
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Judging the feasibility of TiO2 as photocatalyst for chemical energy conversion by quantitative reactivity determinants
Martin
Dilla
a,
Nikolaos G.
Moustakas
 b,
Ahmet E.
Becerikli
a,
Tim
Peppel
b,
Armin
Springer
d,
Robert
Schlögl
ac,
Jennifer
Strunk
*b and
Simon
Ristig
*a
b,
Ahmet E.
Becerikli
a,
Tim
Peppel
b,
Armin
Springer
d,
Robert
Schlögl
ac,
Jennifer
Strunk
*b and
Simon
Ristig
*a
aMax-Planck-Institut für Chemische Energiekonversion, 45470 Mülheim an der Ruhr, Germany. E-mail: simon.ristig@cec.mpg.de
bLeibniz-Institut für Katalyse e.V., 18059 Rostock, Germany. E-mail: jennifer.strunk@catalysis.de
cFritz-Haber-Institut der Max-Planck-Gesellschaft, 14195 Berlin, Germany
dArbeitsbereich Medizinische Biologie und Elektronenmikroskopisches Zentrum (EMZ), Universitätsmedizin Rostock, 18057 Rostock, Germany
First published on 3rd June 2019
Abstract
In this study we assess the general applicability of the widely used P25-TiO2 in gas-phase photocatalytic CO2 reduction based on experimentally determined reactivity descriptors from classical heterogeneous catalysis (productivity) and photochemistry (apparent quantum yield/AQY). A comparison of the results with reports on the use of P25 for thermodynamically more feasible reactions and our own previous studies on P25-TiO2 as photocatalyst imply that the catalytic functionality of this material, rather than its properties as photoabsorber, limits its applicability in the heterogeneous photocatalytic CO2 reduction in the gas phase. The AQY of IrOx/TiO2 in overall water splitting in a similar high-purity gas-solid process was four times as high, but still far from commercial viability.
Introduction
Within the last decades photocatalysis has attracted lots of attention because it bears potential to contribute to the development of novel strategies for renewable energy sources. TiO2 is still the most frequently applied semiconductor in a manifold of different reactions, mainly due to its sufficiently wide band gap (>3.0 eV), its availability and its physical and chemical stability.1 A commercially available TiO2 composite consisting of approximately 20% rutile and 80% anatase (P25, Evonik Industries) is often considered as the standard or benchmark material in photocatalytic research.2 One of the most prominent photocatalytic reactions on semiconducting materials is the reduction of CO2 to CH4 utilizing H2O splitting as hydrogen source (eqn (1)).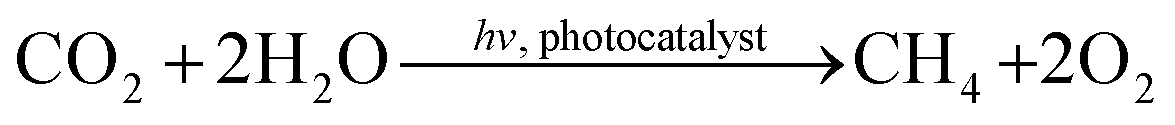 | (1) |
Our groups have also devoted considerable research effort on this research topic with the main focus on carrying out the reaction under high-purity conditions in the absence of carbon-containing impurities. As main conclusions from our work we were able to prove true CH4 formation from CO2 as a reactant, although we were never able to detect gas-phase oxygen as a byproduct (eqn (1)) with pure P25-TiO2 as the photocatalyst.20–23 The variation of reactant concentration and light intensity revealed that the formation of CH4 actually results from a photo-induced process on TiO2 and that maximum CH4 formation was achieved at low reactant concentrations. We have yet to demonstrate the possibility to obtain quantitative activity determinants under high-purity conditions, enabling a final conclusion on the potential of P25-TiO2 in photocatalytic CO2 reduction. In accordance with earlier work24,25 it seems desirable to apply different quantitative descriptors. The apparent quantum yield, as outlined below, is an appropriate measure to describe the light-based efficiency of the process, but it does not take into account the applied photocatalyst quantity. On the other hand, typical measures from heterogeneous catalysis, such as the productivity, do not consider light intensity or light absorption.
The rate of a photocatalytic reaction is strongly influenced by the intensity and energy of the incident light beam, the charge carrier dynamics and the illuminated portion of surface area.26 As a consequence the validity of comparing rate constants of photocatalysts (at a given temperature and pressure) will be limited. To circumvent this, a generally accepted approximation is to describe the activity of a photocatalyst by regarding the quantum yield (QY).27 The QY is defined as the ratio between the amount of charge carriers consumed by product formation and the amount of photons absorbed by the photocatalyst (eqn (2)) which consequently excite an electron–hole-pair. These charge carriers will be consumed by the photochemical process, if they escape from recombination and arrive at the active sites on the surface of the photocatalyst where they undergo a charge transfer reaction with an adsorbed reactant.
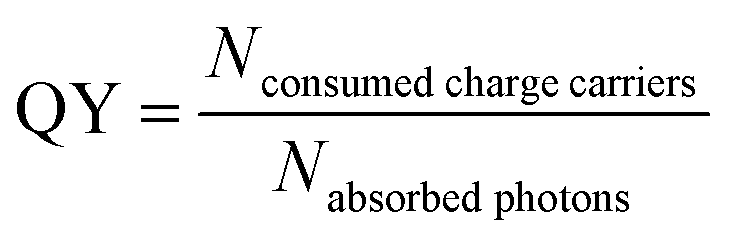 | (2) |
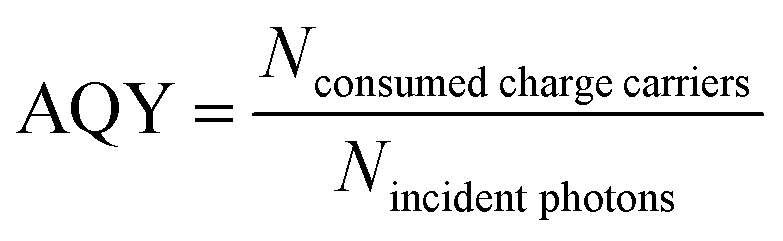 | (3) |
In classical heterogeneous catalysis, the comparison of different catalysts is often performed on the basis of mass-related production rates in μmol g−1 h−1 as a measure for the productivity of a certain catalyst. For photocatalytic reactions, calculation of this quantity requires that the entire sample mass is exposed to light and thus in principle able to participate in the reaction.
A critical point in the literature dealing with CO2 reduction in both, the QY and the productivity estimation, is the ambiguity if the experiments were performed in absence of carbonaceous impurities. It is important to notice that in reactions with such low product yields particular caution has to be exercised concerning impurities on the photocatalyst, as adsorbates from prior air contact or remains from syntheses can contribute to the products formed during the reaction.31–33 Thus, it is clear that both determinations are meaningless without adequate purification steps before the CO2 reduction experiment, as products formed from such species overestimate the number of consumed charge carriers and the amount of formed products. Due to an insufficient removal of carbonaceous species, the sensitivity of the photochemical performance of TiO2 to the illumination properties and the diverse approaches of performing QY analysis, it is obvious that the obtained values often differ by orders of magnitude.34,35
In this work, both reactivity determinants, the AQY and the productivity, are determined under suitable conditions for each quantity. Thin films deposited on glass surfaces were used to study the effect of the deposited mass on the production of hydrocarbons. We show that all of the deposited titania in these films participates in the photoreaction which allows an accurate determination of the productivity. Due to the low product yields, these experiments had to be performed under batch conditions. Under steady methane formation in continuous flow we show an approach to determine the AQY of powdered bare TiO2 (P25) in the gas-phase photocatalytic CO2 reduction. Furthermore the AQY of IrOx modified P25 (IrOx/P25) in the gas-phase photocatalytic H2O splitting was analysed under similar flow conditions. For the first time the AQY of both, the CO2 reduction and H2O splitting is determined under exclusion of contaminants in order to ensure a proper analysis of the activity of P25 and IrOx/P25. These numbers can serve as a reference for others.
Based on the results presented here, we strongly suggest that P25-TiO2 is not suitable as photocatalyst for gas-phase CO2 reduction because (i) the reaction is most likely not a full catalytic cycle with the oxygen probably being consumed by titania and (ii) both, productivity and AQY are far below any commercial viability.
Results and discussion
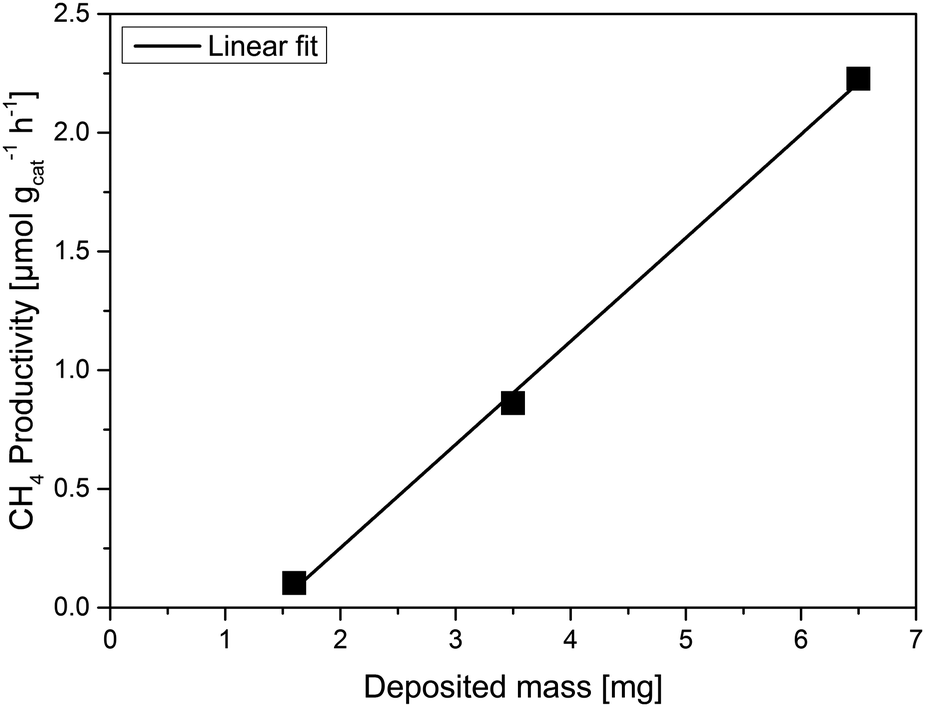
In order to study the effect of the mass of the P25 photocatalyst in CO2 reduction under high-purity conditions, three films were synthesised either by doctor blading (DB) or airbrush technique (AB: air brush; TAB: thick air brush). The latter was employed as it does not require any organic solvents to deposit the P25 particles on the glass surface. The SEM images (Fig. 1) exemplarily illustrate that the TAB film exhibits a rather uniform distribution of the P25 material. This is not the case for the AB sample. In order to produce thinner and more uniform films doctor blading was used. Even though the used paste includes organic solvents, the calcination of the deposited film and the extensive photocatalytic cleaning of the samples inside the reactor ensure that any remaining carbon-containing impurities are successfully removed. In the case of the DB and TAB samples, a calculation of the average film thickness was possible (Table 1). Due to the short airbrush deposition time, the AB film was not uniform exhibiting thicker and thinner domains of deposited P25 (Fig. 1). | ||
| Fig. 1 SEM images for the three tested films DB (A, left), AB (B, middle), TAB (C, right). | ||
| Sample | Average film thickness/μm | Mass/mg | CH4 yield/μmol | CH4 productivity/μmol gcat−1 h−1 |
|---|---|---|---|---|
| DB | 5.4 | 1.6 | 1.5 × 10−3 | 0.103 |
| AB | — | 3.5 | 2.7 × 10−2 | 0.861 |
| TAB | 19.9 | 6.5 | 1.3 × 10−1 | 2.227 |
| TiO2 (P25) powder | — | 50.0 | 3.4 × 10−1 | 0.781 |
Thus, an accurate calculation of the AB film thickness was not possible. Based on the mass of the deposited P25 (Table 1) and the fact that the geometric dimensions were the same for all three films (2.5 × 2.5 cm), it can be assumed that the average film thickness of the AB sample is higher than that of DB and lower than the one of the TAB film.
Each one of the films was subjected to photocatalytic CO2 reduction experiments in batch mode. Excessive cleaning took place inside the reactor until the sample was considered sufficiently clean (no or negligibly low amounts of C-containing impurities). The main product formed in all three cases was CH4 while only traces of ethane were identified for the AB and TAB films. While other studies also report the formation of CO or CH3OH, we could not identify these species as products. The CH4 yields for the three tested films are presented in Fig. 2. After the end of the experiments, the deposited P25 photocatalyst was mechanically removed to quantify the respective deposited masses. Under equal conditions, P25 in powder form (50 mg) was tested as a reference (Fig. 2). All results from the batch experiments are summed up in Table 1.
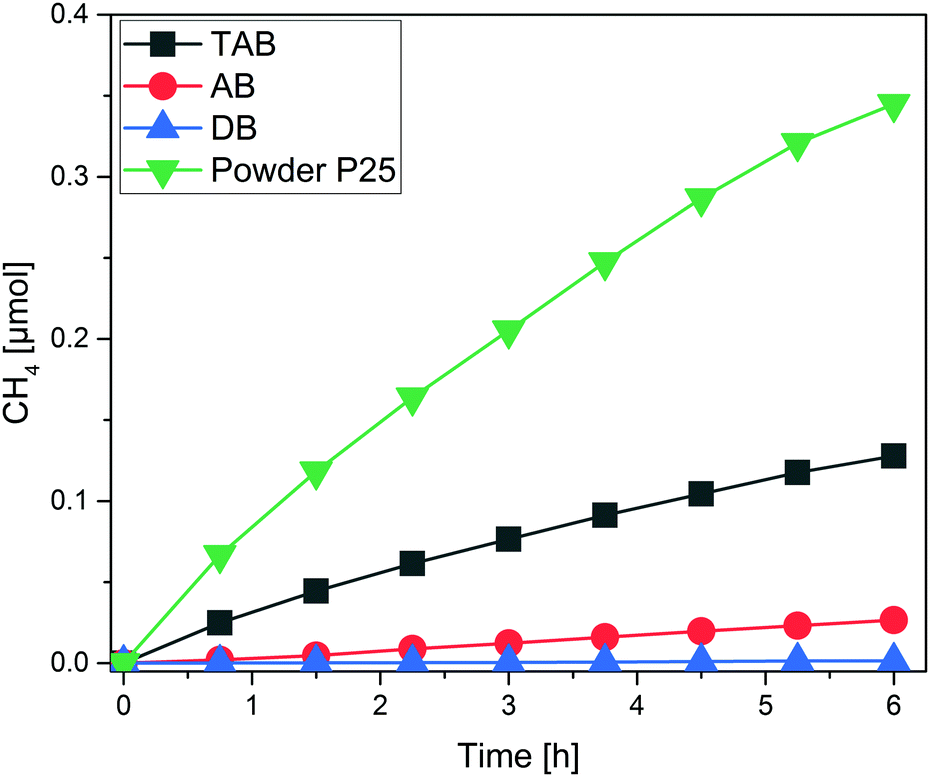 | ||
| Fig. 2 CH4 yield for the DB, AB and TAB thin films. P25 powder was used under the same conditions as a reference. | ||
Based on these results, a calculation of the CH4 productivity (μmol gcat−1 h−1) is possible. As it can be seen from Table 1 there is a linear increase in the deposited mass of the three films (approximate ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:4 DB:AB:TAB) while the CH4 productivity also increases linearly (Fig. 3). The linear correlation evidences that indeed all of the deposited titania in the three films participates in the reaction.25 It is expected, though, that after a certain film thickness and consequently deposited mass is reached, the increase in the CH4 productivity should be marginal or non-existent.24 Either the reactants CO2 and H2O will not be able to reach the lower layers of the film, or scattering and absorption phenomena will hinder the light from irradiating the entire film. The TAB has a much higher CH4 productivity than the other three measured samples, including the powder sample with roughly eightfold titania mass. Based on the observed correlation, we can estimate the necessary titania mass of a film to reach the CH4 yield (μmol) of the powdered P25, amounting to approximately 10 mg on the same 2.5 × 2.5 cm glass substrate. Thus it is clear that a major part of the powdered P25 is inactive in these experiments. The results also show that the mass of all three films is still below a certain threshold where the product yield is independent of the mass of the used photocatalyst. This is not an appropriate basis for the calculation of AQYs.24
:2:4 DB:AB:TAB) while the CH4 productivity also increases linearly (Fig. 3). The linear correlation evidences that indeed all of the deposited titania in the three films participates in the reaction.25 It is expected, though, that after a certain film thickness and consequently deposited mass is reached, the increase in the CH4 productivity should be marginal or non-existent.24 Either the reactants CO2 and H2O will not be able to reach the lower layers of the film, or scattering and absorption phenomena will hinder the light from irradiating the entire film. The TAB has a much higher CH4 productivity than the other three measured samples, including the powder sample with roughly eightfold titania mass. Based on the observed correlation, we can estimate the necessary titania mass of a film to reach the CH4 yield (μmol) of the powdered P25, amounting to approximately 10 mg on the same 2.5 × 2.5 cm glass substrate. Thus it is clear that a major part of the powdered P25 is inactive in these experiments. The results also show that the mass of all three films is still below a certain threshold where the product yield is independent of the mass of the used photocatalyst. This is not an appropriate basis for the calculation of AQYs.24
 | ||
| Fig. 3 CH4 yield for the DB, AB and TAB thin films. P25 powder was used under the same conditions as a reference. | ||
It should be also noted that no gas-phase O2 was observed in any of the tested samples, inconsistent with the predicted stoichiometry of eqn (1) but consistent with our previous observations.20,22
As in batch mode the reactants and the products of the CO2 reduction remain in the reactor until the experiment ends, produced CH4 molecules and other intermediate C-containing species could re-adsorb to the surface of the photocatalyst or react again in further cycles. The adsorbed CH4 molecules could also facilitate the adsorption or reaction of more CO2 molecules, thus improving the production of more C-containing molecules.21 For the aforementioned reasons, it is necessary to perform CO2 reduction experiments under flow conditions where formed hydrocarbons are continuously removed from the reaction chamber, allowing the calculation of more reliable AQYs. As demonstrated above, the mass of the used P25 will not be relevant, as long as it is above 10 mg. In order to compare the results to our own previous flow mode experiments, we decided to use 70 mg of P25.
The photocatalytic CO2 reduction with P25 was tested in a combined experiment with two reaction steps. In the first step carbonaceous impurities were removed and in the second step the activity in CO2 reduction was studied to determine the AQY. Only such a procedure can guarantee that product formation is depending solely on the presence of CO2. As described in our previous study20 the detection of CH4 and CO2 during the first three hours results from the cleaning procedure. The consistent observation in our previous studies20,22 was that a residual CH4 baseline could not be avoided, even after extended cleaning procedures. The only potential reason for this observation not previously excluded is a diffusion of carbonaceous impurities from the bulk of P25. Fig. 4A–C display the result of blank experiments which were conducted to disprove this hypothesis. In all three cases P25 was photocatalytically cleaned for a certain period of time, followed by a dark period to allow diffusion of carbonaceous species from the bulk to the surface, before the photocatalytic cleaning was continued. In the dark period, the He gas flow of 5 mL min−1 was maintained. After the dark period, the illumination was started again, but in contrast to the activity test in Fig. 3 without dosing of CO2.
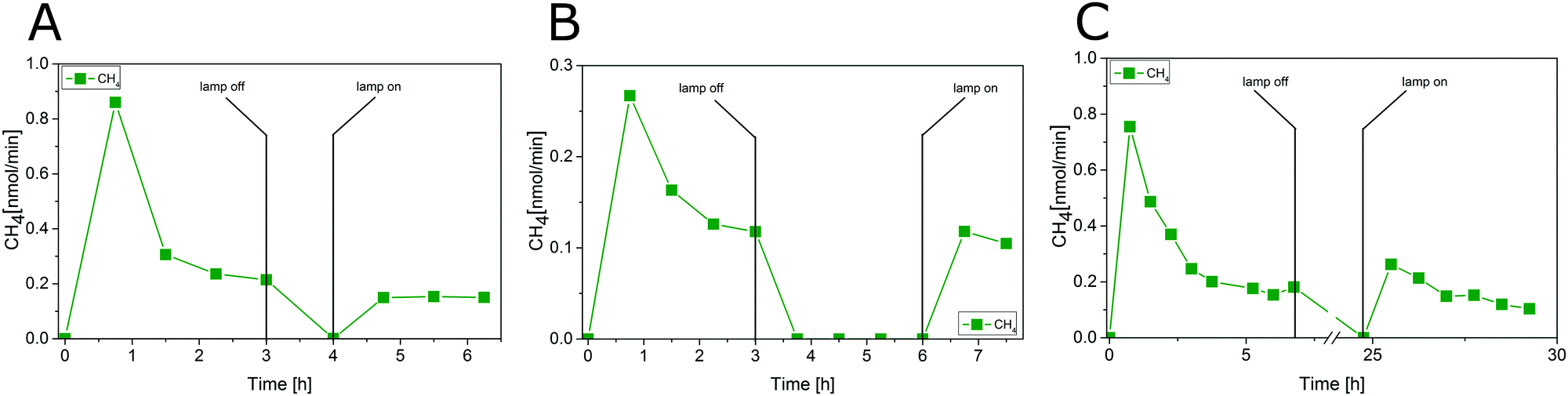 | ||
| Fig. 4 Removal of carbonaceous impurities with intermediate illumination interruptions, (A) 3 h cleaning, 1 h without illumination, resuming cleaning; (B) 3 h cleaning, 3 h without illumination, resuming cleaning, (C) 6 h cleaning, 18 h without illumination, resuming cleaning. | ||
The CH4 formation increases only to the same baseline rate as observed before turning off the illumination (Fig. 4A and B). This result illustrates that no carbonaceous impurities from the bulk are diffusing to the surface of P25 when the light was turned off. Hence, a contribution of impurities from the bulk to the activity in CH4 formation during the CO2 reduction experiment in Fig. 3 can be ruled out. Even when pausing the illumination for 18 h, only a slight increase of the CH4 formation rate can be observed (Fig. 4C).
Regarding the results presented above the CH4 formed after initiating the photocatalytic CO2 reduction experiment at 3.75 h (Fig. 5) truly originates from CO2 as carbon source. A formation of other carbon containing products such as CO, CH3OH, C2H6, C3H6, which are frequently mentioned as products of the photocatalytic CO2 reduction was not observed. Fig. 5 shows that the CH4 formation rate increased from 3.75 to 6 h. A stable CH4 formation rate was observed after 6 h. Since the application of the high-purity reactor was essential for this work, the limited interaction between the reactant gas mixture and the solid P25 in the overflow geometry had to be accepted. As a consequence, the complete mixing of CH4 in the gas flow to detect a stable formation rate is likely delayed.
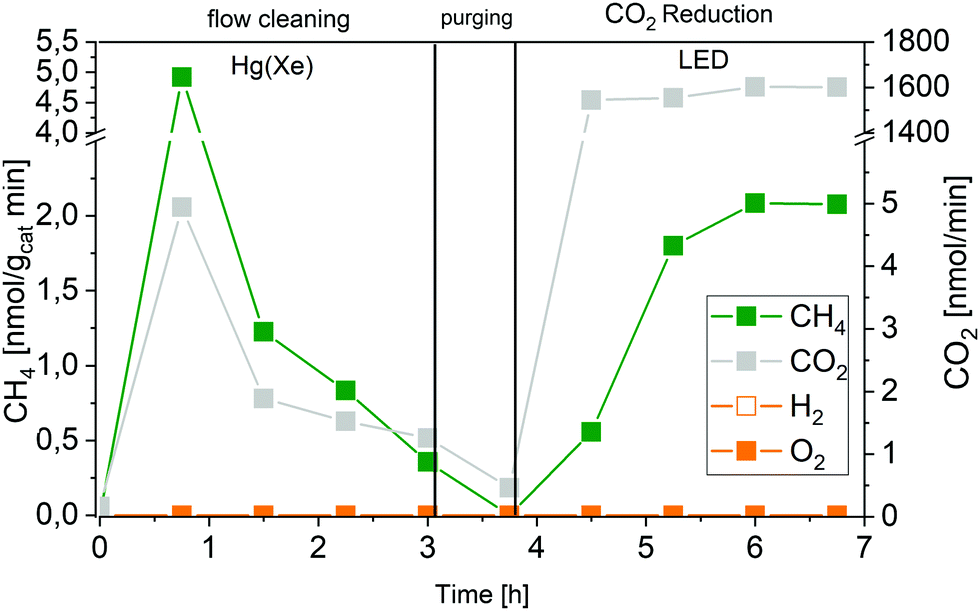 | ||
| Fig. 5 Approximation of the apparent quantum yield of P25 in the photocatalytic CO2 reduction. Removal of carbonaceous impurities with Hg(Xe) lamp (0 to 3 h). Purging with He (3 to 3.75 h), no illumination. Photocatalytic CO2 reduction (3.75 to 6.75 h) with 2 W LED (365 nm). | ||
The stable formation rate of CH4 was used as the basis for the AQY determination. The formation of CH4 from CO2 requires that two C–O double bonds are cleaved and 4 C–H bonds are formed. In this process eight electrons need to be transferred to the carbon atom. On this account the factor eight was used in the numerator of eqn (4) to obtain the number of electrons from the molar amount of CH4 formed.
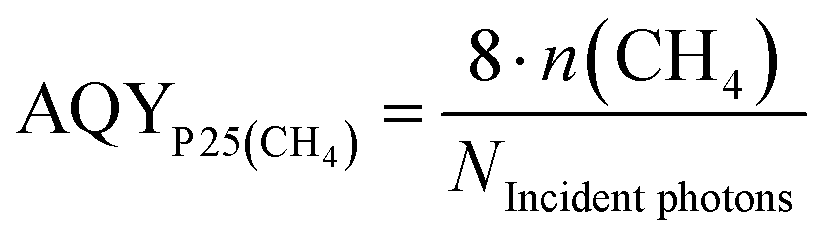 | (4) |
Furthermore, comparing the AQY of CO2 reduction with studies on thermodynamically more favoured reactions, such as the decomposition of less stable organic compounds on TiO2, for instance phenol,36 methylene blue37 or PCBs38 shows that the values can reach 1% up to 20%. The fact that the AQY reaches higher values in these reactions when TiO2 is used as the photocatalyst implies that a significant amount of electrons and holes is available for charge transfer reactions. This indicates that TiO2 sufficiently functions as photoabsorber and creates charge carriers, but that it is not a suitable catalyst for the formation of CH4 in gas-phase CO2 reduction, either because suitable active sites are lacking, or because most charge carriers lack a sufficient thermodynamic driving force to create relevant intermediates.
An additional shortcoming of P25 identified in our previous studies20–23 is its inability to liberate gaseous O2 as the stoichiometric byproduct. The often-represented reaction (1) supposes the formation of CH4 and O2 in a 1:2 ratio. However, under the applied reaction conditions, it was not possible to detect any O2, although the generation of this product would be an important prerequisite for a complete reaction cycle. The formation of O2 is often neglected in the literature. It is either not analysed or cannot be found in the products of photocatalytic CO2 reduction.41,42 With some exceptions8,43–46 the research effort is focused on the formation of carbon related products, since they are the desired products for storing chemical energy.
The absence of O2 can have various reasons: (i) the consumption of O-derived species in the backward reaction (CH4 oxidation), (ii) limitations in H2O oxidation kinetics, so that the reaction stops at adsorbed intermediates, or (iii) the replenishment of defects, namely oxygen vacancies (Ov) on TiO2.47,48 This leads to the suggestion that the oxidation half reaction in photocatalytic CO2 reduction on P25-TiO2 does not occur. Instead, reactions that consume oxygen take place that are not of catalytic but of stoichiometric nature. Recently, we performed studies on Ir-modified P25 in CO2 reduction and H2O splitting. The details are reported in a separate publication,49 but we would like to mention here, that in contrast to the pure P25 overall H2O splitting is possible with such a system. Consequently, using IrOx/P25 gives us the opportunity to comparatively study the AQY of a reaction not limited by a stoichiometric counter reaction.
Fig. 6 demonstrates that IrOx/P25 shows activity in the overall H2O splitting reaction. Both products, H2 and O2 can be detected in a ratio of almost 2:1, according to eqn (5):
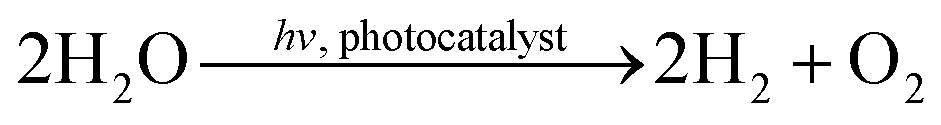 | (5) |
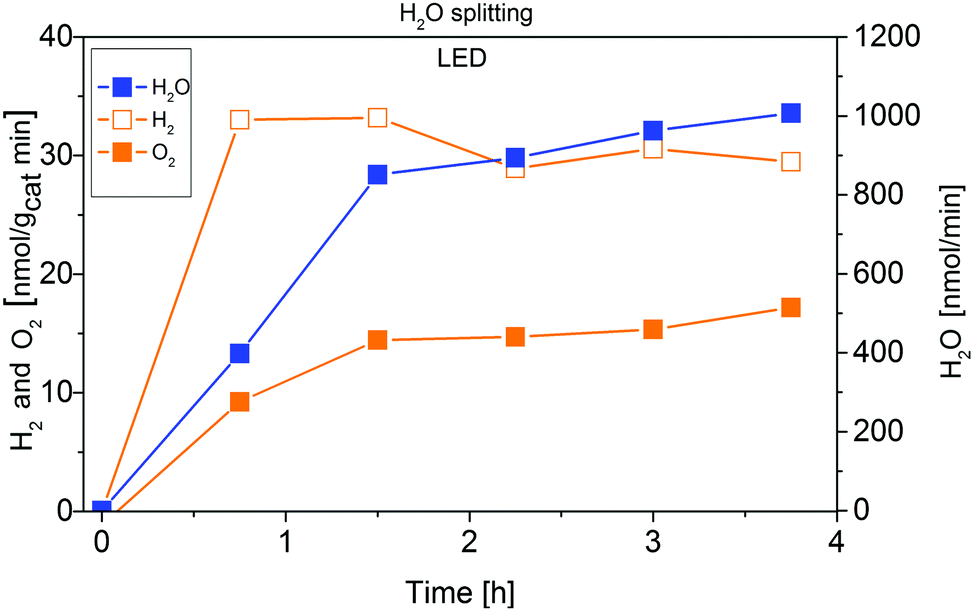 | ||
| Fig. 6 Approximation of the apparent quantum yield of 0.05 wt% IrOx/P25 in the photocatalytic H2O splitting reaction. Illumination with 2 W LED (365 nm). | ||
Using the stable and stoichiometric values of O2 and H2 formation rates, an AQY of 2.5 × 10−3% is obtained. Although this value is almost four times as high as the AQY of CO2 reduction, it is still small. It is important to note, that this value has also been obtained under high-purity conditions, so a falsifying influence of carbonaceous impurities acting as sacrificial reagents can be excluded.
To generally achieve higher yields, it may be beneficial to separate reduction and oxidation reaction more rigorously, for example by employing photoelectrochemical processes, Z-scheme systems, or (nano)membrane-based devices. For the case of separate reduction and oxidation sites, using IrOx as water oxidation catalyst, we already found that CO2 reduction can no longer proceed simultaneous to water splitting,49 so more elaborate concepts need to be implemented.
Experimental
All experiments in this work were carried out in two similar high purity photoreactor set-ups described in detail by Mei et al.50 For the batch experiments, a Shimadzu Tracera GC 2010 plus gas chromatograph equipped with a barrier ionization discharge detector (BID) and a flame ionization detector (FID) was used to quantify CO2, O2, H2O, CH4 and higher hydrocarbons in the single-digit ppm range, as well as H2 and CO from ≥20 ppm. Illumination in the CO2 reduction experiments was carried out with a 10 W 365 nm LED (SeoulViOSys), operated at 7.2 V and 0.92 A with a total output intensity of 755 mW cm−2 (Thorlabs PM100USB actinometer with a S405C measuring cell).For the flow experiments, a Shimadzu Tracera GC 2010 Plus, equipped with a barrier discharge ionization detector (BID) was used which allows quantifying CO2, CH4, H2O, H2 and O2 in the 0.1 ppm range. A detection of CO, CH3OH, C2H6, C3H8 is also feasible. Illumination of the samples for the approximation of the AQY was realised with a 365 nm 2 W high-power LED, operated at 3.5 V and 0.5 A with a total output intensity of 185 mW cm−2 (Thorlabs PM100D actinometer with a S305C measuring cell). Monochromatic LED light sources allow a straightforward determination of the incident photons which can be calculated from the light intensity hitting the sample surface. A transmittivity of the reactor lid window of 86% at 365 nm was accounted for in the calculation of the AQY.
Preparation of P25-TiO2 thin films
To remove any loosely bound carbon-containing impurities the P25 powder (Evonik Industries) was calcined in synthetic air at 400 °C for 3 h prior to use. All thin films were deposited on a 2.5 × 2.5 cm surface of a microscope glass slide. The glass slides were cleaned with a Hellmanex® III cleaning solution, rinsed with de-ionised H2O and then ultrasonicated for 30 min at room temperature (Elmasonic S 60 H).For thin films prepared by doctor blading a P25-TiO2 paste was prepared according to Ito et al.51 with slight modifications. Briefly, 10 mL EtOH were added to a solution of ethyl cellulose (3.5 g, ∼50 cP, 10 wt% in EtOH), P25 (0.8 g) and terpineol (6.5 g, mixture of isomers) in a 50 mL round-bottom flask. The mixture was thoroughly sonicated for 45 min at room temperature in an ultrasonic bath (Elmasonic S 60 H). Afterwards, the solvent was removed stepwise with a rotary evaporator (final T = 40 °C; final p = 7 mbar). The resulting suspension (∼10 wt% P25) was used directly for doctor blading.
For thin films prepared by the air-brush technique, a solution of 0.5 g of P25 in 100 mL de-ionised H2O was sprayed on the surface of the microscope slides. The glass substrates were put on a hot plate at 250 °C to quickly evaporate sprayed H2O leaving behind a P25 coating. In order to get films of different thickness, different deposition times were applied.
After deposition, all thin films were calcined at 300 °C for 3 h to increase the adhesion of the powder to the glass surface and to remove the solvents and other C-containing species from the surface of the samples. SEM images of the films were collected using a Merlin VP compact (Zeiss, Oberkochen, Germany). To estimate the amount of deposited P25, the thin films were mechanically removed from the glass substrates after the CO2 reduction experiments and the received solid was quantified gravimetrically.
Removal of carbonaceous species, photocatalytic CO2 reduction with P25 and approximation of the AQY
70 mg of P25 were used for the approximation of the AQY in the photocatalytic CO2 reduction in flow mode. The sample was calcined at 400 °C for 3 h in synthetic air prior to use. After calcination the sample was further cleaned from residual carbon-containing species before the CO2 reduction was performed. This cleaning step was performed in the photoreactor set-up under continuous flow conditions and at ambient pressure. H2O enriched He 6.0 (99.9999% He) was flushed (5 mL min−1) through the reactor while a 200 W Hg/Xe light source (Oriel Instruments) irradiated the sample. This polychromatic light source was used in order to clean the sample quickly. A GC measurement was performed every 45 minutes to monitor the cleaning progress. As soon as the concentrations of the products were sufficiently low, the purification step was terminated. After purging out the gas-phase in the reactor, the CO2 reduction reaction was initiated. The reactant gas, a diluted CO2 in He mixture (7000 ppm CO2 in He 6.0) was passed through the reactor with a flow rate of 5 mL min−1.For the batch experiments, 15.000 ppm CO2 in He 6.0 and 6000 ppm H2O filled the reactor up to a final pressure of 1500 mbar. Every 45 min a sample was collected to identify the products of the CO2 reduction over the course of 6 h. All results were normalised to take into consideration the resulting pressure drop from each sampling event.
Preparation of IrOx/P25 and approximation of AQY in H2O splitting
The iridium modified sample was prepared by photodeposition in a semi-batch quartz glass reactor. Iridium acetate was used as the precursor for the modification of P25. The nominal loading of iridium was set to 0.05 wt%. On-top illumination was realised by a 1000 W Hg/Xe lamp (LOT quantum design). After 3 h of photodeposition, the illumination was stopped. The obtained powder was dried overnight and calcined at 400 °C for 3 h. For the AQY analysis of photocatalytic H2O splitting, 70 mg of the 0.05 wt% IrOx/P25 were used. Dosing of the reactant H2O was performed from stainless-steel cups, which are temperature-controlled. The temperature of the saturator was adjusted to set the H2O concentration in the gas flow to ∼1000 nmol min−1. The flow rate of the reactant gas flow was also set to 5 mL min−1.Conclusions
As central outcome of this work, reliable values for AQY and productivity of P25-TiO2 in photocatalytic CO2 reduction have been obtained. Thin films composed of less than 10 mg titania are presumably fully illuminated, so that all of the TiO2 contributes to the product formation and reliable values for the productivity can be obtained. Applying a larger amount of P25 allows determination of reliable AQY values. The resulting values and a comparison with thermodynamically more feasible reactions imply that the absorption functionality of P25 would allow higher yields, but the catalytic function is not suitable for CO2 reduction reaction. Nevertheless, P25-TiO2 still represents a valuable material in general photocatalytic research, as it is commercially available and can be used as reference or standard material in other reactions. Implementing the same procedure for overall H2O splitting with IrOx/TiO2 reveals a four times higher AQY value. Additionally, a small amount of O2 missing in comparison to the expected 2:1 ratio (H2:O2) provides further indication that a stoichiometric reaction consuming oxygen species might play an important role in photocatalytic CO2 reduction.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Part of this research was funded by the German Ministry of Education and Research (BMBF) in the scope of the funding scheme CO2Plus, Project-No. 033RC003, PROPHECY. Open Access funding provided by the Max Planck Society.References
- E. V. Kondratenko, et al. , Energy Environ. Sci., 2013, 6, 3112 RSC.
- S. N. Habisreutinger, L. Schmidt-Mende and J. K. Stolarczyk, Angew. Chem., Int. Ed., 2013, 52, 7372–7408 CrossRef CAS PubMed.
- T. Inoue, et al. , Nature, 1979, 277, 637–638 CrossRef CAS.
- K. Mori, H. Yamashita and M. Anpo, RSC Adv., 2012, 2, 3165 RSC.
- I. A. Shkrob, et al. , J. Phys. Chem. C, 2012, 116, 9461–9471 CrossRef CAS.
- J. Yu, et al. , J. Am. Chem. Soc., 2014, 136, 8839–8842 CrossRef CAS PubMed.
- A. Derylo-Marczewska, et al. , Catal. Today, 2006, 114, 293–306 CrossRef CAS.
- L. Liu, et al. , ACS Catal., 2012, 2, 1817–1828 CrossRef CAS.
- C.-C. Yang, et al. , J. Am. Chem. Soc., 2010, 132, 8398–8406 CrossRef CAS PubMed.
- C. P. Sajan, et al. , Nano Res., 2016, 9, 3–27 CrossRef CAS.
- K. Kočí, et al. , Appl. Catal., B, 2009, 89, 494–502 CrossRef.
- Y. T. Liang, et al. , Nano Lett., 2011, 11, 2865–2870 CrossRef CAS PubMed.
- Ş. Neaţu, et al. , J. Am. Chem. Soc., 2014, 136, 15969–15976 CrossRef PubMed.
- I.-H. Tseng, J. C. S. Wu and H.-Y. Chou, J. Catal., 2004, 221, 432–440 CrossRef CAS.
- W. Hou, et al. , ACS Catal., 2011, 1, 929–936 CrossRef CAS.
- Q. Zhai, et al. , Angew. Chem., Int. Ed., 2013, 52, 5776–5779 CrossRef CAS PubMed.
- L. Zhang, et al. , Angew. Chem., Int. Ed., 2015, 54, 15823–15826 CrossRef CAS PubMed.
- X. Liu, et al. , Energy Environ. Sci., 2017, 10, 402–434 RSC.
- W.-J. Ong, et al. , Nano Res., 2014, 7, 1528–1547 CrossRef CAS.
- M. Dilla, R. Schlögl and J. Strunk, ChemCatChem, 2017, 9, 696–704 CrossRef CAS.
- A. Pougin, M. Dilla and J. Strunk, Phys. Chem. Chem. Phys., 2016, 10809 RSC.
- M. Dilla, et al. , ChemCatChem, 2017, 9, 4345–4352 CrossRef CAS.
- M. Dilla, et al. , Photochem. Photobiol. Sci., 2019, 314–318 RSC.
- H. Kisch, Angew. Chem., Int. Ed., 2010, 49, 9588–9589 CrossRef CAS PubMed.
- T. Maschmeyer and M. Che, Angew. Chem., Int. Ed., 2010, 49, 1536–1539 CrossRef CAS PubMed.
- N. Serpone, J. Photochem. Photobiol., A, 1997, 104, 1–12 CrossRef CAS.
- H. Kisch and D. Bahnemann, J. Phys. Chem. Lett., 2015, 6, 1907–1910 CrossRef CAS PubMed.
- N. Serpone, et al. , J. Photochem. Photobiol., A, 1993, 73, 11–16 CrossRef CAS.
- N. Serpone, et al. , J. Photochem. Photobiol., A, 1996, 94, 191–203 CrossRef CAS.
- M. R. Hoffmann, et al. , Chem. Rev., 1995, 95, 69–96 CrossRef CAS.
- T. Yui, et al. , ACS Appl. Mater. Interfaces, 2011, 3, 2594–2600 CrossRef CAS PubMed.
- A. Cybula, M. Klein and A. Zaleska, Appl. Catal., B, 2015, 164, 433–442 CrossRef CAS.
- I. Grigioni, et al. , Catal. Today, 2017, 281, 214–220 CrossRef CAS.
- L. Liu, et al. , ACS Catal., 2016, 6, 1097–1108 CrossRef CAS.
- T.-V. Nguyen and J. C. S. Wu, Appl. Catal., A, 2008, 335, 112–120 CrossRef CAS.
- V. Loddo, et al. , AIChE J., 2006, 52, 2565–2574 CrossRef CAS.
- B. Serrano and H. de Lasa, Ind. Eng. Chem. Res., 1997, 36, 4705–4711 CrossRef CAS.
- C. C. Wong and W. Chu, Chemosphere, 2003, 50, 981–987 CrossRef CAS PubMed.
- Z. He, et al. , Appl. Surf. Sci., 2016, 364, 416–427 CrossRef CAS.
- H. Yaghoubi, et al. , ACS Catal., 2015, 5, 327–335 CrossRef CAS.
- J. Tang, J. R. Durrant and D. R. Klug, J. Am. Chem. Soc., 2008, 130, 13885–13891 CrossRef CAS PubMed.
- F. Saladin and I. Alxneit, J. Chem. Soc., Faraday Trans., 1997, 93, 4159–4163 RSC.
- J.-C. Wang, et al. , ACS Appl. Mater. Interfaces, 2015, 7, 8631–8639 CrossRef CAS PubMed.
- K. Iizuka, et al. , J. Am. Chem. Soc., 2011, 133, 20863–20868 CrossRef CAS PubMed.
- E. Korovin, D. Selishchev and D. Kozlov, Top. Catal., 2016, 59, 1292–1296 CrossRef CAS.
- K. Teramura, et al. , Angew. Chem., Int. Ed., 2012, 51, 8008–8011 CrossRef CAS PubMed.
- Z. Dohnalek, et al. , J. Phys. Chem. B, 2006, 110, 6229–6235 CrossRef CAS PubMed.
- H. Belhadj, et al. , Phys. Chem. Chem. Phys., 2015, 17, 22940–22946 RSC.
- M. Dilla, et al. , Phys. Chem. Chem. Phys., 2019 10.1039/C8CP07765G.
- B. Mei, A. Pougin and J. Strunk, J. Catal., 2013, 306, 184–189 CrossRef CAS.
- S. Ito, et al. , Thin Solid Films, 2008, 516, 4613–4619 CrossRef CAS.
| This journal is © the Owner Societies 2019 |