
Electrochemical reduction of CO2 using palladium modified boron-doped diamond electrodes: enhancing the production of CO†
Yasuaki
Einaga  *ab
*ab
*ab
Abstract
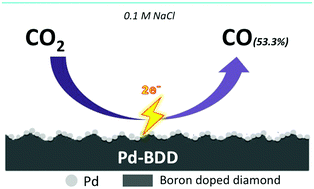
In recent years, boron-doped diamond (BDD) has been utilized as an electrode for the electrochemical reduction of CO2, and several reports have been published on this. The wide potential window of BDD enables the hydrogen evolution reaction, which competes with CO2 reduction, to be suppressed. On the other hand, the high overpotential is still a problem. We attempted to overcome this problem by depositing metal on the BDD electrode. Pd metal was chosen to modify the surface of the BDD electrode (PdBDD). Employing this electrode at a lower potential of −1.6 V vs. Ag/AgCl, we increased the production of CO (53.3% faradaic efficiency) from the reduction of CO2. We present various attempts made to improve the CO production.
- This article is part of the themed collection: 2019 PCCP HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

