
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructures, dipole moments and excited state lifetime of isolated 4-cyanoindole in its ground and lowest electronically excited singlet states†
Marie-Luise
Hebestreit
 a,
Michael
Schneider
a,
Hilda
Lartian
a,
Vivienne
Betz
a,
Michael
Heinrich
a,
Mirko
Lindic
a,
Myong Yong
Choi
b and
Michael
Schmitt
*a
a,
Michael
Schneider
a,
Hilda
Lartian
a,
Vivienne
Betz
a,
Michael
Heinrich
a,
Mirko
Lindic
a,
Myong Yong
Choi
b and
Michael
Schmitt
*a
aHeinrich-Heine-Universität, Institut für Physikalische Chemie I, D-40225 Düsseldorf, Germany. E-mail: mschmitt@uni-duesseldorf.de; Fax: +49 211 8113689; Tel: +49 211 81 12100
bDepartment of Chemistry (BK21+) and Research Institute of Natural Science, Gyeongsang National University, Jinju 52828, Republic of Korea
First published on 21st June 2019
Abstract
The rotationally resolved electronic spectrum of 4-cyanoindole and some N-D and C-D deuterated isotopologues has been measured and analyzed. Dipole moments in the ground and electronically excited state have been determined, using electronic Stark spectroscopy. From the geometry changes upon excitation, orientation of the transition dipole moment, and the values for the permanent dipole moments, the lowest excited singlet state could be shown to be of La symmetry. The excited state lifetime of isolated 4-cyanoindole has been determined to be 11 ns, while for the ringdeuterated isotopologues lifetimes between 5 and 6 ns have been found. The different behavior of 3-, 4-, and 5-cyanoindole is discussed on the basis of the different electronic nature of the electronically excited singlet states.
1 Introduction
It is a well-accepted paradigm, that nature selects chromophores as building blocks, which efficiently dissipate the absorbed photon energy due to their short excited state lifetime. This lifetime strongly depends on the direct molecular surrounding and is therefore strongly different for isolated molecules in the gas phase and in solution of different solvents.1–3The radiative lifetime of isolated molecules can be calculated from the Lorentz contribution to the total linewidth of the transitions in rovibronically resolved fluorescence spectra. For isolated molecules this lifetime depends on the fluorescence rate constant kF and the coupling to the dense manifold of dark background states, which carry no oscillator strengths. For background states of the same multiplicity we deal with internal conversion (IC), while different multiplicities give rise to intersystem crossing (ISC). Thus, the energetic position of the (dark) triplet manifold with respect to the initially excited (bright) singlet state determines the ISC rate kISC. The fluorescence lifetime taking IC and ISC into account is τ = 1/(kF + kIC + kISC). When talking about IC, conical intersections (CIs) of the primarily photo-excited state with other states of proper symmetry, have to be considered.4 These conical intersections are by no way rare events, as stated by Truhlar and Mead: “(…) if one encounters a very small electronic energy gap along a path through configuration space, it is much more likely to be associated with the neighborhood of a conical intersection than with an avoided intersection.”5 Even if the primarily excited electronic state is not directly linked to the ground state through a single CI, a sequence of CIs, involving one repelling state, which is coupled to the ground state has strong effects of the lifetime of the primarily excited state. Its lifetime is then strongly affected by the relative positions of these CIs.2,6–8 Even well-separated ππ* states show CIs, that affect their photophysical properties.9–13
Upon solvation, a plethora of nonradiative channels open, which are closed for the isolated molecule. In most cases, this leads to drastically shorter lifetimes in solution. The experiment of choice in solution is time-correlated single photon counting (TCSPC).
A special class of indole derivatives, the cyanotryptophans are used as dual infrared and fluorescence spectroscopic labels to assess structural dynamics in proteins.14 Markiewicz et al. have shown that 5-cyanotryptophan fluorescence can be utilized as a sensitive probe of protein hydration.15
The fluorescence lifetime of the different cyanoindoles in solution has been found to depend considerably on the solvent.16,17 The reason for these differing photophysical properties, depending on the position of the cyano substituent is due to the relative energetic positions of the lowest excited ππ* singlet states. These states can be classified in the nomenclature of Platt for cata-condensed aromatics as 1La and 1Lb states18 a scheme, which was later extended to indole by Weber.19
Experimental evidence for the relative position of the 1La and 1Lb states in the indole chromophore has been collected for decades.20–28 Also theory provided important details for the classification of these states.7,10,11,29–32 Domcke and Sobolewski have shown that the photophysics of indoles can be influenced by the existence of a πσ* state, which is dissociative along the NH coordinate.2,6,7,33
Much less fundamental investigations on the 1La and 1Lb states has been performed for the cyanoindoles. Rotationally resolved electronic spectra of 5-cyanoindole have been presented.34–36 From the comparison of the experimental findings to quantum chemical calculations at the coupled cluster level of theory, the lowest excited singlet state could be shown to be the 1La in contrast to most other indole derivatives, which have the 1Lb state as lowest excited state. 5-Cyanoindole(H2O)n with n = 0–2 has been investigated using UV–UV hole burning and IR-dip spectroscopy by Min et al.37 They found a different photodissociation pattern of the 5-cyanoindole–water clusters compared to the respective 3-cyanoindole–water clusters.38,39 Using TD-DFT with the LC-BLYP, M06-2X, and B3LYP functionals, the same energetic order of the lowest ππ* states was found compared to calculations using the second-order approximate coupled cluster singles and doubles model (CC2).34
In 3-cyanoindole, the order of the 1La and 1Lb states is reversed again, the 1Lb state being the lower one as determined by rotationally resolved electronic Stark spectroscopy.40 The water clusters of 3-cyanoindole(H2O)n with n = 1, 2 have been studied in the group of Choi by UV–UV hole burning and IR-dip spectroscopy.38,39 Based on the comparison of the IR-dip spectra with the results of normal mode analyses and a Franck–Condon analysis of the mass-selected R2P spectra of 3-cyanoindole(H2O)n with n = 1, 2 they deduced a linearly bound water moiety with an N–H⋯OH2 binding motif. This structure was confirmed by the rotational constants from rotationally resolved electronic spectroscopy.40 The excited state lifetime of 3-cyanoindole has been determined to be 9.8 ns, while that of the 3-cyanoindole(H2O)1 cluster shows a considerably shorter lifetime of 3.6 ns. Interestingly, the fluorescence lifetime of 3-cyanoindole in water solution is the shortest of all cyanoindoles.16
Other 4-substituted indoles show a different behavior concerning their lifetimes, compared to indole. 4-Hydroxyindole has an unusually short excited state lifetime of 200 ps.41 Also 4-methoxyindole (5.0 ns) and 4-fluoroindole (7.4 ns) exhibit shorter lifetimes than indole (17.6 ns) and other monosubstituted indoles, whose lifetimes lie in a range of 10–18 ns.41 In general, the lifetime of the deuterated compounds is higher. For monodeuterated 4-hydroxyindole (deuteration at the OH site) an increase of more than one order of magnitudes is observed (4.3 ns).41 In contrast, the lifetime of 4-methylindole (16.2 ns) is close to the indole value.41
In the present study, we investigate the electronic nature of the lowest excited state of 4-cyanoindole and some deuterated species, their excited state lifetimes and dipole moments in both ground and excited singlet states, using a combination of rotationally resolved fluorescence spectroscopy, rotationally resolved electronic Stark spectroscopy, time-correlated single photon counting, and ab initio quantum chemical calculations for a deeper understanding of the photophysics of electronically excited cyanoindoles.
2 Experimental section
2.1 Experimental procedures
2.2 Quantum chemical calculations
Structure optimizations were performed employing a Dunning's correlation-consistent polarized valence triple zeta (cc-pVTZ) basis set from the TURBOMOLE library.47,48 The equilibrium geometries of the electronic ground and the lowest excited singlet states were optimized using the approximate coupled cluster singles and doubles model (CC2) employing the resolution-of-the-identity (RI) approximation.49–51 For the structure optimizations spin-component scaling (SCS) modifications to CC2 were taken into account.52 Vibrational frequencies and zero-point corrections to the adiabatic excitation energies were obtained from numerical second derivatives using the NumForce script.532.3 Fits of the rovibronic spectra using evolutionary algorithms
The evaluation of the molecular parameters in the Hamiltonian is performed in an automated process based on evolutionary algorithms.54–57 They provide a straightforward approach to find the global minimum in a multi-parameter optimization. Here, we apply the covariance matrix adaptation evolution strategy (CMA-ES), which is described in detail elsewhere.58,593 Results
3.1 Computational results
The CC2/cc-pVTZ optimized structure of 4-cyanoindole in the ground and lowest excited singlet state is planar in both states. The Cartesian coordinates of all optimized states are given in the ESI.† Contrary to 5-cyanoindole, generic CC2 and its spin component scaled variant SCS-CC2 yield the same state as lowest excited singlet state.34 The lowest excited singlet state for both CC2 and SCS-CC2 is the 1La state, what can be inferred from the calculated transition dipole moment orientation (cf.Table 1 and Fig. 1) and the orbital contributions to the excitation, given in Fig. S7 of the ESI.† The lowest singlet state is reached by an almost pure LUMO ← HOMO transition, an indication for an 1La state, while the adiabatically second state is composed of LUMO ← HOMO−1 and LUMO+1 ← HOMO contributions typical for an 1Lb state.| Theory SCS-CC2 | Experiment | |||||||
|---|---|---|---|---|---|---|---|---|
| 4-CI | 4-CI(1-d1) | 4-CI(2-d1) | 4-CI(3-d1) | 4-CI | 4-CI(1-d1) | 4-CI(2-d1) | 4-CI(3-d1) | |
| A′′/MHz | 1790.15 | 1789.75 | 1748.49 | 1746.22 | 1795.11(3) | 1791.56(3) | 1747.85(2) | 1748.40(4) |
| B′′/MHz | 1138.03 | 1110.06 | 1120.49 | 1137.99 | 1146.83(2) | 1116.74(2) | 1116.90(1) | 1145.11(3) |
| C′′/MHz | 695.74 | 685.12 | 682.88 | 688.99 | 699.79(1) | 687.96(2) | 681.52(1) | 691.89(2) |
| ΔI′′/amu Å2 | 0.00 | 0.00 | 0.00 | 0.00 | −0.02 | −0.03 | −0.07 | 0.04 |
| μ a′′/D | ±6.13 | — | — | — | ±6.08(1) | — | — | — |
| μ b′′/D | ±1.82 | — | — | — | ±1.68(1) | — | — | — |
| μ′′/D | 6.39 | — | — | — | 6.31(1) | — | — | — |
| θ D′′/° | −16.5 | — | — | — | −15.45 | — | — | — |
| A′/MHz | 1769.07 | 1768.29 | 1726.20 | 1727.76 | 1778.80(4) | 1774.93(4) | 1733.54(3) | 1734.43(5) |
| B′/MHz | 1123.71 | 1096.29 | 1107.20 | 1123.59 | 1131.14(3) | 1101.72(3) | 1101.71(3) | 1129.24(4) |
| C′/MHz | 687.20 | 676.73 | 674.54 | 680.83 | 691.51(2) | 679.85(3) | 673.71(2) | 683.95(3) |
| ΔI′/amu Å2 | 0.00 | 0.00 | 0.00 | 0.00 | −0.06 | −0.09 | −0.11 | −0.01 |
| μ a′/D | ±9.42 | — | — | — | ±8.92(1) | — | — | — |
| μ b′/D | ±0.10 | — | — | — | ±0.29(3) | — | — | — |
| μ′/D | 9.42 | — | — | — | 8.92(1) | — | — | — |
| θ D′/° | 0.60 | — | — | — | 1.86 | — | — | — |
| ΔA/MHz | −21.08 | −21.46 | −22.29 | −18.46 | −16.31(1) | −16.63(1) | −14.35(1) | −13.98(1) |
| ΔB/MHz | −14.32 | −13.77 | −13.29 | −14.40 | −15.69(1) | −15.02(1) | −15.20(1) | −15.87(1) |
| ΔC/MHz | −8.45 | −8.39 | −8.34 | −8.16 | −8.28(1) | −8.11(1) | −7.81(1) | −7.94(1) |
| ΔνGauss/MHz | — | — | — | — | 15.17 | — | — | — |
| ΔνLorentz/MHz | — | — | — | — | 14.41(2) | 31.70(3) | 25.72(1) | 27.23(1) |
| τ/ns | — | — | — | — | 11.04(3) | 5.0(1) | 6.19(1) | 5.8(1) |
| θ/° | +14.35 | +13.94 | +11.93 | +14.55 | ±30.66(1) | ±28.72(1) | ±35.40(7) | ±29.46(4) |
| θ T/° | +1.3 | — | — | — | +2.23 | — | — | — |
| ν 0/cm−1 | 34![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 776 776 |
— | — | — | 33038.55(10) | 33034.00(15) | 33039.87(19) | 33044.60(1) |
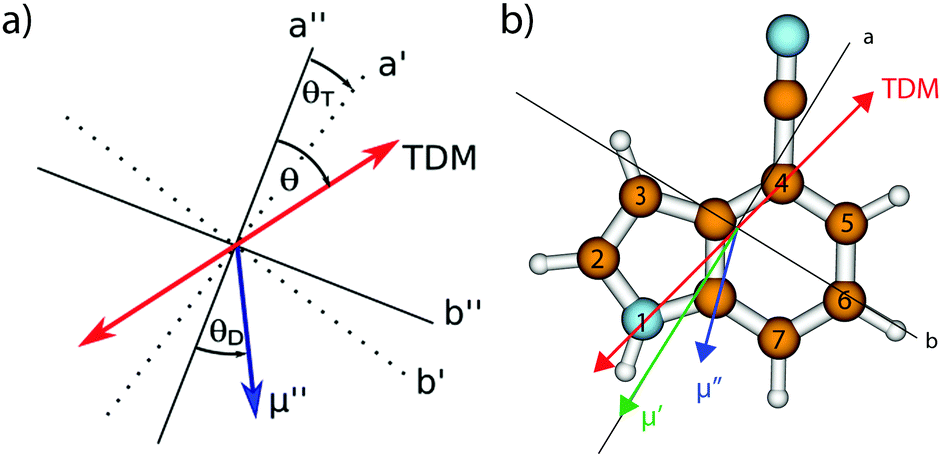 | ||
| Fig. 1 (a) Definitions of the axis reorientation angle θT, the angle of the transition dipole moment with the inertial a-axis θ, and the angle of the permanent dipole moment with the inertial a-axis θD. The doubly primed axes refer to the ground state, the singly primed to the electronically excited state. A positive sign of θT refers to a clockwise rotation of the inertial axis system upon electronic excitation, positive signs of θ and θD refer to a clockwise rotation of the inertial a-axis onto the dipole/TDM vector. (b) Optimized ground state structure, atomic numbering, inertial axes, transition dipole moment for excitation to the lowest excited singlet state (red), and the orientation of the permanent dipole moment in the ground (blue) and first excited state (green) of 4-cyanoindole. | ||
The molecular parameters (rotational constants A, B, and C in both electronic states, the inertial defects ΔI, the angle θ of the transition dipole moment with the inertial a axis, the angle θD of the permanent dipole moment with the inertial a-axis, and the zero-point corrected origin frequency ν0) for all isotopologues studied, are compiled in Table 1 and are compared to the respective experimental results, which are described in detail in Section 3.2.
Table 2 gives the relative vertical energies of the lowest ππ* and πσ* states at the ground state geometry of 4-cyanoindole, along with their oscillator strengths and the dipole moments of the respective state. We included the excitation energies of indole, 3-cyanoindole and 5-cyanoindole for comparison. Interestingly, at the ground state geometry, the Lb state is always the S1, while adiabatically it changes to the S2 for 4- and 5-cyanoindole.
| Indole | 3-Cyanoindole | 4-Cyanoindole | 5-Cyanoindole | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| E (cm−1) | μ (D) | f | E (cm−1) | μ (D) | f | E (cm−1) | μ (D) | f | E (cm−1) | μ (D) | f | |
| ππ* (Lb) | 0 | 2.2 | 0.04 | 0 | 5.4 | 0.05 | −925 | 6.77 | 0.05 | −2972 | 7.62 | 0.01 |
| ππ* (La) | 3534 | 5.0 | 0.09 | 1828 | 6.3 | 0.12 | 0 | 9.60 | 0.14 | 0 | 11.04 | 0.02 |
| πσ* | 13727 |
7.3 | 0.00 | 12552 |
7.6 | 0.00 | 15456 |
8.20 | 0.00 | 16911 |
10.18 | 0.00 |
Hougen and Watson60 have shown that a molecule of low enough symmetry, which undergoes an electronic transition, will change its geometry, leading to the consequence that these two geometries have to be described by two different molecule-fixed axis systems. They can be interconverted by a 3 × 3 rotational matrix, which is called the axis reorientation matrix.‡ For a molecule, which is planar in both electronic states a single axis reorientation angle θT is needed to fully describe the axis reorientation.
From the SCS-CC2 optimized structures, the axis reorientation angle of the inertial axis system upon electronic excitation θT can be determined using the relation for planar molecules given by Hougen and Watson:60
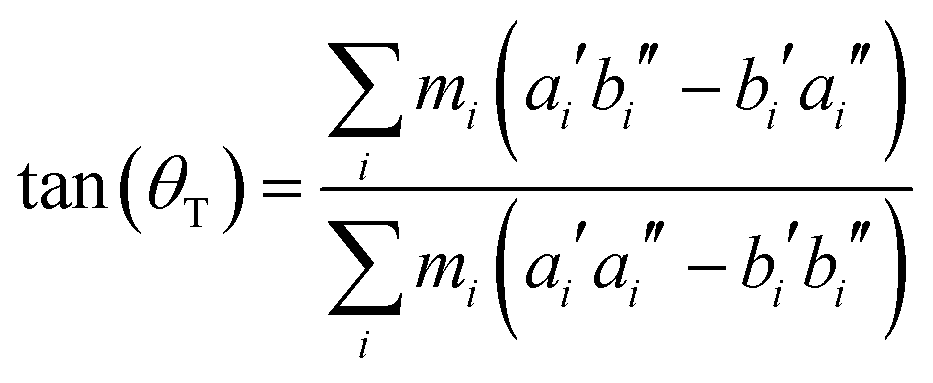 | (1) |
Another consequence of the inertial axis rotation is a rotational Duschinsky mixing of the vibrational modes of 4-cyanoindole. The vibrational modes of the electronically excited state can be expressed as linear combination of the ground state modes using the following linear orthogonal transformation, first given by Duchinsky:61
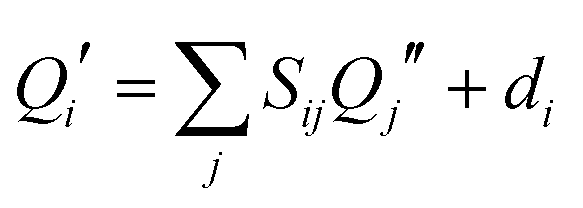 | (2) |
The amount of mode mixing, which is contained in the Duschinsky matrix can easily be visualized graphically. Fig. 2 shows a representation the size of the elements in this matrix. Black squares refer to a matrix element of 1, white squares of 0, intermediate values are shown grey coded. Modes, which show no mixing are represented by a black square (not necessarily on the diagonal, because the order of the vibrations might interchange upon electronic excitation). Similar graphs for 3-cyanoindole and 5-cyanoindole are given in the ESI† (Fig. S1 and S2).
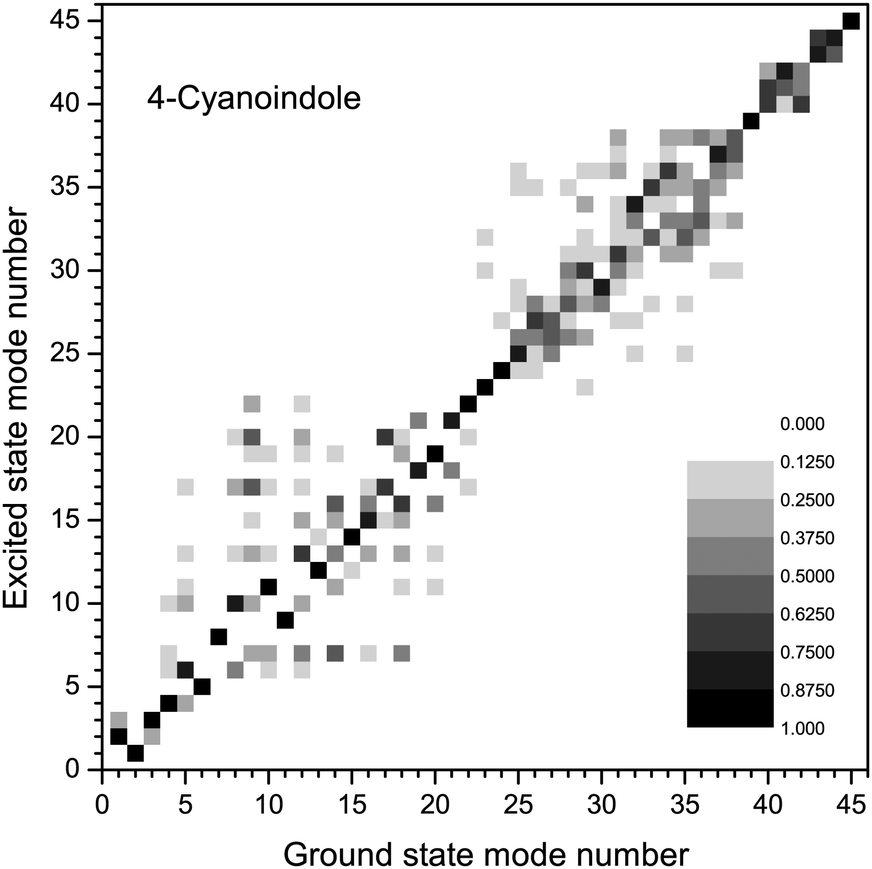 | ||
| Fig. 2 Duschinsky matrix of the 45 vibrational modes of 4-cyanoindole. | ||
3.2 Experimental results
Fig. 3a shows the rotationally resolved electronic spectrum of the origin of 4-cyanoindole at 33038.55 cm−1 (0 on the scale of the figure) along with the best fit at zero field (trace b) and at a field strength of 425.48 V cm−1 (trace c). The electric field in the chosen set-up is parallel to the polarization of the plane of the exciting light, thus ΔM = 0 selection rules for the Stark spectrum hold. The spectrum was fit using a CMA-ES; the molecular parameters from the fit are summarized in Table 1 and are compared to the results of the SCS-CC2/cc-pVTZ calculations. The spectrum is an ab-hybrid, which is mainly polarized along the a-axis.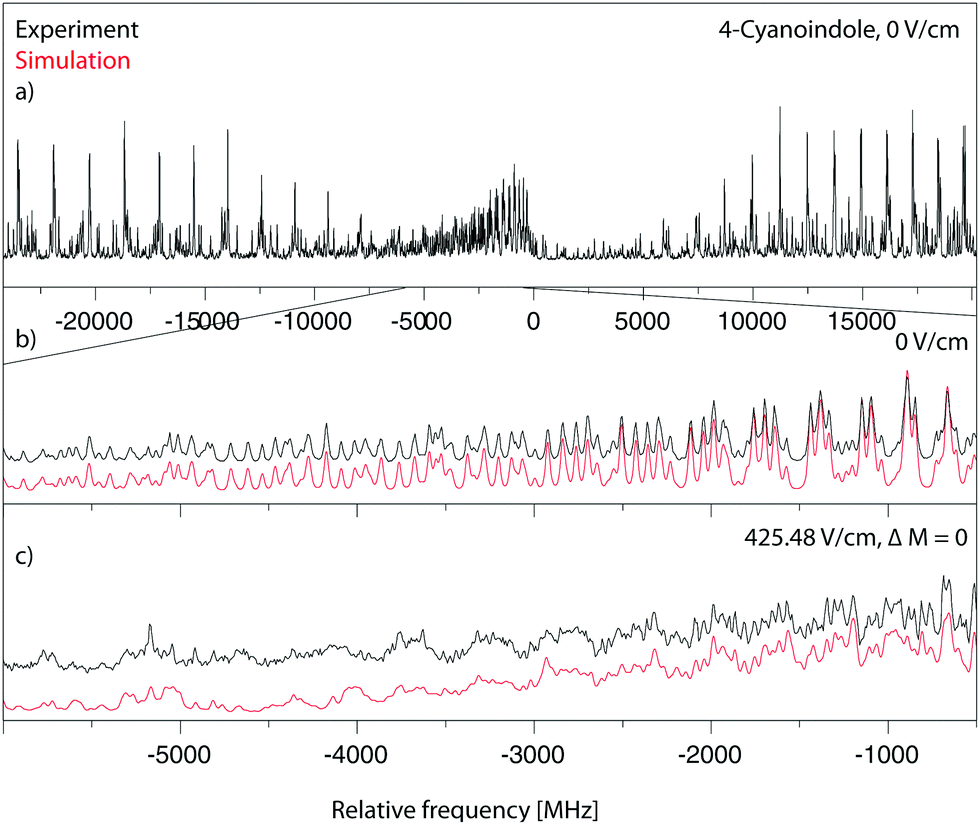 | ||
| Fig. 3 Rotationally resolved electronic spectrum of the electronic origin of 4-cyanoindole, along with a simulation with the best CMA-ES fit parameters. | ||
The rotationally resolved spectra of the electronic origins of the 1-, 2- and 3-deuterated isotopologues at 33034.00 cm−1, 33039.87 cm−1 and 33044.60 cm−1 are shown in the ESI† (Fig. S4–S6). The molecular parameters, obtained from the CMA-ES fits are compiled in Table 1. The angle θ of the TDM in 4-cyanoindole(d1) is slightly smaller than in 4-cyanoindole.
Additionally, we performed lifetime measurements of 3-cyanoindole, 4-cyanoindole, and 5-cyanoindole and indole in different solvents using TCSPC. Fig. 4 presents the decay curves for 4-cyanoindole and 4-cyanoindole(d1) in ethyl acetate and H2O (D2O). The excited state lifetimes of isolated indole, 3-cyanoindole, 4-cyanoindole, and 5-cyanoindole and their N-deuterated isotopologues in ethyl acetate (EA) and H2O (D2O for the deuterated compounds) are summarized in Table 3.
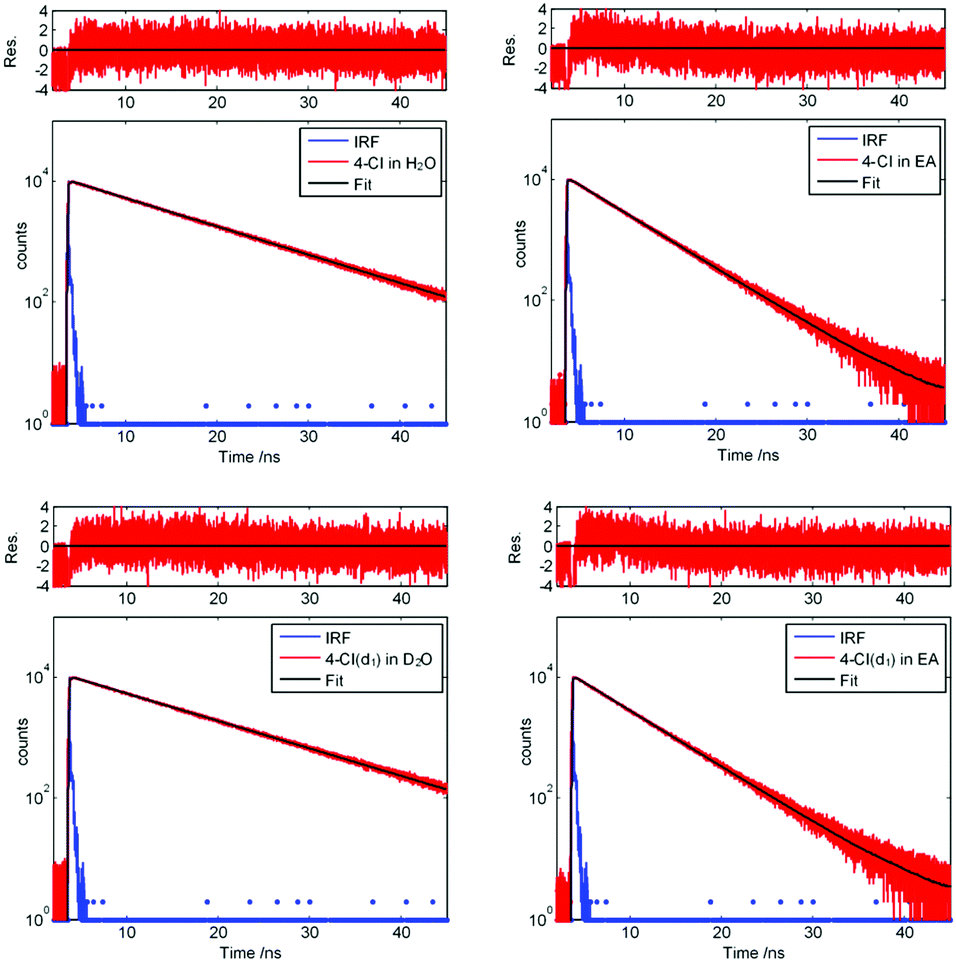 | ||
| Fig. 4 TCSPC traces of 4-cyanoindole in H2O, 4-cyanoindole in EA, 4-cyanoindole(d1) in D2O, and 4-cyanoindole(d1) in EA, along with the instrument response function and the best fit to a single exponential decay. | ||
| In | In(d1) | In-H2O | 3-CI | 3-CI(d1) | 3-CI-H2O | 4-CI | 4-CI(1d1) | 4-CI(2d1) | 4-CI(3d1) | 5-CI | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a Excitation of 3-cyanoindole and 3-cyanoindole(d1) in ethyl acetate (EA) at 283 nm. b 3-CI(d1) has been measured in D2O with excitation at 283 nm. c Excitation of indole in EA at 284 nm. d Excitation of 5-cyanoindole in EA at 295 nm. e Ref. 62. f Ref. 16. g Ref. 34. h Ref. 63. i Ref. 64. j Ref. 40. | |||||||||||
| θ/° | +38e | — | — | +35j | +36j | +51j | −14 | −16 | −9 | −15 | −12g |
| τ/ns | 16.4e | 17.2e | 4h/21i | 9.8j | 14.8j | 3.9j | 11.0 | 5.0 | 5.5 | 5.8 | 12g |
| τ(EA)solv./ns | 3.1c | 3.5 | — | 2.6a | 2.7a | — | 4.7 | 4.7 | 4.7 | 4.7 | 3.6d |
| τ(H2O)solv./ns | 4.5f | 6.1 | — | <0.05f | <0.02b | — | 9.2 | 9.5 | 9.5 | 9.5 | 0.3f |
4 Discussion
4.1 Structural changes upon electronic excitation
From the rotational constants of the normal and the 1-, 2- and 3-deuterated isotopologues, the position of the hydrogen atoms in 1-, 2- and 3-position of the pyrrole ring have been determined on the basis of Kraitchman's equations65 for planar asymmetric molecules in the ground and the electronically excited states, respectively. The |a| and |b| components of the Cartesian coordinates in the coordinate system of the reference isotopologue in the electronic ground and excited states are given in Table 4 along with the center of mass (COM) distances of the respective atom.| 1-Position | 2-Position | 3-Position | ||||
|---|---|---|---|---|---|---|
| |a| | |b| | |a| | |b| | |a| | |b| | |
| S0/pm | 344.13 | 77.46 | 335.42 | 285.96 | 78.98 | 274.93 |
| COM/pm | 352.74 | 440.77 | 286.06 | |||
| S1/pm | 344.91 | 81.58 | 337.65 | 282.29 | 84.11 | 270.36 |
| COM/pm | 354.42 | 440.11 | 283.14 | |||
4.2 Permanent dipole moments
The experimentally determined dipole moment increases from 6.31 D to 8.92 D upon electronic excitation. This is a clear indication, that the excited state is of 1La character. Such an increase of nearly 2 D is not observed for excitation to 1Lb states, in which the permanent dipole moment in general is equal or even smaller than in the electronic ground state. For 3-cyanoindole, for which the lowest state is of 1Lb character, we found a decrease from 5.89 D to 5.39 D upon electronic excitation,40 while for 5-cyanoindole (lowest excited singlet state is 1La) an increase of 1 D is found.66 Thus, the change of permanent dipole moment upon excitation of the singlet state can be used as a sensitive indicator of the electronic nature of this excited state.4.3 Transition dipole moment and the electronic structure of the excited state
The sign of the angle θ between the TDM and the a-axis cannot be determined directly from rotationally resolved spectra. Only the projection of the TDM onto the main inertial axes is obtained from the intensities of the rovibronic transitions. However, there are two methods to obtain also the sign of the TDM angle. Pratt and coworkers have shown, that axis reorientation mixes the rotational wave functions of the ground and excited rovibronic state, and causes a quantum interference effect, which modulates the intensities of the rovibronic transitions.67 These interferences, which cause mixing of the wave functions and therefore generate anomalous intensities of hybrid bands can be considered in two ways. The original method of Hougen and Watson rotates the wave functions of the excited state into the coordinate system of the ground state after diagonalization of the respective Hamiltonian.60 Held et al. applied an equivalent but computationally faster method. They express the excited state Hamiltonian in the coordinate system of the ground state by means of a similarity transformation, i.e. they rotate the Hamiltonian prior to diagonalization.67 It has been shown that in the case of axis reorientation, the rovibronic line intensities depend on the relative sign of the axis reorientation angle θT and the TDM angle θ, which therefore can be determined experimentally.24,28,67,68
Fig. 5 shows a zoomed part of the fit of the rotationally resolved electronic spectrum of the electronic origin of 4-cyanoindole using an axis reorientation Hamiltonian and different combinations of θT and θ signs. Obviously, the best agreement is obtained for ++ (−−) sign combination, while the fit using +− (−+) combinations has larger intensity deviations. Equally, a fit without axis reorientation (θT = 0) is inferior with respect to the agreement between experimental and simulated intensities.
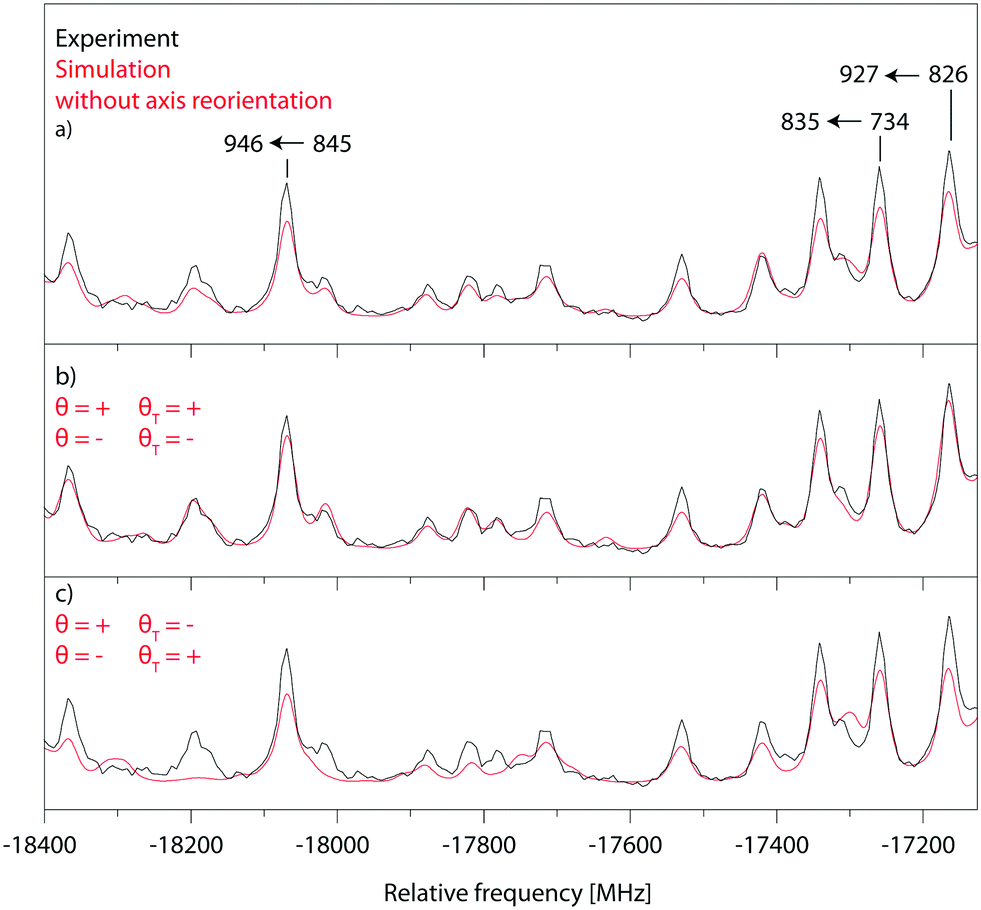 | ||
| Fig. 5 Comparison of a zoomed part of the P-branch of the electronic origin spectrum of 4-cyanoindole, with fits using an axis reorientation Hamiltonian. (a) Experiment vs. fit without axis reorientation (θT = 0). (b) Experiment vs. fit with axis reorientation (θ = + and θT = + or θ = − and θT = −). (c) Experiment vs. fit with axis reorientation (θ = + and θT = − or θ = − and θT = +). | ||
Since the ab initio calculations yielded a positive value of θT (cf. Section 3.1), the sign of θ must also be positive. This means that the rotation of the a-axis onto the TDM is clockwise, as shown in Fig. 1. This orientation of the TDM belong to an La state.
Independently, the orientation of the TDM can be obtained from the comparison of the spectra of the normal and the 2- and 3-deuterated isotopologues.69 Deuteration rotates the inertial a-axis clockwise, as can be seen from Fig. 1. If the rotation of the a-axis onto the TDM is also clockwise (positive), the angle θ will become smaller upon N-deuteration, otherwise it will become larger. Table 1 shows that θ decreases from 30.7° to 28.7°, confirming the positive sign of θ from the axis reorientation.
In order to facilitate the comparison of TDM orientations between the different conformers, the angle of the TDM is given with respect to the pseudo-C2 axis of indole,§ in spite of the inertial a-axis, which will rotate with the change of the substitution position. These projections have been made in Table 3. Clearly, indole (+38°) and 3-cyanoindole (+35°) have similar TDM orientations, belonging to an 1Lb state, while 4-cyanoindole (−14°) and 5-cyanoindole (−12°) among each other have similar 1Lb like TDM orientations (Fig. 6).
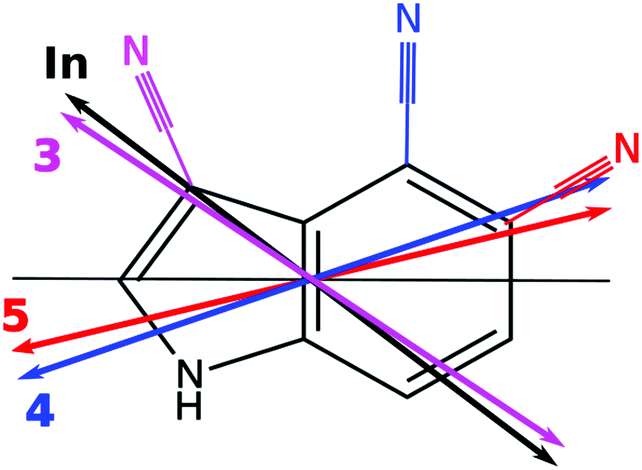 | ||
| Fig. 6 Orientation of the TDM of indole (black double arrow), 3-cyanoindole (magenta double arrow), 4-cyanoindole (blue double arrow), and 5-cyanoindole (red double arrow), relative to the pseudo-C2 axis of indole (black line). | ||
What is now the reason that the lowest electronically excited state in 4- and 5-cyanoindole is of 1La character, while it is 1Lb like in 3-cyanoindole? Looking to the electron density difference plots upon S1 ← S0 excitation, shown in the ESI† (Fig. S8) reveals, that only in 4- and 5-cyanoindole the electron density at the cyano group changes upon excitation.
4.4 Excited state lifetimes
For indole, the chromophore of 4-cyanoindole, the lifetime increases slightly from 16.4 to 17.2 ns upon N(H)-deuteration.62 In isolated 3-cyanoindole an increase from 9.8 ns to 14.8 ns is observed.40 For aniline it has been found that the excited state lifetime of 8.51 ns decreases to 8.48 ns upon N-d2 substitution, but increases to 9.13 ns upon C-d5 substitution.70
In phenol, the effect is more pronounced: OH-deuteration increases the excited state lifetime from 2.4 ns to 13.3 ns. The effect of C-deuteration on the lifetime strongly depends on the position of deuteration. Deuteration in para-position with respect to the hydroxy group leads to a lifetime of 2.1 ns, while combined O(H)-deuteration in ortho- or meta-position leads to substantially higher lifetimes above 30 ns.71
In any case, deuteration (CD, ND, and OD) at the chromophore leads in the majority of cases to an increase of the excited state lifetime, in contrast to the findings for 4-cyanoindole. Even in the chemically similar 3-cyanoindole a 50% increase of excited state lifetime was observed. The reason for the obvious deviation in the case of 4-cyanoindole is subject to further investigation.
4.5 Fluorescence quantum yields
The pure radiative lifetime of a transition from initial state i to final state f can be approximated by: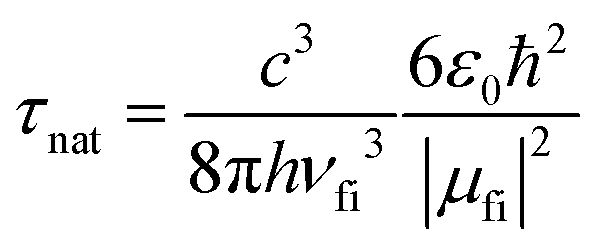 | (3) |
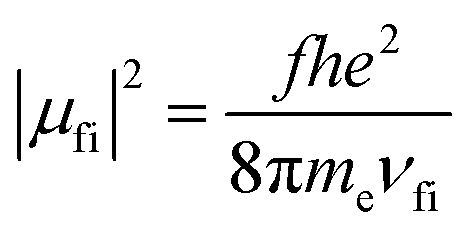 | (4) |
The ratio of the experimental lifetime τexp, which is the inverse of the sum of all radiative and the nonradiative deactivation paths and the natural life time τnat, as the inverse of the fluorescence rate constant kf yields the fluorescence quantum yield:
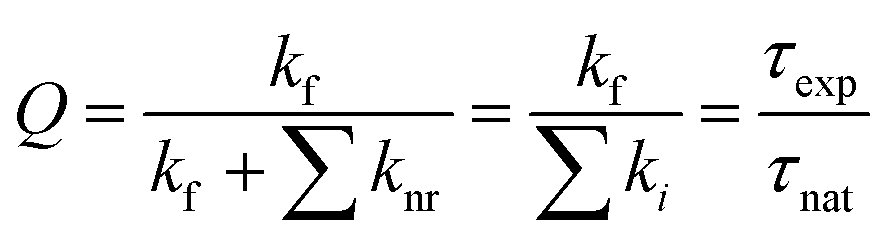 | (5) |
Using the experimental transition frequencies and lifetimes from Table 1 and the dimensionless oscillator strengths f from the ab initio calculations given in Table 2, the fluorescence quantum yield of isolated 4-cyanoindole is calculated to be 0.42. For 3-cyanoindole a similar value of 0.43 has been found using the parameters from ref. 40, while for 5-cycanoindole a substantially smaller quantum yield of 0.10 is found with the parameters from ref. 34. In aqueous solution, that quantum yield of 4-cyanoindole has been determined to 0.85 at an excitation wavelength of 270 nm.17
5 Conclusions
The lowest electronically excited singlet state of 4-cyanoindole is the 1La state, as in the case of 5-cyanoindole, but contrary to 3-cyanoindole. This assignment has been made from the experimentally determined orientations of the transition dipole moment as well as the orientations and absolute values of the permanent dipole moments in the ground and excited state. Also the fluorescence lifetime could be extracted from the Lorentz contribution to the Voigt profile of the rovibronic lines. Thus, rotationally resolved electronic Stark spectroscopy is not only capable of determining structural parameters of electronically excited states, but also to assign the electronic nature of the excited state and obtain information about dynamical processes in the excited state.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support of the Deutsche Forschungsgemeinschaft via grant SCHM1043/12-3 is gratefully acknowledged. Computational support and infrastructure was provided by the “Center for Information and Media Technology” (ZIM) at the Heinrich-Heine-University Düsseldorf (Germany).References
- P. R. Callis, Methods Enzymol., 1997, 278, 113–150 CAS.
- A. L. Sobolewski, W. Domcke, C. Dedonder-Lardeux and C. Jouvet, Phys. Chem. Chem. Phys., 2002, 4, 1093–1100 RSC.
- P. R. Callis, Methods Enzymol., 2011, 487, 1–38 CAS.
- W. Domcke, D. Yarkony and H. Köppel, Conical Intersections: Electronic Structure, Dynamics & Spectroscopy, World Scientific Publishing, Singapore, 2004 Search PubMed.
- D. G. Truhlar and C. Mead, Phys. Rev. A: At., Mol., Opt. Phys., 2003, 68, 032501 CrossRef.
- A. L. Sobolewski and W. Domcke, J. Phys. Chem. A, 2001, 105, 9275–9283 CrossRef CAS.
- A. L. Sobolewski and W. Domcke, Chem. Phys. Lett., 1999, 315, 293–298 CrossRef CAS.
- A. L. Sobolewski and W. Domcke, Eur. Phys. J. D, 2002, 20, 369–374 CrossRef CAS.
- P. R. Callis, J. Chem. Phys., 1991, 95, 4230 CrossRef CAS.
- C. Brand, J. Küpper, D. W. Pratt, W. L. Meerts, D. Krügler, J. Tatchen and M. Schmitt, Phys. Chem. Chem. Phys., 2010, 12, 4968–4997 RSC.
- P. R. Callis, J. T. Vivian and L. S. Slater, Chem. Phys. Lett., 1995, 244, 53 CrossRef CAS.
- L. S. Slater and P. R. Callis, J. Phys. Chem., 1995, 99, 8572 CrossRef CAS.
- M. Böhm, J. Tatchen, D. Krügler, K. Kleinermanns, M. G. D. Nix, T. A. LeGreve, T. S. Zwier and M. Schmitt, J. Phys. Chem. A, 2009, 113, 2456–2466 CrossRef PubMed.
- L. J. G. W. van Wilderen, H. Brunst, H. Gustmann, J. Wachtveitl, J. Broosc and J. Bredenbeck, Phys. Chem. Chem. Phys., 2018, 20, 19906–19915 RSC.
- B. N. Markiewicz, D. Mukherjee, T. Troxler and F. Gai, J. Phys. Chem. B, 2016, 210, 936–944 CrossRef PubMed.
- M. R. Hilaire, D. Mukherjee, T. Troxler and F. Gai, Chem. Phys. Lett., 2017, 685, 133–138 CrossRef CAS PubMed.
- M. R. Hilaire, I. A. Ahmed, C.-W. Lin, H. Jo, W. F. DeGrado and F. Gai, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 6005–6009 CrossRef CAS PubMed.
- J. R. Platt, J. Chem. Phys., 1949, 17, 484–495 CrossRef CAS.
- G. Weber, Biochem. J., 1960, 75, 335–345 CrossRef CAS PubMed.
- B. Albinsson and B. Nordén, J. Phys. Chem., 1992, 96, 6204 CrossRef CAS.
- D. M. Sammeth, S. Yan, L. H. Spangler and P. R. Callis, J. Phys. Chem., 1990, 94, 7340 CrossRef CAS.
- T. L. O. Barstis, L. I. Grace, T. M. Dunn and D. L. Lubman, J. Phys. Chem., 1993, 97, 5820 CrossRef CAS.
- L. A. Philips and D. H. Levy, J. Chem. Phys., 1986, 85, 1327–1332 CrossRef CAS.
- G. Berden, W. L. Meerts and E. Jalviste, J. Chem. Phys., 1995, 103, 9596–9606 CrossRef CAS.
- C. Kang, T. M. Korter and D. W. Pratt, J. Chem. Phys., 2005, 122, 174301 CrossRef PubMed.
- J. Zuclich, J. U. von Schütz and A. H. Maki, J. Am. Chem. Soc., 1974, 96, 710–714 CrossRef CAS PubMed.
- B. J. Fender, K. W. Short, D. K. Hahn and P. R. Callis, Int. J. Quantum Chem., 1999, 72, 347–356 CrossRef CAS.
- J. Küpper, D. W. Pratt, W. L. Meerts, C. Brand, J. Tatchen and M. Schmitt, Phys. Chem. Chem. Phys., 2010, 12, 4980–4988 RSC.
- L. Serrano-Andrés and B. O. Roos, J. Am. Chem. Soc., 1996, 118, 185–195 CrossRef.
- A. C. Borin and L. Serrano-Andrés, Chem. Phys., 2000, 262, 253–265 CrossRef CAS.
- L. Serrano-Andrés and A. C. Borin, Chem. Phys., 2000, 262, 267–283 CrossRef.
- X. Meng, T. Harricharran and L. J. Juszczak, Photochem. Photobiol., 2013, 89, 40–50 CrossRef PubMed.
- W. Domcke and A. L. Sobolewski, Science, 2003, 302, 1693–1694 CrossRef CAS PubMed.
- O. Oeltermann, C. Brand, B. Engels, J. Tatchen and M. Schmitt, Phys. Chem. Chem. Phys., 2012, 14, 10266–10270 RSC.
- C. Brand, B. Happe, O. Oeltermann, M. Wilke and M. Schmitt, J. Mol. Struct., 2013, 1044, 21–25 CrossRef CAS.
- B. Stuhlmann, A. Gräßle and M. Schmitt, Phys. Chem. Chem. Phys., 2014, 16, 899–905 RSC.
- A. Min, C. J. Moon, A. Ahn, J. H. Lee, S. K. Kim and M. Y. Choi, Chem. Phys. Lett., 2016, 658, 63–70 CrossRef CAS.
- A. Ahn, A. Min, C. J. Moon, J. H. Lee and M. Y. Choi, Chem. Phys. Lett., 2014, 616-617, 55–60 CrossRef CAS.
- A. Min, A. Ahn, C. J. Moon, J. H. Lee, M. Y. Choi and S. K. Kim, Chem. Phys. Lett., 2014, 614, 263–268 CrossRef CAS.
- M. Schneider, M.-L. Hebestreit, M. M. Lindic, H. Parsian, A. Y. Torres-Boy, L. Álvarez Valtierra, L. Meerts, R. Kühnemuth and M. Schmitt, Phys. Chem. Chem. Phys., 2018, 20, 23441–23452 RSC.
- Y. Huang and M. Sulkes, Chem. Phys. Lett., 1996, 254, 242–248 CrossRef CAS.
-
S. Gerstenkorn and P. Luc, Atlas du spectre d'absorption de la molécule d'iode 14800–20000 cm−1, CNRS, Paris, 1986 Search PubMed.
- M. Schmitt, Habilitation, Heinrich-Heine-Universität, Math. Nat. Fakultät, Düsseldorf, 2000 Search PubMed.
- M. Schmitt, J. Küpper, D. Spangenberg and A. Westphal, Chem. Phys., 2000, 254, 349–361 CrossRef CAS.
- J. Wilke, M. Wilke, W. L. Meerts and M. Schmitt, J. Chem. Phys., 2016, 144, 044201 CrossRef PubMed.
- K. Wohlfart, M. Schnell, J. U. Grabow and J. Küpper, J. Mol. Spectrosc., 2014, 247, 119–121 CrossRef.
- R. Ahlrichs, M. Bär, M. Häser, H. Horn and C. Kölmel, Chem. Phys. Lett., 1989, 162, 165–169 CrossRef CAS.
- J. T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef.
- C. Hättig and F. Weigend, J. Chem. Phys., 2000, 113, 5154–5161 CrossRef.
- C. Hättig and A. Köhn, J. Chem. Phys., 2002, 117, 6939–6951 CrossRef.
- C. Hättig, J. Chem. Phys., 2002, 118, 7751–7761 CrossRef.
- A. Hellweg, S. Grün and C. Hättig, Phys. Chem. Chem. Phys., 2008, 10, 1159–1169 RSC.
- P. Deglmann, F. Furche and R. Ahlrichs, Chem. Phys. Lett., 2002, 362, 511–518 CrossRef CAS.
- W. L. Meerts, M. Schmitt and G. Groenenboom, Can. J. Chem., 2004, 82, 804–819 CrossRef CAS.
- W. L. Meerts and M. Schmitt, Phys. Scr., 2005, 73, C47–C52 CrossRef.
- W. L. Meerts and M. Schmitt, Int. Rev. Phys. Chem., 2006, 25, 353–406 Search PubMed.
- M. Schmitt and W. L. Meerts, in Handbook of High Resolution Spectroscopy, ed. M. Quack and F. Merkt, John Wiley and Sons, 2011 Search PubMed.
- A. Ostenmeier, A. Gawelcyk and N. Hansen, in Parallel Problem Solving from Nature, PPSN III, ed. Y. Davidor, H.-P. Schwefel and R. Männer, Springer, Berlin/Heidelberg, 1994 Search PubMed.
- N. Hansen and A. Ostermeier, Evol. Comput., 2001, 9, 159–195 CrossRef CAS PubMed.
- J. T. Hougen and J. K. G. Watson, Can. J. Phys., 1965, 43, 298–320 CrossRef CAS.
- F. Duschinsky, Acta Physicochim. URSS, 1937, 7, 551–577 Search PubMed.
- G. A. Bickel, D. R. Demmer, E. A. Outhouse and S. C. Wallace, J. Chem. Phys., 1989, 91, 6013 CrossRef CAS.
- T. M. Korter, D. W. Pratt and J. Küpper, J. Phys. Chem. A, 1998, 102, 7211–7216 CrossRef CAS.
- S. Arnold and M. Sulkes, J. Phys. Chem., 1992, 96, 4768 CrossRef CAS.
- J. Kraitchman, Am. J. Phys., 1953, 21, 17 CrossRef CAS.
- J. Wilke, M. Wilke, C. Brand, W. L. Meerts and M. Schmitt, ChemPhysChem, 2016, 17, 2736–2743 CrossRef CAS PubMed.
- A. Held, B. B. Champagne and D. W. Pratt, J. Chem. Phys., 1991, 95, 8732 CrossRef CAS.
- C. Brand, O. Oeltermann, M. Wilke, J. Tatchen and M. Schmitt, ChemPhysChem, 2012, 13, 3134–3138 CrossRef CAS PubMed.
- M. Schmitt, D. Krügler, M. Böhm, C. Ratzer, V. Bednarska, I. Kalkman and W. L. Meerts, Phys. Chem. Chem. Phys., 2006, 8, 228–235 RSC.
- R. Scheps, D. Florida and S. A. Rice, J. Chem. Phys., 1974, 61, 1730–1747 CrossRef CAS.
- C. Ratzer, J. Küpper, D. Spangenberg and M. Schmitt, Chem. Phys., 2002, 283, 153–169 CrossRef CAS.
- A. Kahan, A. Wand, S. Ruhman, S. Zilberg and Y. Haas, J. Phys. Chem. A, 2011, 115, 10854–10861 CrossRef CAS PubMed.
- I. Burghardt and J. T. Hynes, J. Phys. Chem. A, 2006, 115, 11411–11423 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cp01618j |
| ‡ In the original publication the term axis switching was used instead of axis reorientation. |
| § This axis is close to the inertial a-axis in indole. It would be of exact C2-symmetry axis in the hydrocarbon indane. Since the La and Lb nomenclature of Platt refers to the TDM vector intersecting the atoms or bonds, it is convenient to use this pseudo-C2 axis of indole for a better comparison. |
| This journal is © the Owner Societies 2019 |