
How flexible is the disulfide linker? A combined rotational–computational investigation of diallyl disulfide†

Abstract
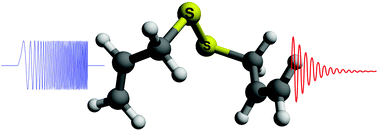
The symmetrically substituted diallyl disulfide adopts a non-symmetric conformation in the gas-phase, as observed with supersonic-jet rotational spectroscopy. The determination of the equilibrium structure with a predicate mixed regression illustrates both the benefits of the mass-dependent method for moderately large molecules and the structural peculiarities of the disulfide bridge.
- This article is part of the themed collection: 2019 PCCP HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

