
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTracking amyloid oligomerization with monomer resolution using a 13-amino acid peptide with a backbone-fixed spin label†
E.
Zurlo
a,
I.
Gorroño Bikandi
a,
N. J.
Meeuwenoord
b,
D. V.
Filippov
 b and
M.
Huber
*a
b and
M.
Huber
*a
aDepartment of Physics, Huygens-Kamerlingh Onnes Laboratory, Leiden University, 2300 RA Leiden, The Netherlands. E-mail: huber@physics.leidenuniv.nl
bLeiden Institute of Chemistry, Gorlaeus Laboratoria, Leiden University, 2300 RA Leiden, The Netherlands
First published on 7th November 2019
Abstract
Amyloid oligomers are suspected as toxic agents in neurodegenerative disease, and are transient and often heterogeneous, making them difficult to detect. Here we show an approach to track the development of amyloid oligomers in situ by room temperature, continuous wave (cw) 9 and 95 GHz EPR. Three amyloid peptides with the 2,2,6,6-tetramethyl-N-oxyl-4-amino-4-carboxylic acid (TOAC) spin label were synthesized by solid phase peptide synthesis: T0EZ (TKVKVLGDVIEVGG) with TOAC (T) at the N-terminus, T5EZ with TOAC in the middle (KVKVTGDVIEVG) and T12EZ with TOAC at the C-terminus (KVKVLGDVIEVTG). These sequences are derived from the K11V (KVKVLGDVIEV) amyloid peptide, which self-aggregates to oligomers with a β-sheet configuration (A. Laganowsky, et al., Science, 2012, 335, 1228–1231). To monitor oligomerization, the rotational correlation time (τr) is measured by cw-EPR. For the backbone-fixed TOAC label that is devoid of local mobility τr should reflect the rotation and thereby the size of the peptide, resp. oligomer. For T5EZ a good match between the measured τr and the size of the peptide is obtained, showing the validity of the approach. One of the three peptides (T0EZ) aggregates (circular dichroism), whereas the other two do not. Since also the respective MTSL (S-(1-oxyl-2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrol-3-yl)methyl methanesulfonothioate) labelled peptides fail to aggregate, molecular crowding due to the label, rather than the helix-inducing properties of TOAC, seems to be responsible. Following in situ oligomer formation of T0EZ by the change in rotational correlation time, two oligomers are observed, a 5–6 mer and a 15–18 mer. The EPR approach, particularly 95 GHz EPR, enables following oligomerization of one monomer at a time, suggesting that the cw-EPR approach presented is a novel tool to follow amyloid oligomerization with high resolution.
Introduction
Amyloid aggregation is a central factor in amyloidogenic neurodegenerative diseases. Such diseases are widespread and so far most of them have not been cured. One obstacle is the gap in knowledge about the physical chemistry of amyloid aggregation. Amyloid aggregation is the process in which the amyloid peptide self-assembles. The thermodynamic end-point of the aggregation is the β-sheet fibril, the main component of the plaques in the brains of patients, so far the main diagnostic feature for neurodegenerative diseases.1,2 Meanwhile, evidence is growing that amyloid oligomers, not fibrils, are the more toxic species.3–8 Oligomers are aggregates of up to tens to hundred monomers. They can be on-pathway to fibrils, i.e. nuclei of fibrilization or off-pathway, meaning that they do not directly participate in fibril formation. Oligomers can differ in size, i.e. the number of peptides, structure, stability and physico-chemical properties. Their transient nature and heterogeneity make the oligomers difficult to track.1,5,9 Harmful oligomers are targets for drugs against neurodegenerative diseases, generating a pressing need to design methods to determine the oligomers’ mode of formation and structure. In particular, methods are needed to detect oligomers in situ, i.e. methods that show which oligomers are formed, their relative concentration and how these parameters develop over time.Here we explore the potential of liquid-solution, room temperature Electron Paramagnetic Resonance (EPR) at standard (9 GHz) and high (95 GHz) EPR frequencies to attain this goal.
These techniques make it possible to follow oligomer formation by detecting the size of the oligomers: for continuous-wave EPR (cw EPR) in liquid solution, the spectral lineshape is a sensitive indicator for the rotational correlation time (τr) of an object, and it covers the time range from 0.03 ns to several ns.11–15 As will be described below, this τr range matches well with the τr expected for the sizes of typical amyloid-forming oligomer peptides and their aggregates at room temperature in aqueous solution. Therefore, we expect to be able to track oligomer formation via the increase in τr.
To perform EPR on amyloid peptides, the peptides need to be spin labeled. Conventionally, spin labels, like the S-(1-oxyl-2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrol-3-yl)methyl methanesulfonothioate (MTSL) label, are introduced via spin-label site-directed mutagenesis, which results in structures like Fig. 1c.11,13–16 While this approach is universal, the local mobility is a disadvantage in the study of amyloid aggregation: rotation about the single bonds linking the nitroxide to the protein backbone dominates the motion and makes the measurement insensitive to peptide/oligomer size. We therefore use the backbone-fixed TOAC spin label (Fig. 1d), which is incorporated into the peptide during solid phase peptide synthesis (SPPS).17–26 In these constructs, the nitroxide, containing the unpaired electron, is directly linked to the protein backbone, and therefore its τr will follow the peptide rotation much more closely than the nitroxide in MTSL.
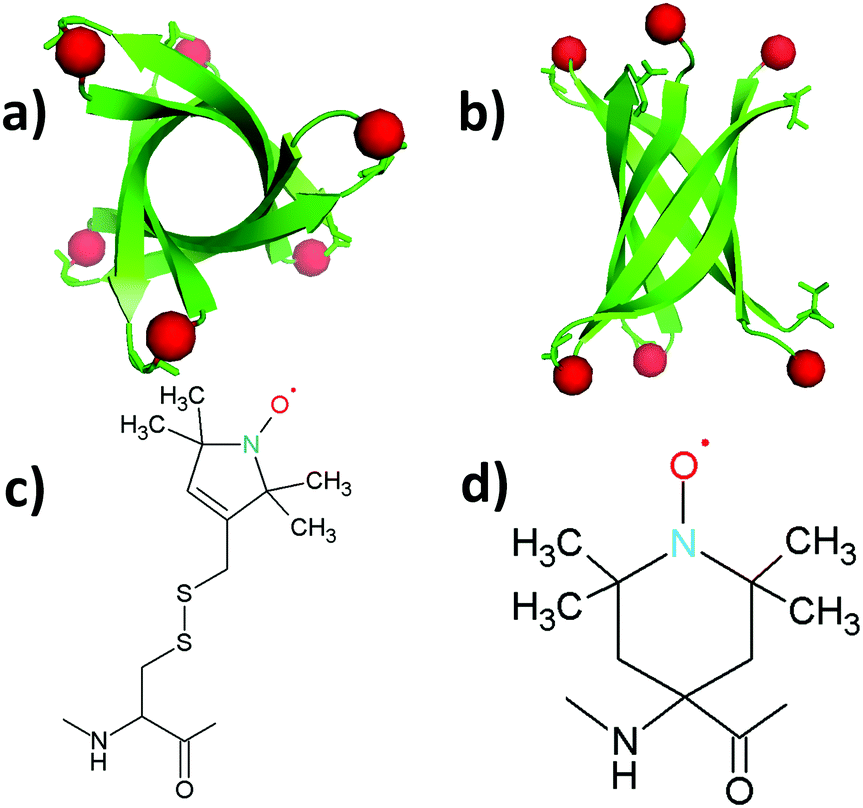 | ||
| Fig. 1 Structure of the K11V oligomer and relevant spin labels. (a and b) Oligomer structure of K11V (PDB 3SGO),10 showing the 2,2,6,6-tetramethyl-N-oxyl-4-amino-4-carboxylic acid (TOAC) location for T0EZ (red dots). For details, see the text. View in (b) is rotated 90° with respect to (a). (c) Molecular structure of the MTSL spin label attached to a cysteine side-chain. (d) Molecular structure of the TOAC spin label. | ||
As a model system we use the K11V peptide (Fig. 1a and b), a short peptide, the sequence of which is derived from the αB crystalline protein.10 Such peptides, comprising 5–15 amino acids, have proven extremely useful for mechanistic studies. They show the essential properties of the protein they are derived from, but their smaller size is helpful to model their aggregation by MD methods, and, pioneered by the Eisenberg group, enable investigation by X-ray crystallography.27–29 The K11V peptide was shown to form toxic amyloid oligomers that could be crystallized. The X-ray structure shows a hexamer in the β-sheet conformation, a β-barrel10 (Fig. 1a and b). Apparently, K11V forms fibrils much more slowly than other amyloid peptides, making it ideal to study oligomerization.
We synthesized four variants of the K11V peptide, a wild type analog (EZ) and three variants with the TOAC spin label in different positions (Table 1). We show that in spite of the α-helix inducing property of TOAC17–19,26,30–32 one of the three TOAC constructs aggregates to oligomers. We demonstrate that our approach enables aggregation to be followed in time with, in principle, monomer-size resolution.
| Sequence | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| K11V | K | V | K | V | L | G | D | V | I | E | V | |||
| T0EZ | T | K | V | K | V | L | G | D | V | I | E | V | G | |
| T5EZ | K | V | K | V | T | G | D | V | I | E | V | G | ||
| T12EZ | K | V | K | V | L | G | D | V | I | E | V | T | G | |
| EZ | K | V | K | V | L | G | D | V | I | E | V | G | G | |
Materials and methods
Synthesis and characterization of peptides
All chemicals were commercial products of the best quality available and, unless otherwise indicated, they were used without any further purification. The EZ peptide was purchased from tebu-bio (Heerhugowaard, The Netherlands).9-Fluorenylmethoxycarbonyl (Fmoc)-amino acids, Fmoc-Gly-Wang Tentagel resin and the other chemicals used for the solid phase peptide synthesis were purchased from Sigma Aldrich. 2,2,6,6-Tetramethylpiperidine-N-oxyl-4-(9-fluorenylmethyloxycarbonyl-amino)-4-carboxylic acid (Fmoc-TOAC-OH) and H-Gly-Wang resin were supplied by Iris Biotech (Marktredwitz, Germany).
The peptide sequences were assembled on an AB433A Peptide Synthesizer (Applied Biosystems, Foster City, CA, USA), using 0.05 mmol of Gly-Wang resin (substitution 0.5 mmol g−1). For all amino acids except TOAC we use 5 equivalents (0.25 mmol) of each AA for the synthesis. Deprotection by Fmoc was done 4 times (3 minutes each) by adding 2.5 mL of a solution of 20% piperidine (PIP) in N-methylpyrrolidone (NMP). Couplings were performed using 2-(6-chloro-1H-benzotriazole-1-yl)-1,1,3,3-tetramethylaminium hexafluorophosphate (HCTU) as an activator and N,N-diisopropylethylamine (DIPEA) as base. We used 1 equivalent of HCTU and 2 equivalents of DIPEA for 1 equivalent of AA. 1 mL of a solution of HCTU (0.25 M in NMP), 0.5 mL of a solution of DIPEA (1 M in NMP) and 0.5 mL of NMP were added to the resin for the coupling reactions. Each coupling reaction lasted for 2 h.
The TOAC spin label was treated differently. Only 2 equivalents of AA were added for 1 equivalent of resin. Instead of HCTU, 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate (HATU) was used as an activator and 400 μL of its solution (0.5 M in NMP) were put directly inside the cartridge with the spin label together with 600 μL of NMP. The coupling in this case lasted for 4 h. For the AA introduced immediately after the TOAC we used a double coupling, keeping the same conditions as for the rest of the sequence. The Fmoc absorption at 301 nm was followed to check the status of the synthesis after each coupling step.
At the end of the synthesis the resin was dried by washing with dichloromethane (DCM). To cleave the peptide from the resin 2 mL of a 95% trifluoroacetic acid (TFA) + 5% water solution was used. A small amount of peptide was cleaved from the resin and characterized by LC-MS. Unless otherwise indicated, the peptides were purified by semi-preparative HPLC.
Analytical HPLC separation was carried out on a LCQ Advantage Thermo Finnigan LC-MS system with UV-vis and Ion-trap mass detectors. The column used was a C-18 Gemini (4.6 × 50 mm, 3 μm particle size) from Phenomenex (Torrance, California). The mobile phases A (H2O), B (acetonitrile, MeCN) and C (aqueous 1% TFA) were used for preparing ternary gradients. Elution conditions: A 80% B 10% C 10–90%, linear gradient B 18–33% in 10 min. Flow rate 1 mL min−1.
Crude peptide purifications were performed on a Gilson HPLC preparative system with a semipreparative Gemini C18 column (10 × 250 mm) from Phenomenex with a UV-vis detector. The mobile phases A (H2O) and B (acetonitrile, MeCN) were used for preparing binary gradients. Elution conditions: A 82% B 18%, linear gradient B 18–33% in 10 min, flow rate 5 mL min−1.
The lyophilization was done on a Christ Alpha 2-4 LO lyophilizer (Salm&Kipp, Breukelen, Netherlands) with a Christ RVC 2-25 rotor. All TOAC peptides had a high level of purity as shown by single band elution and mass spectrometry.
T5EZ: yield 2.0%; LC-MS (C18) tR 4.56 min; purity >95%; mass: calculated for C61H108N15O18 [M + H]+ 1339.61, found: 1339.47.
T0EZ: yield 10.8%; LC-MS (C18) tR 4.68 min; purity >95%; mass: calculated for C69H122N17O20 [M + H]+ 1453.79, found: 1453.53.
T12EZ: yield 1.4%; LC-MS (C18) tR 4.85 min; purity >95%; mass: calculated for C69H122N17O20 [M + H]+ 1453.79, found: 1453.60.
Protocol for the aggregation experiments
Samples were prepared as follows: the powder of the lyophilized spin-labelled EZ peptides was dissolved in Milli-Q water, in order to get a peptide concentration of 500 μM by weight. Aggregation experiments were carried out for one week. After an initial measurement taken at the time when the spin-labelled-peptide powder was diluted in Milli-Q water (t = 0), samples with a total volume of 560 μL in 1.5 mL Eppendorf tubes were aggregated on a thermomixer (Eppendorf, Thermomixer comfort, Waltham, MA, USA) with a speed of 1000 rpm at 293 K. At each time point a 20 μL sample was drawn for an EPR measurement, one of 40 μL for CD and 10 μL were frozen for future experiments, e.g. 95 GHz EPR and ThioT fluorescence. The time points were: one hour, four hours, one day (24 h), two days (48 h), three days (72 h) and seven days (168 h).Thioflavin T fluorescence
The samples were monitored by the standard thioflavin T (ThioT) fluorescence assay.33 In summary 5 μL of sample solution were dissolved in 2 mL of a solution of 5 μM of ThioT and mixed for 30 seconds. The sample was excited at 457 nm and the fluorescence was observed in the range of 475 nm to 600 nm (Varian Cary Eclipse, San Jose, CA, USA). The fluorescence increase was measured with respect to the ThioT blank without the peptide.EPR measurement conditions
The 9 GHz, continuous-wave EPR spectra were recorded using an ELEXSYS E680 spectrometer (Bruker, Rheinstetten, Germany). The measurements were done under the following conditions: room temperature, a microwave power of 0.63 mW and a modulation amplitude of 0.15 mT at a modulation frequency of 100 kHz. The time expended on each measurement was adapted according to the spectral lineshape, i.e., the aggregation time. For the starting point of the aggregation 30 min were expended, and up to 5 h for samples at the end of the aggregation series. Glass micropipettes of a volume of 50 μL (Blaubrand Intramark, Wertheim, Germany) were filled with 20 μL of the sample for each measurement. The spin concentration was determined by comparing the double integral of the EPR spectra with the double integral of a reference sample (MTSL, 100 μM). The spin concentrations were ≈100 μM for a total concentration of the peptide of 500 μM.The 95 GHz EPR spectra were recorded at room temperature on a Bruker ELEXSYS E680 spectrometer using a home-built probehead with a single-mode cavity specially designed for cw measurements. Acquisition parameters: microwave frequency 94.04 GHz, microwave power 0.63 μW, modulation amplitude 1 mT, and modulation frequency 6 kHz. Total measurement time: approximately 5 hours.
Simulations of EPR spectra
MATLAB (version 9.4.0.813654, R2018a, The MathWorks, Inc., Natick, MA, USA) and the EasySpin package (5.2.4) were used for simulations of the EPR spectra.34 The parameters of the simulations were manually adjusted to agree best with the experimental spectra. For all simulations, an isotropic rotation of the nitroxide (S = 1/2) was utilized. The following g-tensor values were used: g = [2.0086 2.0059 2.0020]. These values were obtained from the simulation of the 275 GHz EPR spectrum of a frozen solution (100 K) of the peptide, using the “Pepper” algorithm in EasySpin, and we used these values for all other simulations. The principal values of the 14N hyperfine coupling tensor were Axx = Ayy = 13 MHz and Azz = 110 MHz.35 The spectra were simulated with a superposition of three components: a fast fraction using the “Garlic” function, a medium and a slow fraction using the “Chili” function. For the 9 GHz spectra, a Gaussian component with a linewidth of 0.12 mT was used for the fast component, and 1 mT for the medium and slow components. For the 95 GHz spectra, a Gaussian line with a width of 0.5 mT was used for the fast component, whereas for the medium and slow components a mix of Gaussian and Lorentzian lineshape with a width of 0.1 mT was applied. The τr of the fast component was chosen by simulating the narrow lines of the t = 0 measurements and then kept constant for all other simulations. Optimal τr values of the medium and slow components were derived from later time-point spectra and then kept constant for the entire series. For each time point, the relative contribution of the three components was optimized considering both 9 and 95 GHz spectra, for more details see the ESI.†Interpretation of τr values and molecular volumes
We used the Stokes–Einstein equation to interpret the τr values. This implies a spherical approximation for the particles: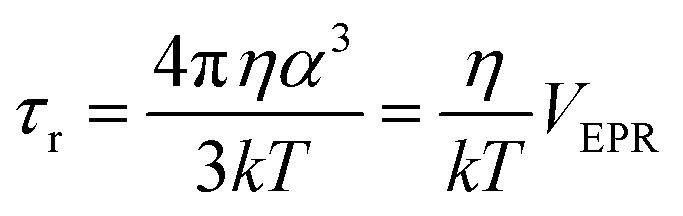 | (1) |
The experimentally determined values of the volume (VEPR) have to be compared to the volumes of the peptide and its oligomers. There are two approaches to derive such estimates:
Method (i) uses the molecular weight (MW) and derives the volume assuming a certain density of the proteins using eqn (2), and we refer the volume obtained as VMW. The second approach (ii) uses the dimension of the K11V oligomer (cylindrin) to derive the to-be-expected volume of the peptide. In the following we describe these two approaches.
Method (i):
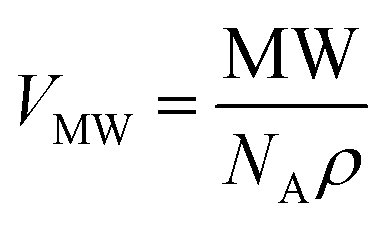 | (2) |
| MWapp,i = VEPRiNAρ | (3a) |
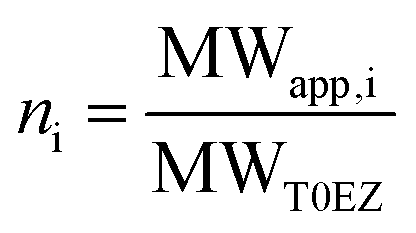 | (3b) |
The density of proteins of MW > 20 kDa is generally assumed to be ρ = 1.35 g cm−3,36 however also other values were reported for smaller proteins, such as 1.50 g cm−3.37 In the ESI,† the influence of different ρ values is discussed and the resulting spread in ni is included in the error estimation. The molecular weight of a monomer of T0EZ and of T12EZ is 1452.60 g mol−1 and that for T5EZ is 1338.47 g mol−1.
Method (ii) uses the dimension of the oligomer of the K11V peptide that had been determined by X-ray crystallography.10
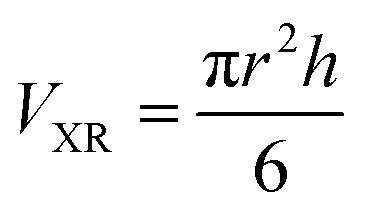 | (4) |
Results
To determine the aggregation of the different TOAC constructs, Circular Dichroism (CD) spectroscopy was performed for samples taken at different time points of the aggregation. Initially all constructs display random coil CD spectra and T0EZ shows increasing β-sheet character over time, whereas T5EZ and T12EZ remain unchanged. More details of CD spectra on T0EZ are given in the ESI,† including the analysis using BeStSel.38,39 To test for the formation of fibrils, ThioT fluorescence33 was measured. None of the samples showed ThioT activity. This is to be expected as ThioT fluorescence requires linear cross-beta sheet structures of minimally six peptides in a row,40 a requirement that the highly curved β-sheet in the β-barrel oligomer (Fig. 1) does not fulfill. Therefore, the absence of ThioT fluorescence shows only that no fibrils are formed, however it does not exclude oligomers. For T0EZ these findings are consistent with a β-sheet oligomer: it displays a β-sheet structure in CD and does not show ThioT fluorescence, which excludes that β-sheet fibrils are formed. In the following, we first describe the EPR properties and the aggregation behavior of T0EZ and then the properties of the other two constructs, T5EZ and T12EZ.Fig. 2 shows the EPR spectra of T0EZ at 9 and 95 GHz obtained at two time points of aggregation; at the start (Fig. 2a and b) and after 48 hours of aggregation (Fig. 2c and d), the spectra of all time points are collected in Fig. S1 (ESI†). The 9 GHz EPR spectra are dominated by three narrow lines (see the inset) of a nitroxide in fast rotation. The arrow marks an additional broad component, representing a component with lower mobility. In the 95 GHz EPR spectra (Fig. 2b and d), taken at around ten times higher field, this component is better separated from the three-line pattern than at 9 GHz EPR. The spectra at the two time points differ mostly in the relative amplitude of the broad component with respect to the narrow component. The broad component has a higher amplitude in Fig. 2c and d than that in Fig. 2a and b. To quantitate the changes in the EPR spectra, we performed spectral simulations.
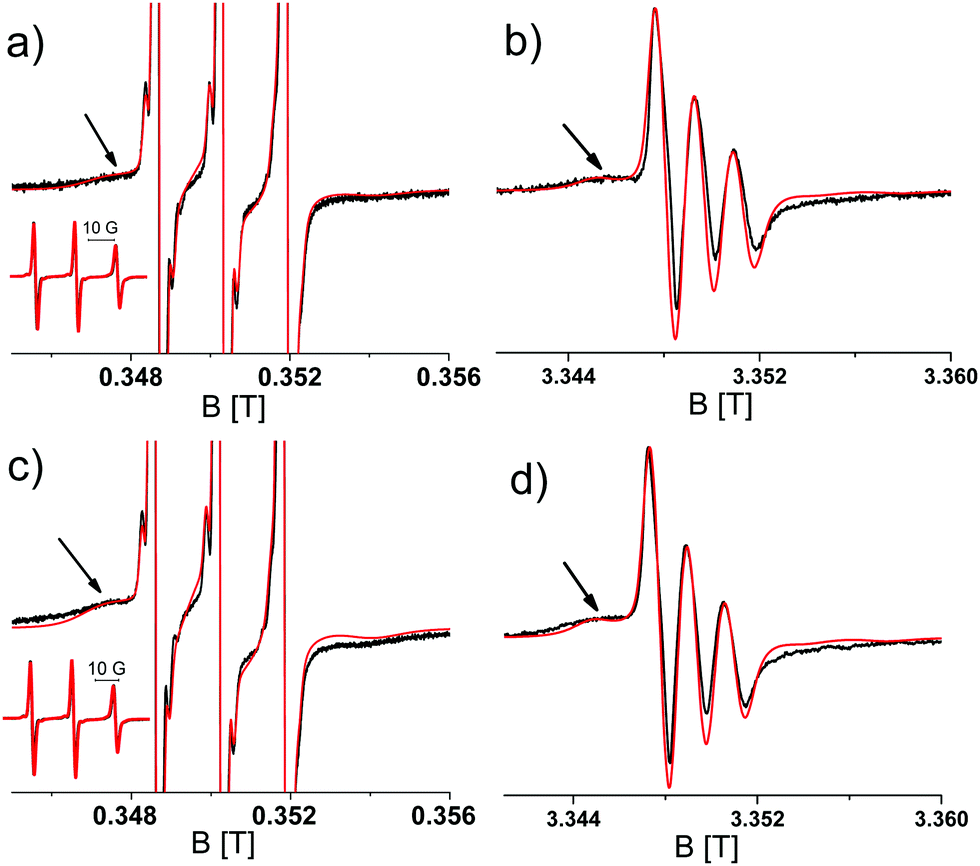 | ||
| Fig. 2 Room temperature 9 and 95 GHz EPR spectra of T0EZ at different time points of aggregation. (a and c) 9 GHz EPR spectra. Full spectra: inset. Zoomed-in spectra: amplitude expanded ten-fold with respect to the inset. (b and d) 95 GHz spectra. Spectra (a and b): start of aggregation (t = 0); (c) and (d) at 48 hours of aggregation. Black: experimental spectra. Red: simulated spectra. Note that at 95 GHz the signal appears at around ten times higher field B0. | ||
Simulations required minimally three components with different rotational correlation times (τr). Fig. 3 shows the shape of these components for the spectra shown in Fig. 2c and d, i.e. at 48 hours of aggregation. The contribution with the longest τr (light blue) gives rise to broad features marked in Fig. 2 by the arrows. The τr values are given in Table 2. To simulate the entire series of time points, the τr of each component was kept constant and only the relative amount of the components was varied throughout the time series. Furthermore, the 9 and 95 GHz EPR spectra for the same time point were simulated with the same relative amounts of the components. The higher resolution of 95 GHz EPR makes it easier to detect the slow components (compare Fig. 2b and d (95 GHz) with Fig. 2a and c (9 GHz)). The slow and medium components are related to aggregates and the amount which they contribute to the spectra in time increases as shown in Fig. 4.
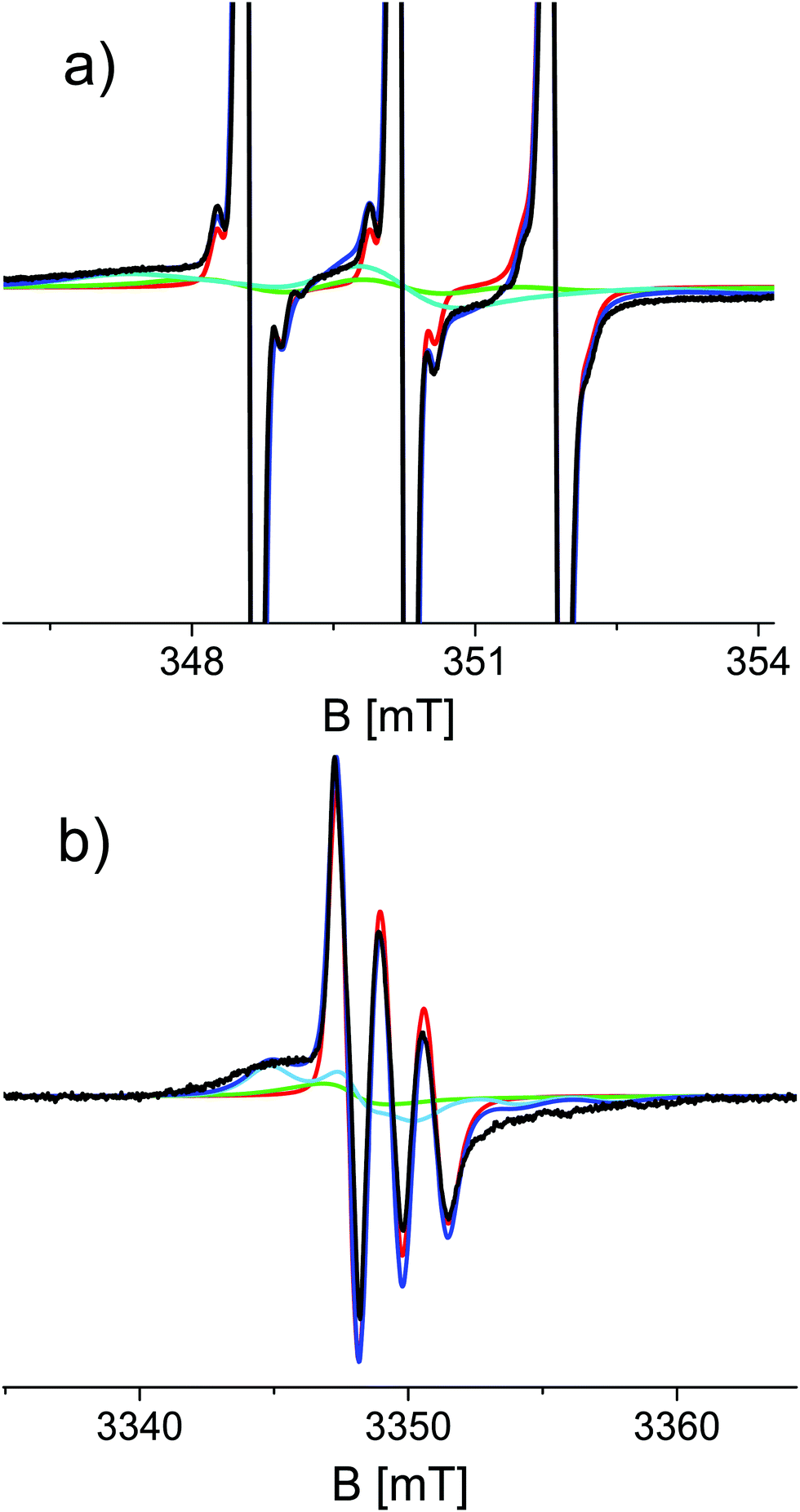 | ||
| Fig. 3 Spectral components used in the simulation of the 9 and 95 GHz EPR spectra of T0EZ (48 hours of aggregation). Experimental spectra (black), fast component (red), medium component (green), and slow component (light blue). Total simulation (dark blue). For details see the text. | ||
| Components | τ r [ns] | V EPR [nm3] | Number of monomers in aggregate | |
|---|---|---|---|---|
| From MWb | From XRc | |||
| a From Stokes–Einstein (eqn (1)). b From protein density (eqn (2) and (3)) using ρ = 1.35 g cm−3, for further uncertainties in these numbers, see the text. c From X-ray crystallography (eqn (4)), for further uncertainties in these numbers, see the text. | ||||
| Fast | 0.16 ± 0.004 | 0.65 ± 0.02 | — | — |
| Medium | 2.00 ± 0.20 | 8.08 ± 0.81 | 5 ± 1 | 6 ± 1 |
| Slow | 6.31 ± 0.70 | 25.49 ± 2.83 | 15 ± 2 | 18 ± 2 |
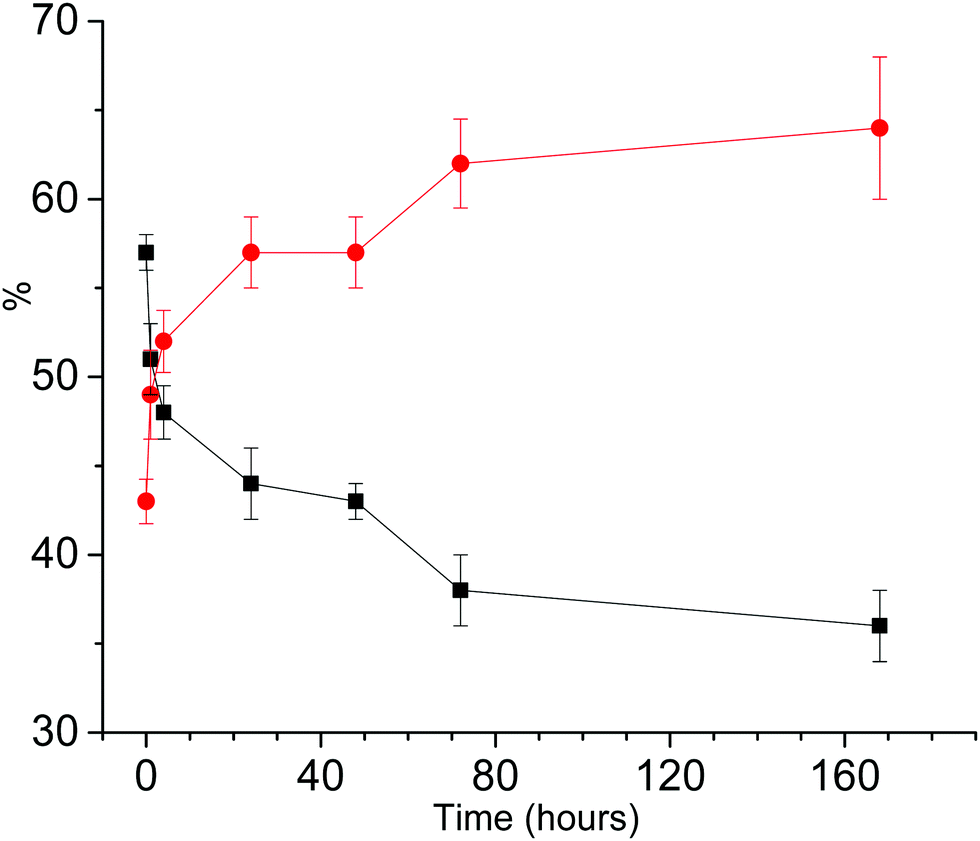 | ||
| Fig. 4 Aggregation of T0EZ as a function of time. Amount of aggregates (red, slow and medium EPR components combined); amount of monomers (black, fast EPR component). The lines are a guide to the eye. | ||
Using the Stokes–Einstein equation (eqn (1)) the τr values can be related to the volumes of the observed species (Table 2). These volumes suggest that the medium fraction is a pentamer or hexamer, and the slow fraction a 15–18 mer.
Fig. 4 shows that the amount of combined slow and medium components increases for the first 70 hours of aggregation, afterwards it stays constant within the error margins, indicating that a steady state is reached. Also at the earliest time points some aggregates are present.
Finally, in Table 3, the τr values of all TOAC-EZ constructs in their monomeric state are given. The peptides T0EZ and T12EZ have the same composition, and T5EZ has one amino-acid residue less. Thus from the volume of the peptide one would expect that T0EZ and T12EZ have longer τr values than T5EZ, however the opposite is true. This suggests that the TOAC in T0EZ and T12EZ has residual freedom to move making the observed τr shorter than expected from the molecular weight.
| Peptides | τ R monomers [ns] | V EPR [nm3] | V MW [nm3] |
|---|---|---|---|
| T0EZ | 0.16 ± 0.004 | 0.65 ± 0.02 | 1.78 ± 0.17 |
| T5EZ | 0.40 ± 0.02 | 1.61 ± 0.07 | 1.65 ± 0.15 |
| T12EZ | 0.31 ± 0.02 | 1.25 ± 0.06 | 1.78 ± 0.17 |
Discussion
In this study we use cw, room temperature EPR at 9 and 95 GHz to determine the size-development of amyloid oligomers in time. The size of the aggregates can be tracked in situ, in the aggregation solution. The method is demonstrated with an amyloid peptide based on K11V10 into which we incorporated the backbone-fixed spin label TOAC.Is TOAC a good monitor for peptide size?
Even though TOAC is directly linked to the peptide backbone (Fig. 1d), local backbone motion could still uncouple it partly from the peptide-oligomer-overall rotation. To assess this factor we consider the τr values for the monomeric peptides (Table 3). The τr values are related to the volume of the object via the Stokes–Einstein relation (eqn (1)) resulting in VEPR. This volume is compared to the molecular volume of the peptides obtained from the molecular weight (eqn (2)), VMW. In Table 3 these volumes are given. For T5EZ, VEPR is close to the VMW, showing that for this peptide, τr is a good measure for the peptide size. The peptides T0EZ and T12EZ have smaller VEPR values than T5EZ, although their molecular volumes are larger. This shows that the motion of the spin label at the N- and C-terminus is partially uncoupled from that of the peptide, in other words, the backbone section to which the TOAC is attached displays local mobility. Comparing all three peptides, the mobility is higher at the N-terminus than at the C-terminus (T0EZ has a shorter τr than T12EZ), and the smallest in the center (T5EZ). The case of T5EZ shows that the concept of using TOAC to monitor the molecular volume works well, at least in a situation where the TOAC is well embedded in the object. The τr values of all TOAC constructs are longer than those of the MTSL analogues, see the ESI,† Table S2, as expected, given that in MTSL the nitroxide is not rigidly linked to the protein backbone.Influence of the TOAC position on the aggregation of the EZ peptides
Two of the three TOAC constructs (T5EZ and T12EZ) do not aggregate. For T5EZ, with the TOAC in the middle of the β-sheet region, two factors combine to inhibit aggregation: crowding in the middle of the β-barrel (see Fig. S3, ESI†) and the α-helix-inducing properties of TOAC, which could inhibit the formation of the β-sheet and thereby of the oligomer. To test whether the α-helix-inducing character of TOAC is the dominant factor inhibiting aggregation, we also tested the MTSL-labelled counterparts (see the ESI†). Also, the equivalent constructs to T5EZ and T12EZ with an MTSL label failed to aggregate. As MTSL labels do not break β-sheets, it seems evident that crowding is more important than β-sheet breaking. More puzzling is the question of why the TOAC at the C-terminus inhibits aggregation, whereas at the N-terminus it does not. Here the difference in the τr values of T0EZ and T12EZ could give a hint. Apparently, the N-terminus has a higher local mobility than the C-terminus, and that, in turn, may enable TOAC at the N-terminus to avoid molecular crowding in the oligomer, enabling T0EZ to aggregate to oligomers.Oligomerization of T0EZ as followed by EPR
Having established that TOAC enables tracking the molecular size, i.e. the number of peptides per oligomer, we use it to analyse the aggregation of T0EZ. The size and the amount of the oligomers increase with time (EPR) (Fig. 4 and Fig. S2, ESI†), the aggregates have a β-sheet structure (CD) and do not convert to fibrils on the time scale of the experiment (ThioT). With respect to the size of oligomers, an initial fraction of smaller oligomers (medium fraction by EPR), some of them present even at the earliest time point taken and a later fraction with larger aggregates (slow fraction) are observed (Fig. S2, ESI†). These are two distinct populations, as two spectral components, medium and slow, are needed to represent them. The number of monomers in the two fractions, penta-to-hexamers and 15–18 mers (see Table 2), has a large uncertainty as several approximations enter in the estimation. In the ESI† these are described and evaluated quantitatively. The impact of some approximations can be determined quantitatively: the two different ways to estimate monomer volumes (Table 2) show that for the smaller oligomers (medium EPR fraction) differences of one monomer unit and for the larger oligomers (slow EPR fraction) differences of three monomer units result. Other factors can be qualitatively assessed: the local mobility of the TOAC in the oligomer leads to an underestimation of the size of the oligomer, thus the oligomer size determined by EPR is a lower limit (for details, see the ESI†). Overall, due to the approximations described in the ESI,† the relative oligomer sizes are more reliable than the absolute values. To determine the absolute number of monomers in the oligomers, dedicated experiments are possible (see below).The oligomers are most likely heterogeneous, i.e. the two oligomer fractions do not consist exclusively of oligomers of one particular size. A homogeneous population would have a distinctive lineshape as shown in Fig. S6 (ESI†), and it is likely that also the superposition of two homogenous populations would still give spectra that are better resolved than those experimentally observed. More details could be obtained by increasing the time resolution, i.e. by measuring more time points. Such experiments could also reveal whether the oligomers grow by adding monomers or whether smaller oligomers assemble into larger ones, however such experiments are beyond the scope of the present study.
Additional EPR experiments could determine the number of monomers in specific oligomers,41,42 as an independent check point, however, these methods rely on frozen solutions and thereby lack the power of the in situ measurement we present here.
Aggregation of T0EZ
The investigation presented gives clear evidence that the aggregation of T0EZ proceeds in several steps. It reveals an initial fraction, the EPR medium fraction, which, considering the approximations described in the ESI,† may be an hexamer similar to that observed by Laganowsky et al.10 The β-barrel structure is confirmed by CD, its hexameric nature fits well with the rotation correlation time derived from EPR. The exact shape, e.g. the antiparallel arrangement of the peptides (Fig. 1), would have to be confirmed by more extensive EPR investigations, see below. While these oligomers are the end point of aggregation under the conditions employed by A. Laganowsky et al.,10 the present study shows further growth of the aggregates over time, to larger oligomers, as proven by the size observed by EPR (15–18 peptides). The absence of ThioT activity rules out the fact that these latter objects are fibrils, in particular, it shows that the oligomers do not have the non-curved β-sheets required for ThioT binding. Laganowsky et al. show that K11V forms fibrils when exposed to vigorous shaking at 50 °C over a period of 7 days. The lower temperature of the present experiments was chosen to enhance oligomers; therefore, in the present study, high temperature and vigorous shaking were avoided. Our finding of two different types or groups of oligomers that differ in size suggests that the respective oligomers can have different physiological/disease effects and that their properties must be studied individually for a full understanding of their relevance.Also other studies have shown that oligomers can increase in size over time.43,44 In particular, ion-mobility mass spectrometry (IM-MS) showed that other short amyloid peptides, when investigated under conditions that are not optimized for crystallization, also go through a series of oligomers, the size of which increases over time.45–48
Overall we observe the aggregation of T0EZ into oligomers with a β-sheet structure. The absence of ThioT fluorescence confirms that, at least within the time scale investigated (12 days), no fibrils are formed. Therefore, these oligomers are unlikely to be seeds for fibrilization and should be considered off-pathway oligomers. More details could be determined by kinetic analysis as pioneered by the Knowles group.44 The approach of the present study is of course not limited to this particular peptide: other short peptides, especially those that aggregate to fibrils,27–29 could be investigated. In these cases, besides the end-product, the fibrils that should be ThioT active, on- and off-pathway oligomers are expected. Comparing the properties of these oligomers to those of the EZ oligomers would give further insight into the aggregation process that from the physical-chemistry point of view is still far from being understood.
Conclusions and outlook
Here we show that peptides with TOAC at strategic positions combined with high-field EPR open a new way to study amyloid aggregation: the real-time measurement of rotational motion reveals directly the size of the oligomer at specific time points. While, in the model system we investigate, only two chief fractions are observed that differ by approximately 12 monomers, the approach itself can follow the development with single-monomer resolution in the otherwise difficult to differentiate43 oligomer size distribution from two to 15 monomers, see the ESI.† Our approach is therefore one of the few methods presently available to track oligomers as they develop. In contrast to IM-MS, a method that provides the molecular mass and information about the outer size of the oligomers after gas-phase ionization, the high-field EPR approach presented here determines the oligomers in solution, avoiding chemical separation, vacuum-desorption and gas-phase, ionization steps. It therefore perfectly complements the existing methods and is an excellent new tool in the quest for molecular information on amyloid aggregation. We show here that the approach can be applied to one particular peptide, however, the principle shown is universal for other peptides of interest, as long as they are amendable to SPPS to incorporate the TOAC, either for the full peptide or a fragment that can be fused to a protein that provides the full-length sequence of interest. We also demonstrate that in spite of the adverse properties of TOAC, aggregating peptides can be generated. Of course, the presence of TOAC and its eventual effects on the aggregation should be tightly monitored. These disadvantages clearly outweigh the merits of the TOAC approach, as can be seen in the poor size differentiation obtained from the conventionally labelled peptides in the ESI.†Finally, once the intermediates are identified and their size development over time is established by the approach presented here, the structure of each of the identified states can be investigated: by distance determination using EPR via spin–spin interaction49 and other techniques, inter- and intramolecular interactions can be determined, yielding structural constraints for modelling. Small angle X-ray scattering (SAXS) can identify the overall shape, and CD and FTIR can yield the internal structure.
Furthermore, for many systems the present details would already yield new insights: on- and off-pathway oligomers could be tracked in their time development.2,50 These experiments would identify states that warrant more detailed investigation such as determining their individual mode of toxicity and applying further structural methods.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr Peter Gast and Dr Pravin Kumar for their contribution to data analysis and measurements. The contribution of Dr József Kardos to the CD analysis is gratefully acknowledged. We thank Dr Federica Galli for exploratory AFM experiments. This work was funded by Netherlands Organization for Scientific Research (NWO) (Grant No. 711.014.003) and FOM foundation grant 10SMPA04.References
- F. Chiti and C. M. Dobson, Annu. Rev. Biochem., 2006, 75, 333–366 CrossRef CAS PubMed.
- T. C. T. Michaels, A. Šarić, J. Habchi, S. Chia, G. Meisl, M. Vendruscolo, C. M. Dobson and T. P. J. Knowles, Annu. Rev. Phys. Chem., 2018, 69, 273–298 CrossRef CAS PubMed.
- W.-F. Xue, A. L. Hellewell, W. S. Gosal, S. W. Homans, E. W. Hewitt and S. E. Radford, J. Biol. Chem., 2009, 284, 34272–34282 CrossRef CAS PubMed.
- R. Kodali and R. Wetzel, Curr. Opin. Struct. Biol., 2007, 17, 48–57 CrossRef CAS PubMed.
- B. Caughey and P. T. Lansbury Jr, Annu. Rev. Neurosci., 2003, 26, 267–298 CrossRef CAS PubMed.
- M. D. Kirkitadze, G. Bitan and D. B. Teplow, J. Neurosci. Res., 2002, 69, 567–577 CrossRef CAS PubMed.
- C. Haass and D. J. Selkoe, Nat. Rev. Mol. Cell Biol., 2007, 8, 101–112 CrossRef CAS PubMed.
- F. van Diggelen, A. W. J. W. Tepper, M. M. Apetri and D. E. Otzen, Isr. J. Chem., 2017, 57, 699–723 CrossRef CAS.
- F. Chiti and C. M. Dobson, Nat. Chem. Biol., 2009, 5, 15–22 CrossRef CAS PubMed.
- A. Laganowsky, C. Liu, M. R. Sawaya, J. P. Whitelegge, J. Park, M. Zhao, A. Pensalfini, A. B. Soriaga, M. Landau and P. K. Teng, et al. , Science, 2012, 335, 1228–1231 CrossRef CAS PubMed.
- W. L. Hubbell and C. Altenbach, Curr. Opin. Struct. Biol., 1994, 4, 566–573 CrossRef CAS.
- E. Bordignon and H.-J. Steinhoff, Membrane Protein Structure and Dynamics Studied by Site-Directed Spin-Labeling ESR, in ESR Spectroscopy in Membrane Biophysics, ed. M. A. Hemminga, L. J. Berliner, Springer US, Boston, MA, 2007, pp. 129–164 Search PubMed.
- H. S. Mchaourab, M. A. Lietzow, K. Hideg and W. L. Hubbell, Biochemistry, 1996, 35, 7692–7704 CrossRef CAS PubMed.
- W. L. Hubbell, D. S. Cafiso and C. Altenbach, Nat. Struct. Biol., 2000, 7, 735–739 CrossRef CAS PubMed.
- W. L. Hubbell, H. S. Mchaourab, C. Altenbach and M. A. Lietzow, Structure, 1996, 4, 779–783 CrossRef CAS PubMed.
- P. G. Fajer, J. Phys.: Condens. Matter, 2005, 17, S1459–S1469 CrossRef CAS.
- J. C. McNulty, J. L. Silapie, M. Carnevali, C. T. Farrar, R. G. Griffin, F. Formaggio, M. Crisma, C. Toniolo and G. L. Millhauser, Biopolymers, 2000, 55, 479–485 CrossRef CAS PubMed.
- A. D. Milov, Y. D. Tsvetkov, F. Formaggio, M. Crisma, C. Toniolo and J. Raap, J. Am. Chem. Soc., 2001, 123, 3784–3789 CrossRef CAS PubMed.
- M. Di Valentin, M. Albertini, E. Zurlo, M. Gobbo and D. Carbonera, J. Am. Chem. Soc., 2014, 136, 6582–6585 CrossRef CAS PubMed.
- J. Petrlova, H.-S. Hong, D. A. Bricarello, G. Harishchandra, G. A. Lorigan, L.-W. Jin and J. C. Voss, Proteins, 2011, 79, 402–416 CrossRef CAS PubMed.
- R. Bartucci, R. Guzzi, M. De Zotti, C. Toniolo, L. Sportelli and D. Marshz, Biophys. J., 2008, 94, 2698–2705 CrossRef CAS PubMed.
- P. Hanson, D. J. O. E. Anderson, G. Martinez, G. Millhauser, F. Formaggio, M. Crisma, C. Toniolo and C. Vita, Mol. Phys., 1998, 95, 957–966 CAS.
- S. Schreier, S. R. Barbosa, F. Casallanovo, R. D. F. F. Vieira, E. M. Cilli, A. C. M. Paiva and C. R. Nakaie, Biopolymers, 2004, 74, 389–402 CrossRef CAS PubMed.
- S. Schreier, J. C. Bozelli, N. Marín, R. F. F. Vieira and C. R. Nakaie, Biophys. Rev., 2012, 4, 45–66 CrossRef CAS PubMed.
- R. Latajka, M. Jewginski, M. Makowski, M. Pawełczak, T. Huber, N. Sewald and P. Kafarski, J. Pept. Sci., 2008, 14, 1084–1095 CrossRef CAS PubMed.
- V. Monaco, F. Formaggio, M. Crisma, C. Toniolo, P. Hanson and G. L. Millhauser, Biopolymers, 1999, 50, 239–253 CrossRef CAS PubMed.
- J. A. Rodriguez, M. I. Ivanova, M. R. Sawaya, D. Cascio, F. E. Reyes, D. Shi, S. Sangwan, E. L. Guenther, L. M. Johnson and M. Zhang, et al. , Nature, 2015, 525, 486 CrossRef CAS PubMed.
- M. Landau, M. R. Sawaya, K. F. Faull, A. Laganowsky, L. Jiang, S. A. Sievers, J. Liu, J. R. Barrio and D. Eisenberg, PLoS Biol., 2011, 9, e1001080 CrossRef CAS PubMed.
- R. Nelson, M. R. Sawaya, M. Balbirnie, A. Ø. Madsen, C. Riekel, R. Grothe and D. Eisenberg, Nature, 2005, 435, 773 CrossRef CAS PubMed.
- C. Toniolo, M. Crisma and F. Formaggio, Pept. Sci., 1998, 47, 153–158 CrossRef CAS.
- V. Monaco, F. Formaggio, M. Crisma, C. Toniolo, P. Hanson, G. Millhauser, C. George, J. R. Deschamps and J. L. Flippen-Anderson, Bioorg. Med. Chem., 1999, 7, 119–131 CrossRef CAS PubMed.
- P. Hanson, G. Millhauser, F. Formaggio, M. Crisma and C. Toniolo, J. Am. Chem. Soc., 1996, 118, 7618–7625 CrossRef CAS.
- H. Naiki, K. Higuchi, M. Hosokawa and T. Takeda, Anal. Biochem., 1989, 177, 244–249 CrossRef CAS PubMed.
- S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 42–55 CrossRef CAS PubMed.
- M. Hashemi Shabestari, M. Son, A. Moretto, M. Crisma, C. Toniolo and M. Huber, Pept. Sci., 2014, 102, 244–251 CrossRef CAS.
- L. R. Murphy, N. Matubayasi, V. A. Payne and R. M. Levy, Folding Des., 2005, 3, 105–118 CrossRef.
- H. Fischer, I. Polikarpov and A. F. Craievich, Protein Sci., 2004, 13, 2825–2828 CrossRef CAS PubMed.
- A. Micsonai, F. Wien, É. Bulyáki, J. Kun, É. Moussong, Y.-H. Lee, Y. Goto, M. Réfrégiers and J. Kardos, Nucleic Acids Res., 2018, 46, W315–W322 CrossRef CAS PubMed.
- A. Micsonai, F. Wien, L. Kernya, Y.-H. Lee, Y. Goto, M. Réfrégiers and J. Kardos, Proc. Natl. Acad. Sci. U. S. A., 2015, 112(24), E3095–E3103 CrossRef CAS PubMed.
- M. Biancalana and S. Koide, Biochim. Biophys. Acta, Proteins Proteomics, 2010, 1804, 1405–1412 CrossRef CAS PubMed.
- A. D. Milov, A. B. Ponomarev and Y. D. Tsvetkov, Chem. Phys. Lett., 1984, 110, 67–72 CrossRef CAS.
- B. E. Bode, D. Margraf, J. Plackmeyer, G. Dürner, T. F. Prisner and O. Schiemann, J. Am. Chem. Soc., 2007, 129, 6736–6745 CrossRef CAS PubMed.
- L. Gu, C. Liu, J. C. Stroud, S. Ngo, L. Jiang and Z. Guo, J. Biol. Chem., 2014, 289, 27300–27313 CrossRef CAS PubMed.
- S. I. A. Cohen, S. Linse, L. M. Luheshi, E. Hellstrand, D. A. White, L. Rajah, D. E. Otzen, M. Vendruscolo, C. M. Dobson and T. P. J. Knowles, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 9758–9763 CrossRef CAS PubMed.
- T. D. Do, W. M. Kincannon and M. T. Bowers, J. Am. Chem. Soc., 2015, 137, 10080–10083 CrossRef CAS PubMed.
- C. Bleiholder, N. F. Dupuis, T. Wyttenbach and M. T. Bowers, Nat. Chem., 2011, 3, 172 CrossRef CAS PubMed.
- J. Seo, W. Hoffmann, S. Warnke, X. Huang, S. Gewinner, W. Schöllkopf, M. T. Bowers, G. von Helden and K. Pagel, Nat. Chem., 2017, 9, 39 CrossRef CAS PubMed.
- W. Hoffmann, K. Folmert, J. Moschner, X. Huang, H. von Berlepsch, B. Koksch, M. T. Bowers, G. von Helden and K. Pagel, J. Am. Chem. Soc., 2017, 140, 244–249 CrossRef PubMed.
- G. Jeschke, Annu. Rev. Phys. Chem., 2012, 63, 419–446 CrossRef CAS PubMed.
- A. G. Kreutzer and J. S. Nowick, Acc. Chem. Res., 2018, 51, 706–718 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: CD data, diamagnetic dilution, simulation fits, and approximations used in oligomer size estimation. See DOI: 10.1039/c9cp01060b |
| This journal is © the Owner Societies 2019 |