
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAdsorption site, orientation and alignment of NO adsorbed on Au(100) using 3D-velocity map imaging, X-ray photoelectron spectroscopy and density functional theory
Saada
Abujarada
ab,
Alex S.
Walton
 ab,
Andrew G.
Thomas
bc,
Urslaan K.
Chohan
c and
Sven P. K.
Koehler
*d
ab,
Andrew G.
Thomas
bc,
Urslaan K.
Chohan
c and
Sven P. K.
Koehler
*d
aSchool of Chemistry, The University of Manchester, Oxford Road, Manchester, M13 9PL, UK
bPhoton Science Institute, The University of Manchester, Oxford Road, Manchester, M13 9PL, UK
cSchool of Materials, The University of Manchester, Oxford Road, Manchester, M13 9PL, UK
dSchool of Science and the Environment, Manchester Metropolitan University, Chester Street, Manchester, M1 5GD, UK. E-mail: s.koehler@mmu.ac.uk
First published on 7th May 2019
Abstract
Nitric oxide adsorption on a Au(100) single crystal has been investigated to identify the type of adsorption, the adsorption site, and the orientation and alignment of the adsorbed NO relative to the surface. This was done using a combination of 3D-surface velocity map imaging, near-ambient pressure X-ray photoelectron spectroscopy, and density functional theory. NO was observed to be molecularly adsorbed on gold at ∼200 K. Very narrow angular distributions and cold rotational distributions of photodesorbed NO indicate that NO adsorbs on high-symmetry sites on the Au crystal, with the N–O bond axis close to the surface normal. Our density functional theory calculations show that NO preferentially adsorbs on the symmetric bridge (2f) site, which ensures efficient overlap of the NO π* orbital with the orbitals on the two neighbouring Au atoms, and with the N–O bond axis aligned along the surface normal, in agreement with our conclusions from the rotational state distributions. The combination of XPS, which reveals the orientation of NO on gold, with 3D-surface velocity map imaging and density functional theory thus allowed us to determine the adsorption site, orientation and alignment of nitric oxide adsorbed on Au(100).
Introduction
Nitric oxide (NO) is a diatomic radical with one unpaired electron, which causes its notable reactivity.1 It is known for its toxicity, has diverse physiological functions,2 serves as a signalling molecule (neurotransmitter) in the nervous system,3 and is also one of the gases contributing to acid rain. NO is a molecule of industrial importance, e.g. in the liquid-phase catalytic hydrogenation of nitric oxide or nitrate to hydroxylamine.4,5 It is a key intermediate in many industrial processes, including the reduction of some nitrogen compounds such as nitrates and nitrites to ammonia or N2O, and in the oxidation of ammonia.6–8 The exact pathway for ammonia oxidation on metal surfaces is still under investigation, not only with regards to the intermediates formed but also the mechanism, with both Langmuir–Hinshelwood and Eley–Rideal mechanisms being suggested.6,9,10NOx is produced in fuel combustion engines, and its reduction to nitrogen by three-way-catalysts is crucial in reducing air pollution.11 Catalytic convertors based on platinum group metals (PGMs) including platinum, rhodium and palladium play a crucial role in the clean-up of car emissions. Dispersed gold as nano-sized particles on transition metal oxides has demonstrated significant catalytic activity in the conversion of toxic car emissions including carbon monoxide, incompletely burnt hydrocarbons and nitrogen oxides.12 The potential of using gold as a substitute of PGMs for car emission control is justified by its low-temperature activity and its thermal stability. Hence the interaction of NOx with gold metal surfaces is of considerable importance. The complexity of the adsorption behaviour of NO on metal surfaces – due to the unpaired electron in the antibonding 2π* orbital – leads to various adsorbate species potentially being formed including NO itself, N, O, (NO)2, N2O, and NO2. The coexistence of these species on some metal surfaces complicates the experimental characterisation and identification of individual chemical species.
Ueda and Haruta investigated the catalytic reduction of NO on a supported gold catalyst and concluded that N2 and N2O were the only products.13 Thermal desorption spectroscopy (TDS) provided an estimate of the NO chemisorption strength on Au(100) of 57 ± 4 kJ mol−1. Bukhtiyarov et al. investigated the adsorption of NO on gold particles deposited on an alumina film in the temperature range 300–500 K using in situ X-ray photoelectron spectroscopy (XPS).14 Two nitrogen-containing adsorbed species were found to exist on the surface of gold particles. The first was identified as adsorbed nitrogen atoms characterised by a N 1s binding energy (BE) of 399.4 eV. The second species was identified as a surface complex with N2O stoichiometry characterised by a BE of 402.7 eV and found to be thermally stable only in a narrow temperature range of 325–425 K. Identification of these species was aided by a separate experiment in which NO was adsorbed onto a stepped Au(533) crystal. The assignments of the recorded BEs seems to be in agreement with Vinod et al. who investigated the adsorption of NO on the (310) face of a gold single crystal in the temperature range of 80–220 K.15
Bukhtiyarov et al. employed XPS to investigate the decomposition of NO on flat Au(111) and stepped Au(310) and (533) surfaces at elevated NO pressures up to 7 Pa and temperature range between 300 and 500 K.16 NO was found not to chemisorb on Au(111), however, dissociation of NO was observed on both Au(310) and (533) surfaces. The authors concluded that Au step-sites are responsible for adsorption and dissociation of NO. The study revealed that while NO dissociation on Au(310) exclusively forms Nads on the surface whose coverage decreases with increasing surface temperature, NO dissociation on Au(533) initially forms N2Oads on the surface that is replaced by more stable Nads at increasing NO exposure and/or surface temperature. Fajin et al. used density functional theory (DFT) to show that the presence of hydrogen was necessary for the dissociation of NO on the stepped Au(321) surface.17 Employing XPS and TDS experiments, Vinod et al. observed the formation of N2O and Oads following adsorption of NO on a stepped Au(310) surface even at temperatures as low as 80 K.15 A common theme amongst many of these studies is the importance of low coordinated Au atoms acting as active sites for adsorption.18–22 Hussain et al. employed DFT to study NO adsorption on Au(111), (100), (110) and (310) surfaces, as well as on adatoms on Au(100).23 The adsorption energy of the gaseous molecules was found to increase considerably with increasing coordinative unsaturation of the gold atoms. These results are consistent with the view that defects, steps and kinks on the surface determine the activity of gold catalysts. For low index surfaces, the bridge site is favoured, however top coordination is preferred at the stepped (310) surface. In all adsorption configurations, the N–O molecular axis is perpendicular to the surfaces except for Au(310).
Wu et al. investigated the low-temperature adsorption and decomposition of NO on low-coordinated Au(997) and Au(110)-1 × 2 surfaces employing TDS and XPS measurements as well as DFT calculations.24 Although the lowest coordinated Au atoms on both surfaces are 7-coordinated, their surface chemistry towards NO adsorption and decomposition was found to be different. On both surfaces, (NO)2 dimer species and not NOads were found to be the active surface species for the NO decomposition into O adatoms and N2O. However, different types of (NO)2 dimers were found to form depending on the Au surface structure. (NO)2 dimer species adsorbed on the 7-coordinated ridge Au atoms on both surfaces decompose into O adatom and N2O upon heating. This type of dimer dominates on Au(997). On the Au(110)-1 × 2 surface, another more abundant and less stable (NO)2 dimer was found to easily decompose into O adatoms and N2O during NO exposure at 105 K.
We have previously studied the dynamics of the laser-induced desorption of adsorbed NO from Au(100) surfaces using 3D-surface velocity map imaging (3D-surface VMI) which allowed us to capture all desorbed NO molecules from the gold surface within a sold angle of 5.6 sr. While this previous work focussed on the differentiation of two different desorption mechanisms, a direct and a thermal one depending on desorption wavelength, we here report on the use of 3D-surface VMI in combination with near-ambient pressure-XPS (NAP-XPS) and condensed-phase DFT simulations to ascertain the adsorption site, orientation and alignment of the adsorbed nitric oxide. 3D-VMI in particular delivers crucial information about the adsorption site of NO (through the narrow angular distribution of the desorbed NO) and the alignment of the N–O (through rotational state distributions), supported by the NAP-XPS and DFT results.
Methods
3D-surface velocity map imaging
The 3D-VMI apparatus has been described elsewhere;25 here only brief description will be given. The spectrometer consists of two chambers, a molecular beam chamber and a UHV chamber that contains the Au(100) single crystal and the twelve plates VMI optics. A base pressure of 5 × 10−10 Torr is achieved using a 1000 L s−1 turbomolecular pump backed by a mechanical pump. A K-type thermocouple attached to the Au(100) crystal measures its temperature. The crystal is mounted on a 3-axes/rotational manipulator which allows for heating and liquid nitrogen cooling. Pure NO gas is introduced into the chamber for adsorption on the Au(100) crystal through a molecular beam valve at a background pressure of ∼5 × 10−6 Torr. Desorption of NO from the Au(100) crystal is initiated by a frequency-tripled Nd:YAG laser (Quanta Ray DCR-11) with a pulse duration of 7 ns and a pulse energy of ∼1.2 mJ. The beam was directed at the crystal surface at an angle of 45°. The desorbed NO molecules may fly in all directions within a hemisphere above the surface. The desorbed NO molecules are detected state selectively in a 1 + 1 resonance-enhanced multi-photon ionisation (REMPI) scheme via the A ← X (0,0) transition by employing a frequency-doubled Radiant Dyes NarrowScan laser, operating on Coumarin 460, pumped by a Continuum Powerlite 8020 Nd:YAG laser. NO+ ions are accelerated down the time-of-flight tube (for mass separation) onto the position-sensitive multichannel plate (MCP) detector. The ions’ arrival positions are imaged onto a phosphor screen and captured by a synchronized CCD camera (FO124TB). Time-of-flight spectra were recorded by varying the time delay between the desorption and the probe laser in discrete steps, and were converted into velocity distributions that provide the velocity component along the surface normal (vz) only. For each time delay (and hence vz), we recorded VM images which correspond to the remaining two velocity components vx and vy parallel to the gold surface.Near-ambient pressure X-ray photoelectron spectroscopy
The near-ambient pressure X-ray photoelectron spectroscopy (NAP-XPS) system employed in this work is equipped with a SPECS Focus 500 monochromatic Al Kα source (photon energy 1486.6 eV), which can be focused to a spot size of 300 μm. The analyser is a SPECS 150 mm Phoibos 150 NAP, fitted with a three-stage, differentially pumped electrostatic lens that allows working pressures up to 25 mbar. NAP-XPS measurements are performed in a specially designed cell which couples to the entrance cone of the analyser lens system. The Au(100) crystal surface was cleaned by Ar+ ion sputtering cycles. The UHV chamber was left to pump down to a base pressure better than 10−9 Torr, after which the crystal was moved to the NAP-cell and the gate between the cell section and the UHV chamber was closed. XP spectra were recorded with NO flowing into the cell at 0.75 Torr and varying substrate temperatures. However, we failed to detect any NO adsorbed on Au(100) even at a base temperature of 220 K after the substrate had been prepared by sputtering and annealing, hence our XP spectra at 220 K and 266 K are all recorded on a sputtered-only gold substrate.The binding energies of the photoelectron spectra were calibrated to the bulk Au 4f7/2 peak at 84 eV. All spectra were normalised using the baseline intensity and background subtraction and fitted with an appropriate number of Gaussian functions using CasaXPS.26
Condensed-phase density functional theory
We performed first principles quantum chemical calculations based on density-functional theory. We employed the Cambridge Serial Total Energy Package (CASTEP) which solves the Kohn–Sham equations with a plane wave basis set employing three-dimensional periodic boundary conditions.27,28 The Perdew–Burke–Ernzerhof (PBE) version of the generalized gradient approximation (GGA) was used to represent the exchange and correlations, and on-the-fly ultrasoft pseudopotentials for Au, N and O. The Monkhorst–Pack (MP) algorithm was selected for k point sampling, and a cut-off energy of 750 eV was employed.An eight-layer slab was employed to describe the (100) surface of a fcc gold single crystal. We used the BFGS algorithm to relax the positions of the Fe atoms in the top four layers, whilst the bottom four layers were constrained in position.
Adsorption energies were calculated via the relation
| Eads = Eslab+NO − (Eslab + ENO) | (1) |
Most importantly for this work, core level shifts were calculated using the Slater−Janak transition state approximation,29 which then allows convenient comparison with the NAP-XPS data.
Results and discussion
3D-VM images
Fig. 1 shows a raw velocity map image recorded for NO molecules which were photodesorbed from the surface with a speed along the surface normal (vz) of 420 m s−1. This and similar images were converted to velocity space and then deliver the velocity distribution in the two dimensions, vx and vy, which are parallel to the Au(100) surface. The raw image is composed of single images from 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 laser shots.
000 laser shots.
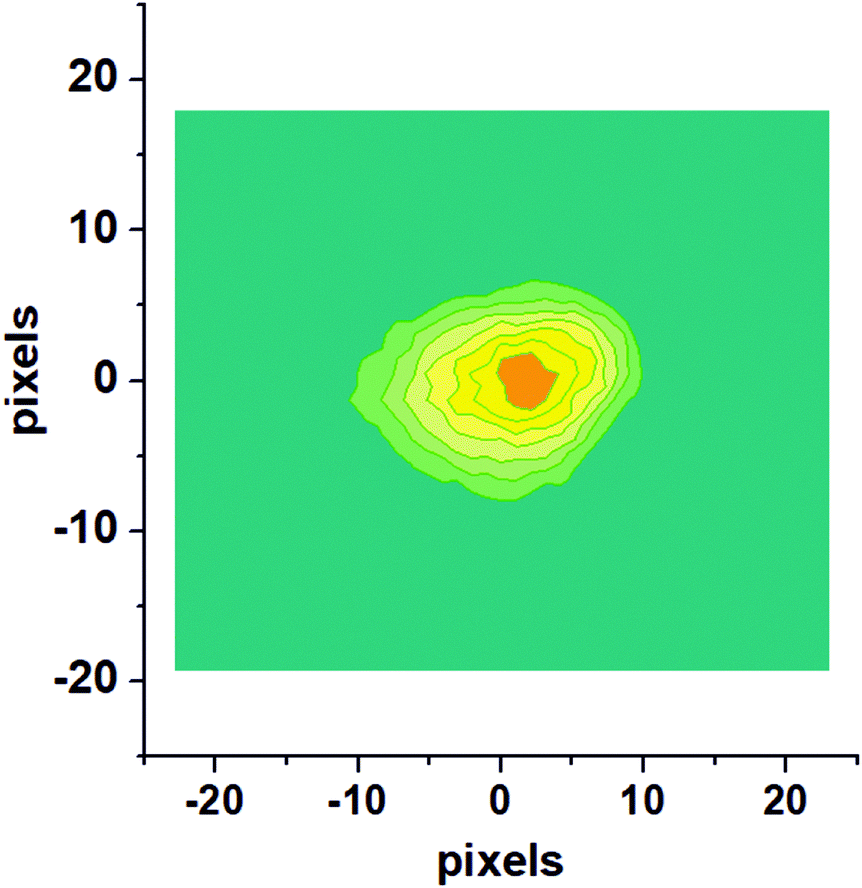 | ||
| Fig. 1 Raw velocity map image for NO molecules photodesorbed from the Au(100) surface with a velocity along the surface normal, vz, of 420 m s−1. | ||
Recording similar images for a range of velocities along the surface normal (vz) allows us to extract a complete velocity distribution with the velocity components in all three dimensions in space obtained independently. This can then be converted to an angularly-resolved speed distribution of NO after 355 nm photodesorption from Au(100), see Fig. 2. It can be clearly seen that the angular distribution is (1) along the surface normal, and (2) extremely narrow, with all NO molecules desorbing off the surface within a very narrow angular distribution with a polar angle relative to the surface normal of <20°, and much narrower for most molecules. The results from these VMI experiments indicate that NO is either adsorbed on-top of a gold atom, or at another high symmetry site such as a 2f or 4f site; this is because repulsive forces between gold and the NO molecule during the desorption process from these high-symmetry sites are along the surface normal; for example, the repulsive forces parallel to the surface from the four Au atoms in case of adsorption on the 4f site cancel each other out, while forces along the surface normal are accumulative; equally, in case of on-top adsorption, the NO molecules fly along the Au–N bond and hence along the surface normal, yielding a narrow angular distributions;30 adsorption at a non-symmetry site would on the other hand result in the angular distribution being tilted away from the surface normal due to non-cancelling lateral forces. This example shows the potential of 3D-VMI to learn about adsorption sites and surface structure through the measurement of angular distributions in laser desorption experiments.
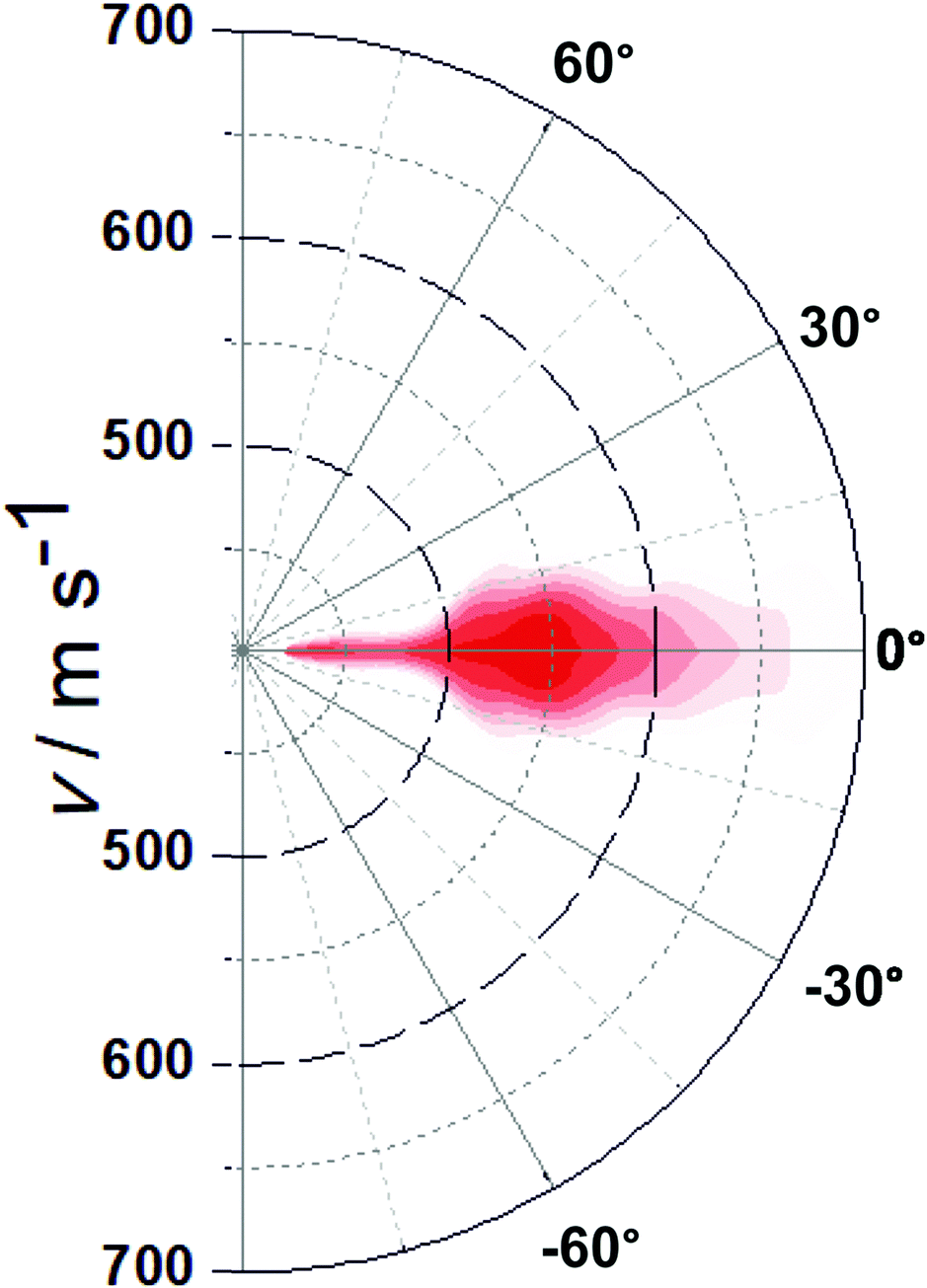 | ||
| Fig. 2 Plot of the NO flux as a function of overall speed and angle after 355 nm photodesorption. Colour coding from dark red (most intense) to white (zero intensity). | ||
Rotational state distributions
In order to record rotational state distributions, we fixed the delay time between the pump and the probe laser to correspond to the specific velocity of the vz distribution of 560 m s−1, which is the maximum in the velocity distribution. The probe laser was scanned over an appropriate wavelength range in steps of 0.005 nm to obtain REMPI spectra for NO. A rotationally resolved (1 + 1) REMPI spectrum of 355 nm photodesorbed NO recorded on the A2Σ1/2 ← X2Π3/2 (0,0) transition is shown in Fig. 3. Rotational state distributions were extracted by comparison with thermal nitric oxide REMPI spectra, and rotational temperatures were fitted using Boltzmann plots. We want to stress that rotational distributions of laser-desorbed nitric oxide are not necessarily thermal ensembles, and rotational temperatures were merely fitted to obtain a single value for ease of interpretation. The rotational temperature based on analysis of a few isolated rotational lines of the O12 branch was found to be (206 ± 5) K. While slighter hotter than the surface temperature, this is surprisingly low considering the excess energy from the laser photon. We interpret this cold rotational temperature to mean that the N–O bond is aligned along the surface normal. We concluded in the previous section that the repulsive forces between the gold surface and the N atom are along the surface normal. If the N–O bond was not along the surface normal (i.e. the angle between AuAS–N–O, where AuAS is the surface adsorption site, would deviate significantly from 180°), but the N would be repelled away from the surface along the normal, then this would impart a significant torque onto the NO molecule, causing it to tumble and rotate, which would lead to a much higher rotational temperature. A laser-desorbed N–O molecule which adsorbs with its molecular axis along the surface normal would on the other hand be rotationally cold, as in our case here.31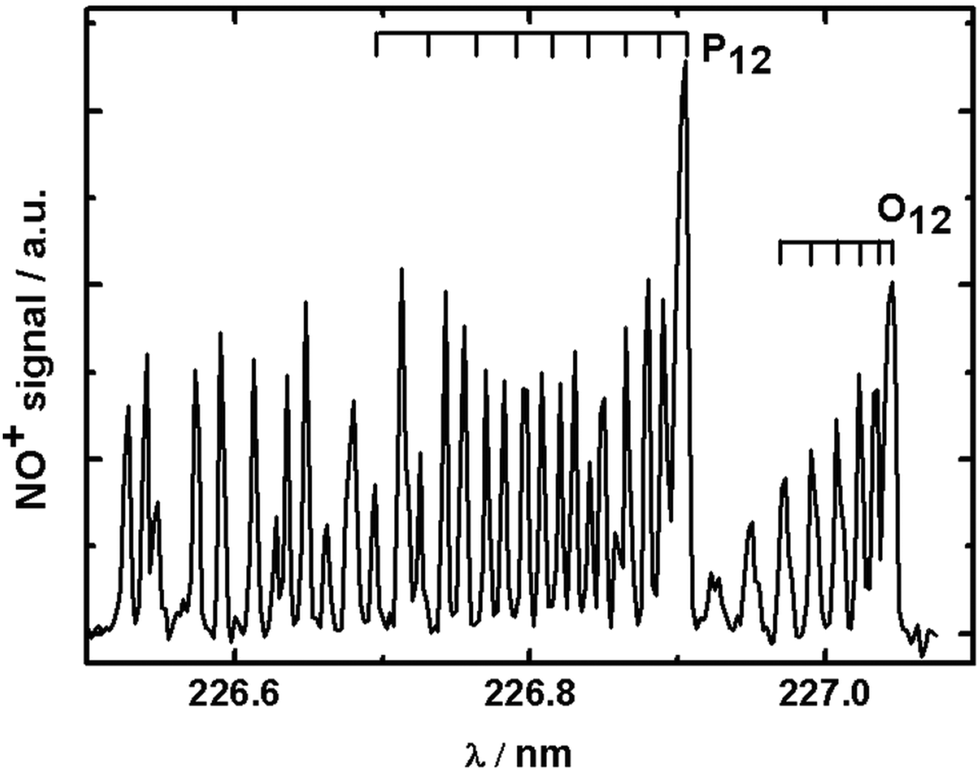 | ||
| Fig. 3 Typical (1 + 1) REMPI spectrum of NO desorbed from gold using 355 nm photons at a time delay corresponding to NO molecules with vz = 560 m s−1. | ||
NAP-XPS results
XPS is considered by many as the standard for the identification of surface species, and one of the goals in this study was to support our conclusions from the 3D-VMI experiments using more established surface analysis techniques such as XPS. In particular, XPS would be able to reveal whether NO adsorbs molecularly or atomically on Au(100) under our conditions (backing-up the conclusions above from the detection of intact nitric oxide in the gas-phase), to reveal the alignment of NO (N–O parallel to the surface would shift the XPS peaks for N and O roughly equally, whereas N–O normal to the surface would shift the XPS peak of the atom closer to the surface more than the respective other atom, supporting our conclusions from the rotational distributions), and possibly to reveal the adsorption site.Exposure of Au(100) to NO at 298 K
Fig. 4 shows XPS spectra recorded during 1 mbar NO exposure of the surface at room temperature. The N 1s spectrum (Fig. 4a) shows two peaks at binding energies of 406.1 eV and 407.5 eV, with the former being more intense. In accordance with the results obtained by Shimada et al. and Yu et al., these peaks are attributed to gas-phase NO molecules.32,33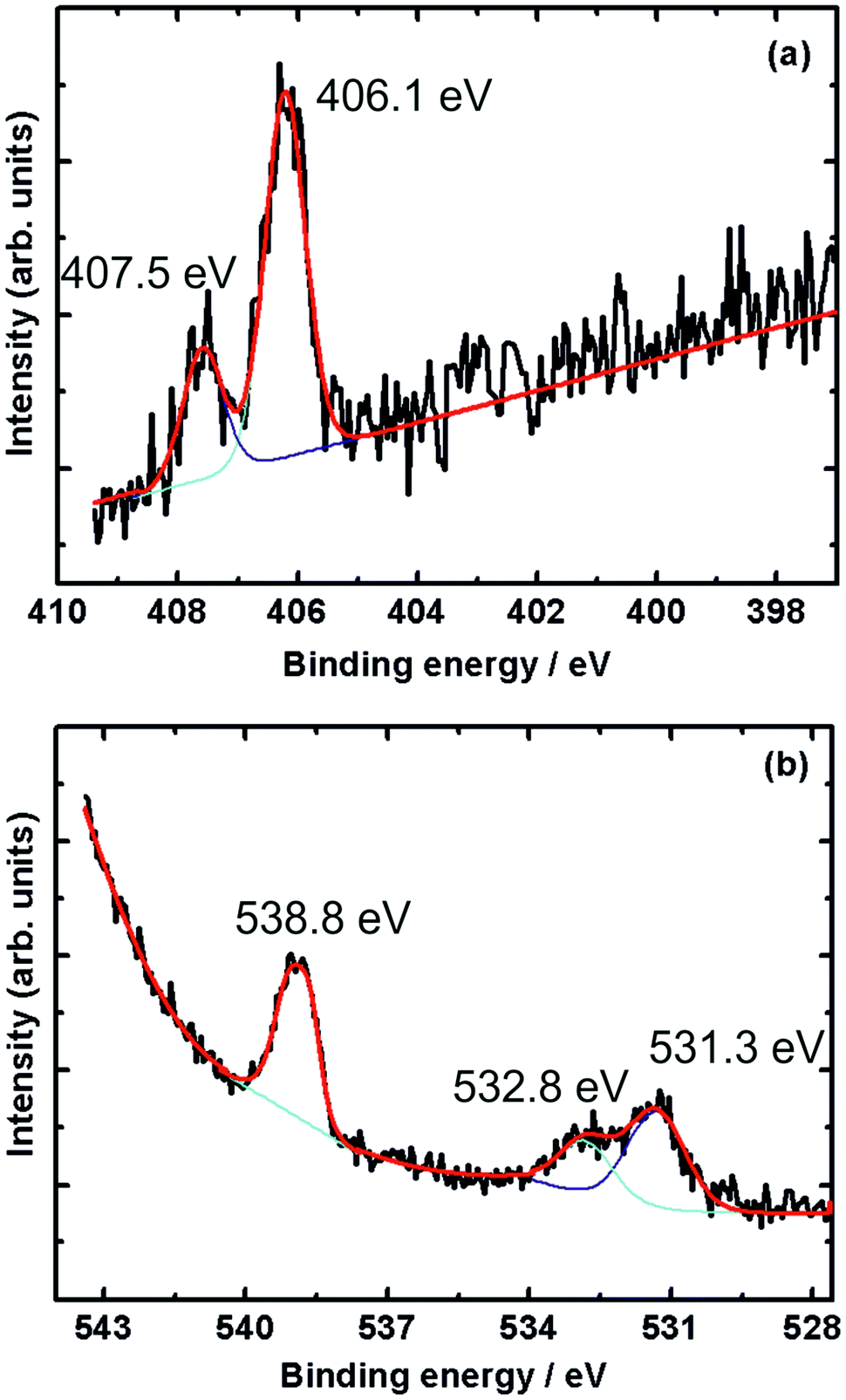 | ||
| Fig. 4 (a) N 1s and (b) O 1s X-ray photoelectron spectra during exposure of Au(100) to NO at 1 mbar pressure and 298 K. The black line is the recorded XP spectrum, while the red line is a fit, deconvoluted into the purple and turquoise contributions. | ||
The O 1s spectrum shows three peaks at 531.3, 532.8 and 538.8 eV. While the peak at 538.8 eV is due to the oxygen atom in gas-phase NO, the twin peaks appearing at 531.3 and 532.8 eV can be attributed to residual CO adsorbed on the surface. The twin O 1s peaks appearing in this energy region were also reported in CO adsorption on Pt(111).34,35 The above results show that NO does not adsorb on Au(100) under these conditions of temperature and pressure.
Adsorption at 220 K
As mentioned previously, XPS spectra recorded at 220 K for the ordered sputtered and annealed Au(100) surface show no adsorption of NO (data not shown, but spectra qualitatively similar to Fig. 4) as indicated by the absence of N 1s and O 1s peaks at ∼400 eV and ∼531 eV, respectively.16 As in the case of room temperature measurements, only gas-phase NO peaks were observed.We hence studied NO adsorption on a sputtered (but not annealed) gold surface. The surface was sputtered for 30 min followed by exposure to NO.
Fig. 5 shows the N 1s and O 1s XPS core level spectra of nitric oxide adsorbed on gold at 1 mbar NO pressure and 220 K, the lowest temperature reached in our NAP-spectrometer, but ∼40 K higher than the base temperature in our 3D-VMI spectrometer (178 K).
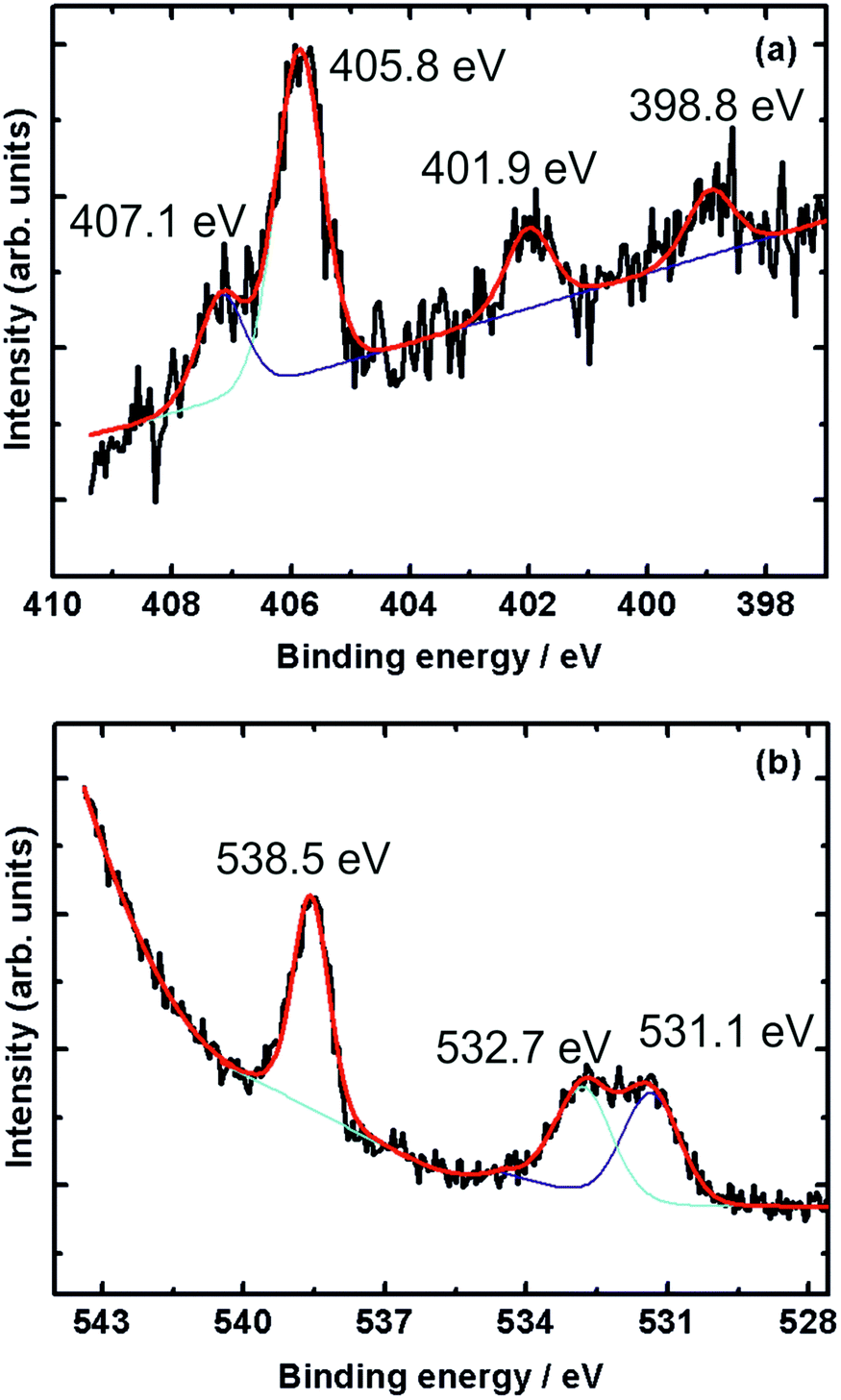 | ||
| Fig. 5 (a) N 1s and (b) O 1s XPS core-level spectra of NO adsorbed on sputtered gold surface taken at 220 K and 1 mbar NO pressure. The black line is the recorded XP spectrum, while the red line is a fit, deconvoluted into the purple and turquoise contributions. | ||
The N 1s spectrum (Fig. 5a) shows three distinct peaks at 398.8, 401.9 and 405.8 eV, and another peak at 407.1 eV appearing as a shoulder of the 405.8 eV peak. The two peaks at 405.8 eV and 407.1 eV are due to gaseous NO, as shown in Fig. 4. The other two peaks at 398.8 eV and 401.9 eV point to the presence of adsorbed nitrogen species on the sputtered gold surface.
Rienks et al. reported three main peaks at 394.7, 398.1 and 401.3 eV in the N 1s region recorded during the exposure of Pt(100) to NO at 200 K.36 The peak at 394.7 eV – not observed in the current study – was attributed to atomic nitrogen due to dissociation of adsorbed NO. The peaks at 398.1 eV and 401.3 eV, and hence close to those observed in the current study, were attributed to two different molecularly adsorbed NO species on the surface. Further support of molecularly adsorbed NO comes from a study by Sugai et al. who observed a peak at 401.5 eV due to molecularly adsorbed NO on Pt(100) and (310),37 and a study by Kiskinova et al. who observed a N 1s peak at 400.7 eV due to molecularly adsorbed NO on Pt(111).38
In case of dissociative adsorption, one might also expect a feature in the O 1s region just below 530 eV characteristic of atomic oxygen,39,40 but this was not observed on our sputtered gold surface.
Instead, a peak at 538.5 eV and two partially overlapping peaks at 531.1 and 532.7 eV appear in the O 1s region of the spectrum, see Fig. 5b. All three peaks also appear in the room temperature spectra shown in Fig. 4, but in particular the latter two are now more intense. The higher binding energy peak is due to gas-phase NO, while the two peaks around 532 eV are likely due to molecularly adsorbed NO, possibly with some contribution from adsorbed residual CO. Kiskinova et al. assigned these two peaks in the case of NO adsorption on Pt(111) to bridge-bonded (lower BE, 530.6 eV) and on-top (higher BE, 532.5 eV) NO.38 While we are working with a sputtered gold surface, we nonetheless conclude that we observe NO on adsorption sites that resemble bridge sites (398.1 eV in the N 1s spectrum and 531.1 eV in the O 1s spectrum) and on-top sites (401.3 eV in the N 1s spectrum and 532.7 eV in the O 1s spectrum). The fact that the difference in binding energy between the bridge and on-top site is greater in the N 1s region than it is for the oxygen indicates that N is facing the surface, in agreement with previous work for NO on Pt(100) and Pt(111).36,38
Adsorption at 266 K
After adsorption at 220 K and recording of the XPS spectra, the temperature of the surface was gradually raised to 266 K while keeping the NO flowing into the cell, and the spectra were recorded again, shown in Fig. 6. Once again only peaks related to gaseous NO are observed. The two peaks associated with the N surface species have disappeared. This shows that NO has desorbed from the surface at this temperature in agreement with our previous 3D-VMI results.31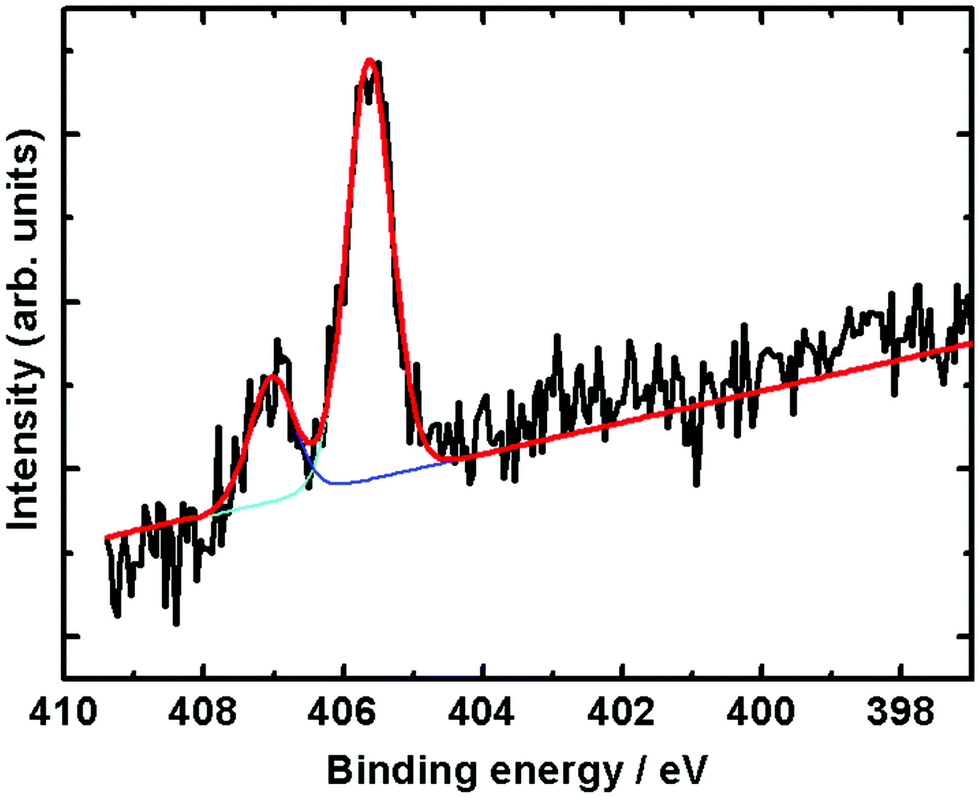 | ||
| Fig. 6 N 1s XPS core-level spectra during exposure of NO at 266 K and 1 mbar pressure. | ||
DFT results
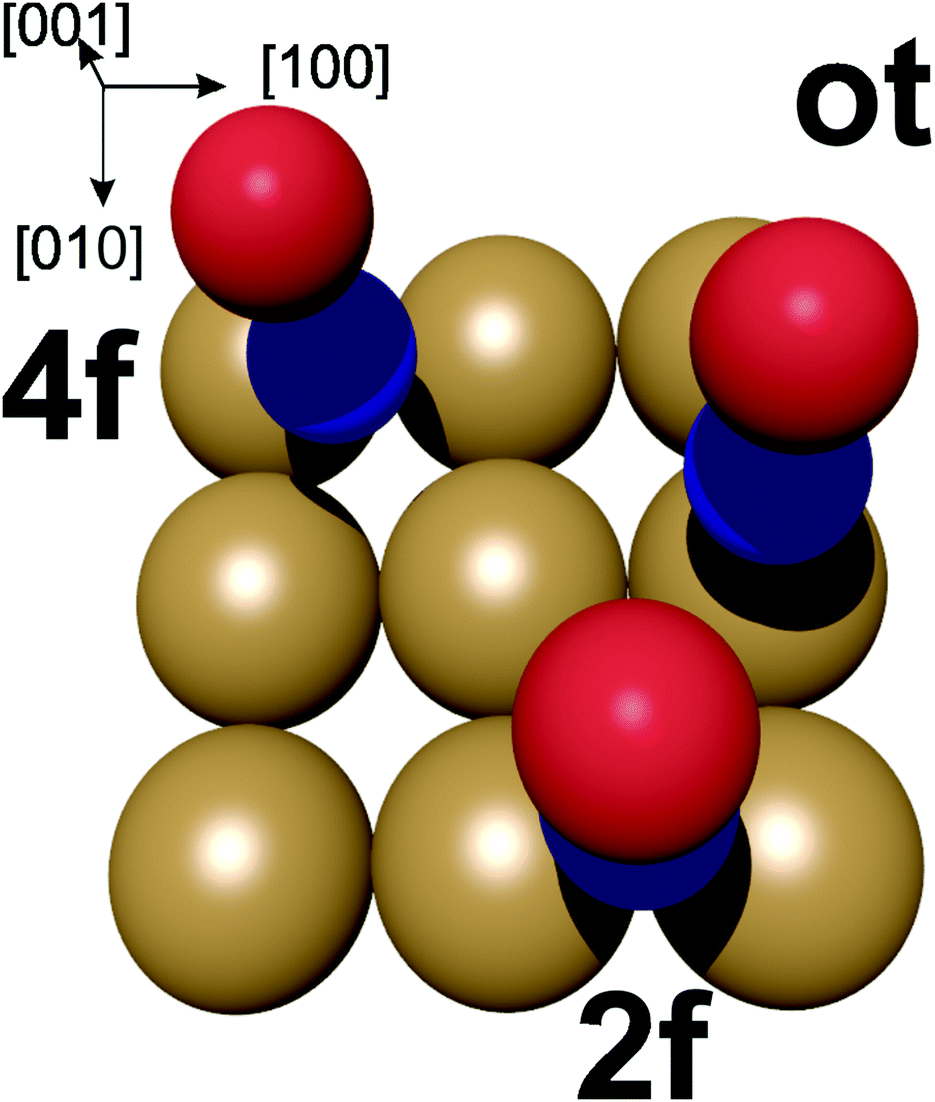 | ||
| Fig. 7 Potential adsorption sites for NO on the Au(100) surface. ot = on-top, 2f = bridge site, 4f = hollow site. Oxygen in red, nitrogen in blue. | ||
In the energetically more favourable 2f site, the single electron in the two NO π* orbitals (belonging to the nitrogen atom) can efficiently overlap with the orbitals on two neighbouring Au atoms at the surface between which the NO is located, resulting in the N–O bond being aligned along the surface normal. In contrast, our calculations yield a small tilt angle of 0.2° of the N–O bond axis with respect to the surface normal in the case of the energetically slightly less-favourable on-top adsorption. This is due to the overlap of the π* orbital with the orbitals on a single Au atom. This tilting geometry of NO adsorbed on Au(111) was also calculated by Zhang et al.44 In agreement with our 3D-surface VMI results, the cold rotational state distribution is due to the majority of the NO being aligned along the normal of the surface, with the slightly tilted NO at the on-top side not noticeably contributing to the low rotational excitation.
Conclusions
In this paper, we have presented the results of NO adsorption on a Au(100) single crystal surface using 3D-VMI, NAP-XPS and condensed-phase DFT calculations. The combined results of those three different techniques point to a molecular adsorption of NO on the Au(100) surface at low temperatures. The VMI and DFT results show that NO adsorbs onto the gold surface from its N end with the O atom pointing away from the surface. The novelty of this work is that it demonstrates the capability of the 3D-VMI technique not only in elucidating the photodesorption dynamics of pre-adsorbed molecules on metal substrates, but also in revealing the adsorption site, orientation and alignment of adsorbates in a fashion not too dissimilar to early electron-stimulated desorption ion angular distribution studies (ESDIAD) by Madey, Yates, and co-workers.46,47Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. A. is grateful to the Libyan government for funding. We also thank Rosemary Jones and Khadisha Zahrab for help in the interpretation of the photoelectron spectra.Notes and references
- G. Czapski, J. Holcman and B. H. J. Bielski, J. Am. Chem. Soc., 1994, 116, 11465–11469 CrossRef CAS.
- I. M. Goldstein, P. Ostwald and S. Roth, Vision Res., 1996, 36, 2979–2994 CrossRef CAS PubMed.
- J. V. Esplugues, Br. J. Pharmacol., 2002, 135, 1079–1095 CrossRef CAS PubMed.
- J. Ritz, H. Fuchs and H. G. Perryman, Hydroxylamine. In Ulmann's Encyclopedia of Industrial Chemistry, Wiley, Chichester, 6th edn, 2000 Search PubMed.
- C. G. M. van de Moesdijk, The catalytic reduction of nitrate and nitric oxide to hydroxylamine: kinetics and mechanism, PhD thesis, Eindhoven University of Technology, 1979 Search PubMed.
- S. Horold, K.-D. Vorlop, T. Tacke and M. Sell, Catal. Today, 1993, 17, 21–30 CrossRef.
- V. Rosca, G. L. Beltramo and M. T. M. Koper, Langmuir, 2005, 21, 1448–1456 CrossRef CAS PubMed.
- N. I. ll’chenko and G. I. Golodets, J. Catal., 1975, 39, 57–72 CrossRef.
- A. Cuesta and M. Escudero, Phys. Chem. Chem. Phys., 2008, 10, 3628–3634 RSC.
- A. Clayborne, H.-J. Chun, R. B. Rankin and J. Greeley, Angew. Chem., Int. Ed., 2015, 54, 8255–8258 CrossRef CAS PubMed.
- B. E. Nieuwenhuys, Adv. Catal., 1999, 44, 259 CAS.
- Y. Zhang, R. W. Cattrall, I. D. Mckelvie and S. D. Kolev, Gold Bull., 2011, 44, 145 CrossRef CAS.
- A. Ueda and M. Haruta, Gold Bull., 1991, 32, 3–11 CrossRef.
- A. V. Bukhtiyarov, A. V. Nartova and R. I. Kvon, Kinet. Catal., 2011, 52, 756–760 CrossRef CAS.
- C. P. Vinod, J. W. H. Niemantsverdriet and B. E. Nieuwenhuys, Appl. Catal., A, 2005, 291, 93–97 CrossRef CAS.
- A. V. Bukhtiyarov, R. I. Kvon, A. V. Nartova, I. P. Prosvirin and V. I. Bukhtiyarov, Surf. Sci., 2012, 606, 559–563 CrossRef CAS.
- J. L. C. Fajín, M. Natália, D. S. Cordeiro and J. R. B. Gomes, J. Phys. Chem. C, 2009, 113, 8864–8877 CrossRef.
- R. Zanella, S. Giorgio, C. H. Shin, C. R. Henry and C. Louis, J. Catal., 2004, 222, 357–367 CrossRef CAS.
- C. Lemire, R. Meyer, S. Shaikhutdinov and H.-J. Freund, Surf. Sci., 2004, 552, 27–34 CrossRef CAS.
- X. Deng, B. K. Min, A. Guloy and C. M. Friend, J. Am. Chem. Soc., 2005, 127, 9267–9270 CrossRef CAS PubMed.
- T. V. W. Janssens, A. Carlsson, A. Puig-Molina and B. S. Clausen, J. Catal., 2006, 240, 108–133 CrossRef CAS.
- T. V. W. Janssens, B. S. Clausena, B. Hvolbækb, H. Falsigc, C. H. Christensen, T. Bligaard and J. K. Nørskovb, Top. Catal., 2007, 44, 15–26 CrossRef CAS.
- A. Hussain, D. Curulla Ferré, J. Gracia, B. E. Nieuwenhuys and J. W. Niemantsverdriet, Surf. Sci., 2009, 603, 2734–2741 CrossRef CAS.
- Z. Wu, L. Xu, W. Zhang, Y. Mac, Q. Yuan, Y. Jin, J. Yang and W. Huang, J. Catal., 2013, 304, 112–122 CrossRef CAS.
- M. Reid and S. P. K. Koehler, Rev. Sci. Instrum., 2013, 84, 044101 CrossRef PubMed.
- N. Fairley, CasaXPS Manual 2.3.15 Introduction to XPS and AES, Casa Software, 2009 Search PubMed.
- S. J. Clark, I. Matthew, D. Segall, C. J. Pickard, P. J. Hasnip, M. I. J. Probert, K. Refson and M. C. Payne, Z. Kristallogr. – Cryst. Mater., 2005, 220, 567–570 CAS.
- U. K. Chohan, E. Jimenez-Melero and S. P. K. Koehler, Appl. Surf. Sci., 2016, 387, 385–392 CrossRef CAS.
- J. F. Janak, Phys. Rev. B: Condens. Matter Mater. Phys., 1978, 18, 7165 CrossRef CAS.
- S. Abujarada, H. AlSalem, U. K. Chohan, G. L. Draper and S. P. K. Koehler, J. Chem. Phys., 2016, 145, 184201 CrossRef PubMed.
- S. Abujarada, C. Flathmann and S. P. K. Koehler, J. Phys. Chem. C, 2017, 121, 19922–19929 CrossRef CAS.
- T. Shimada, B. S. Mun, I. F. Nakai, A. Banno, H. Abe, Y. Iwasawa, T. Ohta and H. Kondoh, J. Phys. Chem. C, 2010, 114, 17030–17035 CrossRef CAS.
- S.-W. Yu, W. C. Stolte, R. Guillemin, G. Öhrwall, I. C. Tran, M. N. Piancastelli, R. Feng and D. W. Lindle, J. Phys. B: At., Mol. Opt. Phys., 2004, 37, 3583–3592 CrossRef CAS.
- P. N. Norton, J. W. Goodale and E. B. Selkirk, Surf. Sci., 1979, 83, 18 CrossRef.
- M. Kiskinova, G. Pirug and H. P. Bonzel, Surf. Sci., 1983, 133, 321–343 CrossRef CAS.
- E. D. L. Rienks, J. W. Bakker, A. Baraldi, S. A. C. Carabineiro, S. Lizzit, C. J. Weststrate and B. E. Nieuwenhuys, Surf. Sci., 2002, 516, 109–117 CrossRef CAS.
- S. Sugai, K. Takeuchi, T. Ban, H. Miki, K. Kawasaki and T. Kioka, Surf. Sci., 1993, 282, 67–75 CrossRef CAS.
- M. Kiskinova, G. Pirug and H. P. Bonzel, Surf. Sci., 1984, 136, 285–295 CrossRef CAS.
- M. V. Ganduglia-Pirovano, M. Scheffler, A. Baraldi, S. Lizzit, G. Comelli, G. Paolucci and R. Rosei, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 205415 CrossRef.
- J. F. Zhu, M. Kinne, T. Fuhrmann, R. Denecke and H. P. Steinruck, Surf. Sci., 2003, 529, 384–396 CrossRef CAS.
- O. Olvera-Neria, V. Bertin and E. Poulain, Int. J. Quantum Chem., 2011, 111, 2054 CrossRef CAS.
- E. D. L. Rienks, G. P. van Berkel, J. W. Bakker and B. E. Nieuwenhuys, Surf. Sci., 2004, 571, 187–193 CrossRef CAS.
- Y. Wang, D. Zhang and C. Liu, Sci. China: Chem., 2011, 54, 194 CAS.
- W. Zhang, Z. Li, Y. Luo and J. Yang, J. Chem. Phys., 2008, 129, 134708 CrossRef PubMed.
- Z.-H. Zeng, J. L. F. Da Silva, H.-Q. Deng and W.-X. Li, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 205413 CrossRef.
- T. E. Madey and J. T. Yates Jr., Surf. Sci., 1975, 49, 465–496 CrossRef CAS.
- T. E. Madey and J. T. Yates Jr., Surf. Sci., 1977, 63, 203–231 CrossRef.
| This journal is © the Owner Societies 2019 |