P. K. Mykhailiuk, S. L. Grage and A. S. Ulrich, Trifluoromethyl-Substituted α-Amino Acids as Solid State 19F-NMR Labels for Structural Studies of Membrane-Bound Peptides, in Fluorine in Pharmaceutical and Medicinal Chemistry: From Biophysical Aspects to Clinical Applications, ed. V. Gouverneur and K. Müller, Imperial College Press, 2012, pp. 91–138 Search PubMed.
- I. V. Komarov, S. Afonin and A. S. Ulrich, 19F-Labeled amino acids for NMR structure analysis of membrane bound peptides, in Fluorine in Life Sciences: Pharmaceuticals, Medicinal Diagnostics, and Agrochemicals, ed. G. Haufe and F. R. Leroux, Academic Press, 2019, 349–395 Search PubMed.
- B. Bechinger and S. Opella, J. Magn. Reson., 1991, 95, 585–588, DOI:10.1016/0022-2364(91)90173-Q.
- A. S. Ulrich, Prog. Nucl. Magn. Reson. Spectrosc., 2005, 46, 1–21, DOI:10.1016/j.pnmrs.2004.11.001.
- S. H. Park and S. J. Opella, J. Mol. Biol., 2005, 350, 310–318, DOI:10.1016/j.jmb.2005.05.004.
- T. Kim and W. Im, Biophys. J., 2010, 99, 175–183, DOI:10.1016/j.bpj.2010.04.015.
- E. Strandberg, S. Esteban-Martín, A. S. Ulrich and J. Salgado, Biochim. Biophys. Acta, Biomembr., 2012, 1818, 1242–1249, DOI:10.1016/j.bbamem.2012.01.023.
- A. J. de Jesus and T. W. Allen, Biochim. Biophys. Acta, Biomembr., 2013, 1828, 851–863, DOI:10.1016/j.bbamem.2012.09.012.
- E. Strandberg, D. Tiltak, S. Ehni, P. Wadhwani and A. S. Ulrich, Biochim. Biophys. Acta, Biomembr., 2012, 1818, 1764–1776, DOI:10.1016/j.bbamem.2012.02.027.
- E. Strandberg and A. S. Ulrich, Biochim. Biophys. Acta, Biomembr., 2015, 1848, 1944–1954, DOI:10.1016/j.bbamem.2015.02.019.
- D. Marsh, Biophys. J., 2007, 93, 3884–3899, DOI:10.1529/biophysj.107.107938.
- A. Kessel, N. Ben-Tal and S. May, Biophys. J., 2001, 81, 643–658, DOI:10.1016/S0006-3495(01)75729-3.
- N. P. Franks, J. Mol. Biol., 1976, 100, 345–358, DOI:10.1016/S0022-2836(76)80067-8.
- D. L. Worcester and N. P. Franks, J. Mol. Biol., 1976, 100, 359–378, DOI:10.1016/S0022-2836(76)80068-X.
- L. J. Libertini, A. S. Waggoner, P. C. Jost and O. H. Griffith, Proc. Natl. Acad. Sci. U. S. A., 1969, 64, 13–19, DOI:10.1073/pnas.64.1.13.
- Y. Gambin, M. Reffay, E. Sierecki, F. Homblé, R. S. Hodges, N. S. Gov, N. Taulier and W. Urbach, J. Phys. Chem. B, 2010, 114, 3559–3566, DOI:10.1021/jp911354y.
- J. A. Killian, FEBS Lett., 2003, 555, 134–138, DOI:10.1016/S0014-5793(03)01154-2.
- A. Holt and J. A. Killian, Eur. Biophys. J., 2010, 39, 609–621, DOI:10.1007/s00249-009-0567-1.
- H. W. Huang and Y. Wu, Biophys. J., 1991, 60, 1079–1087, DOI:10.1016/S0006-3495(91)82144-0.
- D. Maisch, P. Wadhwani, S. Afonin, C. Böttcher, B. Koksch and A. S. Ulrich, J. Am. Chem. Soc., 2009, 131, 15596–15597, DOI:10.1021/ja9067595.
- T. Nagao, D. Mishima, N. Javkhlantugs, J. Wang, D. Ishioka, K. Yokota, K. Norisada, I. Kawamura, K. Ueda and A. Naito, Biochim. Biophys. Acta, Biomembr., 2015, 1848, 2789–2798, DOI:10.1016/j.bbamem.2015.07.019.
- D. W. Urry, Proc. Natl. Acad. Sci. U. S. A., 1971, 68, 672–676, DOI:10.1073/pnas.68.3.672.
- R. R. Ketchem, B. Roux and T. A. Cross, Structure, 1997, 5, 1655–1669, DOI:10.1016/S0969-2126(97)00312-2.
- B. Roux, Acc. Chem. Res., 2002, 35, 366–375, DOI:10.1021/ar010028v.
- M.-C. Gagnon, E. Strandberg, A. Grau-Campistany, P. Wadhwani, J. Reichert, J. Bürck, F. Rabana, M. Auger, J.-F. Paquin and A. S. Ulrich, Biochemistry, 2017, 56, 1680–1695, DOI:10.1021/acs.biochem.6b01071.
- J. A. Killian, Biochim. Biophys. Acta, Rev. Biomembr., 1998, 1376, 401–416, DOI:10.1016/S0304-4157(98)00017-3.
- J. Ren, S. Lew, Z. Wang and E. London, Biochemistry, 1997, 36, 10213–10220, DOI:10.1021/bi9709295.
- J. Ren, S. Lew, J. Wang and E. London, Biochemistry, 1999, 38, 5905–5912, DOI:10.1021/bi982942a.
- E. R. Stockwald, L. M. E. Steger, S. Vollmer, C. Gottselig, S. L. Grage, J. Bürck, S. Afonin, J. Fröbel, A.-S. Blümmel, J. Setzler, W. Wenzel, M. Müller, A. S. Ulrich and T. H. Walther, submitted.
- A. H. Clayton and W. H. Sawyer, Eur. Biophys. J., 2002, 31, 9–13, DOI:10.1007/s002490100182.
- M. Bulacu, X. Périole and S. J. Marrink, Biomacromolecules, 2012, 13, 196–205, DOI:10.1021/bm201454j.
- N. J. Gleason, V. V. Vostrikov, D. V. Greathouse, C. V. Grant, S. J. Opella and R. E. Koeppe, II, Biochemistry, 2012, 51, 2044–2053, DOI:10.1021/bi201732e.
- K. A. Sparks, N. J. Gleason, R. Gist, R. Langston, D. V. Greathouse and R. E. Koeppe, II, Biochemistry, 2014, 53, 3637–3645, DOI:10.1021/bi500439x.
- V. Kubyshkin and N. Budisa, Org. Biomol. Chem., 2019, 17, 2502–2507, 10.1039/c9ob00070d.
Footnotes |
| † Electronic supplementary information (ESI) available: Further details on peptide synthesis; analytical chromatograms and 1H NMR spectra for peptides. See DOI: 10.1039/c9cp02996f |
| ‡ The ρ angle can be visualized as the angle needed to turn the helix around its axis until the tilt vector points towards the 5th Cα-carbon. The tilt vector is orthogonal to the helix axis and points to the direction in which the C-terminus is slanted, see Fig. 4A for graphical representation. |
| § We note at this point that for a peptide in an upright orientation, the splitting value should remain the same regardless of the helical length. |
| ¶ Here, we would like to note that the 19F NMR splittings in the SM state of peptides 1, 12 (three residues added at the N-terminal section), and 13 (three residues added at the C-terminal section) are the same. This observation directly confirms the 3.0 residue/turn periodicity of the PII helix in the SM state(s) of the peptides. |
| This journal is © the Owner Societies 2019 |