
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
London dispersion effects in the coordination and activation of alkanes in σ-complexes: a local energy decomposition study†
Qing
Lu
 ,
Frank
Neese
and
Giovanni
Bistoni
*
,
Frank
Neese
and
Giovanni
Bistoni
*
Max-Planck-Institut für Kohlenforschung, Kaiser-Wilhelm-Platz 1, 45470, Mülheim an der Ruhr, Germany. E-mail: giovanni.bistoni@kofo.mpg.de
First published on 2nd April 2019
Abstract
Local energy decomposition (LED) analysis decomposes the interaction energy between two fragments calculated at the domain-based local pair natural orbital CCSD(T) (DLPNO-CCSD(T)) level of theory into a number of chemically meaningful contributions. Herein, this scheme is applied to the interaction between the transition metal (TM) and the alkane in σ-complexes. It is demonstrated that the often-neglected London dispersion (LD) energy is a fundamental component of the TM–alkane interaction for a wide range of experimentally characterized σ-complexes. LD effects determine the structure and the thermodynamic stability of σ-complexes and influence the selectivity of CH activation reactions. The magnitude of the LD energy can be modulated by increasing the size of the alkane and of the ancillary ligands on the TM. These results provide further evidence on the fundamental role that London dispersion plays in organometallic chemistry.
1 Introduction
The London dispersion (LD) energy constitutes the attractive part of the van der Waals potential and it is omnipresent in chemistry. In the Symmetry Adapted Perturbation Theory (SAPT) framework,2 the leading dipole–dipole term of the LD energy grows with the polarizabilities of the interacting fragments and decays asymptotically as R−6, with R being the interfragment distance.3 Hence, as the molecular system size increases, the magnitude of the LD energy increases, often making it stronger than covalent or electrostatic interactions.4 Due to its size dependence, LD is responsible for the stability of seemingly too crowded molecules, such as diamantyl,5 all-meta tBu-triphenylmethane,6 and all-meta tBu-hexaphenylethane dimers.7 Moreover, its importance has been widely recognized in biochemistry,8 material science,9 and organic chemistry.10On the other hand, LD is typically considered weaker than covalent or other non-covalent interactions, especially for small systems.11 In particular, although recent studies have underlined its importance in organometallic chemistry,4,10,12–22 its contribution to coordinate covalent bonds is still often ignored or assumed to be negligible. In fact, the interaction between a transition metal (TM) and a ligand (L) is still often described as a simple donor–acceptor interaction and the structure, the catalytic behaviour and the spectroscopic properties of TM complexes are in most cases discussed using orbital models such as the popular Dewar–Chatt–Duncanson (DCD) bonding model.23,24
Despite the success of these simple interpretative frameworks, a unified understanding of intermolecular interactions in the various fields of chemical research would require models that explicitly include LD as one of the components of the chemical bond, irrespective of the intermolecular distance or of the interaction strength. Unfortunately, rigorous perturbative approaches like SAPT are not applicable to strong TM–L interactions.
More in general, the LD energy, as well as all the other physical components of the interaction typically discussed in chemical models, is not a quantum mechanical observable. Hence, its theoretical definition is ambiguous, especially when the interacting fragments are close to each other and their electron density significantly overlap.3
Another major problem is that the computational modeling of organometallic complexes requires the accurate inclusion of electron correlation for systems with many electrons and complicated electronic structures. The most commonly used computational methodology to practically address this problem is Density Functional Theory (DFT). Although it is well-known that DFT does not properly describe LD forces,25 several strategies have been suggested to deal with this shortcoming,26,27 thus allowing the calculation of accurate structures and energies for organometallic complexes. In particular, a force-field like correction term (the -D correction) is typically added to the DFT energy to account for long-range LD effects, which are not or poorly described by the exchange–correlation functional. Hence, the magnitude of the -D correction can be used as a qualitative estimate for the LD energy, albeit with caution in those cases when short- to mid-range LD effects are important.13
Recently,28–30 we proposed a new strategy called Local Energy Decomposition (LED) analysis, which provides a decomposition of interaction energies computed at the domain-based local pair natural orbital coupled cluster DLPNO-CCSD(T)31–33 level into various terms representing the most important chemical components of the interaction. This scheme allows for a simple definition of the LD energy at the DLPNO-CCSD(T) level29,34,35 and has been already applied in a wide range of different chemical situations.36 Importantly, the LED analysis can be applied for obtaining a consistent quantification of the LD energy for virtually any system within the range of applicability of DLPNO-CCSD(T) scheme, going from weak noncovalent interactions29 to strong ionic and covalent bonds.13,34,37
Herein, the DLPNO-CCSD(T)/LED approach is used to quantify the LD component of TM–L bonds of various strength and to elucidate the factors that contribute to its magnitude in organometallic complexes. As a case study, we consider the bond between the TM and the CH bond of an alkane in σ-complexes. These complexes have been suggested as crucial intermediates for alkane C–H activation reactions,38–44 which are of paramount importance in various chemical industries. In particular, the development of chemical processes for the conversion of alkanes from fossil sources to more-valuable compounds has attracted great attention over past decades.44–48 In the simplest mechanistic scenario, the CH bond initially coordinates to the TM to form a σ-complex (intermediate 2 in Scheme 1a), eventually leading to the cleavage of the CH bond through oxidative addition to the TM.
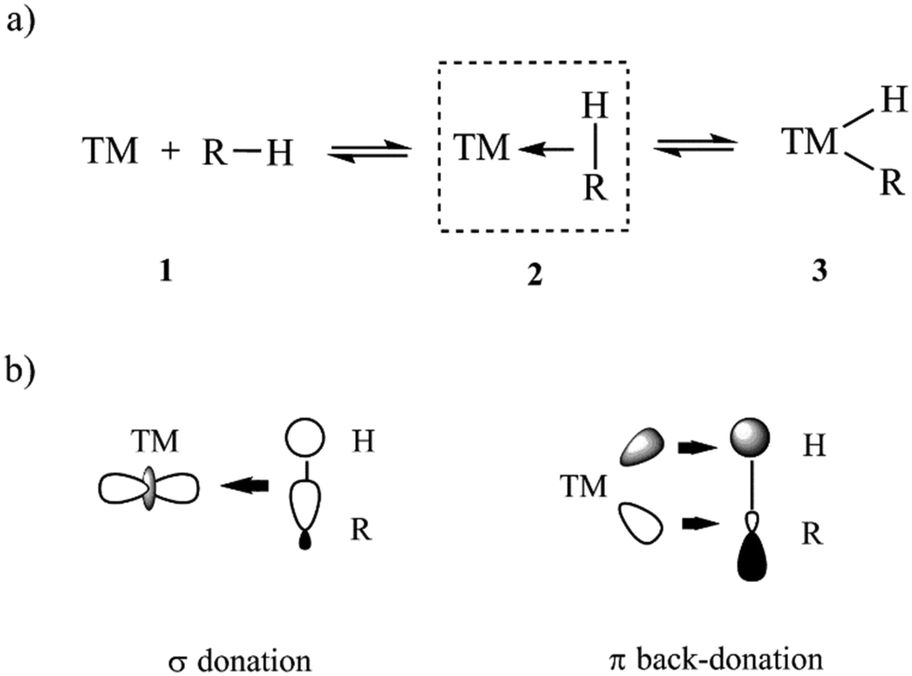 | ||
| Scheme 1 (a) Oxidative addition of an alkane to a TM. R denotes an alkyl group. (b) Simple orbital representation describing the donor acceptor interaction between the R–H bond and the TM in alkane σ-complexes. | ||
The chemical bond between the TM and the CH bond in σ-complexes is typically described as a donor–acceptor in the framework of the DCD model (Scheme 1b). In this model, the C–H group formally donates electrons from its filled σ orbital to the metal center (TM ← HC σ-donation) while at the same time the TM back-donates d electrons into the empty C–H σ* orbital in the alkane (TM → HC π-backdonation).49–51 This model can be considered as an extension of the three-center two-electron (3c-2e) model initially adopted by Brookhart52 and later corroborated by energy decomposition schemes at the density functional theory (DFT) level of theory.42,53 Both σ-donation and π-backdonation are expected to decrease the CH bond order, thus favoring the oxidative addition to the TM.54
Although alkane σ-complexes have been proposed for decades, their direct experimental characterization has constituted one of the major challenges for organometallic chemists.54,55 The few experimental characterized σ-complexes can be grouped into three families, differing for the complex structure and their synthetic route. Herein, we base our analysis on a series of σ-complexes that includes representative systems of each class.
The first class of σ-complexes investigated herein is a series of group VII TM complexes that were previously characterized mainly by means of IR spectroscopy. Their structure is shown in Scheme 2a. The series includes neutral complexes differing for the size of the alkane such as [CpMn(CO)2(alkane)] (alkane = C2H6, n-C7H16)56–59 and [CpRe(CO)2(alkane)] (alkane = CH4, n-C5H12, n-C7H16)56,57,60 as well as the positively charged [(HEB)Re(CO)2(n-C5H12)]+ (HEB = hexaethylbenzene).61 Note that for the longer alkanes many conformers are possible, depending on their relative orientation with the TM. As case studies, two conformers are investigated for each complex, in which the TM interacts with a CH bond located on either the methyl or the methylene carbon. Conformers of the first type are denoted hereafter as “head-on” structures, whilst the latter are denoted as “side-on” structures.
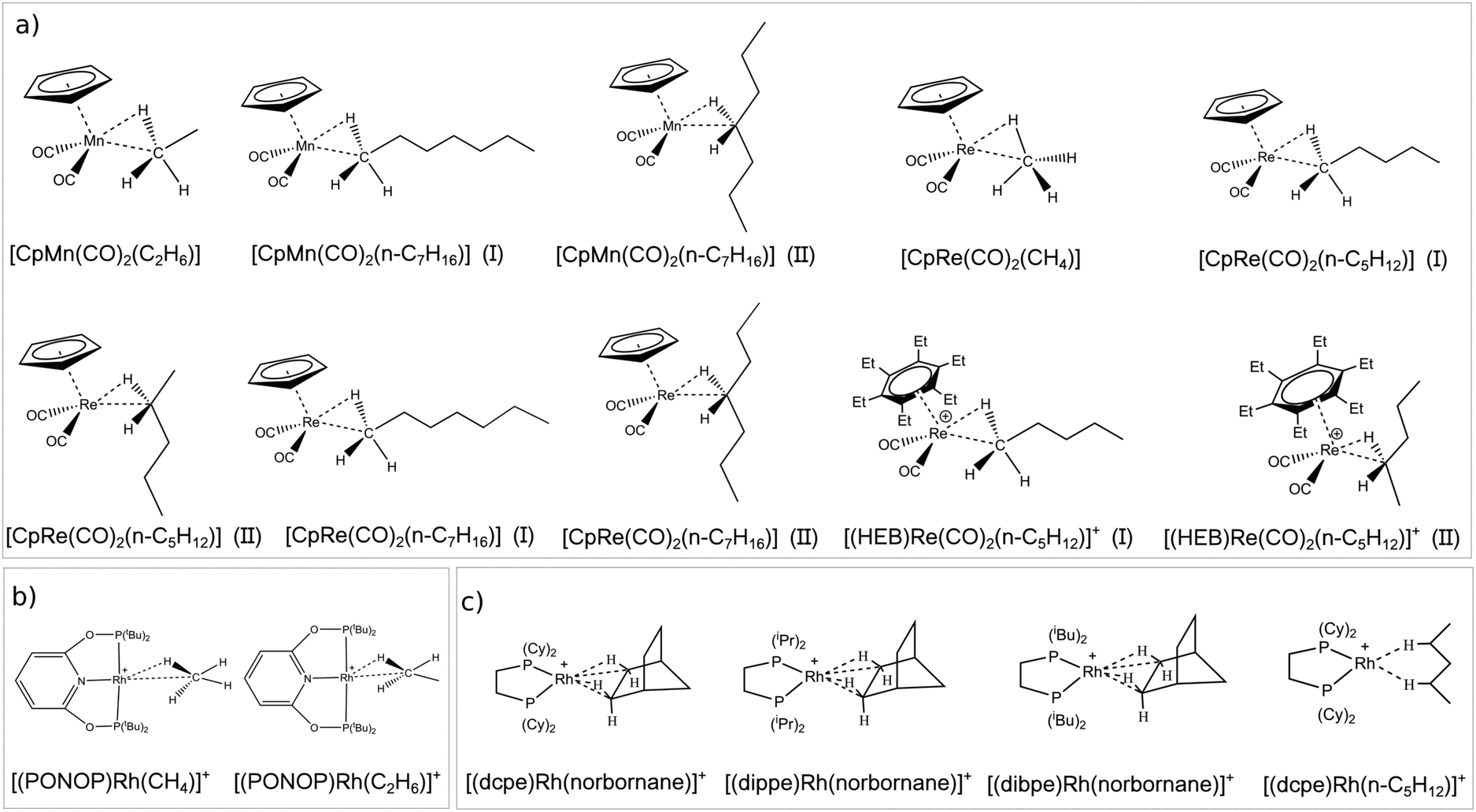 | ||
| Scheme 2 TM–alkane complexes investigated and nomenclature used in this work. | ||
The second family of σ-complexes investigated herein is shown in Scheme 2b and includes two Rh complexes bearing the same tridentate ligand (PONOP = 2,6-(tBu2PO)2C5H3N) but different alkanes [(PONOP)Rh(alkane)]+ (alkane = CH4, C2H6). These complexes were characterized by means of NMR spectroscopy.62,63
Finally, the last class includes Rh complexes of norbornane bearing different diphosphine ligands [(L)Rh(norbornane)]+ (L = dpce, dippe, dibpe) as well as the [(dcpe)Rh(n-C5H12)]+ complex. All these complexes were fully characterized by X-ray crystallography and solid state NMR.49,64–66 Their structures are shown in Scheme 2c.
For all these complexes, the main factors contributing to their formation, the coordination preference of the alkane and the activation of the CH bond are discussed in the framework of the LED analysis and the role played by LD in all these situations is elucidated. Moreover, the factors that contribute to the magnitude of the LD energy are identified and designing principles for σ-complexes with tailored bonding features are given.
2 Method
Geometry optimizations were carried out using the TPSS functional67 and including the -D3 dispersion correction term68 with Becke–Johnson damping parameters.69 The def2-TZVP basis set70 was used. The def2-ECP effective core potential was used for Re and Rh atoms. Frequencies were calculated at the same level of theory to verify the nature of minima and transition states and to compute free energies. The optimized geometries and vibrational frequencies obtained with these computational settings were found to be in good agreement with available X-ray structures and IR data, as shown in Table S1 in the ESI.† Moreover, these settings were shown to provide geometries and IR frequencies in good agreement with experimental data for the closely related agostic complexes.12Single point energies at the DLPNO-CCSD(T) level were computed using TightPNO settings.31–33 The def2-TZVPP basis set was used in conjunction with the def2-TZVPP/C auxiliary basis set. Table S2 (ESI†) shows that the DLPNO-CCSD(T) results are at convergence with respect to the basis set size. The RIJCOSX approximation71 was used for the calculation of Coulomb and exchange integrals in the HF reference. The def2/J auxiliary basis set was used. The COSX grid was set to GridX5.
Binding energies (ΔE) at the DLPNO-CCSD(T) level were calculated as the electronic energy difference between the σ-complex and its constituting fragments (the alkane and the remaining metallic moiety) at their equilibrium TPSS-D3 geometry. The binding energies were decomposed into dispersive (Edisp) and non-dispersive (ΔEno-disp) contributions by means of the LED technique.28
The dominant term in Edisp is the DLPNO-CCSD contribution EC-CCSDdisp, which was calculated by summing up all the instantaneous dipole–dipole excitations between the two fragments. It includes contributions from both strong and weak pairs, as detailed in ref. 29. For the sake of simplicity, an effective triples correction contribution to the LD energy was also included. It was estimated by multiplying the intermolecular component of the perturbative triples correction, i.e., the contribution coming from triples of occupied orbitals located on different fragments E(X,Y)C-(T),28 by a correction factor γ. The γ correction was defined as the ratio between the dispersion contribution from the strong pairs and the total intermolecular strong pair contribution. Hence, the total LD energy reads:
| Edisp = EC-CCSDdisp + γE(X,Y)C-(T) | (1) |
The remaining part of the correlation contribution to the binding energy is denoted as ΔEC-CCSD(T)no-disp. The sum of this term with the overall HF contribution to the binding energy ΔEHF gives the overall non-dispersive contribution to the binding energy, i.e., ΔEno-disp.
In order to provide an in-depth analysis of the factors contributing to the LD energy, the Dispersion Interaction Density (DID) plot29,72 were employed. They provide a useful spatial analysis of the LD energy and were calculated as detailed in ref. 29.
Finally, the orbital relaxation contribution to the binding energy was calculated as detailed in ref. 29. It provides a useful estimate for the charge transfer and polarization energy at the reference level of theory, consistent with that provided by standard energy decomposition schemes.73
3 Results and discussion
3.1. The London dispersion component of the TM–CH4 bond in [CpRe(CO)2(CH4)]
We first describe here a detailed investigation of the Re⋯CH bond in the [CpRe(CO)2(CH4)] complex. The analysis will be then extended to the whole series of σ-complexes shown in Scheme 2 in the next section.The Re⋯CH4 binding energy profile as a function of the Re⋯C distance calculated at the DLPNO-CCSD(T) level of theory is shown in the upper panel of Fig. 1. In the same panel, the decay of the LD energy (Edisp) and of the non-dispersive component of the binding energy (ΔEno-disp) is also reported. As expected, the binding energy is dominated by LD forces in the long range where no significant charge transfer occurs between the fragments. As the fragments approach each other, the relative contribution of Edisp to the binding energy slightly decreases. At the equilibrium geometry, Edisp amounts to −46.7 kJ mol−1, which is about the 67% of the overall binding energy (−69.8 kJ mol−1). The remaining 33% comes from ΔEno-disp. It describes the balance between the orbital relaxation energy at the HF level (−135.2 kJ mol−1) and the other components of the interaction (+112.1 kJ mol−1), such as permanent electrostatics, electronic preparation and the correlation correction for these terms ΔEC-CCSD(T)no-disp.29 For the sake of simplicity, only Edisp and ΔEno-disp will be discussed in the following. Additional information on the various terms of the decomposition can be found in the ESI.†
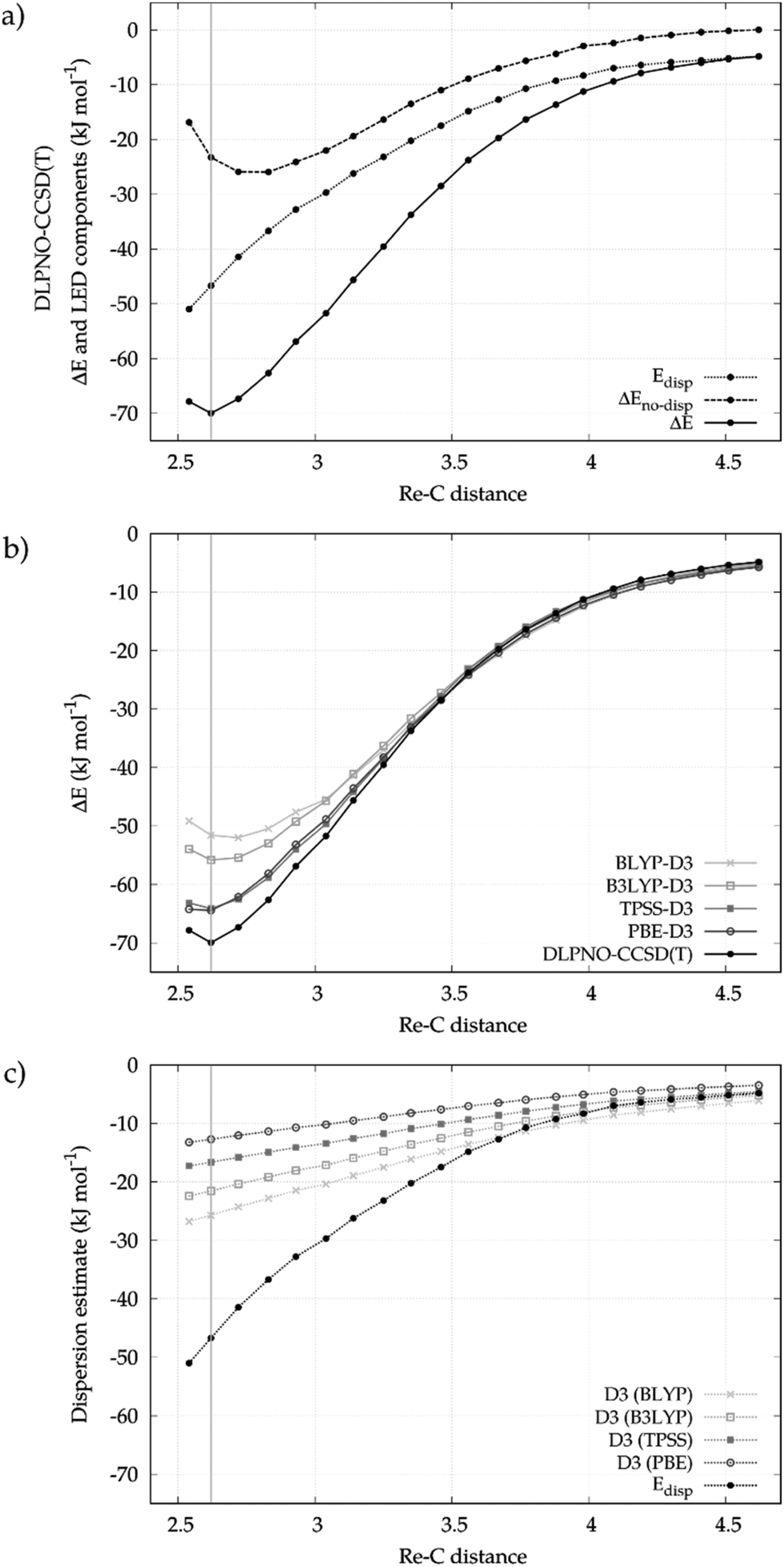 | ||
| Fig. 1 (a) DLPNO-CCSD(T) binding energy decay with the Re⋯CH4 distance in [CpRe(CO)2(CH4)]. Its decomposition into dispersive (Edisp) and non-dispersive (ΔEno-disp) contributions is also shown. (b) Binding energy profiles with different computational methods. (c) Comparison of LED (Edisp) and -D3 estimate for the London dispersion. The vertical line marks the equilibrium geometry. | ||
Although Re⋯CH4 is a relatively weak coordination bond, the fact that the Edisp amounts to the 67% of the overall binding energy is surprising. These results are consistent with the ones recently12 found in the context of agostic interactions, i.e. the intramolecular interaction between a C–H bond and a coordinatively unsaturated TM. It was found that short-range LD forces between the agostic C–H bond and the metal center are largely responsible for the stability of a wide range of agostic complexes, consistent with a large number of experimental findings, including the fact that many agostic complexes do not show evidence of significant C–H bond activation.74
In order to compare different computational methodologies, the binding energy profile obtained with different functionals is compared with the DLPNO-CCSD(T) one in the central panel of Fig. 1. In the bottom panel of the same figure the decay of the -D3 correction for different functionals is compared with the Edisp values extracted from the LED scheme.
In the long range, all methods provide similar Re⋯CH4 binding energies. At the equilibrium geometry, the difference between the various functionals increases, with binding energies being −51.6, −55.8, −64.2 and −64.5 kJ mol−1 for BLYP-D3, B3LYP-D3, TPSS-D3 and PBE-D3, respectively. The corresponding -D3 correction follows the opposite trend, being −25.7, −21.6, −16.6 and −12.7 kJ mol−1. The differences between the -D3 estimates are mostly due to the effect of the damping functions, as also recently discussed by Grimme et al.13 By comparing these figures with the Edisp value (−46.7 kJ mol−1), one finds that the functionals that better reproduce the DLPNO-CCSD(T) binding energy (e.g. PBE) are those for which the -D3 correction is smaller and far from the accurate LED values. Hence, in the following only DLPNO-CCSD(T)/LED results will be discussed.
In summary our results clearly show that LD is fundamental for both the formation and the thermodynamic stability of [CpRe(CO)2(CH4)]. In fact, it dominates the TM–alkane interaction in the long range and significantly contributes to the magnitude of the binding energy at the equilibrium geometry.
3.2. London dispersion in σ-complexes
In order to test the generality of our findings for the Re⋯CH4 interaction in [CpRe(CO)2(CH4)], geometries, binding energies and the corresponding LD contributions were calculated for the whole series of σ-complexes investigated in this work. The results obtained are summarized in Fig. 2. Note that, for the sake of simplicity, only electronic energies are discussed in this section, while the free energies at experimental conditions are reported in Table S4 (ESI†). | ||
| Fig. 2 Comparison of TM⋯alkane binding energies (ΔE) and the associated LD contribution (Edisp) for the systems investigated in this work. The distance between the TM and the closest carbon atom on the alkane r(TM–C) is also reported. | ||
Binding energies span a broad range of values, going from −57.6 to −127.8 kJ mol−1 for [CpMn(CO)2(C2H6)] and [(dcpe)Rh(norbornane)]+, respectively. Similarly, the LD energy ranges from −46.7 to −111.3 kJ mol−1 for [CpRe(CO)2(CH4)] and [(dcpe)Rh(norbornane)]+, respectively.
Remarkably enough, Edisp amounts to at least the 67% of the binding energy for all complexes. For [CpMn(CO)2(C7H6)] (II) and [(dibpe)Rh(norbornane)]+, its magnitude exceeds the binding energy. Hence, without this important component of the interaction, these experimentally characterized complexes would not be stable. These results are consistent with our previous findings on the agostic EtTiCl3(dmpe) complex.12
Having established the fundamental importance of LD for the thermodynamic stability of σ-complexes, we aim at identifying the most important factors contributing to its magnitude in a view of aiding to the development of designing principle for stable TM–alkane adducts.
As mentioned in the introduction, LD increases with the polarizability of the interacting fragments and decreases with the intermolecular distance. Hence, the size of the ancillary ligands and of the alkane, the nature and the oxidation state of the TM, and the coordination mode of the alkane (i.e. whether it forms head-on or side-on structures) are all expected to contribute to its magnitude.
A revealing insight into this aspect comes from the analysis of the DID plots (Fig. 3), which provide a simple mean of visualizing the most important contacts contributing to the magnitude of the LD energy of the TM⋯alkane interaction.72
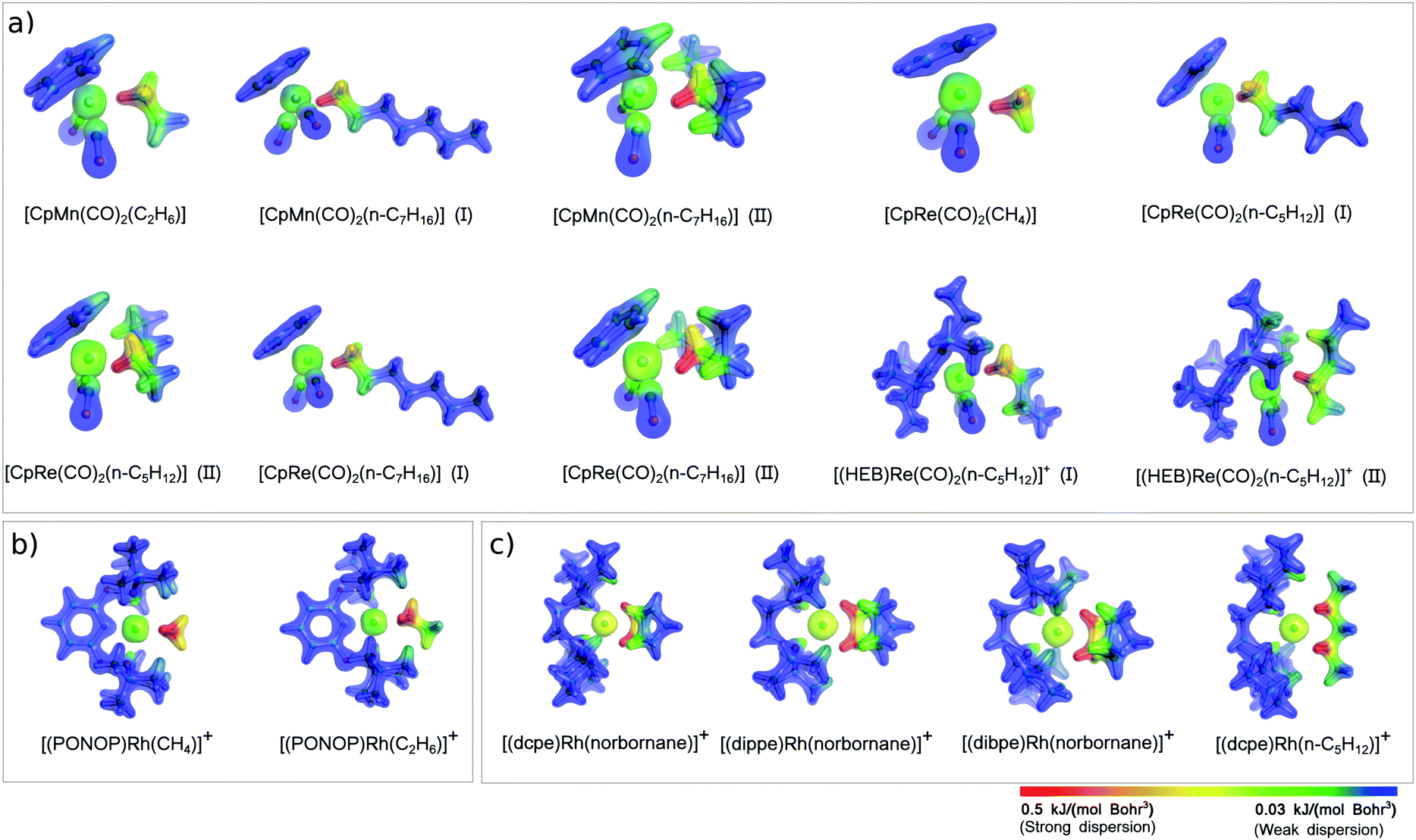 | ||
| Fig. 3 Dispersion interaction density (DID) plots for selected TM–alkane σ-complexes experimentally characterized (see Scheme 2). The density isosurfaces were generated for a contour value of 0.1 e Bohr−3. The DID color scheme ranges from highest value (red) to zero dispersion energy (blue). | ||
The first eye-catching feature of these plots is that the most significant contribution to the LD energy originates from short TM⋯H–C contacts, as evidenced by the red color of the density isosurfaces around the C–H groups in close proximity of the TM. In Mn and Re complexes, only one C–H group strongly interacts with the TM, whilst all Rh complexes (Fig. 2b and c) are stabilized by two short TM⋯H–C contacts. For this reason, the LD energy in Rh complexes is typically larger than in the other σ-complexes investigated in this work.
The size of the alkane also plays an important role in affecting the magnitude of the LD contribution. By comparing [CpRe(CO)2(CH4)], [CpRe(CO)2(n-C5H12)] (I), and [CpRe(CO)2(n-C7H16)] (I), in which alkanes of different size interact through their methyl carbon with the same TM fragment, it can be found that the LD increases with the size of the alkane, in line with the above considerations. However, [CpRe(CO)2(n-C5H12)] (I) and [CpRe(CO)2(n-C7H16)] (I) have essentially the same amount of LD energy. This occurs because the tail of the heptane chain in [CpRe(CO)2(n-C7H16)] is too far away from the Re atom to affect the LD energy, as shown by the blue color that the DID plot assumes in this region. The same trend can also be observed for other pair of complexes, i.e., [CpMn(CO)2(C2H6)] vs. [CpMn(CO)2(n-C7H16)] (I) and [(PONOP)Rh(CH4)]+vs. [(PONOP)Rh(C2H6)]+. As these variations in the LD energy correlate well with the corresponding variations in the binding energies, it can be concluded that the size of the alkane does not significantly affect the other components of the interaction.
Similarly, the use of bulky ligands on the TM also increases the LD contribution to the binding energy. In fact, going from [(dippe)Rh(norbornane)]+ to [(dbpe)Rh(norbornane)]+, Edisp increases from 103 to 110 kJ mol−1 as a consequence of the substitution on the four iPr groups on the diphosphine ligand with the more polarizable tBu groups. However, these figures do not correlate with the corresponding variations in the binding energy. This is probably due to steric effects associated with the inclusion of bulkier tBu groups, as shown by the fact that r(TM–C) distance slightly increases going from [(dippe)Rh(norbornane)]+ (2.40 Å) to [(dbpe)Rh(norbornane)]+ (2.42 Å).
As mentioned above, different coordination modes (e.g. side-on vs. head-on) are possible for the longer alkanes. To investigate the effect of LD on the coordination preference of the alkane, it is useful to compare binding and LD energies for the pairs [CpMn(CO)2(n-C7H16)] (I/II), [CpRe(CO)2(n-C5H12)] (I/II), [CpRe(CO)2(n-C7H16)] (I/II) and [(HEB)Re(CO)2(n-C5H12)]+.
In all cases, LD is larger for side-on than for head-on structures. In fact, the former are more compact and feature many TM⋯HC contacts. Consistent with this observation, the TM–alkane binding energy is always slightly larger for side-on structures, with the only exception being the positively charged [(HEB)Re(CO)2(n-C5H12)]+. In this case, the positive charge on the metal probably increases the relative importance of the non-dispersive interaction components, such as polarization and TM ← CH σ-donation. Indeed, by subtracting the LD energy from the binding energy, one finds that the non-dispersive contributions favour head-on structures over side-on structures in all cases. In fact, due to the less congested environment around the TM center, the head-on structures feature slightly smaller r(TM–C) values (Fig. 2), and charge transfer and polarization effects decay quickly with the intermolecular distance.29,73 This aspect will be further discussed in Section 3.3 while investigating the physical components affecting the selectivity of CH activation reactions.
These results demonstrate that two opposing effects, i.e., dispersive and non-dispersive energy contributions, determine the coordination preference of σ-complexes. Although LD typically dominates in σ-complexes, the non-dispersive contributions are also important and particularly significant in charged species. These results are consistent with previous experimental NMR studies showing that [CpRe(CO)2(n-C5H12)] has a slight preference to form side-on structures,1,75 whilst [(HEB)Re(CO)2(n-C5H12)]+ favors head-on structures.76
In summary, LD is fundamental for the thermodynamic stability of σ-complexes. It favors the formation of σ-complexes of long alkanes, influences the alkane coordination preference and favors the formation of compact structure with many TM⋯CH contacts.
3.3. Activation of alkanes in σ-complexes
The finding that LD forces determine to a large extent the coordination preference of the alkane, favoring the formation of compact side-on structures, is in apparent contrast with the fact that the majority of alkane CH activation reactions take place at the methyl carbon, which is usually qualitatively explained in terms of steric effects.77,78To provide an insight into this aspect, we investigated a model oxidative addition step in which n-pentane interacts with CpRe(CO)2 to form the oxidative addition product via the formation of the σ-complex intermediate [CpRe(CO)2(n-C5H12)] (Scheme 2a). This reaction has been recently studied by means of DFT calculations, but the role played by the LD energy in this context was not discussed.53 The computed mechanism at the DLPNO-CCSD(T) level of theory is shown in Fig. 4.
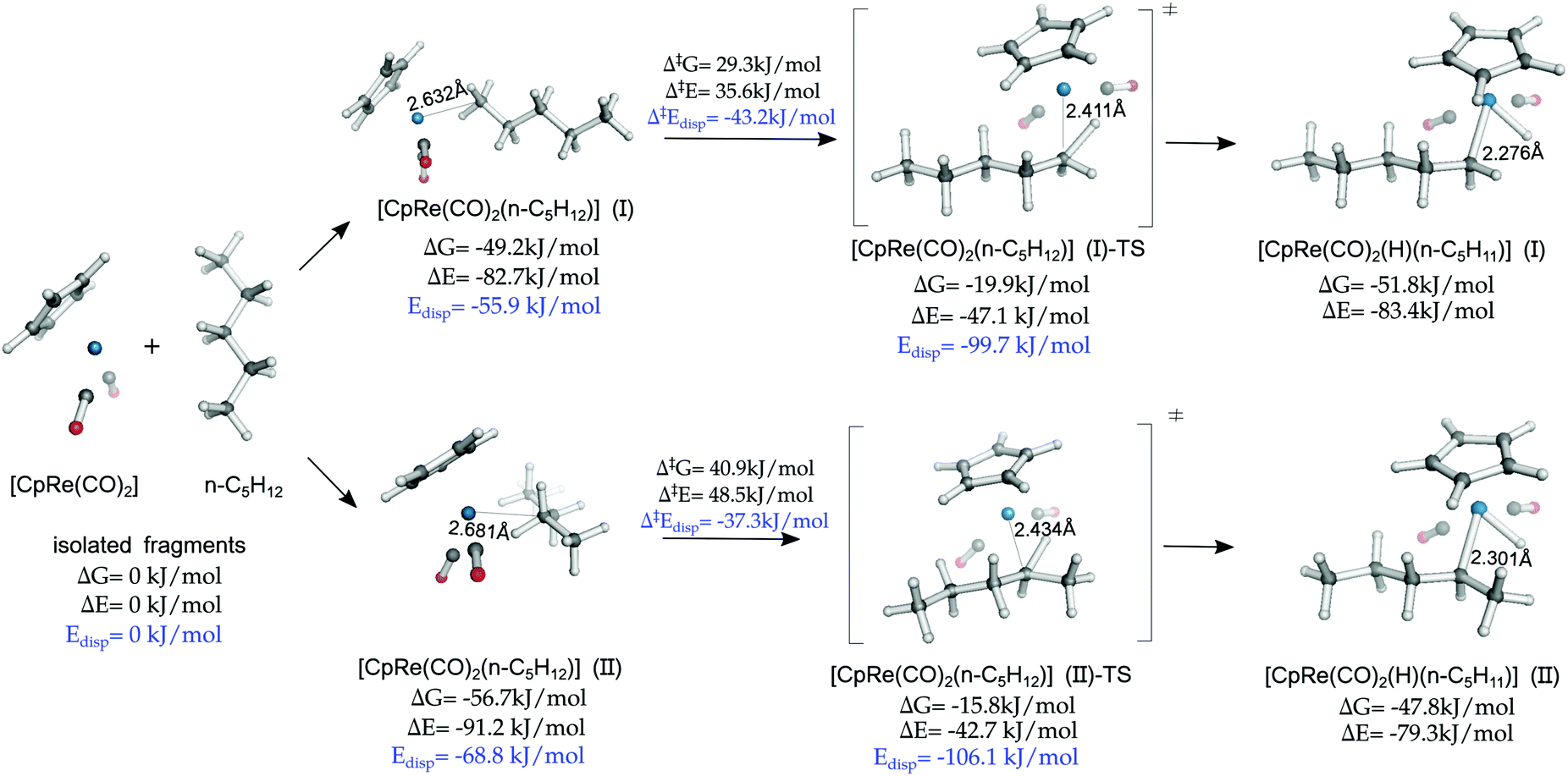 | ||
| Fig. 4 Oxidative addition of pentane to the CpRe(CO)2 fragment. Electronic energies (ΔE) and free energies (ΔG) at 190 K are reported relative to the isolated fragments. The distance between the TM and the closest carbon atom on the alkane r(TM–C) is also reported. | ||
As [CpRe(CO)2(n-C5H12)] (II) is the absolute minimum in the section of the potential energy surface investigated, the oxidative addition is thermodynamically disfavored. This is consistent with the fact that the [CpRe(CO)2(n-C5H12)] intermediate can be characterized by NMR spectroscopy.1
Although the side-on [CpRe(CO)2(n-C5H12)] (II) is thermodynamically more stable than the head-on [CpRe(CO)2(n-C5H12)] (I) conformer, consistent with the above mentioned experimental observation, the cleavage of the coordinated CH bond in the former requires a larger free energy barrier (ΔG‡ = 40.9 kJ mol−1) than in the latter (ΔG‡ = 29.3 kJ mol−1). These results are consistent with previous experimental findings on closely related systems.79
These results can be explained by considering that the CH bond has to be in close proximity to the TM in order to be activated towards the oxidative addition. However, the equilibrium TM⋯C distance in [CpRe(CO)2(n-C5H12)] (II) is significantly longer than that of [CpRe(CO)2(n-C5H12)] (I) (2.68 vs. 2.63 Å) and this difference is partially retained also in the corresponding transition states (2.43 vs. 2.41 Å) and products (2.30 vs. 2.28 Å). This effect probably originates from significant steric effects in the side-on conformation, due to the congested environment around the TM center.
Consistent with these observations, the non-dispersive contribution to the binding energy amounts to −22.4 and −26.8 kJ mol−1 for [CpRe(CO)2(n-C5H12)] (II) and [CpRe(CO)2(n-C5H12)] (I), respectively, and to +63.4 and +52.6 kJ mol−1 for the corresponding transition states. Hence, polarization and charge transfer effects seem to favor the activation of the CH bond at the methyl carbon.
In summary, even though LD determines to a large extent the thermodynamic stability of σ-complexes, the activation barrier associated with the oxidative addition step is determined by an interplay of LD, steric and charge transfer effects.
4 Conclusions
The recently introduced DLPNO-CCSD(T)/LED scheme was used to compute TM⋯alkane binding energies for a broad range of experimentally characterized σ-complexes and to provide an accurate quantification of the London dispersion component of the coordination bond.It was found that London dispersion significantly contributes to the TM⋯alkane interaction and is largely responsible for the thermodynamic stability of σ-complexes. Its magnitude can be modulated by modifying the size of the substituents on the TM fragment or by increasing the number of carbons in the alkane.
Moreover, LD influences the coordination preference of the alkane to the TM. As LD increases with the number of TM⋯CH contacts, side-on structures are generally favored over head-on structures, consistent with previous experimental findings. On the other hand, due to the steric effects, head-on structures feature a shorter distance between the TM and the coordinated carbon, leading to larger polarization and charge transfer interactions.
As charge transfer effects weaken the CH bond, activating it towards the cleavage, the oxidative addition of methyl CH bonds to the TM is expected to be kinetically preferred, as shown in a prototype oxidative addition reaction and consistent with previous experimental findings.
As LD is ubiquitous in nature, it is expected that our findings concerning its importance in organometallic chemistry can be applied to other TM–L interactions, such as the wide range of σ-complexes that serve as intermediates in CH activation reactions.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the Priority Program “Control of Dispersion Interactions in Molecular Chemistry” (SPP 1807) of the Deutsche Forschungsgemeinschaft for financial support. Open Access funding provided by the Max Planck Society.References
- G. E. Ball, C. M. Brookes, A. J. Cowan, T. A. Darwish, M. W. George, H. K. Kawanami, P. Portius and J. P. Rourke, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 6927 CrossRef CAS PubMed.
- B. Jeziorski, R. Moszynski and K. Szalewicz, Chem. Rev., 1994, 94, 1887 CrossRef CAS.
- A. Stone, The theory of intermolecular forces, OUP Oxford, 2013 Search PubMed.
- J. P. Wagner and P. R. Schreiner, Angew. Chem., Int. Ed., 2015, 54, 12274 CrossRef CAS PubMed.
- A. A. Fokin, T. S. Zhuk, S. Blomeyer, C. Perez, L. V. Chernish, A. E. Pashenko, J. Antony, Y. V. Vishnevskiy, R. J. F. Berger, S. Grimme, C. Logemann, M. Schnell, N. W. Mitzel and P. R. Schreiner, J. Am. Chem. Soc., 2017, 139, 16696 CrossRef CAS PubMed.
- S. Rosel, H. Quanz, C. Logemann, J. Becker, E. Mossou, L. Canadillas-Delgado, E. Caldeweyher, S. Grimme and P. R. Schreiner, J. Am. Chem. Soc., 2017, 139, 7428 CrossRef PubMed.
- S. Grimme and P. R. Schreiner, Angew. Chem., Int. Ed., 2011, 50, 12639 CrossRef CAS PubMed.
- C. N. Pace, J. M. Scholtz and G. R. Grimsley, FEBS Lett., 2014, 588, 2177 CrossRef PubMed.
- S. Parveen, S. Rana and R. Fangueiro, J. Nanomater., 2013, 19 Search PubMed.
- D. J. Liptrot and P. P. Power, Nat. Rev. Chem., 2017, 1, 0004 CrossRef.
- M. Strauss and H. A. Wegner, Eur. J. Org. Chem., 2019, 295 CrossRef CAS.
- Q. Lu, F. Neese and G. Bistoni, Angew. Chem., Int. Ed., 2018, 57, 4760 CrossRef CAS PubMed.
- M. Bursch, E. Caldeweyher, A. Hansen, H. Neugebauer, S. Ehlert and S. Grimme, Acc. Chem. Res., 2019, 52, 258 CrossRef CAS PubMed.
- P. Sliwa and J. Handzlik, Chem. Phys. Lett., 2010, 493, 273 CrossRef CAS.
- M. S. G. Ahlquist and P. O. Norrby, Angew. Chem., Int. Ed., 2011, 50, 11794 CrossRef CAS PubMed.
- Y. Minenkov, G. Occhipinti, W. Heyndrickx and V. R. Jensen, Eur. J. Inorg. Chem., 2012, 1507, DOI:10.1002/ejic.201100932,.
- A. Hansen, C. Bannwarth, S. Grimme, P. Petrovic, C. Werle and J. P. Djukic, ChemistryOpen, 2014, 3, 177 CrossRef CAS PubMed.
- L. P. Wolters, R. Koekkoek and F. M. Bickelhaupt, ACS Catal., 2015, 5, 5766 CrossRef CAS.
- G. Lu, R. Y. Liu, Y. Yang, C. Fang, D. S. Lambrecht, S. L. Buchwald and P. Liu, J. Am. Chem. Soc., 2017, 139, 16548 CrossRef CAS PubMed.
- T. R. Cundari, B. P. Jacobs, S. N. MacMillan and P. T. Wolczanski, Dalton Trans., 2018, 47, 6025 RSC.
- E. Detmar, V. Muller, D. Zell, L. Ackermann and M. Breugst, Beilstein J. Org. Chem., 2018, 14, 1537 CrossRef CAS PubMed.
- A. A. Thomas, K. Speck, I. Kevlishvili, Z. H. Lu, P. Liu and S. L. Buchwald, J. Am. Chem. Soc., 2018, 140, 13976 CrossRef CAS PubMed.
- J. Dewar, Bull. Soc. Chim. Fr., 1951, 18, C71 Search PubMed.
- J. Chatt and L. Duncanson, J. Chem. Soc., 1953, 2939 RSC.
- S. Kristyán and P. Pulay, Chem. Phys. Lett., 1994, 229, 175 CrossRef.
- S. Grimme, A. Hansen, J. G. Brandenburg and C. Bannwarth, Chem. Rev., 2016, 116, 5105 CrossRef CAS PubMed.
- R. Peverati and D. G. Truhlar, Philos. Trans. R. Soc., A, 2014, 372, 20120476 CrossRef PubMed.
- W. B. Schneider, G. Bistoni, M. Sparta, M. Saitow, C. Riplinger, A. A. Auer and F. Neese, J. Chem. Theory Comput., 2016, 12, 4778 CrossRef CAS PubMed.
- A. Altun, F. Neese and G. Bistoni, J. Chem. Theory Comput., 2019, 15, 215 CrossRef CAS PubMed.
- A. Altun, M. Saitow, F. Neese and G. Bistoni, J. Chem. Theory Comput., 2019, 15, 1616–1632 CrossRef CAS PubMed.
- C. Riplinger and F. Neese, J. Chem. Phys., 2013, 138, 034106 CrossRef PubMed.
- C. Riplinger, B. Sandhoefer, A. Hansen and F. Neese, J. Chem. Phys., 2013, 139, 134103 CrossRef PubMed.
- C. Riplinger, P. Pinski, U. Becker, E. F. Valeev and F. Neese, J. Chem. Phys., 2016, 144, 024109 CrossRef PubMed.
- G. Bistoni, A. A. Auer and F. Neese, Chem. – Eur. J., 2017, 23, 865 CrossRef CAS PubMed.
- A. Altun, F. Neese and G. Bistoni, Beilstein J. Org. Chem., 2018, 14, 919 CrossRef CAS PubMed.
- F. Neese, M. Atanasov, G. Bistoni, D. Maganas and S. Ye, J. Am. Chem. Soc., 2019, 141, 2814–2824 CrossRef CAS PubMed.
- V. M. Schmiedel, Y. J. Hong, D. Lentz, D. J. Tantillo and M. Christmann, Angew. Chem., Int. Ed., 2018, 57, 2419 CrossRef CAS PubMed.
- C. L. Gross and G. S. Girolami, J. Am. Chem. Soc., 1998, 120, 6605 CrossRef CAS.
- D. D. Wick, K. A. Reynolds and W. D. Jones, J. Am. Chem. Soc., 1999, 121, 3974 CrossRef CAS.
- W. H. Bernskoetter, S. K. Hanson, S. K. Buzak, Z. Davis, P. S. White, R. Swartz, K. I. Goldberg and M. Brookhart, J. Am. Chem. Soc., 2009, 131, 8603 CrossRef CAS PubMed.
- T. A. Mobley, C. Schade and R. G. Bergman, J. Am. Chem. Soc., 1995, 117, 7822 CrossRef CAS.
- E. A. Cobar, R. Z. Khaliullin, R. G. Bergman and M. Head-Gordon, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 6963 CrossRef CAS PubMed.
- S. E. Bromberg, H. Yang, M. C. Asplund, T. Lian, B. K. McNamara, K. T. Kotz, J. S. Yeston, M. Wilkens, H. Frei, R. G. Bergman and C. B. Harris, Science, 1997, 278, 260 CrossRef CAS.
- M. Lersch and M. Tilset, Chem. Rev., 2005, 105, 2471 CrossRef CAS PubMed.
- B. G. Hashiguchi, S. M. Bischof, M. M. Konnick and R. A. Periana, Acc. Chem. Res., 2012, 45, 885 CrossRef CAS PubMed.
- W. D. Jones, Inorg. Chem., 2005, 44, 4475 CrossRef PubMed.
- Y. Qin, L. H. Zhu and S. Z. Luo, Chem. Rev., 2017, 117, 9433 CrossRef CAS PubMed.
- T. Brent Gunnoe, in Alkane C-H Activation by Single-Site Metal Catalysis, ed. P. Pedro, Springer, Dordrecht, Heidelberg, New York, London, 2012, pp. 1–15 Search PubMed.
- F. M. Chadwick, T. Kramer, T. Gutmann, N. H. Rees, A. L. Thompson, A. J. Edwards, G. Buntkowsky, S. A. Macgregor and A. S. Weller, J. Am. Chem. Soc., 2016, 138, 13369 CrossRef CAS PubMed.
- M. L. H. Green and G. Parkin, Struct. Bonding, 2017, 171, 79 CrossRef CAS.
- J. M. Buchanan, J. M. Stryker and R. G. Bergman, J. Am. Chem. Soc., 1986, 108, 1537 CrossRef CAS.
- W. Lamanna and M. Brookhart, J. Am. Chem. Soc., 1981, 103, 989 CrossRef CAS.
- M. Thenraj and A. G. Samuelson, J. Comput. Chem., 2015, 36, 1818 CrossRef CAS PubMed.
- D. Balcells, E. Clot and O. Eisenstein, Chem. Rev., 2010, 110, 749 CrossRef CAS PubMed.
- C. Hall and R. N. Perutz, Chem. Rev., 1996, 96, 3125 CrossRef CAS PubMed.
- A. J. Cowan, P. Portius, H. K. Kawanami, O. S. Jina, D. C. Grills, X. Z. Sun, J. McMaster and M. W. George, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 6933 CrossRef CAS PubMed.
- G. I. Childs, C. S. Colley, J. Dyer, D. C. Grills, X. Z. Sun, J. X. Yang and M. W. George, J. Chem. Soc., Dalton Trans., 2000, 1901–1906, 10.1039/b001987i.
- O. Torres, J. A. Calladine, S. B. Duckett, M. W. George and R. N. Perutz, Chem. Sci., 2015, 6, 418 RSC.
- F. P. A. Johnson, M. W. George, V. N. Bagratashvili, L. N. Vereshchagina and M. H. Palmer, Mendeleev Commun., 1991, 1, 26 CrossRef.
- X. Z. Sun, D. C. Grills, S. M. Nikiforov, M. Poliakoff and M. W. George, J. Am. Chem. Soc., 1997, 119, 7521 CrossRef CAS.
- H. M. Yau, A. I. McKay, H. Hesse, R. Xu, M. He, C. E. Holt and G. E. Ball, J. Am. Chem. Soc., 2016, 138, 281 CrossRef CAS.
- W. H. Bernskoetter, C. K. Schauer, K. I. Goldberg and M. Brookhart, Science, 2009, 326, 553 CrossRef CAS.
- M. D. Walter, P. S. White, C. K. Schauer and M. Brookhart, J. Am. Chem. Soc., 2013, 135, 15933 CrossRef CAS PubMed.
- S. D. Pike, A. L. Thompson, A. G. Algarra, D. C. Apperley, S. A. Macgregor and A. S. Weller, Science, 2012, 337, 1648 CrossRef CAS PubMed.
- F. M. Chadwick, N. H. Rees, A. S. Weller, T. Kramer, M. Iannuzzi and S. A. Macgregor, Angew. Chem., Int. Ed., 2016, 55, 3677 CrossRef CAS PubMed.
- S. D. Pike, F. M. Chadwick, N. H. Rees, M. P. Scott, A. S. Weller, T. Kramer and S. A. Macgregor, J. Am. Chem. Soc., 2015, 137, 820 CrossRef CAS PubMed.
- J. M. Tao, J. P. Perdew, V. N. Staroverov and G. E. Scuseria, Phys. Rev. Lett., 2003, 91, 146401 CrossRef.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef.
- A. D. Becke and E. R. Johnson, J. Chem. Phys., 2005, 123, 154101 CrossRef.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- F. Neese, F. Wennmohs, A. Hansen and U. Becker, Chem. Phys., 2009, 356, 98 CrossRef CAS.
- A. Wuttke and R. A. Mata, J. Comput. Chem., 2017, 38, 15 CrossRef CAS.
- M. P. Mitoraj, A. Michalak and T. Ziegler, J. Chem. Theory Comput., 2009, 5, 962 CrossRef CAS PubMed.
- W. Scherer and G. S. McGrady, Angew. Chem., Int. Ed., 2004, 43, 1782 CrossRef CAS PubMed.
- D. J. Lawes, S. Geftakis and G. E. Ball, J. Am. Chem. Soc., 2005, 127, 4134 CrossRef CAS.
- R. D. Young, D. J. Lawes, A. F. Hill and G. E. Ball, J. Am. Chem. Soc., 2012, 134, 8294 CrossRef CAS.
- A. Sivaramakrishna, P. Suman, E. V. Goud, S. Janardan, C. Sravani, T. Sandeep, K. Vijayakrishna and H. S. Clayton, J. Coord. Chem., 2013, 66, 2091 CrossRef CAS.
- J. A. Labinger and J. E. Bercaw, Nature, 2002, 417, 507 CrossRef CAS.
- M. C. Asplund, P. T. Snee, J. S. Yeston, M. J. Wilkens, C. K. Payne, H. Yang, K. T. Kotz, H. Frei, R. G. Bergman and C. B. Harris, J. Am. Chem. Soc., 2002, 124, 10605 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cp01309a |
| This journal is © the Owner Societies 2019 |