
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBlock copolymers as bile salt sequestrants: intriguing structures formed in a mixture of an oppositely charged amphiphilic block copolymer and bile salt†
Karin
Schillén
 *a,
Luciano
Galantini
*b,
Guanqun
Du
a,
Alessandra
Del Giudice
b,
Viveka
Alfredsson
a,
Anna M.
Carnerup
a,
Nicolae V.
Pavel
b,
Giancarlo
Masci
b and
Bo
Nyström
c
*a,
Luciano
Galantini
*b,
Guanqun
Du
a,
Alessandra
Del Giudice
b,
Viveka
Alfredsson
a,
Anna M.
Carnerup
a,
Nicolae V.
Pavel
b,
Giancarlo
Masci
b and
Bo
Nyström
c
aDivision of Physical Chemistry, Department of Chemistry, Lund University, P.O. Box 124, SE-221 00 Lund, Sweden. E-mail: Karin.Schillen@fkem1.lu.se
bDepartment of Chemistry, Sapienza University of Rome, P.O. Box 34-Roma 62, Piazzale A. Moro 5, I-00185 Roma, Italy. E-mail: luciano.galantini@uniroma1.it
cDepartment of Chemistry, University of Oslo, P.O. Box 1033, Blindern N-0315, Oslo, Norway
First published on 20th May 2019
Abstract
To study the formation and characterize the structure of mixed complexes of oppositely charged block copolymers and surfactants are of great significance for practical applications, e.g., in drug carrier formulations that are based on electrostatically assisted assembly. In this context, biocompatible block copolymers and biosurfactants (like bile salts) are particularly interesting. In this work, we report on the co-assembly in dilute aqueous solution between a cationic poly(N-isopropyl acryl amide) (PNIPAM) diblock copolymer and the oppositely charged bile salt surfactant sodium deoxycholate at ambient temperature. The cryogenic transmission electron microscopy (cryo-TEM) experiments revealed the co-existence of two types of co-assembled complexes of radically different morphology and inner structure. They are formed mainly as a result of the electrostatic attraction between the positively charged copolymer blocks and bile salt anions and highlight the potential of using linear amphiphilic block copolymers as bile salt sequestrants in the treatment of bile acid malabsorption and hypercholesterolemia. The first complex of globular morphology has a coacervate core of deoxycholate anions and charged copolymer blocks surrounded by a PNIPAM corona. The second complex has an intriguing tape-like supramolecular morphology of several micrometer in length that is striped in the direction of the long axis. A model is presented in which the stretched cationic blocks of several block copolymers interact electrostatically with the bile salt molecules that are associated to form a zipper-like structure. The tape is covered on both sides by the PNIPAM chains that stabilize the overall complex in solution. In addition to cryo-TEM, the mixed system was investigated in a range of molar charge fractions at a constant copolymer concentration by static light scattering, small angle X-ray scattering, and electrophoretic mobility measurements.
Introduction
Mixtures of polyelectrolytes and oppositely charged surfactants are important in general for biological and technological applications as they give rise to complexes with unique structures and functionality, which can be controlled by varying the charge molar fractions of the components and their individual characteristics.1–5 The strong attractive electrostatic interaction in these kinds of mixtures often leads to an associative phase separation over a wide range of concentrations, the driving force of which is an entropic effect arising from the counter ion release and it depends on both the surfactant hydrophobic–hydrophilic balance and the polymer molecular weight.6–8 The polyion–surfactant ions aggregate to form a concentrated phase, while the dilute phase contains mainly water and counterions. However, the phase separation becomes microscopic by using charged block copolymers containing one (or several) water-soluble nonionic block(s), or by adding nonionic surfactants, see e.g.ref. 9–12 and the references therein.The self-assembly of the block copolymer can thus be promoted by complexation via an electrostatically driven co-assembly with oppositely charged components, such as surfactants, block copolymers, charged linear polymers or proteins. The resultant nanostructures sometimes with a liquid crystalline interior13 are referred to as polyion complex micelles, block ionomer complexes, inter-polyelectrolyte complexes, complex coacervate core micelles or colloidal complexes (in this work mixed complexes).14–21 Interestingly, the solution behavior of these nanostructures is much more versatile than the one of those formed by self-assembly of traditional amphiphiles (surfactants or amphiphilic block copolymers). This is because the electrostatic driving force is tunable through variables such as mixing ratio and added salt and also affected by the presence of counterions and additional hydrophobic interactions to list a few. In order to precisely control the mechanism of self-organization in oppositely charged polymer–surfactant systems, it is thus crucial to characterize the morphology and inner structure of the different mixed complexes formed in solution as well as their solution behavior.
The aim of this work was to explore the co-assembly of the bile salt (BS) surfactant, sodium deoxycholate (NaDC), and an oppositely charged diblock copolymer constituted of one poly(N-isopropyl acryl amide) (PNIPAM) block and a cationic polymer block of poly(3-acrylamidopropyl)-trimethylammonium chloride (PAMPTMA(+)), denoted PNIPAMm=120-b-PAMPTMA(+)n=30, where m and n denote the degrees of polymerization. The synthesis and aqueous solution properties of this type of copolymers have been reported in ref. 22 and 23. The effect of temperature on self-assembly in water of block copolymers with one PNIPAM block and one charged block have been extensively studied in the literature.22–31 The main characteristic feature of the aqueous phase behavior of thermo-responsive polymers, such as PNIPAM, is the existence of a lower critical solution temperature (LCST) above which the system enters a two-phase region for the reason that the polymer is no longer soluble in the solvent. This behavior regulates the temperature-dependent self-assembly of block copolymers carrying one or several PNIPAM blocks. We refer to the excellent review on PNIPAM/water phase diagrams by Halperin et al. for further details.32
BSs, like NaDC, have a steroidal molecular structure, which is completely different from the typical head–tail one of conventional surfactants.33,34 This governs their self-assembly in solution forming structures with specific supramolecular arrangements, e.g., globular, rod-like or tubular morphologies.33,35 as well as hexagonal liquid crystalline phases.36 Because of their molecular architecture, they are also used as substrates for the synthesis of an expanded family of rigid amphiphiles with uncommon aggregation behaviors37–40 and dispersing ability.41 BSs are some of the most important surfactants in many living organisms where they work as dispersing agents of dietary lipids to regulate their uptake in the small intestine.42 Due to this, the aqueous phase and assembly behavior of bile salts and lipids has been studied for decades.43–47 The biosynthesis of BSs or bile acids (BAs) takes place from cholesterol in the liver and they are stored in the gall bladder. Different pathologies are associated to disorders in the bile acid metabolism, such as BA malabsorption cause of chronic diarrhea, liver disease and gallstones.48 The BA biosynthesis is regulated by the Fibroblast growth factor 19 (FGF19), which is produced in the terminal ileum by the binding of BSs to the nuclear farnesoid X receptor FXR.49 In BA diarrhea, a reduced FGF19 production leads to a defective feedback inhibition of the BA biosynthesis and excess BAs will pass into the colon with diarrhea as the result.50,51 It can be treated by using polymeric sequestrants aimed at binding BSs and by that decreasing the level in the intestine.42,52,53 There has been little commercial development of these drugs, whereas instead attention has been drawn to stimulate FGF19 levels in patients with BA diarrhea.54 While lowering the BS concentration, the bile salt sequestrants promote the cholesterol-to-BS transformation thus reducing the low-density lipoprotein (LDL) cholesterol concentration in the human body. Therefore, they are also used in the treatment of hypercholesterolemia.55,56 They can also serve as therapeutic “add-on” for patients with type 2 diabetes mellitus.57 Still, there are problems with the current BS sequestrants, e.g., the water-insoluble cationic hydrogels cholestyramine (Questran®), colestipol (Colestid®) and colesevelam (Cholestagel®),58 that bind bile salts, including rather low efficacy, poor compliance and aggravation of severe constipation.52 There is therefore room for improvements and optimization of BS sequestrants in general.55,59–63
We have in several earlier studies reported on the interaction between the bile salt sodium glycodeoxycholate (NaGDC) and different PEO–PPO–PEO triblock copolymers.64–66 Our studies were driven by the fact that these copolymers could have a potential use as BS sequestrants in the therapy of bile-acid related diseases including hypercholesterolemia and as smart nanocarriers of active molecules.67 In ref. 64–66 we demonstrated that NaGDC associates to PEO–PPO–PEO micelles to form mixed complexes through hydrophobic interactions. However, the bile salt eventually disintegrates the micelles and their “resistance” to disruption follows the same increasing trend as the critical micelle temperature values of the copolymers.66 In this context, although PEO–PPO–PEO copolymers have many advantages, amphiphilic block copolymers carrying a positively charged block are expected to associate more strongly with the anionic BSs and thus being even better candidates than the nonionic ones for being used as BS sequestrants as well as in various biocompatible formulations.
This work was inspired by our previous study on a cationic PNIPAM-based copolymer mixed system, where the interactions between the same copolymer as studied here and a thermo-responsive modified BS was investigated. The results revealed that very interesting supramolecular mixed structures were formed via a combination of attractive electrostatic and hydrophobic intermolecular interactions.68 Here, in this physico-chemical study, we employed static light scattering (SLS) together with measurements of electrophoretic mobility to explore the solution properties of the PNIPAM120-b-PAMPTMA(+)30/NaDC aqueous system as a function of mixture composition. Furthermore, cryogenic transmission electron microscopy (cryo-TEM) and small angle X-ray scattering (SAXS) were utilized for morphological and structural characterization of the mixed PNIPAM120-b-PAMPTMA(+)30–NaDC complexes. Finally, models of their internal structure are proposed. In this work, we focused on the fascinating structures that are formed in these complex mixtures at ambient temperature, since these features are more or less hidden at elevated temperatures due to the emergence of strong association effects generated by the temperature-induced hydrophobicity of the PNIPAM block. Therefore, to avoid these intricate effects, this study was conducted at ambient temperatures well below the phase transition of PNIPAM.
Experimental
Materials
The PNIPAM120-b-PAMPTMA(+)30 diblock copolymer was previously synthesized by atom transfer radical polymerization.22 The copolymer has a number-average molar mass (Mn) of 19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 871 g mol−1 (proton nuclear magnetic resonance) or 20000 g mol−1 (gel permeation chromatography) and a polydispersity index of 1.21. Sodium deoxycholate (Mn = 414.55 g mol−1) was purchased from Sigma Aldrich (assay ≥99%) and used as received. The chemical structures of the two oppositely charged species are illustrated in Fig. 1.
871 g mol−1 (proton nuclear magnetic resonance) or 20000 g mol−1 (gel permeation chromatography) and a polydispersity index of 1.21. Sodium deoxycholate (Mn = 414.55 g mol−1) was purchased from Sigma Aldrich (assay ≥99%) and used as received. The chemical structures of the two oppositely charged species are illustrated in Fig. 1.
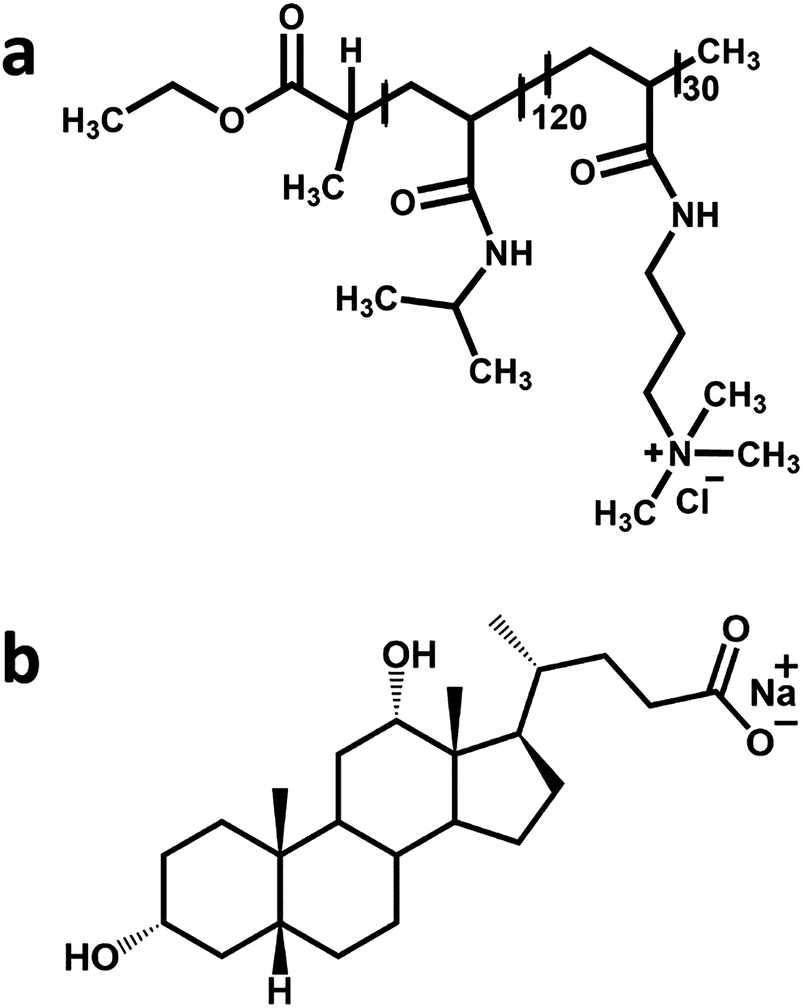 | ||
| Fig. 1 The chemical structure of (a) the PNIPAM120-b-PAMPTMA(+)30 diblock copolymer and (b) sodium deoxycholate. | ||
The aqueous mixed solutions of PNIPAM120-b-PAMPTMA(+)30 and NaDC were prepared from stock solutions keeping the block copolymer concentration constant. Water used for the sample preparation was purified using a Milli-Q system (Millipore Corporation, Bedford, MA, U.S.). The solution composition is expressed in molar fraction of negative charge, defined as X = n−/(n− + n+), where n− and n+ are the number of negative charges (= number of moles of bile salt) and number of moles of positive charges (= 30 × number of moles of PNIPAM120-b-PAMPTMA(+)30), respectively. Initially nine samples were prepared with the following X = 0.06, 0.11, 0.20, 0.33, 0.50, 0.67, 0.80, 0.89, and 0.94 (corresponding to bile salt-to-copolymer molar charge ratio CR = nNaDC/np = 1/16, 1/8, 1/4, 1/2, 1, 2, 4, 8, and 16, respectively). The copolymer–bile salt mixed solutions used for light scattering and electrophoretic mobility measurements had a constant copolymer concentration of 0.13 wt% (= 0.065 mM) and were prepared as follows. For each sample, 1.5 mL of a 0.27 wt% PNIPAM120-b-PAMPTMA(+)30 solution and different volumes of stock solution of NaDC were added until the desired charge fraction was obtained. Water was then added up to a total volume of 3 mL. The pH of the pure bile salt and block copolymer solution was 7.6 and 7.2, respectively. The solutions were filtered through 0.45 μm-Millex-HV syringe filters from Millipore (now Merck Millipore) for the light scattering and electrophoretic mobility measurements. The samples for SAXS experiments were prepared using the same protocol but had a higher copolymer concentration (0.50 wt%) and they were not filtered. For comparison, a filtered PNIPAM120-b-PAMPTMA(+)30–NaDC mixed solution (X = 0.5, 0.25 wt% copolymer) was investigated by cryo-TEM, dynamic light scattering (DLS) and SAXS.
Static light scattering and electrophoretic mobility
A Zetasizer Nano ZS instrument from Malvern Instruments (now part of Malvern Panalytical), Malvern, U.K., was utilized to perform SLS measurements in a backscattering geometry at a scattering angle of θ = 173° using DTS1070 disposable folded capillary cells. The measurements were carried out at 22 °C. The total scattering intensity (Itot) without any subtraction of the solvent scattering is presented here as the average of three consecutive measurements together with the estimated standard deviation (std).Measurements of the electrophoretic mobility (μ) at 22 °C were preformed at θ = 13° using the same instrumental setup as for the SLS experiments and with the same type of cells. In this instrument, a laser Doppler electrophoresis technique is combined with a phase analysis light scattering method in which the Doppler effect is exploited to determine the electrophoretic velocity of the charged particles moving in an applied field and which is proportional to μ.2 The presented μ values are the average of three consecutive measurements together with the estimated std.
Cryogenic transmission electron microscopy
Cryo-TEM experiments were carried out on a JEM-2200FS transmission electron microscope (JEOL) specially optimized for cryo-TEM at the National Center for High Resolution Electron Microscopy (nCHREM) at Lund University. It is equipped with a field-emission electron source, a cryo pole piece in the objective lens and an in-column energy filter (omega filter). Zero-loss images were recorded at an acceleration voltage of 200 kV on a bottom-mounted TemCam-F416 camera (TVIPS) using SerialEM under low-dose conditions. Specimens were prepared using an automatic plunge freezer system (Leica Em GP) with the environmental chamber operated at 25 °C and 90% relative humidity. A 4 μL droplet of the sample solution was deposited on a lacey formvar carbon coated grid (Ted Pella) and was blotted with filter paper to remove excess fluid. The grid was then plunged into liquid ethane (around −183 °C) to ensure rapid vitrification of the sample in its native state. The specimens were thereafter stored in liquid nitrogen (−196 °C) and prior to imaging transferred into the microscope using a cryo transfer tomography holder (Fischione, Model 2550).Small angle X-ray scattering
SAXS measurements were performed at the SWING beamline of SOLEIL Synchrotron Facility (Saint-Aubin, France). The sample to detector distance and the X-ray wavelength λ were set to 3.075 m and 0.99 Å. With this setting, the explored q region was 0.023 nm−1 < q < 3.4 nm−1, where q = 4πsin(θ/2)/λ, θ being the scattering angle. Scattering data were collected using a 17 × 17 cm2 low-noise AVIEX CCD detector. The measurements were conducted using an automated robot provided at the beamline.69 A continuous flow of solution (50 μL min−1) in front of the beam reduced possible beam damage. Both the capillary and the sample before the injection were thermostated and kept at the same temperature (25 °C). Additional measurements on a filtered PNIPAM120-b-PAMPTMA(+)30–NaDC mixed solution (X = 0.5) were performed using a laboratory based Xeuss 3.0 HR P300K Q-Xoom system (Xenocs SA, Sassenage, France), having a micro-focus Genix 3D X-ray source with λ = 1.54 Å and a two-dimensional Pilatus3 R 300K detector from Dectris (Dectris Ltd, Baden, Switzerland). The sample was characterized in a sealed quartz capillary with thickness 1.5 mm. Measurements were performed at room temperature (25 ± 1 °C) and in vacuum (pressure below 0.1 mbar), with two different sample-detector distances, in order to record the sample scattering within the q range 0.045 nm−1 < q < 3.4 nm−1. Water was used to collect the background data for substraction. The SAXS data reduction (radial averaging, background subtraction, absolute intensity scaling70) was performed using the FoxTrot software developed at SOLEIL. Pair distance distribution functions were obtained from the scattering profiles by using the BayesApp software.71 Model scattering profiles were calculated and fitted to the data with the software SasView 4.1 20180214 (http://www.sasview.org) and SASfit.72
Results and discussion
The dilute PNIPAM120-b-PAMPTMA(+)30–NaDC mixed solutions were investigated at compositions below and above charge neutralization in the range 0 ≤ X ≤ 0.94 (where X = 0 denotes the pure block copolymer) and at ambient temperature, well below the phase transition of PNIPAM. The results are reported and discussed below in subsections dedicated to the different techniques.Static light scattering measurements
The results from the SLS measurements at 22 °C on different PNIPAM120-b-PAMPTMA(+)30/NaDC mixtures at constant block copolymer concentration (0.13 wt%) are presented in Fig. 2, where the total SLS intensity (i.e., without any solvent scattering subtracted) is reported as a function of the charge fraction in the mixture. As noticed, Itot is confined in between a narrow range of relatively low values (2000–18000 kHz) at X ≤ 0.80, whereas a significant increase is observed X > 0.8 (values >80 000 kHz). In the mixtures, co-assembly is expected to occur due to the attractive electrostatic interaction between the bile salt anions (deoxycholate ion, DC−) and the PAMPTMA(+) blocks. Indeed, the system follows the behavior of the PNIPAM120-b-PAMPTMA(+)30/bile salt system studied in ref. 68. BSs form micelles in a stepwise process and according to the literature the second critical micelle concentration (CMC) of NaDC is 5.4 mM at 25 °C.73 According to this, the concentration of NaDC is significantly higher than CMC only at the two largest X values investigated (15 and 31 mM at X = 0.88 and 0.94, respectively). The steep increase in Itot at high NaDC content suggests that the bile salt is included in the mixed complexes even if present in very large excess. It is noted that the solution properties of the PNIPAM120-b-PAMPTMA(+)30/NaDC system is different from mixtures of a polyacrylamide-b-PAMPTMA(+) block copolymer and sodium dodecyl sulfate, where a peak in turbidity was observed at charge neutralization, which indicated a phase separation in that system74 (a difference which is also revealed in the electrophoretic mobility results, see below).
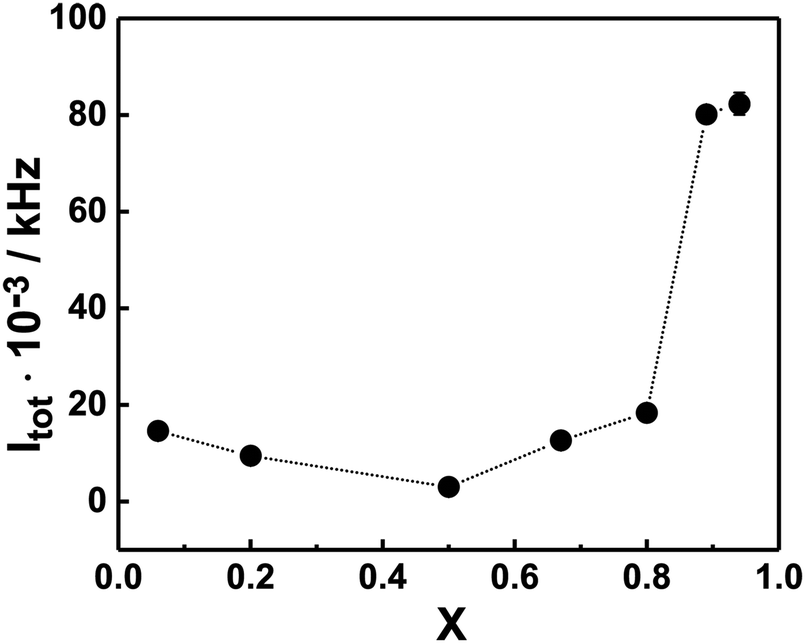 | ||
| Fig. 2 The total SLS intensity (Itot) of the PNIPAM120-b-PAMPTMA(+)30–NaDC solutions as a function of charge fraction (X) at 22 °C. The error bars (given in ±std of three consecutive measurements) are smaller than the symbol size. The line is a guide to the eye. The copolymer concentration was kept constant at 0.13 wt%. | ||
Electrophoretic mobility measurements
In order to gain information about the composition of the mixed complexes at room temperature we examined their mean electrophoretic mobility μ in copolymer–bile salt solutions with different X (Fig. 3). For mixtures with an equimolar charge ratio or with a minor content of bile salt (X ≤ 0.5), μ was close to zero or only slightly positive, suggesting that the bile salt preferentially interacts electrostatically with the cationic PAMPTMA(+) blocks to form mixed complexes of neutral or with slightly positive charge even at off-stoichiometric compositions. We found a similar behavior for mixtures of the same block copolymer but containing a bile salt derivative instead of NaDC.68 Generally, in oppositely charged polymer/surfactant mixtures the surfactant self-assemble into micellar-like aggregates in the vicinity of the polyelectrolyte chain, where the charged surfactant headgroups are neutralized by the polymer ionic units while the counterions are released into the aqueous phase.75,76 The cooperative self-assembly process starts at a critical aggregation concentration, which is less than the CMC of the surfactant. The structural stability and composition of the mixed polymer–surfactant complex formed is dictated by different contributions, e.g., polymer architecture,77 or by addition of nonionic surfactants.11 The electrostatic interactions within the complexes, which are optimized when all the charged groups are reciprocally neutralized, play here an important role.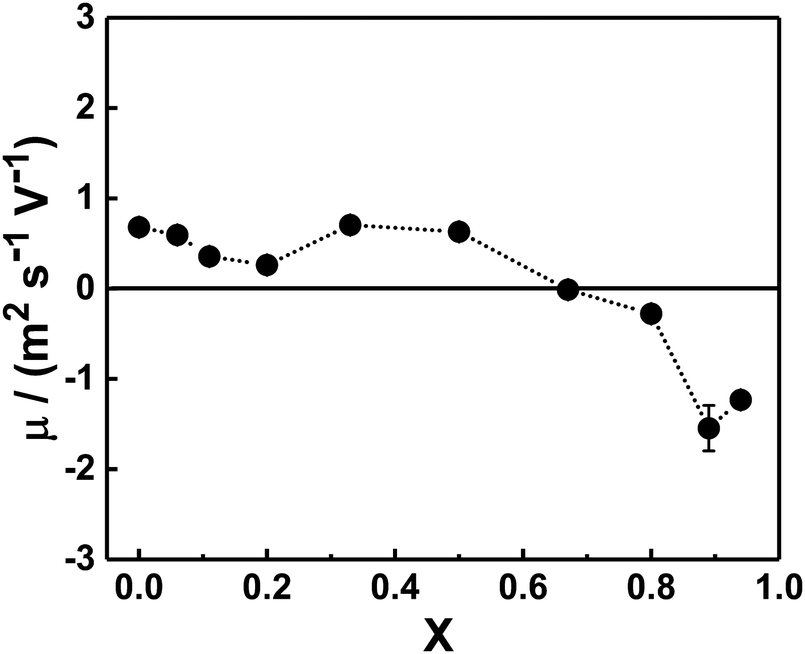 | ||
| Fig. 3 Dependence of the mean electrophoretic mobility (μ) of the mixed PNIPAM120-b-PAMPTMA(+)30–NaDC complexes at different charge fractions (X): 0.06 ≤ X ≤ 0.94. The error bars are given in ±std of three consecutive measurements. The temperature was 22 °C and the copolymer concentration was 0.13 wt%. The interpolating line is a guide to the eye. | ||
In the light of this, given that the block copolymer is readily soluble in water at the temperature of the measurements (i.e., at temperatures below the phase transition temperature of PNIPAM), it is reasonable to expect that when present in excess in terms of charge groups (X ≤ 0.5), the block copolymer molecules participate in the formation of the mixed complex until almost complete neutralization, leaving the surplus dispersed in solution in the form of single copolymer chains. The scattered light intensity of this exceeding fraction of block copolymer is negligible and reasonably not detected in the electrophoretic mobility measurements, which explains why the electrophoretic mobility was consistent with similar slightly positive or almost neutral mixed complexes at X ≤ 0.5.
For samples rich in bile salt (X ≥ 0.80), negative values of electrophoretic mobility were obtained (Fig. 3). This may indicate that mixed complexes able to contain an exceeding fraction of bile salt dominate in these mixtures. The reason for this behavior, contrary to the mixtures with excess copolymer, could be that the amphiphilic bile salt molecules associate to the mixed complexes through hydrophobic interactions in addition to the electrostatic attraction. A similar observation is reported in a study on a mixed system of a random copolymer of methoxy-poly-(ethylene glycol) monomethacrylate and (3-(methacryloylamino)propyl)trimethylammonium chloride and different bile salts, where incorporation of excess bile salt ions into the mixed complexes led to negative electrophoretic mobility values.78 As shown in Fig. 3, the crossover from slightly positive to negative complexes occurred at X values around 0.67 (CR = 2) suggesting that a consistent concentration of free bile salt was in equilibrium with the aggregates.
Cryo-TEM experiments and structural models
Cryo-TEM experiments were performed on a mixed solution at equimolar charge condition and at a copolymer concentration of 0.25 wt% and equilibrated at 25 °C prior to vitrification. The images collected demonstrate that two types of supramolecular co-assembled structures co-exist in the mixture – globular-like objects and long tape-like structures patterned with dark thin stripes (Fig. 4). | ||
| Fig. 4 Cryo-TEM images of a PNIPAM120-b-PAMPTMA(+)30–NaDC mixed solution at X = 0.5 showing (a) coexisting globular and striped tape-like mixed complexes; (b) the striped structure of the tape-like complexes and (c) an intensity profile analysis (Fig. S2, ESI†) from which a mean spacing of 5.5 nm between the dark stripes in the tapes is estimated. The copolymer concentration was 0.25 wt% and the temperature was 25 °C. The scale bar is 200 nm. | ||
The globular objects have a size of 21 ± 3 nm in diameter (based on calculations shown in ESI†). However, this value is only an estimate due to the limitations of the technique, see Fig. S1 in ESI.† Many images revealed the particles to be arranged with a specific interparticle distance (Fig. 4a and Fig. S1, ESI†). However, this is an apparent effect, induced by shear forces during the blotting procedure when the grids are prepared. The ordering does not exist in solution. Nevertheless, the fact that the particle-to-particle distance is constant suggests that the particles are close-packed. The average center-to center distance between two globules was estimated to 40 ± 5 nm obtained from visual image analysis of a collection of 20 globules presented in Fig. S1, ESI.† This suggests that only the most electron-dense part of the particles, here denoted “the core” with a diameter of 21 nm, can be perceived in the images. The contrast of the less electron-dense part, “the corona”, is not discerned from the background. It should be noted, that in other systems, where the contrast difference was sufficient to allow distinction between core and corona, the difference in electron density between these entities was larger than in our system.79,80
We can elaborate further on the composition and internal structure of the globules. As discussed above, DC− molecules are expected to interact preferentially with the cationic PAMPTMA(+) blocks suggesting that attractive electrostatic forces drive the formation of a charge-neutralized complex with a coacervate core surrounded by a PNIPAM corona in a similar way as to what is found for other oppositely charged block copolymer–surfactant systems, see, e.g., ref. 21. In this scenario, the measured interparticle distance of 40 nm could thus be the shortest distance reachable before the globules experience a strong increase in the steric repulsive force as dictated by the overlap of the PNIPAM coronas. In order to verify our hypothesis on the internal structure, we performed some geometrical calculations (see ESI† for details). The number of block copolymer chains in the complex could be estimated assuming, as explained above, that the structures visible in the cryo-TEM images are the cores carrying no water18 (which is a coarse assumption81). Assigning the core diameter Dcore = 21 nm (Fig. S1, ESI†), the core volume Vcore is 4849 nm3. The volume of one “electrostatically neutralized pair” in the core (Vpair) may be set as the sum of the volume of one PAMPTMA(+)30 block and the volume of the associated DC− molecules (see ESI†). It is reasonable to assume that the number of DC− molecules is 30 because the mean electrophoretic mobility measured is close to zero, see Fig. 3. From this, the copolymer aggregation number can be estimated using Nagg = Vcore/Vpair to be 174 copolymers and consequently the core also contains 5220 DC− molecules.
Considering that the core surface accommodates 174 PNIPAM corona chains, an available surface area per chain can be estimated, which in turn leads to a chain-to-chain average distance (s) of 3.2 nm. This value may be compared to that obtained in the case of unperturbed PNIPAM chains. The dimension of an unconstrained PNIPAM120 polymer may be estimated by using the concept of Flory radius RF of a polymer in a thermodynamically good solvent as82–84
| RF = leff·n3/5 | (1) |
Employing eqn (1) with n = 120 and nC–C = 2, gave a RF of 5.44 nm. In this case, the average chain-to-chain distance on the core surface is 10.9 nm (= 2RF), which is remarkably larger than s obtained above. This finding let us to conclude that the PNIPAM blocks are in the brush regime with stretched chains that extend out in the solution. At this condition, the PNIPAM chains are able to stabilize and protect the complexes from association by means of steric repulsive interparticle forces. By using another approach in the comparison of surface densities of PNIPAM led to the same conclusion, see, e.g., ref. 82.
Moreover, the maximum extension of a corona chain cannot be larger than its contour length, Rmax, i.e., the maximum end-to-end distance in all-trans conformation according to
| Rmax = n·nC–C·l·sin(θbond/2) | (2) |
Using eqn (2) with n = 120 and nC–C = 2, Rmax of the PNIPAM block is 30.2 nm. Hence, the maximum total radius of the spherical complexes (i.e., the sum of the core radius from cryo-TEM and the maximum corona thickness) is Rtot ≈ 41 nm. Recalling that the measured average center-to-center distance is 40 nm (Fig. 4), this result implies that the corona chains between two spherical complexes are interdigitated, thus creating a large steric repulsion between them. It is important to stress that a more realistic model that includes some hydration water molecules in the core leads to a slightly lower copolymer aggregation number and thus a smaller corona thickness.
Concerning the tape-like complex, the second type of structure found in the cryo-TEM images (Fig. 4b), a broad distribution of widths was found with a maximum value of about 35 nm (Fig. 4c and Fig. S2, ESI†). An aforementioned pattern of longitudinal regularly separated dark thin stripes is recognizable on the tapes. This structure is similar to the one observed for helical nanoribbons formed by calcium cholate reported in the literature.85 Inspired by the model proposed for those structures, we are proposing a supramolecular inner organization for the tapes formed in the system investigated here at ambient temperature, which is reported in Fig. 5.
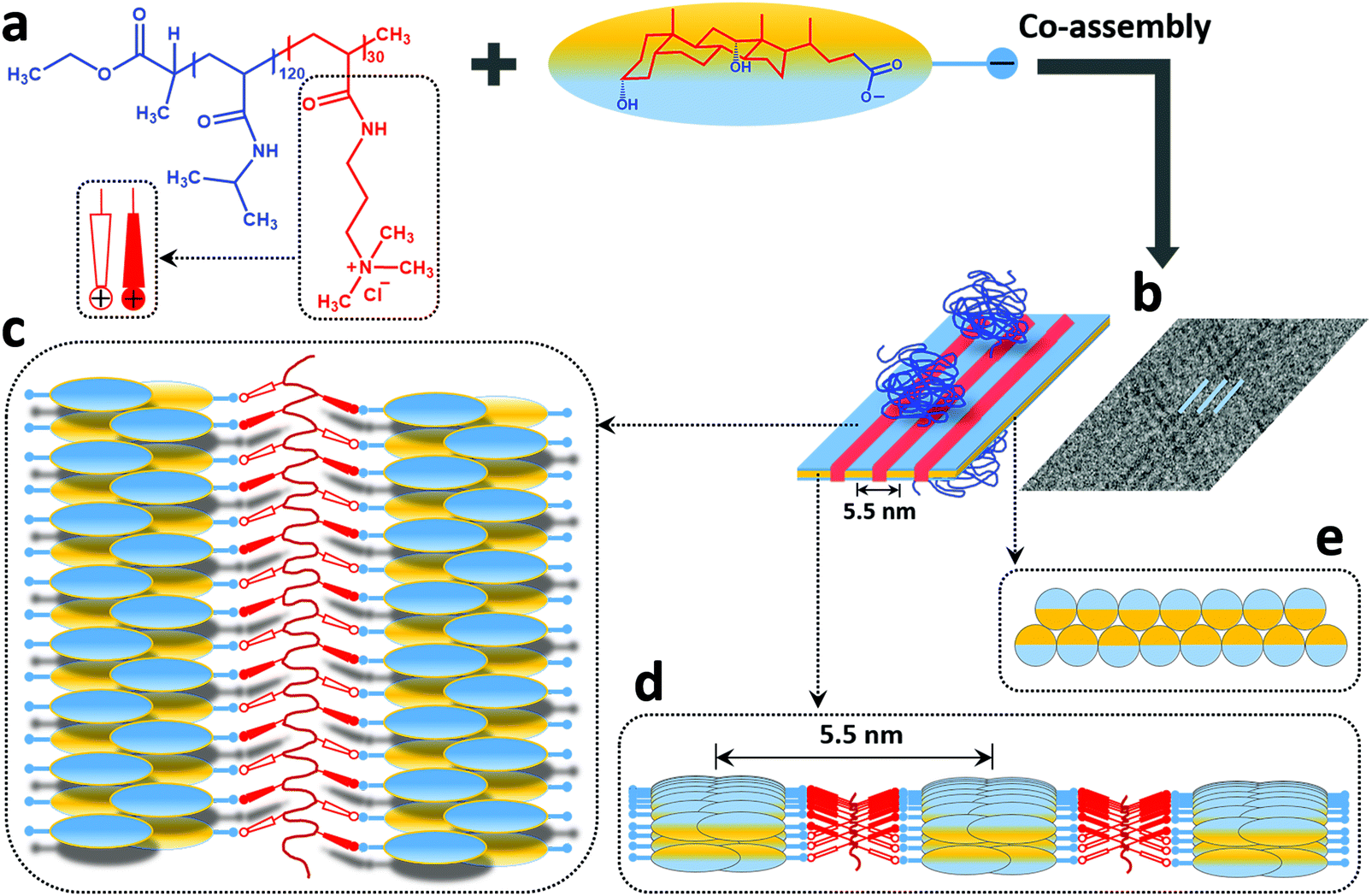 | ||
| Fig. 5 The proposed model of the structure of the tape-like mixed complexes in a 1:1 charge composition mixture of PNIPAM120-b-PAMPTMA(+)30 and NaDC (X = 0.5) at ambient temperature (see text for detailed description). Colors: hydrophobic (yellow) and hydrophilic (blue) face of NaDC, PNIPAM (dark blue) and PAMTPMA(+) (red) block of the copolymer. | ||
In the proposed model, the DC− anions (Fig. 5a) are supposed to lay flat on the surface of the ribbon oriented perpendicular to the elongation direction to create a tape-like flat structure. They adapt a strand consisting of self-assembled oppositely oriented molecules that partly overlap to form a supramolecular “zipper” (Fig. 5b–e). The bile salt strand (marked blue in Fig. 5b) is stabilized by intermolecular hydrophobic interactions and hydrogen bonds as usually observed in bile salt aggregates and crystal structures. It is proposed that these strands correspond to the repeating dark stripes of the ribbon in the cryo-TEM images (Fig. 4b). At the same time, the DC− anions interact with positively charged groups of the PAMPTMA(+) blocks that are proposed to be located in between the dark stripes (marked red in Fig. 5b–d) thus forming a low contrast spacer. Several PNIPAM120-b-PAMPTMA(+)30 copolymers are engaged in the interaction and they extend longitudinally along the ribbon (Fig. 5c). A periodicity of 5.5 nm measured between the dark strands (Fig. 5b and d) suggests that the side groups of the PAMPTMA(+) chains are oriented for an optimal interaction with the DC− anions in two adjacent strands of bile salt (Fig. 5d and e). The PNIPAM chains protrude out from the tape on both sides (Fig. 5b) and by that stabilizing the whole structure and prevent several tapes to stack. As mentioned, a PAMPTMA(+)30 chain is expected to be rather stretched along the tape, but we are not able to describe its conformation in detail. In a fully extended conformation a length of 7.5 nm could be hypothesized from eqn (2) for this block along the tape. According to the model this length would therefore be the periodicity with which the PNIPAM blocks are present along the stripes of the ribbons. Assuming an alternate orientation of these chains on both sides of the tape, they would be located on each side of the ribbon with a longitudinal spacing of sl = 15 nm. In a reference segment of a ribbon with a length ls = 150 nm and containing 5 stripes with a periodicity p = 5.5 nm, the number of PNIMAM chains in each side would then be nchain = 0.5·5ls/sl = 25 having an average area per chain of 5pls/25 = 165 nm2. According to the value of the Flory radius of PNIPAM120 chain (RF = 5.44 nm) a much smaller area (AF = πRF2 = 93 nm2) is needed per chain in order to adopt an unconstrained conformation. This means that an unperturbed chain conformation could be hypothesized for the PNIPAM blocks on each side of the ribbon (Fig. 5b), which therefore is only partially covered by the chains. The degree of coverage could be improved if, reasonably, a conformation less stretched than the fully extended one is considered for the PAMPTMA chains.
SAXS measurements
SAXS measurements were performed at 25 °C on mixed solutions of PNIPAM120-b-PAMPTMA(+)30 and NaDC with X = 0.20, 0.33, 0.50 and 0.80. Solutions of the pure PNIPAM120-b-PAMPMTA(+)30 and NaDC were also investigated (Fig. S3, ESI†).The result of the SAXS experiment on the pure 0.5 wt% PNIPAM120-b-PAMPTMA(+)30 solution (Fig. S3, ESI†) demonstrates that the copolymer is soluble and a portion of the copolymer is present as unimers in the solution at room temperature. The SAXS scattering intensity I(q) can be described as originating from single polymer chains and the downward trend observed in the range 0.1–0.25 nm−1 suggests that repulsive interactions between the positively charged copolymer chains affect the scattering profile. This kind of weak correlation peak is typical in solutions of copolymers of similar kind.28,29 Indeed, it is possible to fit the scattering profile (in the q range 0.1–2.5 nm−1) as the simple product of the scattering form factor of a Gaussian chain with a radius of gyration Rg = 4.4 nm and a hard sphere structure factor representing the intermolecular electrostatic repulsion. The obtained Rg value is in agreement with the order of magnitude of the RF of the whole copolymer chain in a Gaussian coil conformation, which is equal to 6.23 nm (with n = 150 in eqn (1)). The value may also be compared to an apparent hydrodynamic radius RH,app ≈ 5–9 nm inferred by DLS at 1 wt% block copolymer in 0.1 M NaCl at 25 °C.68 According to a DLS measurement, the solution also contained large aggregates formed as a result of the amphiphilic character of PNIPAM86 in agreement with previous work at our laboratories on the same kind of block copolymers.23,30 This could lead to the observed increase of the SAXS curve at q values <0.1 nm−1. It is compatible with the presence of objects larger than the size limit estimated on the basis of the lowest q accessible, i.e., 273 nm. Another explanation could be an effect of the formation of multichain domains as reported from SANS studies on polyelectrolyte solutions at low ionic strengths, e.g., ref. 87.
The mixed samples were inherently complex to analyze due to polydispersity in shapes and sizes of the co-existing complexes (as demonstrated by cryo-TEM on the solution with X = 0.5, Fig. 4). However, some information on the structure of the mixed aggregates could still be obtained. Fig. 6a presents SAXS curves at 25 °C for different mixture compositions. The progressive addition of bile salt dictated significant variations of the SAXS curves, especially at low q values. With the first addition of NaDC (X = 0.2), the correlation peak at intermediate q values disappears, suggesting an interaction between the negatively charged bile salt and the positively charged copolymer that leads to the formation of a mixed complex and a reduction of the electrostatic repulsive contribution in the structure factor. This is in agreement with the electrophoretic mobility measurements, which show that slightly positive or almost neutral mixed complexes are formed under these conditions. A variation in the scattered intensity at low q compared to the pure copolymer solution is also noticed that can be ascribed to the initial formation of mixed complexes. As the complexes gradually replace the block copolymer aggregates no remarkable variation of the q = 0 extrapolated intensity is observed in agreement with the previously shown SLS data. The scattered intensity at low q gradually changes as the NaDC fraction in the solutions increases until the scattering profile in the low q-domain flattens out (X ≥ 0.33), and approaches the Guinier behavior at X = 0.5 as expected for globular objects for which a Rg = 14 ± 1 nm is estimated. This observation suggests that the scattering intensity at X = 0.5 is reasonably dominated in this q range by the contribution of the globular PNIPAM-protected coacervate complex. On the other hand, at intermediate q values, the observed q−2 dependence is consistent with the scattering of Gaussian chains. In this region, we anticipate that contributions of the unconstrained PNIPAM chains located on the surfaces of the flat tape-like complexes dominate the scattering curve at X = 0.5, but also from the chains of the corona of the globular mixed complexes (Fig. 4a). For samples with excess of NaDC, the variation of the scattering curve at low q in comparison to samples with X ≤ 0.5 is modest. This could be explained by the fact that when most of the charges in the system are neutralized the growth of the mixed complexes with further addition of NaDC is governed by an additional hydrophobic interaction that becomes relevant only at very large NaDC fractions (X > 0.8) (note the negative electrophoretic mobility values in Fig. 3).
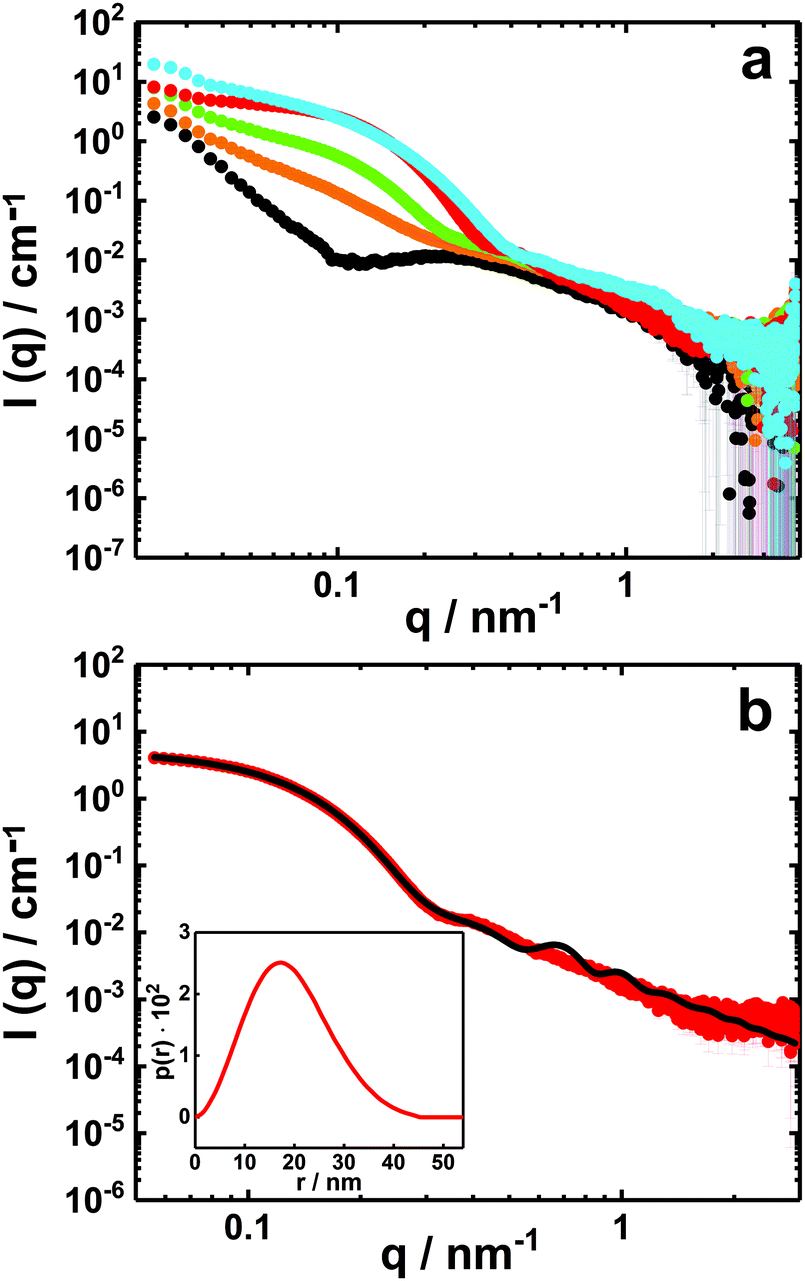 | ||
| Fig. 6 SAXS curves (I(q) vs. q) on absolute scale of PNIPAM120-b-PAMPTMA(+)30–NaDC mixed solutions with a copolymer concentration of 0.5 wt% and various charge fractions, X: (a) X = 0 (black), X = 0.2 (orange), X = 0.33 (green), X = 0.5 (red) and X = 0.80 (blue). (b) Experimental SAXS profile of the PNIPAM120-b-PAMPTMA(+)30–NaDC mixture at X = 0.5 (red symbols) and best fitting curve (black line) for particles with a spherical core and a graded interface of Gaussian chains (see text and Table S1, ESI†). The corresponding pair distance distribution function obtained from IFT analysis is presented in the inset. The temperature was 25 °C. | ||
To describe the globular mixed complexes in the system, an attempt to interpret the scattered intensity behavior was carried out in the case of X = 0.5, for which the support of cryo-TEM experiments is available. As shown in Fig. 6b, the scattering curve of this mixture is well-described by a form factor of a spherical particle with a graded outer interface composed of polymers in a Gaussian chain conformation.28,88 A good fit was achieved for particles with a core diameter of about 18 nm in good agreement with the size inferred from cryo-TEM (Fig. 6b and Table S1, ESI†).
The SAXS profile for the mixture at X = 0.5 was also fitted using the Indirect Fourier Transform (IFT) method.89 The fit provided a pair distance distribution function (p(r)) with a bell shape curve with a low slowly decreasing tail at high distances (Fig. 6b, inset), which vanishes at a maximum distance of 45 nm. This type of distribution is consistent with spherical particles with a radially decreasing density representing well the polymer-brush-protected globular particles. The pair-distance distributions might also be affected by a size polydispersity of the globules.
The presence of the globular complexes clearly dominates the scattering curves of the mixture at X = 0.5 and by that possibly overshadowing the contribution of the tape-like complexes (Fig. 6). A sample with this composition and 0.25 wt% in copolymer was diluted to 0.1 wt% and filtered by using a syringe filter with pores of 0.45 μm and thereafter analyzed by cryo-TEM (Fig. 7a). The results demonstrate that tape-like mixed complexes are reformed, whereas the globular complexes are less prominent or in principle absent and probably redistributed into the tapes after filtration. A SAXS measurement performed on the filtered sample presents a profile with much lower absolute intensity (Fig. 7b) than the unfiltered solution, partly due to the lower copolymer concentration used. More importantly, the SAXS profile is different from that of the corresponding unfiltered sample in Fig. 6b and for q < 0.1 nm−1 it shows a q−1-dependence typical for elongated structures. From the curve, a pair distance distribution function of the cross section (pc(r)) could be inferred by an IFT fit for elongated objects (Fig. 7b, inset). An asymmetric shape of the pc(r) with a maximum distance of around 30 nm was obtained, which correlates well with a rectangular cross section of the elongated structures. Accordingly, the data could be also fitted to a form factor of an infinitely long parallelepiped with a rectangular cross section that is 25 nm wide and 16 nm high (Fig. 7b and Table S2, ESI†). The results from the fits are in good agreement with those inferred from cryo-TEM on the same sample (Fig. 7a). It is also interesting to note that DLS measurements on the same sample and performed simultaneously as the cryo-TEM investigation, revealed two relaxation processes (Fig. S4, ESI†). Both are likely linked to the dynamics of the elongated structures. In addition, we found that the tape-like mixed complexes are remarkably stable as confirmed by DLS measurements repeated 9 months later (Fig. S5, ESI†).
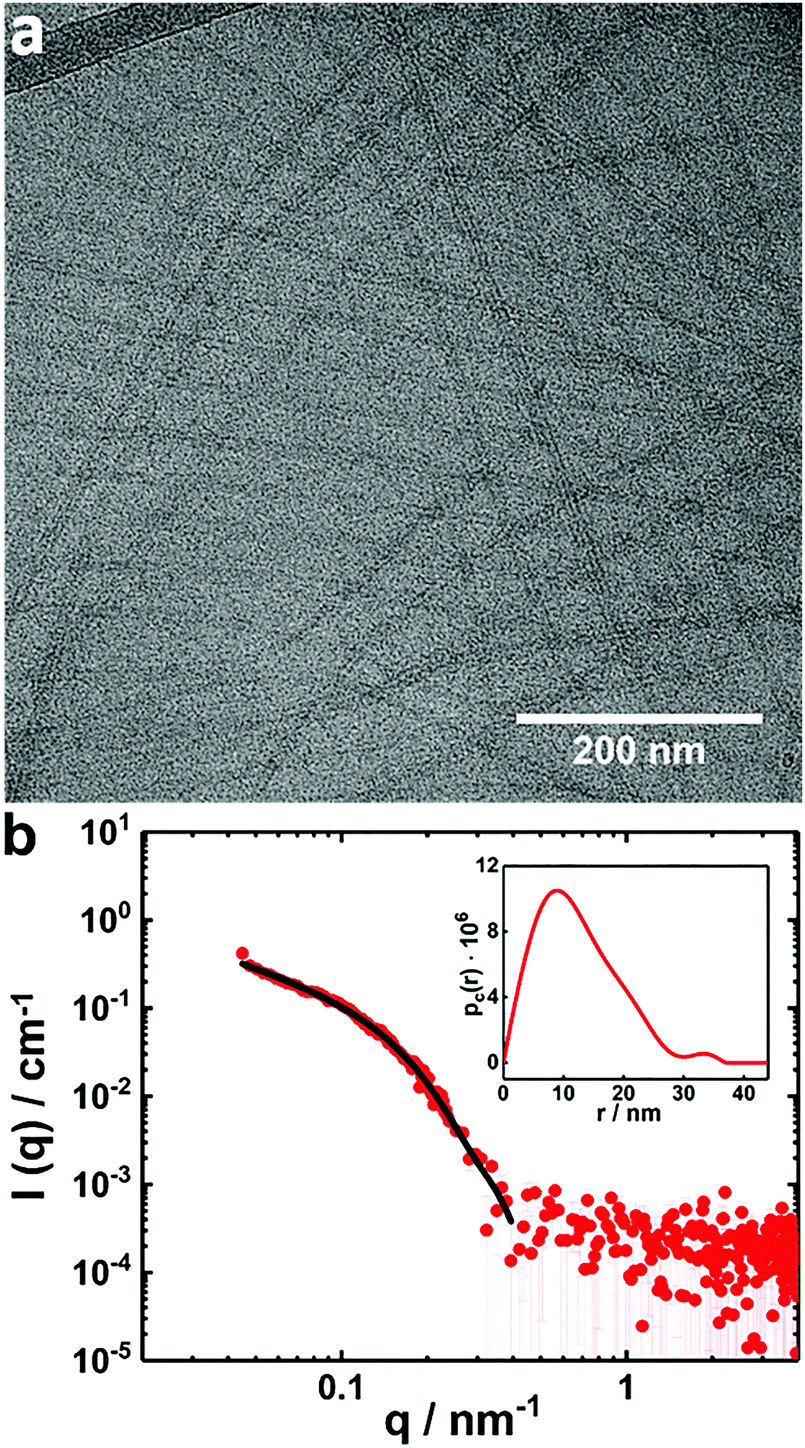 | ||
| Fig. 7 (a) Cryo-TEM image of a filtered PNIPAM120-b-PAMPMTA(+)30–NaDC mixed solution with X = 0.5; (b) a SAXS curve of the same sample as in (a) (red symbols). The black line is a fit to the data based on a form factor of an infinitely long parallelepiped with a rectangular cross section (see text and Table S2, ESI†). The inset shows the corresponding pc(r) from IFT. The copolymer concentration was 0.1 wt% and the temperature was 25 °C. | ||
Conclusions
This study is based on mixtures of biocompatible PNIPAM-b-PAMPTMA(+) block copolymers and bile salts. PNIPAM-based block copolymers and bile salts are both interesting for medical applications.33,90 Indeed, recent studies have shown that PNIPAM-b-PAMPTMA(+) copolymers can be used as DNA and siRNA carriers.91,92 To study the formation of mixed complexes and to characterize the complex structure are of great significance for practical applications as there is a need for developing new systems to be used in drug carrier formulations based on electrostatically assisted assembly. The complexes can also be made thermo-responsive by using temperature-responding polymer components as done in this work. Furthermore, there is a strong interest for a development of BS sequestrants that are based on new biocompatible substances, to be used in treatment of bile acid malabsorption and hypercholesterolemia. From our perspective, linear oppositely charged biocompatible block copolymers could be promising candidates as bile salt sequestrants59,60 as they may have an advantage to the commercial sequestrants, where the large dose required in vivo leads to poor patient compliance. The BS uptake action of the block copolymers studied in this work would be based on electrostatically co-assembly also involving hydrophobic interaction. In this way, considering that the copolymers form water-soluble mixed complexes with bile salt, in addition to that they have a simpler molecular structure compared to the polymeric hydrogels currently used, it could be interesting to test them for this type of application.In this context of applications, we investigated the electrostatically driven co-assembly for an oppositely charged system of the bile salt NaDC and the diblock copolymer PNIPAM120-b-PAMPTMA(+)30 at various compositions. Electrophoretic mobility measurements at room temperature on solutions with a large variation of molar charge fractions showed that the mixed complexes are virtually charge neutral even at off-stoichiometric conditions, except at very high bile salt fractions. This demonstrates that electrostatic intermolecular interactions dominate the co-assembly in the system and the excess of either copolymer or bile salt is not involved in the complex in a wide range of compositions.
Cryo-TEM illustrated that two morphologically different mixed complexes are formed and coexist at ambient temperatures. The first complex has a globular morphology with a coacervate core of deoxycholate anions and PAMPTMA(+) blocks surrounded by a PNIPAM corona. Assuming no hydration of the core, the aggregation numbers of copolymers and DC-molecules in this complex were estimated in a crude calculation to be about 174 and 5220. The second complex has an unusual tape-like supramolecular morphology of several micrometers in length that is striped in the direction on the long axis, which has never been observed before in oppositely charged polymer–surfactant systems. In our model, we propose that the stripes consist of the self-assembled oppositely oriented DC-molecules that partly overlap as a result of the hydrophobic interactions between them. In this way the bile salt molecules are associated and joined by the cationic side chains of the copolymer blocks. The whole tape is build-up of many block copolymers and is covered on both sides by the PNIPAM blocks, probably in a slightly coiled conformation, which are able to prevent aggregation of the tapes under ambient temperature conditions.
The coexistence of two types of complexes may have its origin in the block length of the PNIPAM block of the copolymer. For copolymers with long PNIPAM blocks, due to packing restrictions on a flat surface, complexes with globular morphology may provide more favorable conformational conditions for the PNIPAM chains on the core/corona interface. However, filtration imposes a reorganization of the material leading to a more uniform solution containing tape-like complexes as the dominant structures as evidenced by cryo-TEM and SAXS and also indicated in DLS results.
In forthcoming studies, the thermo-responsiveness of the PNIPAM120-b-PAMPTMA(+)30–NaDC system will be investigated and, in addition, the relevance of the relative block lengths and hydrophobicity of the bile salt on the structure of the mixed complexes.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Matija Tomšič and Jan Skov Pedersen for fruitful and stimulating discussions concerning the SAXS results and Alessio Di Giuseppe for performing part of the light scattering and electrophoretic mobility measurements. The SOLEIL synchrotron is acknowledged for providing beam-time and lab facilities (ID20160186) and we would like to thank Javier Perez for the relevant assistance at the SWING beamline. We acknowledge the SAXSLab Sapienza facility at Sapienza University of Rome where the in-house SAXS experiments were performed. We also thank the nCHREM at Lund University where the cryo-TEM experiments were performed. K. S. gratefully acknowledges funding from the Swedish Research Council (VR) (621-2013-4339), The Magnus Bergvall Foundation (2017-02225) and The Crafoord Foundation (20180549).References
- K. Kogej, Adv. Colloid Interface Sci., 2010, 158, 68–83 CrossRef CAS PubMed.
- J. Janiak, S. Bayati, L. Galantini, N. V. Pavel and K. Schillén, Langmuir, 2012, 28, 16536–16546 CrossRef CAS PubMed.
- L. Piculell, Langmuir, 2013, 29, 10313–10329 CrossRef CAS PubMed.
- G. A. Ferreira and W. Loh, Curr. Opin. Colloid Interface Sci., 2017, 32, 11–22 CrossRef CAS.
- V. S. Meka, M. K. Sing, M. R. Pichika, S. R. Nali, V. R. Kolapalli and P. Kesharwani, Drug Discovery Today, 2017, 22, 1697–1706 CrossRef CAS PubMed.
- B. Lindman and K. Thalberg, in Interactions of surfactants with polymers and proteins, ed. E. Goddard and K. Ananthapadmanabhan, CRC, Boca Raton, 1993, pp. 203–276 Search PubMed.
- L. Piculell and B. Lindman, Adv. Colloid Interface Sci., 1992, 41, 149–178 CrossRef CAS.
- L. Piculell, J. Norrman, A. V. Svensson, I. Lynch, J. S. Bernardes and W. Loh, Adv. Colloid Interface Sci., 2009, 147, 228–236 CrossRef PubMed.
- M. Swanson-Vethamuthu, P. L. Dubin, M. Almgren and Y. Li, J. Colloid Interface Sci., 1997, 186, 414–419 CrossRef CAS PubMed.
- J. Janiak, L. Piculell, G. Olofsson and K. Schillén, Phys. Chem. Chem. Phys., 2011, 13, 3126–3138 RSC.
- J. Janiak, M. Tomšič, D. Lundberg, G. Olofsson, L. Piculell and K. Schillén, J. Phys. Chem. B, 2014, 118, 9745–9756 CrossRef CAS PubMed.
- A. M. Percebom, J. Janiak, K. Schillén, L. Piculell and W. Loh, Soft Matter, 2013, 9, 515–526 RSC.
- N. M. Carneiro, A. M. Percebom and W. Loh, ACS Omega, 2017, 2, 5518–5528 CrossRef CAS.
- A. Harada and K. Kataoka, Macromolecules, 1995, 28, 5294–5299 CrossRef CAS.
- A. V. Kabanov, T. K. Bronich, V. A. Kabanov, K. Yu and A. Eisenberg, Macromolecules, 1996, 29, 6797–6802 CrossRef CAS.
- D. V. Pergushov, E. V. Remizova, J. Feldthusen, A. B. Zezin, A. H. Müller and V. A. Kabanov, J. Phys. Chem. B, 2003, 107, 8093–8096 CrossRef CAS.
- J.-F. Berret, B. Vigolo, R. Eng, P. Hervé, I. Grillo and L. Yang, Macromolecules, 2004, 37, 4922–4930 CrossRef CAS.
- M. Danial, H.-A. Klok, W. Norde and M. A. Cohen Stuart, Langmuir, 2007, 23, 8003–8009 CrossRef CAS PubMed.
- I. K. Voets, A. de Keizer and M. A. C. Stuart, Adv. Colloid Interface Sci., 2009, 147, 300–318 CrossRef PubMed.
- J. Courtois and J.-F. Berret, Langmuir, 2010, 26, 11750–11758 CrossRef CAS PubMed.
- J.-F. Berret, Adv. Colloid Interface Sci., 2011, 167, 38–48 CrossRef CAS PubMed.
- M. L. Patrizi, M. Diociaiuti, D. Capitani and G. Masci, Polymer, 2009, 50, 467–474 CrossRef CAS.
- S. Bayati, K. Zhu, L. T. Trinh, A.-L. Kjøniksen and B. Nyström, J. Phys. Chem. B, 2012, 116, 11386–11395 CrossRef CAS PubMed.
- C. M. Schilli, M. Zhang, E. Rizzardo, S. H. Thang, Y. Chong, K. Edwards, G. Karlsson and A. H. Müller, Macromolecules, 2004, 37, 7861–7866 CrossRef CAS.
- G. Masci, M. Diociaiuti and V. Crescenzi, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 4830–4842 CrossRef CAS.
- A.-L. Kjøniksen, K. Zhu, R. Pamies and B. Nyström, J. Phys. Chem. B, 2008, 112, 3294–3299 CrossRef PubMed.
- A.-L. Kjøniksen, K. Zhu, M. A. Behrens, J. S. Pedersen and B. Nyström, J. Phys. Chem. B, 2011, 115, 2125–2139 CrossRef PubMed.
- M. A. Behrens, M. Lopez, A.-L. Kjøniksen, K. Zhu, B. Nyström and J. S. Pedersen, Langmuir, 2011, 28, 1105–1114 CrossRef PubMed.
- M. A. Behrens, A.-L. Kjøniksen, K. Zhu, B. Nyström and J. S. Pedersen, Macromolecules, 2011, 45, 246–255 CrossRef.
- G. Lazzara, G. Olofsson, V. Alfredsson, K. Zhu, B. Nyström and K. Schillén, Soft Matter, 2012, 8, 5043–5054 RSC.
- E. Karjalainen, N. Chenna, P. Laurinmäki, S. J. Butcher and H. Tenhu, Polym. Chem., 2013, 4, 1014–1024 RSC.
- A. Halperin, M. Kröger and F. M. Winnik, Angew. Chem., Int. Ed., 2015, 54, 15342–15367 CrossRef CAS PubMed.
- L. Galantini, M. C. di Gregorio, M. Gubitosi, L. Travaglini, J. V. Tato, A. Jover, F. Meijide, V. H. S. Tellini and N. V. Pavel, Curr. Opin. Colloid Interface Sci., 2015, 20, 170–182 CrossRef CAS.
- M. C. di Gregorio, L. Travaglini, A. Del Giudice, J. Cautela, N. V. Pavel and L. Galantini, Langmuir, 2018 DOI:10.1021/acs.langmuir.8b02657.
- D. Madenci and S. Egelhaaf, Curr. Opin. Colloid Interface Sci., 2010, 15, 109–115 CrossRef CAS.
- E. F. Marques, H. Edlund, C. La Mesa and A. Khan, Langmuir, 2000, 16, 5178–5186 CrossRef CAS.
- F. Meijide, J. V. Trillo, S. de Frutos, L. Galantini, N. V. Pavel, V. H. Soto, A. Jover and J. V. Tato, Steroids, 2012, 77, 1205–1211 CrossRef CAS PubMed.
- L. Travaglini, A. D’Annibale, M. C. di Gregorio, K. Schillén, U. Olsson, S. Sennato, N. V. Pavel and L. Galantini, J. Phys. Chem. B, 2013, 117, 9248–9257 CrossRef CAS PubMed.
- M. Gubitosi, L. Travaglini, A. D’Annibale, N. V. Pavel, J. Vàzquez Tato, M. Obiols-Rabasa, S. Sennato, U. Olsson, K. Schillén and L. Galantini, Langmuir, 2014, 30, 6358–6366 CrossRef CAS PubMed.
- M. Gubitosi, L. Travaglini, M. C. di Gregorio, N. V. Pavel, J. Vázquez Tato, S. Sennato, U. Olsson, K. Schillén and L. Galantini, Angew. Chem., Int. Ed., 2015, 54, 7018–7021 CrossRef CAS PubMed.
- M. Gubitosi, J. V. Trillo, A. Alfaro Vargas, N. V. Pavel, D. Gazzoli, S. Sennato, A. Jover, F. Meijide and L. Galantini, J. Phys. Chem. B, 2014, 118, 1012–1021 CrossRef CAS PubMed.
- A. F. Hofmann and K. J. Mysels, Colloids Surf., 1987, 30, 145–173 CrossRef.
- D. M. Small and W. Admirand, Nature, 1969, 221, 265–267 CrossRef CAS.
- J. Ulmius, G. Lindblom, H. Wennerström, L. Johansson, K. Fontell, O. Söderman and G. Arvidson, Biochemistry, 1982, 21, 1553–1560 CrossRef CAS PubMed.
- M. Cárdenas, K. Schillén, V. Alfredsson, R.-D. Duan, L. Nyberg and T. Arnebrant, Chem. Phys. Lipids, 2008, 151, 10–17 CrossRef PubMed.
- S. Salentinig, S. Phan, J. Khan, A. Hawley and B. J. Boyd, ACS Nano, 2013, 7, 10904–10911 CrossRef CAS PubMed.
- M. A. Kiselev and D. Lombardo, Biochim. Biophys. Acta, 2017, 1861, 3700–3717 CrossRef CAS.
- M. Camilleri, Gut Liver, 2015, 9, 332 CAS.
- T. Lundåsen, C. Gälman, B. Angelin and M. Rudling, J. Intern. Med., 2006, 260, 530–536 CrossRef PubMed.
- J. R. Walters, A. M. Tasleem, O. S. Omer, W. G. Brydon, T. Dew and C. W. Le Roux, Clin. Gastroenterol. Hepatol., 2009, 7, 1189–1194 CrossRef CAS PubMed.
- I. Johnston, J. Nolan, S. S. Pattni and J. R. Walters, Curr. Gastroenterol. Rep., 2011, 13, 418–425 CrossRef PubMed.
- C. Wilcox, J. Turner and J. Green, Aliment. Pharmacol. Ther., 2014, 39, 923–939 CrossRef CAS PubMed.
- J. R. Walters and S. S. Pattni, Ther. Adv. Gastroenterol., 2010, 3, 349–357 CrossRef PubMed.
- J. R. Walters, Nat. Rev. Gastroenterol. Hepatol., 2014, 11, 426 CrossRef CAS PubMed.
- W. H. Mandeville and D. I. Goldberg, Curr. Pharm. Des., 1997, 3, 15–28 CAS.
- M. Camilleri and G. J. Gores, Am. J. Physiol.: Gastrointest. Liver Physiol., 2015, 309, G209–G215 CrossRef CAS PubMed.
- V. A. Fonseca, J. Rosenstock, A. C. Wang, K. E. Truitt and M. R. Jones, Diabetes Care, 2008, 31, 1479–1484 CrossRef CAS PubMed.
- C. C. Huval, S. R. Holmes-Farley, W. H. Mandeville, R. Sacchiero and P. K. Dhal, Eur. Polym. J., 2004, 40, 693–701 CrossRef CAS.
- N. S. Cameron, A. Eisenberg and G. R. Brown, Biomacromolecules, 2002, 3, 116–123 CrossRef CAS.
- N. S. Cameron, A. Eisenberg and G. R. Brown, Biomacromolecules, 2002, 3, 124–132 CrossRef CAS PubMed.
- P. V. Mendonça, A. C. Serra, C. L. Silva, S. Simões and J. F. Coelho, Prog. Polym. Sci., 2013, 38, 445–461 CrossRef.
- P. V. Mendonça, M. J. Moreno, A. C. Serra, S. Simões and J. F. Coelho, RSC Adv., 2016, 6, 52143–52153 RSC.
- E. Heřmánková, A. Žák, L. Poláková, R. Hobzová, R. Hromádka and J. Širc, Eur. J. Med. Chem., 2018, 144, 300–317 CrossRef PubMed.
- S. Bayati, L. Galantini, K. D. Knudsen and K. Schillén, Langmuir, 2015, 31, 13519–13527 CrossRef CAS PubMed.
- S. Bayati, L. Galantini, K. D. Knudsen and K. Schillén, Colloids Surf., A, 2016, 504, 426–436 CrossRef CAS.
- S. Bayati, C. Haglund Anderberg, N. V. Pavel, L. Galantini and K. Schillén, RSC Adv., 2016, 6, 69313–69325 RSC.
- E. Tasca, A. Del Giudice, L. Galantini, K. Schillén, A. M. Giuliani and M. Giustini, J. Colloid Interface Sci., 2019, 540, 593–601 CrossRef CAS PubMed.
- M. C. di Gregorio, M. Gubitosi, L. Travaglini, N. V. Pavel, A. Jover, F. Meijide, J. V. Tato, S. Sennato, K. Schillén, F. Tranchini, S. De Santis and L. Galantini, Phys. Chem. Chem. Phys., 2017, 19, 1504–1515 RSC.
- G. David and J. Perez, J. Appl. Crystallogr., 2009, 42, 892–900 CrossRef CAS.
- D. Orthaber, A. Bergmann and O. Glatter, J. Appl. Crystallogr., 2000, 33, 218–225 CrossRef CAS.
- S. Hansen, J. Appl. Crystallogr., 2014, 47, 1469–1471 CrossRef CAS.
- I. Breßler, J. Kohlbrecher and A. F. Thünemann, J. Appl. Crystallogr., 2015, 48, 1587–1598 CrossRef PubMed.
- K. Kumar, B. S. Patial and S. Chauhan, J. Chem. Thermodyn., 2015, 82, 25–33 CrossRef CAS.
- M. L. Fielden, P. M. Claesson and K. Schillén, Langmuir, 1998, 14, 5366–5375 CrossRef CAS.
- D. Langevin, Adv. Colloid Interface Sci., 2009, 147, 170–177 CrossRef PubMed.
- L. Chiappisi, I. Hoffmann and M. Gradzielski, Soft Matter, 2013, 9, 3896–3909 RSC.
- A. M. Percebom, L. R. S. Barbosa, R. Itri and W. Loh, Langmuir, 2014, 30, 11493–11503 CrossRef CAS PubMed.
- C. Nisha, S. V. Manorama, J. N. Kizhakkedathu and S. Maiti, Langmuir, 2004, 20, 8468–8475 CrossRef CAS PubMed.
- Y. Zheng, Y.-Y. Won, F. S. Bates, H. T. Davis, L. Scriven and Y. Talmon, J. Phys. Chem. B, 1999, 103, 10331–10334 CrossRef CAS.
- J. J. Crassous, M. Ballauff, M. Drechsler, J. Schmidt and Y. Talmon, Langmuir, 2006, 22, 2403–2406 CrossRef CAS PubMed.
- J. H. Ortony, S.-H. Choi, J. M. Spruell, J. N. Hunt, N. A. Lynd, D. V. Krogstad, V. S. Urban, C. J. Hawker, E. J. Kramer and S. Han, Chem. Sci., 2014, 5, 58–67 RSC.
- K. Schillén, P. M. Claesson, M. Malmsten, P. Linse and C. Booth, J. Phys. Chem. B, 1997, 101, 4238–4252 CrossRef.
- X. Zhu, C. Yan, F. Winnik and D. Leckband, Langmuir, 2007, 23, 162–169 CrossRef CAS PubMed.
- G. Lazzara, R. A. Campbell, S. Bayati, K. Zhu, B. Nyström, T. Nylander and K. Schillén, Colloid Polym. Sci., 2017, 295, 1327–1341 CrossRef CAS PubMed.
- Y. Qiao, Y. Lin, Y. Wang, Z. Yang, J. Liu, J. Zhou, Y. Yan and J. Huang, Nano Lett., 2009, 9, 4500–4504 CrossRef CAS PubMed.
- R. Pelton, J. Colloid Interface Sci., 2010, 348, 673–674 CrossRef CAS PubMed.
- B. D. Ermi and E. J. Amis, Macromolecules, 1998, 31, 7378–7384 CrossRef CAS.
- J. S. Pedersen and M. C. Gerstenberg, Macromolecules, 1996, 29, 1363–1365 CrossRef CAS.
- O. Glatter, J. Appl. Crystallogr., 1977, 10, 415–421 CrossRef.
- H. Wei, S.-X. Cheng, X.-Z. Zhang and R.-X. Zhuo, Prog. Polym. Sci., 2009, 34, 893–910 CrossRef CAS.
- M. T. Calejo, A. M. S. Cardoso, A.-L. Kjøniksen, K. Zhu, C. M. Morais, S. A. Sande, A. L. Cardoso, M. C. P. De Lima, A. Jurado and B. Nyström, Int. J. Pharm., 2013, 448, 105–114 CrossRef CAS PubMed.
- A. M. Cardoso, M. T. Calejo, C. M. Morais, A. L. Cardoso, R. Cruz, K. Zhu, M. C. Pedroso de Lima, A. l. S. Jurado and B. Nyström, Mol. Pharmaceutics, 2014, 11, 819–827 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Structural analysis of PNIPAM120-b-PAMPMTA(+)30–NaDC complexes by cryo-TEM, SAXS and DLS. See DOI: 10.1039/c9cp01744e |
| This journal is © the Owner Societies 2019 |