
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chemical equilibria of aqueous ammonium–carboxylate systems in aqueous bulk, close to and at the water–air interface†
Yina Salamanca
Blanco‡
a,
Önder
Topel§
 a,
Éva G.
Bajnóczi
a,
Josephina
Werner
ab,
Olle
Björneholm
b and
Ingmar
Persson
*a
a,
Éva G.
Bajnóczi
a,
Josephina
Werner
ab,
Olle
Björneholm
b and
Ingmar
Persson
*a
aDepartment of Molecular Sciences, Swedish University of Agricultural Sciences, P.O. Box 7015, SE-750 07 Uppsala, Sweden. E-mail: ingmar.persson@slu.se
bDepartment of Physics and Astronomy, Uppsala University, P.O. Box 516, SE-751 20 Uppsala, Sweden
First published on 22nd May 2019
Abstract
Previous studies have shown that the water–air interface and a number of water molecule layers just below it, the surface region, have significantly different physico-chemical properties, such as lower relative permittivity and density, than bulk water. The properties in the surface region of water favor weakly hydrated species as neutral molecules, while ions requiring strong hydration and shielding of their charge are disfavored. In this study the equilibria NH4+(aq) + RCOO−(aq) ⇌ NH3(aq) + RCOOH(aq) are investigated for R = CnH2n+1, n = 0–8, as open systems, where ammonia and small carboxylic acids in the gas phase above the water surface are removed from the system by a gentle controlled flow of nitrogen to mimic the transport of volatile compounds from water droplets into air. It is shown that this non-equilibrium transport of chemicals can be sufficiently large to cause a change of the chemical content of the aqueous bulk. Furthermore, X-ray photoelectron spectroscopy (XPS) has been used to determine the relative concentration of alkyl carboxylic acids and their conjugated alkyl carboxylates in aqueous surfaces using a micro-jet. These studies confirm that neutral alkyl carboxylic acids are accumulated in the surface region, while charged species, as alkyl carboxylates, are depleted. The XPS studies show also that the hydrophobic alkyl chains are oriented upwards into regions with lower relative permittivity and density, thus perpendicular to the aqueous surface. These combined results show that there are several chemical equilibria between the aqueous bulk and the surface region. The analytical studies show that the release of mainly ammonia is dependent on its concentration in the surface region, as long as the solubility of the carboxylic acid in the surface region is sufficiently high to avoid a precipitation in/on the water–air interface. However, for n-octyl- and n-nonylcarboxylic acid the solubility is sufficiently low to cause precipitation. The combined analytical and surface speciation studies in this work show that the equilibria involving the surface region are fast. The results from this study increase the knowledge about the distribution of chemical species in the surface region at and close to the water–air interface, and the transport of chemicals from water to air in open systems.
Introduction
The effects of atmospheric aerosols are among the major uncertainties for climate modeling.1 Due to the small size of atmospheric aerosol particles, surface effects are important for their properties.2 Two important examples of this are cloud droplet formation from aerosol particles called cloud condensation nuclei,3 and heterogeneous reactions at aerosol surfaces.4 Organic compounds are often surface active, and there is a large variety of oxidized organic compounds present in the atmosphere.5 Many of these originate from reduced organic compounds with low water solubility, which due to the oxidizing properties of the atmosphere are readily oxidized to substantially more water soluble compounds.6 There is very strong evidence that atmospheric particles contain surface active compounds accumulated in the surface region lowering the surface tension.7–13 The accumulation of neutral organic compounds in the surface region may also have an impact on transport across the surface region. Evidences have been presented that accumulation of neutral organic compounds in the surface region hamper water evaporation from atmospheric droplets.14,15 Oxidized organic compounds are readily accumulated in atmospheric water droplets,16 seen in their relatively large Henry's law constants.17 The water droplets in clouds are small with a mean diameter of 0.010–0.015 mm,18 with consequently large surface area![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :bulk volume ratio. Surfaces and interfaces, where species from the two media are exchanged and react with each other, are well known for their special chemistry,19–21 and surface propensive compounds will have increasing effect of the physico-chemical properties of atmospheric water droplets with decreasing size. This means that accumulation or depletion of specific compounds becomes increasingly important with decreasing droplet size, as well as for the chemical reactions preferably taking place in the surface region or at the water–air interface. It has for example been shown that the hygroscopicity and cloud formation potential of secondary organic aerosols (SOA) change with the oxidation state of the surface species,2,3,22,23 and SOA coatings on aerosols and cloud droplets/crystals can modify their chemical and physical properties.24
:bulk volume ratio. Surfaces and interfaces, where species from the two media are exchanged and react with each other, are well known for their special chemistry,19–21 and surface propensive compounds will have increasing effect of the physico-chemical properties of atmospheric water droplets with decreasing size. This means that accumulation or depletion of specific compounds becomes increasingly important with decreasing droplet size, as well as for the chemical reactions preferably taking place in the surface region or at the water–air interface. It has for example been shown that the hygroscopicity and cloud formation potential of secondary organic aerosols (SOA) change with the oxidation state of the surface species,2,3,22,23 and SOA coatings on aerosols and cloud droplets/crystals can modify their chemical and physical properties.24
In a series of studies using X-ray photoelectron spectroscopy (XPS) on liquid micro-jets, the surface propensity of various compounds in aqueous solution have been studied.25–34 It is evident that the surface propensity of carboxylic acids, alkyl amines and alcohols increases with increasing length and branching of the alkyl chain.26–28,32–34 This implicates that a number of equilibria of carboxylic acids and carboxylate ions are involved in the distribution of chemical species in the aqueous bulk, and between aqueous bulk and surface region, surface region and gas phase and surface region and floating solids, Fig. 1. The reported surface propensity studies indicate clearly that the physico-chemical properties of water as solvent in the surface region are significantly different from those in the aqueous bulk including disrupted hydrogen bonding between water molecules, necessary very close to the water–air interface, and lower relative permittivity and density. It has been shown by ellipsometric35 and X-ray reflectivity36,37 measurements that the surface region, defined as the region of the water surface with a density range of 0.1–0.9 g cm−3, is ca. 8 Å at room temperature. Theoretical simulations have reported somewhat smaller thickness.38–42 As the density increases from close to zero in the air–water interface to 1.00 g cm−3 in the aqueous bulk, it can be assumed that the permittivity increases gradually in the same way from 1.0006 in air,43 to 78.32 in pure water at room temperature (298.15 K).44 This means that the surface region from the water–air interface (even though the water surface is rough) with a relative permittivity close to 1.0 at the water–air interface to the aqueous bulk should be in the order of 10–15 Å based on experimental data. As a consequence, neutral molecules prefer to be present in the surface region (sr, a number of water molecule layers just below the water–air interface), while ions seem not to be able to reach close to the water–air interface unless ion-pairs are formed shielding their effective charge. Nevertheless, ion-pairs are not capable to reach as close to the water–air interface as neutral molecules.29–31
 | ||
| Fig. 1 Chemical equilibria, here represented by a carboxylic acid, in the aqueous bulk and its coupled equilibria to the aqueous surface, the water–air interface and air. In the aqueous bulk there may be a solubility equilibrium when the compound is not miscible with water, (solubility product with salt). As the surface region has different physico-chemical properties than bulk water there is an equilibrium between these two regions, Ksrd. For compounds which are accumulated in the surface region Ksrd is larger than unity, while the opposite is true for surface depleted compounds. There is also an equilibrium between the surface region and the gas phase above it, normally air. Ksd, The product of the equilibria Ksrd and Ksd is known as the Henry's law constant, KH. The Henry's law constant is accurately determined for a vast number of compounds, ref. 17, while the individual values Ksrd and Ksd are normally not. For compounds with low solubility, and lower density than aqueous bulk and the surface region, the solubility, ssd, is expected to be different from the solubility in aqueous bulk. The short-dashed line represents the border where water has water bulk properties, and the long-dashed line the estimated level in the surface region to which monovalent ions can reach. | ||
Of the equilibria in Fig. 1, the solubility, s, of a neutral compound, or the solubility product, Ks, of a salt, in the aqueous bulk, have been determined with very high accuracy, as well as their temperature and ionic medium dependence. The Henry's law constants, KH, HA(g) ⇌ HA(aq), are almost equally well characterized.17 However, they are a combination of at least two equilibria, HA(aq) ⇌ HA(sr) and HA(sr) ⇌ HA(g) of which individual values have not been determined. The equilibrium constants between aqueous bulk and surface region are possible to estimate from e.g. surface tension or surface propensity studies using XPS.27,28,32,33 It has been shown in a recent study that surface tension can be studied in very small aqueous droplets.45 It is shown for very small water droplets that organic compounds accumulated in the surface region decrease the surface tension significantly, while the presence of inorganic salts as sodium chloride increases it.45
Most chemical species require water molecules in all three dimensions for efficient hydration, such as the carboxylate, phosphate and sulfate ions,46,47 and they will be increasingly depleted from the water–air interface with increasing hydration strength as approaching this region decreases the possibility for complete hydration,26,48 mainly due to entropy effects, and thereby chemical stability. However, ions with strong hydration in only two dimensions, as the guanidinium and benzoate ions, can enter a region closer to the water–air interface being oriented parallel to it.28,30,49 However, they still need to be accompanied by a counter ion to be able to reach a position in the surface region close to the water–air interface, and they never seem to reach as close to the water–air interface as the conjugated neutral form.50
Different composition of the surface region and the aqueous bulk is of great importance especially to physically small systems, such as liquid/aqueous atmospheric aerosols. Atmospheric water droplets contain often both inorganic and organic compounds.51,52 It has been shown that the concentration of inorganic salts has a strong influence on the surface propensity of organic acids, such as succinic acid.32 XPS studies have shown that neutral molecules, e.g. alcohols or carboxylic acids, yield much higher concentration in the surface region than the solubility or actual concentration in the aqueous bulk.27,32–34,53 The main reasons are that water loses its well-defined rigid three-dimensional structure close to the water–air interface and thereby many of its physico-chemical properties, and that alkyl and aryl group(s) of surface active compounds can be oriented into air and/or integrated with single water molecules from weakly hydrogen-bound surface water molecules instead of forcing the aqueous bulk structure to rearrange around them as in bulk water. This causes that weakly hydrated molecules can be accumulated at rather high concentrations in the surface region compared to the aqueous bulk. Furthermore, it is proven that carboxylic acids with an alkyl group with at least seven carbons can self-assemble into vesicle structures,54 and it is possible that carboxylic acids with even shorter alkyl chains can form micelle-like structures in or close to the surface region.
Two important classes of compounds known to be present in atmospheric aerosols are the corresponding acid–base pairs: carboxylic acid–carboxylate ion and ammonium ion-ammonia/amine. These can participate in the following proton-transfer reaction:
| NH4+(aq) + RCOO−(aq) ⇌ NH3(aq) + RCOOH(aq); K ≈ 10−4.5 | (1) |
The proton-transfer reactions of eqn (1) have equilibrium constants in aqueous solution of ca. 10−4.5, using molar concentrations, calculated from the acidic constants of the ammonium ion and the carboxylic acids at 25 °C;62 the acidity constants of the alkyl carboxylic acids are summarized in Table S1 (ESI†). Thus, less than one percent of the charged reactants of eqn (1) will be converted to neutral products at equilibrium in aqueous solutions containing equal start concentrations of the reactants. The large Henry's law constants of ammonia and carboxylic acids, KH = 60 and >103 M atm−1, respectively, Table 1, make the expected equilibrium concentrations of ammonia and carboxylic acid in the gas phase very small above a 0.10 mol dm−3 solution, ca. 9 × 10−4 and 1.3 × 10−5% of the total ammonia/ammonium and acetic acid/acetate concentration, respectively, Table 1 and Table S1 (ESI†). Consequently, gaseous carboxylic acids and ammonia in the atmosphere will to a large extent dissolve in water droplets and react almost completely with each other, more than 99.4%, will form ammonium and carboxylate ions dissolved in water according to the physico-chemical data recorded in aqueous bulk, reverse of eqn (1), Table 1. The short-chained carboxylic acids, formic, acetic, propionic and butyric acid, are fully miscible with water, while the solubility of carboxylic acids with longer alkyl chains decreases sharply with increasing alkyl chain length, Table 1. Carboxylic acids, especially those with long alkyl chains, have strong surface propensity and are accumulated in the surface region.25–28,32,53 The carboxylic acid group is only weakly hydrated as the charge densities on the hydrogen and oxygen atoms are low, and the number of hydrating water molecules is significantly lower than for the conjugated carboxylate ion.63 The heat of hydration of acetic acid is only −24.87 kJ mol−1 (calculated from the heat of solution in water, −1.51 kJ mol−1,64 and heat of vaporization of acetic acid, +23.36 kJ mol−1,64 respectively) while it is −369 kJ mol−1 for the acetate ion (calculated from the lattice energy of NaCH3COO, −761 kJ mol−1,65 the heat of solution in water of NaCH3COO, −17.3 kJ mol−1,64 and the heat of hydration of the sodium ion, −409 kJ mol−165). The same pattern in heats of hydration is found for ammonia and the ammonium ion, −30.5 and −329 kJ mol−1, respectively.64,66
| Common name | IUPAC name | Formula | K H , | Solubility |
|---|---|---|---|---|
| a Ref. 17. b Selected mean value. | ||||
| Formic acid | Methanoic acid | HCOOH | 8.9 × 103 | Miscible |
| Acetic acid | Ethanoic acid | CH3COOH | 4.1 × 103 | Miscible |
| Propionic acid | Propanoic acid | C2H5COOH | 4.0 × 103 | Miscible |
| Butyric acid | Butanoic acid | n-C3H7COOH | 2.7 × 103 | Miscible |
| Valeric acid | Pentanoic acid | n-C4H9COOH | 1.8 × 103 | 0.487 |
| Caproic acid | Hexanoic acid | n-C5H11COOH | 1.5 × 103 | 0.0856 |
| Enanthic acid | Heptanoic acid | n-C6H13COOH | 1.3 × 103 | 0.0198 |
| Caprylic acid | Octanoic acid | n-C7H15COOH | 1.1 × 103 | 0.0051 |
| Pelargonic acid | Nonanoic acid | n-C8H17COOH | 7.0 × 102 | 0.0018 |
| Ammonia | Ammonia | NH3 | 60 | 5.55 |
Sum Frequency Generation (SFG) spectroscopic studies have shown that ammonia forms weak hydrogen bonds to surface water molecules in aqueous solution, and that the OH stretching of water is suppressed at high ammonia concentrations.67,68 Furthermore, a series of theoretical simulations has shown that ammonia is hydrogen bound and present in the surface region of aqueous soutions.69–71 This supports that the ammonia concentration is enhanced in the surface region in comparison to the aqueous bulk.
In this study the NH4+(aq) + RCOO−(aq) ⇌ NH3(aq) + RCOOH(aq) equilibria have been studied as open systems at 298 K for carboxylates with 0–8 carbons in the alkyl chain to get increased understanding of the chemical equilibria involved in the transport of chemical species in eqn (1) from the aqueous bulk to the surface region, and further into the gas phase with the help of analytical (pH) and XPS measurements. The applied open system mimics the transport from water droplets into air by constant removal of gaseous or solid products, in this case ammonia and carboxylic acids. One shall of course keep in mind that in a real system there can be a similar transport in the opposite direction when air contains e.g. ammonia and carboxylic acid. XPS studies on aqueous surfaces have been performed to get increased understanding how close to the water–air interface different chemical species can reach, whether chemical compounds are accumulated in or depleted from the surface region of an aqueous solution, and the orientation of the molecules in the surface region.
Experimental
Chemicals
Nitric acid (Merck, 65%), ammonium nitrate, NH4NO3 (Merck, ≥99%), sodium hydroxide, NaOH (Sigma, >99.5%), sodium formate, NaHCOO (Sigma, capillary GC, ≥99%), formic acid, HCOOH (Merck, >98%), acetic acid, CH3COOH, propionic acid, C2H5COOH, butyric acid, n-C3H7COOH, pentanoic acid (valeric acid), n-C4H9COOH, hexanoic acid (caproic acid), n-C5H11COOH, heptanoic acid (enanthic acid), n-C6H13COOH, octanoic acid (caprylic acid), n-C7H15COOH, and nonanoic acid (pelargonic acid), n-C8H17COOH, (all Sigma-Aldrich, ≥99%) were used as purchased. The corresponding sodium carboxylates except sodium formate were synthesized in our laboratory, vide infra. The carboxylic acids purchased were all found to be pure according to the given specifications, while sodium carboxylates, as e.g. sodium butyrate, often contained significant amounts of impurities not possible to remove by recrystallization.Preparation of sodium carboxylates
Sodium carboxylates (R-COONa, R = CnH2n+1, n = 0–8) were prepared by adding a 5.00 mol dm−3 sodium hydroxide solution drop-wise to an aqueous solution of the corresponding carboxylic acid. The pH in the reaction mixture was followed by an Orion Star A211 pH meter, equipped with an Orion Ross Combination electrode. The equivalence point was calculated from previously recorded pH titration curves to determine the actual pKa value at the very same conditions as in the preparation titration. The resulting sodium carboxylates were precipitated by evaporating off all water and they were finally dried in an oven at 333 K. The purity was verified using 1H-NMR. All solids were kept in a dry atmosphere in desiccator until use.Preparation of aqueous solutions
For each compound, three stock solutions, 0.20, 0.50 and 1.00 mol dm−3, were prepared by dissolution in de-ionized water further purified by a Milli Q Plus Ultrapure water system. Solutions for the experiments were prepared by mixing equal volumes of sodium carboxylate and ammonium nitrate stock solutions with the same concentration. For the sodium alkyl carboxylates with a solubility lower than 1.00 mol dm−3, the 0.50 mol dm−3 solutions were prepared by adding the appropriate amount of ammonium nitrate.Experimental set-up and instruments for the pH measurements
The experimental set-up is shown in Fig. 2. It consists of three jacketed glass vessels (Methrohm) through which thermostated water (298.15 ± 0.07 K) continuously flowed, and the cell compartments had in-let and out-let for a 4.0 mL s−1 continuous flow of nitrogen gas, controlled by a Kojima Kofloc M2204 inlet pressure regulator valve (Kojima Instruments Inc., Japan), which was applied over the solutions with the aim to transport away the formed gaseous compounds, i.e. ammonia or small carboxylic acids. The nitrogen gas was bubbled through a washing flask (vessel 1) with a solution of ammonium nitrate and sodium chloride with the same concentrations of ammonium nitrate and sodium alkyl carboxylate as in vessel 2 before entering vessel 2 to secure that the moisture level of the incoming nitrogen gas was the same as the one leaving vessel 2 to avoid evaporation. Vessel 2 contained the studied samples of 0.10, 0.25 or 0.50 mol dm−3 aqueous solutions of ammonium carboxylate and sodium nitrate. The ammonia and carboxylic acid formed in vessel 2 was trapped in 0.004 mol dm−3 nitric acid or 0.002 mol dm−3 aqueous solution of sodium carbonate, respectively, in vessel 3 to form non-volatile salts. To monitor the amount of ammonia or carboxylic acid trapped in vessel 3 the pH was recorded continuously using a conventional ROSS pH electrode (Thermo Scientific) attached to a Orion Star A211 (Thermo Scientific) pH-meter with read-outs at every fifth minute. The electrode was calibrated using buffer reference standards of pH 4.01 ± 0.01, 7.00 ± 0.01 and 10.00 ± 0.01 (Sigma-Aldrich) at 298 K. Gentle magnetic stirring was applied with an arbitrary but constant speed (vide infra) in all vessels to ensure proper mixing. 20.0 cm3 solution was applied in each vessel. The reactions were followed for 24 hours or longer. To assess the reproducibility of the studied reactions and to evaluate the limitations of the method at least three independent measurements with the same concentration were performed. The reproducibility was better than 2%, Fig. 3A. | ||
| Fig. 2 The applied experimental setup. Vessel no. 1 always contained an appropriate salt solution to saturate the moisture content of the N2 gas, no. 2 contained the studied solution and the pH was measured in the trapping vessel, no. 3. | ||
 | ||
| Fig. 3 (A) Formation of ammonia as function of time for the ammonium–octanoate system at a starting concentration of 0.50 mol dm−3 in four separate experiments. (B) Formation of ammonia as function of time for the ammonium–butyrate system at a start concentrations of 0.10, 0.25 and 0.50 mol dm−3. | ||
A set of blank background experiments were also performed, where the reaction vessel (no. 2) was empty, the water-saturated N2 bubbled directly to vessel 3, where the pH was measured. These blank experiments were necessary to perform as the change in the measured pH due to the diffusion through the porous membrane of the pH electrode cannot be neglected during long experiments lasting up to a day or longer.
Optimization of gas flow and stirring parameters
The experimental set-up parameters for the open system, the nitrogen flow rate and the stirring rate in vessel 2, were optimized in several steps. Three different flow rates were tested, and it was found that the nitrogen flow rate affects the rate of the amount of ammonia transported from the reaction vessel to a large extent. A higher flow rate pushes the ammonia and carboxylic acid formed more efficiently from vessel 2 to vessel 3. A nitrogen flow rate with 4.00 mL s−1 was chosen as working flow. This standard flow rate secured that a continuous flow was achieved and the volumes in the vessels were constant during experiments lasting up to ten days. It is of utmost importance to have a constant nitrogen flow rate as only small variations in the flow rate will affect the reproducibility of the measurements. A stable gas flow was assured by an inlet pressure regulator valve, vide supra. The effect of the stirring rate on the system was also studied at two nitrogen flow rates. The amount of formed ammonia or carboxylic acid was found to be independent of the stirring rate.XPS experiments
The XPS experiments were performed at the soft X-ray beamline I411 at the Swedish national synchrotron facility MAX-lab, Lund University. Details of the experimental setup and conditions can be found elsewhere.26,31,33,34,72,73 The carboxylic acids and their corresponding sodium salts were dissolved in demineralized water (the electrical resistivity of the water was 18.2 MΩ cm, Millipore DirectQ). Prior to each experiment the sample solutions were filtered (Whatman Puradisc FP30 syringe filters, 1.2 μm pore size) to remove dust particles, which may disturb the liquid micro-jet and cause the injection system to fail.In all experiments, the carboxylic acid abundance was monitored via the intensity of the carbon 1s photoemission line using a photon energy of 360 eV, at which also the 1b1 valence spectrum of liquid water was recorded. The latter was used to calibrate the kinetic energy scale by aligning the 1b1 photoemission line of liquid water to 11.16 eV.74 At the chosen photon energy, the kinetic energy of the emitted C 1s electrons is between 65 and 70 eV. This results in highly surface sensitive XPS experiments, due to short mean effective attenuation length of the emitted photoelectrons reaching the detector (∼1 nm).75 Furthermore, the valence spectrum of the cleaning solution (50 mmol dm−3 aqueous solution of NaCl) between each sample was recorded and relative deviations in the intensity of the 1b1 line were used to quantify instabilities of the experimental setup and the X-ray source. For constant reference intensities (with max. deviations of a few %), the recorded XPS intensities of different sample solutions can be compared. The C 1s signal of the formate ion was used as reference between different experimental runs.
Generally, the recorded intensity in the XPS measurements at a given kinetic energy of the emitted photoelectrons can be directly related to the concentration of a chemical species in the surface region and the photoionization cross section of the given species, but exponentially attenuated along its path, which can be quantified by the photoelectron's effective attenuation length. Due to these dependencies, the obtained signal is dominated by the species present in the air–water interface and the upper part of the surface region, but it also contains small contributions from the species in the aqueous bulk. The absolute amount of a certain species in the detected volume is generally not available from XPS spectra. However, qualitative information can be obtained from ratios of XPS intensities of the same element. In this study the C 1s intensities of the different carboxylic acids and the conjugated carboxylates are related to one another. For this purpose all C 1s spectra were intensity normalized against the photon flux and acquisition time. The spectral intensities were also normalized by dividing them by the bulk concentration and number of carbon atoms in the alkyl chain of the investigated chemical species for a proper comparison.
Results
Evaporation of ammonia and carboxylic acids from aqueous solutions
To investigate the development of the coupled equilibria NH4+(aq) + RCOO−(aq) ⇌ NH3(aq) + RCOOH(aq), NH3(aq) ⇌ NH3(g) and RCOOH(aq) ⇌ RCOOH(g) the amounts of evaporated ammonia and carboxylic acid over time were determined during 24 hours, and in some cases up to 10 days. In some cases the change in concentration of alkyl carboxylic acid in the aqueous solution was also checked by 1H NMR in the aqueous bulk after completed experiment. The first step was to check the long-term stability of the measurement procedure. The performed experiments show clearly the importance of blank experiments including drift of the pH electrode over time. Experiments with the reaction vessel empty showed a small but significant drift in the recorded pH due to the diffusion through the porous membrane of the pH electrode, which cannot be neglected during such long measurements as in this case, and it needs to be adjusted for in the data analyses, Fig. 4 (black symbols). The observed increase in pH in the nitric acid solution due to the drift of the pH electrode was almost linear with time, Fig. 4A, while in the sodium carbonate solution a slight non-linearly decrease in pH with time was observed, Fig. 4C. Thus, the drift of the pH electrode causes an observed increase in pH on the acidic side and a decrease on the alkaline side with time. The blank experiments of released ammonia per hour show a decrease with time according to an exponential function, but after ca. 10 hours a steady-state situation with constant release per hour has been established, Fig. 4A and B. For acetic acid the evaporation is very small and becomes linear with time, Fig. 4C and D. The amount of the released acetic acid is ∼0.15% per day of the initial acetic acid content. The amounts of released ammonia and acetic acid were determined on the same solutions and the same conditions but with different trapping solution (vessel 3). The amount of acetic acid evaporating from the aqueous solution is approximately the 1/8 of the amount of released ammonia. This is a larger fraction than expected from the values of the Henry's law constant, and may depend on different kinetic behavior.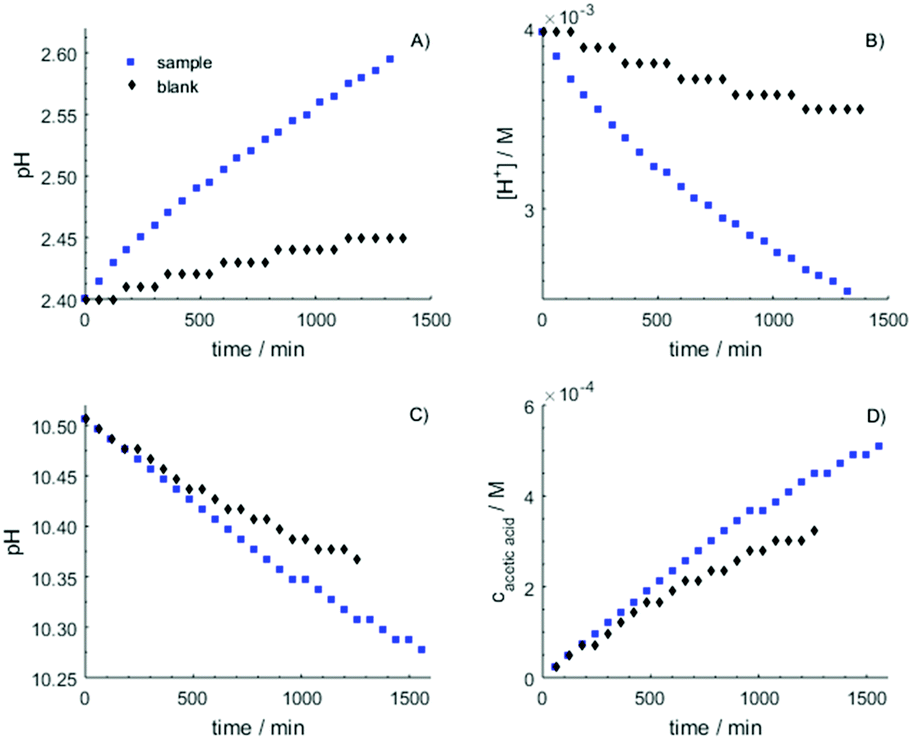 | ||
| Fig. 4 A complete set of measurements for the detection of the forming ammonia and carboxylic acid demonstrated on the ammonium acetate system. (A) and (C) show the measured pH values as function of time, while (B) and (D) present the respective calculated concentrations in vessel no. 3. | ||
Reactions between ammonium and n-alkyl carboxylate ions in an open system
The reactions between n-alkyl carboxylate, R-COO−, R = CnH2n+1, n = 3 and 7, and ammonium ions were followed via the measurement of released ammonia for four and seven days, respectively, to determine the required time to reach a steady state situation in an open system, when the amounts of formed and released ammonia and carboxylic acid reach a steady-state situation, thus the amount released is only dependent on time and possibly total concentration in the aqueous bulk. For the remaining systems, n = 0–2 and 4–6, the reactions were followed during 24 hours. To reach a steady state situation with a constant amount of ammonia formed with time takes less than 1 and 4 days for the ammonium–butyrate and –n-octanoate systems, respectively, Fig. 3A and B. The amounts of ammonia formed after one day in an open system are as expected much larger than the amounts formed in a closed, equilibrated system. This shows that the equilibria in the surface region and over the water surface region–air interface are fast, and the transport of chemicals to and from the surface region can be significant.In the case of the ammonium n-octanoate and n-nonanoate systems the rate of the ammonia release was significantly faster than for the systems involving alkyl carboxylates with six or less carbon atoms in the alkyl chain. Furthermore, the reaction rates increase exponentially with increasing concentration, except for the lowest concentration of the n-octanoate system, which behaves as the other carboxylate systems studied, Table S2 (ESI†). The solubilities of n-octanoic and n-nonanoic acid are very low in water, Table 1. After saturation of the surface region only very small amounts can be dissolved in the aqueous bulk, and thereafter they start to precipitate on the aqueous surface. This means that both ammonia and n-octanoic/n-nonanoic acid will leave the system pushing eqn (1) strongly to the right. A crust of n-octanoic acid was visible after a couple of days, and after a week the entire water surface was covered, Fig. S1 (ESI†). The formation of solid n-octanoic and n-nonaoic acid takes obviously place when both the surface region and the aqueous bulk are saturated. At these conditions when both ammonia and carboxylic acid leave the aqueous system, a much higher reaction rate is observed in comparison when only one compound, e.g. ammonia, leaves the system in significant amounts, Fig. 5.
 | ||
| Fig. 5 The initial reaction rate of the ammonia release as function of initial ammonium carboxylate and the number of carbon atoms in the alkyl chain at 298.15 K and at 4.0 mL s−1 N2 flow rate. | ||
The principal reaction behavior of eqn (1) can be divided into two major groups, alkyl carboxylates with six or less carbon atoms in the alkyl chain, and those with longer alkyl chains. For the alkyl carboxylates with short alkyl chains the reaction rates are proportional to the total concentration, while for the n-octanoate and n-nonanoate the reaction rate increase exponentially with concentration. The initial reaction rates are given in Fig. 5, with the reaction rate after 24 hours in Fig. S2 (ESI†); the reaction rates are summarized in Table S2 (ESI†). The ammonium formate system displays the slowest reaction rate, 0.6% of the ammonium is converted to ammonia in 24 hours, indicating that formic acid is less prone than the other carboxylic acids to enter the surface region. The ammonium acetate, propionate, butyrate, pentanoate, hexanoate and heptanoate systems have similar behavior with 1.0–1.7% of the total amount of ammonium leaving the system as ammonia within the first 24 hours, and a minor fraction carboxylic acid as well. The carboxylic acids have been shown in previous XPS studies to have strong surface propensity and are accumulated in the surface region. Thus, the Ksrd constant is larger than unity, and it can be expected that it increases with increasing length of the alkyl chain, while the Ksd constant is expected to decrease, ending up with slightly decreasing Henry's law constants,17 with increasing length of the alkyl chain, Table 1. The amounts of carboxylic acid leaving the systems are only small fractions of the total amount carboxylic acid/carboxylate, and the amounts of ammonia leaving seem to be proportional to the total concentration of ammonium carboxylate. This will result in a build-up of carboxylic acid in the surface region until it is saturated, and thereafter all newly formed carboxylic acid will contribute to an increased concentration in the aqueous bulk, as proved by 1H NMR, Table S2 (ESI†), as long as ammonia is leaving the system and eqn (1) is kept to be pushed to the right in the aqueous bulk.
XPS experiments of relative signal intensity as function of alkyl chain length of n-alkyl carboxylic acid
In order to qualitatively compare the surface propensities of the different species and their orientation in the surface region, surface sensitive XPS measurements have been carried out. The C 1s XPS spectra of formate and butyrate ions, and acetic, butyric, valeric and caproic acid in aqueous solution at different bulk concentrations are depicted in Fig. 6. The XPS intensities were normalized by bulk concentrations and number of carbon atoms in the alkyl chain. The intensity of the peaks at 290.3 eV (alkyl carbons) was divided by the number of carbons in the alkyl chain for the comparison. This scale factor has to be included for a proper comparison of the C 1s signal intensities from the different carboxylic acids and carboxylate ions. It can be clearly seen that the XPS intensities of valeric (C4H9COOH) and caproic (C5H11COOH) acid are in principle equal and much more intense than that of butyric acid (C3H7COOH), while the XPS intensities of acetic acid, butyrate and formate ions are hardly seen on the same scale, Fig. 6. Generally, the observed intensities from surface sensitive XPS experiments depend on the solution's real density profile as a function of depth from the water–air interface, and exponentially attenuated along its path. This means that it contains both information on the surface propensity of a compound residing at or close to the water–air interface and a possibly increased surface region concentration. Note that absolute concentrations cannot be obtained directly from XPS results, but qualitative trends of similar compounds can be discussed without further assumptions. The relative intensities of the signals from the alkyl chain, I290, and the carboxylic acid or carboxylate group, I294, may also reflect how compounds are structurally organized at the water–air interface and in the surface region, e.g. an I290/I294corr ratio significantly larger than one strongly indicates that the alkyl chain is closer to the surface than the carboxylic acid or carboxylate group. The ratio I290/I294corr for the carboxylic acids show a clear trend with largest ratio for valeric and caproic acid, ca. 1.6 and the lowest for acetic acid, 1.2. The large I290/I294corr ratio and the strong intensity of valeric and caproic acid show that the alkyl chain is closer to the surface, or most likely partly in air. Thus, at the studied concentrations carboxylic acids with at least 4 carbon atoms long alkyl chains are, at least partly, sticking up in air while the attached carboxylic group is in contact with water molecules residing in the surface region. On the other hand, for acetic acid the I290/I294corr ratio is significantly smaller, 1.25, and the signal intensity is low. This shows that the methyl group of acetic acid is preferentially oriented upwards into a volume with lower relative permittivity and density than the more strongly hydrated carboxylic acid group, but less significant than carboxylic acids with longer alkyl chains. A comparison of butyric acid and the butyrate ion show that butyric acid is much closer to the aqueous surface than the butyrate ion, but they have the same I290/I294corr ratio, 1.4, showing that they are oriented in the same way but on somewhat different levels in the surface region. The intensity of the carboxylate signal of the formate and butyrate ion are identical, Fig. 6, showing they are positioned at the same level, in spite of the butyrate ion having an alkyl group which the formate ion lacks. This strongly indicates that the carboxylate group is positioned in the lower part of the surface region close to the aqueous bulk with sufficient density of water for effective hydration. The formate ion is usually regarded as strongly depleted from the interfacial region.9 This is the first experimental evidence that carboxylate ions cannot be present close to the air–water interface where the permittivity and water density are too low to allow sufficient hydration of the carboxylate group. | ||
| Fig. 6 C 1s XPS spectra of sodium formate and butyrate, and acetic, butyric, valeric and caproic acid in aqueous solution measured as a liquid micro-jet and normalized for concentration and number of carbon atoms in the alkyl chain. This normalization causes that the intensity of the peak at 294.3 eV (carboxylic acid or carboxylate carbon) is inverse to the number of carbons in the alkyl chain. | ||
Discussion
The results of the analytical experiments show that ammonia, and to a smaller extent carboxylic acids, leave an open system with different reaction rates depending on the length of the attached alkyl chains and the initial bulk concentration. That the formation of ammonia is proportional to the ammonium and alkyl carboxylate concentration in the aqueous bulk, as found for the alkyl carboxylates with six or less carbon atoms in the alkyl chain, seems reasonable for a chemical reaction following first order kinetics. The slowest transfer of ammonia is observed in the ammonium–formate system, and a small increase in the initial reaction rates can be seen with increasing length of the alkyl chain of the carboxylic acid, Fig. 5. This depends most likely on that formic acid lacks a hydrophobic group and therefore has the smallest Ksrd value, Fig. 1, and the Ksrd value is expected to increase slightly with increasing alkyl chain length. It seems therefore likely that the transport rate of ammonia is proportional to its concentration in the surface region, which in turn is dependent on mainly the Ksrd value for the system. This is in agreement with an ammonia release following a first order reaction. On the other hand, the transfer rates of the n-octyl and n-nonyl carboxylate systems increase exponentially with concentration when a solid phase is formed in/on the water surface. In this case the transfer rate depends also on the concentration of carboxylic acid in the surface region, and as the solubility of n-octyl- and n-nonylcarboxylic acid is low and low Ssd values, a more or less immediate precipitation will take place. As the solubilities of n-octyl- and n-nonylcarboxylic acid are expected to be even lower in the aqueous bulk than in the surface region this should result in a promoted reaction between ammonium and n-octyl- or n-nonylcarboxylate ions in the aqueous bulk to compensate for the loss of carboxylic acid in the surface region. This causes formation of ammonia in large amounts at a high rate as observed in the experiments.These results show that neutral molecules are enriched in the surface region, and that the transport of neutral molecules between the aqueous bulk and the surface region is fast. A recent surface tension study of surfaces of small millisecond old droplets, show a significant decrease in surface tension of droplets containing glutaric acid with increasing concentration in comparison with pure water.45 Aqueous solutions of sodium chloride show on the other hand increasing surface tension with increasing concentration. Furthermore, it requires a 16-fold excess of sodium chloride to counter-balance the surface tension decrease of glutaric acid.44 This supports the importance of accumulation of surface active or hydrophobic compounds in the surface region of small droplets for their physico-chemical properties. To compensate for the loss of neutral molecules in the aqueous bulk eqn (1) will be pushed more or less to the right. It shall be noted that the concentration of neutral species in the surface region can largely exceed the concentration in the aqueous bulk,32–34,53 but the total amount of chemical species accumulated in the surface region, as the carboxylic acids in this study, is only a very small fraction of the total amount in a macroscopic system. On the other hand, charged species seem not to be able to reach close to the water–air interface as the physico-chemical properties of the region close to the water–air interface cannot accommodate charged species as the amount of water for efficient hydration is not sufficient due to low density of water in the surface region. However, they can approach somewhat closer to the water–air interface through formation of ion-pairs, and when one ion in the ion-pair has a long alkyl chain, it may come fairly close to the water–air interface, Fig. 1.
The relatively large difference in C 1s signal intensity between the alkyl and carboxylic acid carbons give information about the orientation and the relative position of the carboxylic acids versus the water–air interface, Table 2. It can be seen that the longer the alkyl chain is in the carboxylic acid, the stronger orientation effect. The very strong overall C 1s signal intensities for valeric and caproic acid strongly indicate that they are present in the water–air interface. The rather large ratio I290/I294corr yields most likely from part of their alkyl chains that are sticking up in air. Such a model is fully supported by the fact that relative signal intensity of the alkyl group is significantly stronger than for the carbon in the carboxylic acid group after scaling, Table 2. The latter seems to be hydrogen bound to water molecules close to the water–air interface reducing its ability to evaporate. On the other hand, acetic and butyric acid display low overall signal intensity and a smaller intensity ratio between alkyl and carboxylic acid carbons, but still with the alkyl chains closer to the surface than the carboxylic acid group. This shows that their alkyl chains are in principal oriented perpendicular to the surface (at the studied bulk concentrations) independent of position in the surface region.
| C5H11COOH | C4H9COOH | C3H7COOH | CH3COOH | C3H7COO− | |
|---|---|---|---|---|---|
| I 290/I294 | 8.2 ± 0.2 | 5.9 ± 0.2 | 4.2 ± 0.2 | 1.25 ± 0.15 | 4.1 ± 0.25 |
| I 290/I294corr | 1.6 | 1.5 | 1.4 | 1.25 | 1.4 |
In order for a salt of monovalent ions to dissociate, or to allow monovalent ions to enter a solvent region, a relative permittivity of 15–20 is required, and the ability to be close to the water–air interface increases with decreasing charge density and hydrophilicity of the ion.76 The former has been shown in several studies of the halide ions in water with iodide coming closest to the water–air interface.77–80 At the same time neutral species preferring non-polar solvents are in fact promoted to be at the water–air interface or very close to it.
It is important to stress that the solubility of a salt can be relatively high in solvents with low relative permittivity due formation of ion-pairs or clusters neutralizing the effective charge as alkali metal and magnesium halides and Grignard reagents in diethyl ether.81–85 Similar arrangements in the surface region are harder to imagine as the thickness is only some water molecule layers, and more important, the change in physico-chemical properties is dramatic over the very short distance making up the surface region, a limited number of water molecule layers. As observed in this study it is more likely that molecules and ions with a hydrophobic group are oriented perpendicular to the water surface allowing the hydrophobic group to be in the upper part of the surface region with low relative permittivity and density, while e.g. acid groups and ion-pairs requiring a higher charge shielding are situated in the lower part of the surface region or even in the aqueous bulk. Charged species requiring strong hydration and effective charge neutralization are forced to be in the lower part of the surface region or in the aqueous bulk and possible hydrophobic groups can approach the lower parts of the surface region as the butyrate ion.
The XPS C 1s intensity studies of acetic and butyric acid show that these, and certainly also propionic acid, are accumulated in the surface region as a concentrated mixture with interfacial water with lower density which certainly lacks the rigid hydrogen bound water structure present in the aqueous bulk. To further decrease the charge it is expected that a larger fraction than in the aqueous bulk is present as dimers.86,87 The dimerization in water increases with increasing alkyl chain length and concentration, and dimerization is very prominent in organic solvents with low permittivity.88 Hence, the surface region can become saturated with tightly packed carboxylic acid molecules. When this happens in the open vessel-system, the transport from the aqueous bulk will stop and additionally formed carboxylic acid will remain in the aqueous bulk. However, as ammonia in the gas phase will continue to be removed due to the nitrogen flow, there will be a deficit in both the gas phase and the surface region, and the reaction eqn (1) will continue to be pushed to the right, Fig. 1. The results from our analytical studies show clearly that ammonia and carboxylic acid removed from the system are rapidly replaced showing that the transport to and from the surface region is fast. When the surface region is saturated with carboxylic acid a steady state situation is obtained controlled by the gas flow in the gas phase (removal of ammonia and carboxylic acid) and the rate of the shift of the equilibria NH3(aq) ⇌ NH3(sr), NH3(sr) ⇌ NH3(g), RCOOH(aq) ⇌ RCOOH(sr) and RCOOH(sr) ⇌ RCOOH(g) becomes reasonable constant. The proposed equilibrium scheme for eqn (1) is given in Fig. 7.
 | ||
| Fig. 7 Equilibria of ammonium and carboxylate ions, ammonia and carboxylic acid in the aqueous bulk (bulk), the interface water–air (sr) and open air. The dashed line represent the border for bulk water properties. | ||
The formate and butyrate ions are not able to enter the upper part of the surface region as discussed above, and it can be assumed that neither the valerate, caproate nor enanthante ions can do it as long as they do not form any charge screening ion-pairs. Their alkyl chain can however be oriented upwards into the upper surface region and if it is long enough all the way up to the water–air interface. The XPS C 1s intensity studies of valeric and caproic acid show that they are present at the water–air interface with the alkyl chain at least partly sticking out of the surface and the carboxylic acid group hydrogen bind to water molecules in the surface region. This shows that alkyl carboxylic acids are structurally organized in the surface region. For carboxylic acids with low solubility in the aqueous bulk, as in the case of n-octanoic and n-nonanoic acid, the aqueous bulk of the open-vessel system will be saturated rapidly, and formation of solid carboxylic acid takes place on top of the aqueous solution (hydrophobic and lower density than water). This solid phase continues to form until a thick crust covers the entire surface which eventually will stop the transport of ammonia to the gas phase, Fig. S1 (ESI†). The reaction rate when both ammonia and carboxylic acid are continuously removed is high and increases sharply with increasing concentration, Fig. 5 and Fig. S2, Table S2 (ESI†). The proposed reaction mechanism and structure of the formed solid phase on top of the aqueous solution for n-octanoic and n-nonanoic acid is given in Fig. 8.
 | ||
| Fig. 8 The surface sites, the surface region and the aqueous bulk are saturated and crystallization takes place on top of the aqueous surface; the model of solid n-octanoic acid is taken from ref. 89. | ||
Conclusions
Results from XPS studies of the surfaces of aqueous solutions of carboxylic acids and sodium formate and butyrate show large differences in C 1s signal intensities which can be related to the species’ distance from the water–air interface. The formate and butyrate ions display equally weak signals for the carboxylate carbon. This strongly indicates that they cannot enter the upper parts of the surface region close to the water–air interface where water has different physico-chemical properties, including lower density and relative permittivity, than bulk water. On the other hand, the neutral carboxylic acids are promoted to enter the region close to the water–air interface. It shall be noted that the orientation of alkyl carboxylic acids and carboxylates in the surface region is in principle the same with the alkyl chain oriented upwards into regions with lower relative permittivity and density than the more strongly hydrated carboxylic acid and carboxylate groups, Fig. 6 and Table 2. The carboxylic acids with at least four carbons in the alkyl chain reside in the water–air interface and are probably sticking up their alkyl chain into air, while acetic, propionic and butyric acid form a concentrated aqueous solution with surface water where they possibly partly form dimers to further reduce the local charge.The accumulation of carboxylic acids in the surface region, also when they are miscible with water,26 shows that there are equilibria between the aqueous bulk and the surface region, RCOOH(aq) ⇌ RCOOH(sr), and between the surface region and the gas phase above it, RCOOH(sr) ⇌ RCOOH(g). The Henry's law constant, KH, RCOOH(g) ⇌ RCOOH(aq), is therefore a product of at least two equilibria, Ksrd and Ksd, Fig. 1, with presently unknown values. For carboxylic acids the value of Ksrd is certainly larger than unity with the exception of formic acid, which seems not to be accumulated in the surface region. The value Ksrd can be estimated from e.g. surface tension measurements and surface speciation studies. As a consequence, the values of Ksd for ammonia and carboxylic acids become smaller than the corresponding KH value. The analytical studies show that the release of ammonia is dependent on its concentration in the surface region. This is also true for carboxylic acids as long as their solubility in the surface region is sufficiently high to avoid a precipitation on the water surface. However, for n-octyl- and n-nonylcarboxylic acid the solubility is sufficiently low for precipitation in/on the water–air interface. This causes that the surface region becomes depleted on carboxylic acid which promotes a transfer from the aqueous bulk to the surface region to maintain the equilibrium concentrations. The precipitation of n-octyl and n-nonyl carboxylic acid is therefore also the driving force to a much larger and faster release of ammonia. The combined analytical and surface speciation studies in this work show that the equilibria described by Ksrd and Ksd establish fast as they are detected in the XPS studies. This allows for a fast transport of chemicals from the aqueous bulk to the surface region, and that a minor fraction of the compounds in the surface region is released into air according to the equilibrium constant Ksd. Ksd is significantly larger for ammonia as compared to the small carboxylic acids with less than three carbons in the alkyl chain. The findings in this study are of particular importance for small water droplets with large surface area:bulk volume ratio in air where dynamic equilibria between air and water drops exist.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The financial support the Swedish Research Council is gratefully acknowledged. The authors would like to thank the staff at the MAX IV Laboratory and beamline I411 for smooth operation of the synchrotron ring and experimental facility.References
- Intergovernmental Panel on Climate Change, in Climate Change 2013: The Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change, ed. T. F. Stocker, D. Qin, G.-K. Plattner, M. Tignor, S. K. Allen, J. Boschung, A. Nauels, Y. Xia, V. Bex and P. M. Midgley, Cambridge University Press, Cambridge, UK, and New York, NY, USA, 2013, p. 1535 Search PubMed.
- B. Noziere, Science, 2016, 351, 1396–1397 CrossRef CAS PubMed.
- C. R. Ruehl, J. F. Davies and K. R. Wilson, Science, 2016, 351, 1447–1450 CrossRef CAS PubMed.
- B. J. Finlayson-Pitts, Chem. Rev., 2003, 103, 4801–4822 CrossRef CAS PubMed.
- M. Hallquist, J. C. Wenger, U. Baltensperger, Y. Rudich, D. Simpson, M. Claeys, J. Dommen, N. M. Donahue, C. George, A. H. Goldstein, J. F. Hamilton, H. Herrmann, T. Hoffmann, Y. Iinuma, M. Jang, M. E. Jenkin, J. L. Jimenez, A. Kiendler-Scharr, W. Maenhaut, G. McFiggans, T. F. Mentel, A. Monod, A. S. H. Prévôt, J. H. Seinfeld, J. D. Surratt, R. Szmigielski and J. Wildt, Atmos. Chem. Phys., 2009, 9, 5155–5236 CrossRef CAS.
- J. H. Kroll, N. M. Donahue, J. L. Jimenez, S. H. Kessler, M. R. Canagaratna, K. R. Wilson, K. E. Altieri, L. R. Mazzoleni, A. S. Wozniak, H. Bluhm, E. R. Mysak, J. D. Smith, C. E. Kolb and D. R. Worsnop, Nat. Chem., 2011, 3, 133–139 CrossRef CAS PubMed.
- M. C. Facchini, M. Mircea, S. Fuzzi and R. J. Charlson, Nature, 1999, 401, 257–259 CrossRef CAS.
- S. Frka, J. Dautović, Z. Kozarac, B. Ćosović, A. Hoffer and G. Kiss, Tellus, Ser. B, 2012, 64, 18490 CrossRef CAS.
- N. Sareen, A. N. Schwier, T. L. Lathem, A. Nenes and V. F. McNeill, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 2723–2728 Search PubMed.
- V. F. McNeill, N. Sareen and A. N. Schwier, Atmospheric and Aerosol Chemistry, Springer, Berlin, Heidelberg, 2013, pp. 201–259 Search PubMed.
- B. Nozière, C. Baduel and J. L. Jaffrezo, Nat. Commun., 2014, 5, 3335 CrossRef PubMed.
- V. Gérard, B. Nozière, C. Baduel, L. Fine, A. A. Frossard and R. C. Cohen, Environ. Sci. Technol., 2016, 50, 2974–2982 CrossRef PubMed.
- A. Kroflic, S. Frka, M. Simmel, H. Wex and I. Grgic, Environ. Sci. Technol., 2018, 52, 9179–9187 CrossRef CAS PubMed.
- J. F. Davies, R. E. H. Miles, A. E. Haddrell and J. P. Reid, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 8807–8812 CrossRef CAS PubMed.
- C. R. Ruehl and K. R. Wilson, J. Phys. Chem. A, 2014, 118, 3952–3966 CrossRef CAS PubMed.
- T. Jokinen, T. Berndt, R. Makkonen, V.-M. Kerminen, H. Junninen, P. Paasonen, F. Stratmann, H. Herrmann, A. B. Guenther, D. R. Worsnop, M. Kulmala, M. Ehn and M. Sipilä, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 7123–7128 CrossRef CAS PubMed.
- R. Sander, Atmos. Chem. Phys., 2015, 15, 4399–4981 CrossRef CAS , and references therein.
- J. M. Wallace and P. V. Hobbs, Atmospheric Science – An Introducory Survey, Academic Press, 2nd edn, 2006, ch. 6, ISBN 0-12-732951-2 Search PubMed.
- E. M. Knipping, M. J. Lakin, K. L. Foster, P. Jungwirth, D. J. Tobias, R. B. Gerber, D. Dabdub and B. J. Finlayson-Pitts, Science, 2000, 288, 301–306 CrossRef CAS PubMed.
- S. Rossignol, L. Tinel, A. Bianco, M. Passananti, M. Brigante, D. J. Donaldson and C. George, Science, 2016, 353, 699–702 CrossRef CAS PubMed.
- E. C. Griffith, B. K. Carpenter, R. K. Shoemaker and V. Vaida, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 11714–11719 CrossRef CAS PubMed.
- J. L. Jimenez, N. M. R. Canagaratna, N. M. Donahue, A. S. H. Prevot, Q. Zhang, J. H. Kroll, P. F. DeCarlo, J. D. Allan, H. Coe, N. L. Ng, A. C. Aiken, K. S. Docherty, I. M. Ulbrich, A. P. Grieshop, A. L. Robinson, J. Duplissy, J. D. Smith, K. R. Wilson, V. A. Lanz, C. Hueglin, Y. L. Sun, J. Tian, A. Laaksonen, T. Raatikainen, J. Rautiainen, P. Vaattovaara, M. Ehn, M. Kulmala, J. M. Tomlinson, D. R. Collins, M. J. Cubison, E. J. Dunlea, J. A. Huffman, T. B. Onasch, M. R. Alfarra, P. I. Williams, K. Bower, Y. Kondo, J. Schneider, F. Drewnick, S. Borrmann, S. Weimer, K. Demerjian, D. Salcedo, L. Cottrell, R. Griffin, A. Takami, T. Miyoshi, S. Hatakeyama, A. Shimono, J. Y. Sun, Y. M. Zhang, K. Dzepina, J. R. Kimmel, D. Sueper, J. T. Jayne, S. C. Herndon, A. M. Trimborn, L. R. Williams, E. C. Wood, A. M. Middlebrook, C. E. Kolb, U. Baltensperger and D. R. Worsnop, Science, 2009, 326, 1525–1529 CrossRef CAS PubMed.
- J. Ovadnevaite, A. Zuend, A. Laaksonen, K. J. Sanchez, G. Roberts, D. Ceburnis, S. Decesari, M. Rinaldi, M. C. Facchini, J. H. Seinfeld and C. O’Dowd, Nature, 2017, 546, 637–641 CrossRef CAS.
- J. F. Davies, R. E. H. Miles, A. E. Haddrell and J. P. Reid, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 8807–8812 CrossRef CAS PubMed.
- O. Björneholm, J. Werner, N. Ottosson, G. Öhrwall, V. Ekholm, B. Winter, I. Unger and J. Söderström, J. Phys. Chem. C, 2014, 118, 29333–29339 CrossRef.
- N. Ottosson, E. Wernersson, J. Söderström, W. Pokapanich, S. Kaufmann, S. Svensson, I. Persson, G. Öhrwall and O. Björneholm, Phys. Chem. Chem. Phys., 2011, 13, 12261–12267 RSC.
- J. Werner, I. Persson, O. Björneholm, D. Kawecki, C.-M. Saak, M.-M. Walz, V. Ekholm, I. Unger, C. Valtl, C. Caleman, G. Öhrwall and N. L. Prisle, Phys. Chem. Chem. Phys., 2018, 20, 23281–23293 RSC.
- N. Ottosson, A. O. Romanova, J. Söderström, O. Björneholm, G. Öhrwall and M. V. Fedorov, J. Phys. Chem. B, 2012, 116, 13017–13023 CrossRef CAS PubMed.
- J. Werner, E. Wernersson, V. Ekholm, N. Ottosson, G. Öhrwall, J. Heyda, I. Persson, J. Söderström, P. Jungwirth and O. Björneholm, J. Phys. Chem. B, 2014, 118, 7119–7127 CrossRef CAS PubMed.
- V. Ekholm, M. Vaxdar, P. E. Mason, E. Bialik, M.-M. Walz, G. Öhrwall, J. Werner, J.-E. Rubensson, P. Jungwirth and O. Björneholm, J. Chem. Phys., 2018, 148, 144508 CrossRef PubMed.
- J. Werner, M. Dalirian, M.-M. Walz, V. Ekholm, U. Wideqvist, S. J. Lowe, G. Öhrwall, I. Persson, I. Riipinen and O. Björneholm, Environ. Sci. Technol., 2016, 50, 7434–7442 CrossRef CAS PubMed.
- J. Werner, J. Julin, M. Dalirian, N. L. Prisle, G. Öhrwall, I. Persson, O. Björneholm and I. Riipinen, Phys. Chem. Chem. Phys., 2014, 16, 21486–21495 RSC.
- M.-M. Walz, C. Caleman, J. Werner, V. Ekholm, D. Lundberg, N. L. Prisle, G. Öhrwall and O. Björneholm, Phys. Chem. Chem. Phys., 2015, 17, 14036–14044 RSC.
- M.-M. Walz, J. Werner, V. Ekholm, N. L. Prisle, G. Öhrwall and O. Björneholm, Phys. Chem. Chem. Phys., 2016, 18, 6648–6656 RSC.
- K. Kinosita and H. Yokota, J. Phys. Soc. Jpn., 1965, 20, 1086 CrossRef.
- A. Braslau, M. Deutsch, P. S. Pershan, A. H. Weiss, J. Als-Nielsen and J. Bohr, Phys. Rev. Lett., 1985, 54, 114–117 CrossRef CAS PubMed.
- A. Braslau, P. S. Pershan, G. Swislow, B. M. Ocko and J. Als-Nielsen, Phys. Rev. A Gen. Phys., 1988, 38, 2457–2470 CrossRef.
- R. S. Taylor, L. X. Dang and B. C. Garrett, J. Phys. Chem., 1996, 100, 11720–11725 CrossRef CAS.
- M. Matsumoto and Y. Kataoka, J. Chem. Phys., 1988, 88, 3233–3245 CrossRef CAS.
- J. Alejandre, D. J. Tildesley and G. A. Chapela, J. Chem. Phys., 1995, 102, 4574–4583 CrossRef CAS.
- I. Benjamin, Chem. Rev., 1996, 96, 1449–1476 CrossRef CAS PubMed.
- J. O’Bockris and A. Gonzales-Martin, in Spectroscopic and Diffraction Techniques in Interfacial Electrochemistry, ed. C. Gutierrez, C. Melendres, Kluwer Academic Publishers, Dordrecht, 1990 Search PubMed.
- L. G. Hector and H. L. Schultz, Physics, 1936, 7, 133–136 CrossRef.
- C. G. Malmberg and A. A. Maryott, J. Res. Natl. Bur. Stand., 1956, 56, 2641 Search PubMed.
- R. E. H. Miles, M. W. J. Glerum, H. C. Boyer, J. S. Walker, C. S. Dutcher and B. R. Bzdek, J. Phys. Chem. A, 2019, 123, 3021–3029 CrossRef CAS PubMed.
- I. Persson, M. Trublet and W. Klysubun, J. Phys. Chem. A, 2018, 122, 7413–7420 CrossRef CAS PubMed.
- V. Vchirawongkwin, B. M. Rode and I. Persson, J. Phys. Chem. B, 2007, 111, 4150–4155 CrossRef CAS PubMed.
- C. Caleman, J. S. Hub, P. J. van Maaren and D. van der Spoel, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 6838–6842 CrossRef CAS.
- B. Minofar, P. Jungwirth, M. R. Das, W. Kunz and W. Mahiuddin, J. Phys. Chem. C, 2007, 111, 8242–8247 CrossRef CAS.
- L. O. P. Jungwirth and E. Wernersson, Phys. Chem. Chem. Phys., 2012, 14, 10248–10257 RSC.
- Q. Zhang, J. L. Jimenez, M. R. Canagaratna, J. D. Allan, H. Coe, I. Ulbrich, M. R. Alfarra, A. Takami, A. M. Middlebrook, Y. L. Sun, K. Dzepina, E. Dunlea, K. Docherty, P. F. DeCarlo, D. Salcedo, T. Onasch, J. T. Jayne, T. Miyoshi, A. Shimono, S. Hatakeyama, N. Takegawa, Y. Kondo, J. Schneider, F. Drewnick, S. Borrmann, S. Weimer, K. Demerjian, P. Williams, K. Bower, R. Bahreini, L. Cottrell, R. J. Griffin, J. Rautiainen, J. Y. Sun, Y. M. Zhang and D. R. Worsnop, Geophys. Res. Lett., 2005, 34, L13801 Search PubMed.
- M. Kanakidou, J. H. Seinfeld, S. N. Pandis, I. Barnes, F. J. Dentener, M. C. Facchini, R. Van Dingenen, B. Ervens, A. Nenes, C. J. Nielsen, E. Swietlicki, J. P. Putaud, Y. Balkanski, S. Fuzzi, J. Horth, G. K. Moortgat, R. Winterhalter, C. E. L. Myhre, K. Tsigaridis, E. Vignati, E. G. Stephanou and J. Wilson, Atmos. Chem. Phys., 2005, 5, 1053–1123 CrossRef CAS.
- N. L. Prisle, N. Ottosson, G. Öhrwall, J. Söderström, M. Dal Maso and O. Björneholm, Atmos. Chem. Phys., 2012, 12, 12227–12242 CrossRef CAS.
- C. L. Apel, D. W. Deamer and M. N. Mautner, Biochim. Biophys. Acta, Biomembr., 2002, 1559, 1–9 CrossRef CAS.
- G. Öhrwall, N. L. Prisle, N. Ottosson, J. Werner, V. Ekholm, M.-M. Walz and O. Björneholm, J. Phys. Chem. B, 2015, 119, 4033–4040 CrossRef PubMed.
- M.-T. Lee, F. Orlando, L. Artiglia, S. Chen and M. Ammann, J. Phys. Chem. A, 2016, 120, 9749–9758 CrossRef CAS PubMed.
- B. A. Wellen, E. A. Lach and H. C. Allen, Phys. Chem. Chem. Phys., 2017, 19, 26551–26558 RSC.
- E. M. Adams, B. A. Wellen, R. Thiraux, S. K. Reddy, A. S. Vidalis, F. Paesani and H. C. Allen, Phys. Chem. Chem. Phys., 2017, 19, 10481–10490 RSC.
- S. Li, L. Du, Z. Wei and W. Wang, Sci. Total Environ., 2017, 580, 1155–1161 CrossRef CAS PubMed.
- H. Fan, T. W. Masaya and F. Goulay, Phys. Chem. Chem. Phys., 2019, 21, 2992–3001 RSC.
- N. Rastak, A. Pajunoja, J. C. Acosta Navarro, J. Ma, M. Song, D. G. Partridge, A. Kirkevåg, Y. Leong, W. W. Hu, N. F. Taylor, A. Lambe, K. Cerully, A. Bougiatioti, P. Liu, R. Krejci, T. Petäjä, C. Percival, P. Davidovits, D. R. Worsnop, A. M. L. Ekman, A. Nenes, S. Martin, J. L. Jimenez, D. R. Collins, D. O. Topping, A. K. Bertram, A. Zuend, A. Virtanen and I. Riipinen, Geophys. Res. Lett., 2017, 44, 5167–5177 CrossRef CAS PubMed.
- CRC Handbook of Chemistry and Physics, ed. D. R. Lide, 2015–2016, 96th edn, p. 5-93-102 Search PubMed.
- M. V. Fedotova and S. E. Kruchinin, J. Mol. Liq., 2011, 164, 201–206 CrossRef CAS.
- CRC Handbook of Chemistry and Physics, ed. D. R. Lide, 2015–2016, 96th edn, p. 5–110, 6–131, 12–22 Search PubMed.
- D. W. Smith, J. Chem. Educ., 1977, 54, 540–542 CrossRef CAS.
- D. R. Rosseinsky, Chem. Rev., 1965, 65, 467–490 CrossRef CAS.
- D. Simonelli, S. Baldelli and M. J. Schultze, Chem. Phys. Lett., 1998, 298, 400–404 CrossRef CAS.
- D. Simonelli and M. J. Schultze, J. Chem. Phys., 2000, 112, 6804–6816 CrossRef CAS.
- C.-F. Fu and S. X. Tian, J. Phys. Chem. C, 2013, 117, 13011–13020 CrossRef CAS.
- D. Chakraborty and A. Chandra, J. Chem. Phys., 2011, 135, 114510 CrossRef.
- S. Paul and A. Chandra, J. Chem. Phys., 2005, 123, 174712 CrossRef PubMed.
- B. Winter and M. Faubel, Chem. Rev., 2006, 106, 1176–1211 CrossRef CAS PubMed.
- M. Faubel, K. R. Siefermann, Y. Liu and B. Abel, Acc. Chem. Res., 2012, 45, 120–130 CrossRef CAS PubMed.
- B. Winter, R. Weber, W. Widdra, M. Dittmar, M. Faubel and L. V. Hertel, J. Phys. Chem. A, 2004, 108, 2625–2632 CrossRef CAS.
- H. Nikjoo, S. Uehara, D. Emfietzoglou and A. Brahme, New J. Phys., 2008, 10, 075006 CrossRef.
- Y. Marcus and G. Hefter, Chem. Rev., 2006, 106, 4585–4621 CrossRef CAS PubMed.
- P. Jungwirth and D. J. Tobias, Molecular Structure of Salt Solution: A new view of the interface with implications for heterogenous atmospheric chemistry, J. Phys. Chem., 2001, 105, 10468–10472 CrossRef CAS.
- N. Ottosson, J. Heyda, E. Wernersson, W. Pokapanich, S. Svensson, B. Winter, G. Öhrwall, P. Jungwirth and O. Björneholm, Phys. Chem. Chem. Phys., 2010, 12, 10693–10700 RSC.
- L. Piatkowski, Z. Zhang, E. H. G. Backus, H. J. Bakker and M. Bonn, Nat. Commun., 2014, 5, 4083 CrossRef CAS PubMed.
- J. D. Smith and S. W. Rick, Condens. Matter Phys., 2016, 19, 23002 CrossRef.
- J. Goulon, C. Goulon-Ginet and M. Chabanel, J. Solution Chem., 1981, 10, 649–672 CrossRef CAS.
- A. Ericson and I. Persson, J. Organomet. Chem., 1987, 326, 151–158 CrossRef CAS.
- A. Wellmar and I. Persson, J. Organomet. Chem., 1991, 415, 143–153 CrossRef CAS.
- A. Wellmar and I. Persson, J. Organomet. Chem., 1991, 415, 155–165 CrossRef CAS.
- A. Wellmar, A. Hallberg and I. Persson, J. Organomet. Chem., 1991, 415, 167–180 CrossRef CAS.
- K. Suzuki, Y. Taniguchi and T. Watanabe, J. Phys. Chem., 1973, 77, 1918–1922 CrossRef CAS.
- J. Chen, C. L. Brooks III and H. A. Scheraga, J. Phys. Chem. B, 2008, 112, 242–249 CrossRef CAS PubMed.
- Y. Fujii, H. Yamada and M. Mizuta, J. Phys. Chem., 1988, 92, 6768–6772 CrossRef CAS.
- A. D. Bond, New J. Chem., 2004, 28, 104–114 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Summary of acidic constants of the carboxylic acids, reaction rates after 24 hours (table and figure) and photo of solid n-octanoic acid formed on the aqueous surface. See DOI: 10.1039/c9cp02449b |
| ‡ Present address: Environmental Engineering Program and Chemistry Department, Universidad El Bosque, Carrera 7b Bis No. 132-11, Bogotá D.C., Colombia. |
| § Present address: Department of Chemistry, Faculty of Sciences, Akdeniz University, 07058 Antalya, Turkey. |
| This journal is © the Owner Societies 2019 |