
Scanning probe microscopy for real-space observations of local chemical reactions induced by a localized surface plasmon
 a
and
Yousoo
Kim
*a
a
and
Yousoo
Kim
*a
Abstract
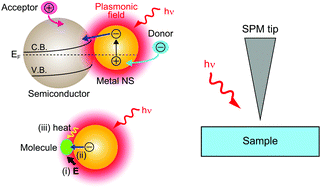
Localised surface plasmon (LSP) resonance has attracted considerable attention in recent years as an efficient driving force for chemical reactions. The chemical reactions induced by LSP are classified into two types, namely, redox reactions based on plasmon-induced charge separation (PICS) and chemical reactions induced by the direct interaction between LSP and molecules (plasmon-induced chemical reactions). Although both types of reactions have been extensively studied, the mechanisms of PICS and plasmon-induced chemical reactions remain unexplained and controversial because conventional macroscopic methods can hardly grasp the local chemical reactions induced by LSP. In order to obtain mechanistic insights, nanoscale observations and investigations are necessary. Scanning probe microscopy (SPM) is a powerful experimental tool to investigate not only the surface morphology but also the physical and chemical properties of samples at a high spatial resolution. In this perspective review, we first explain SPM combined with optical excitation, and then, review the recent studies using SPM techniques for real-space observations of the chemical reactions induced by LSP.
- This article is part of the themed collection: 2019 PCCP HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

