
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Understanding methane/carbon dioxide partitioning in clay nano- and meso-pores with constant reservoir composition molecular dynamics modeling†
Narasimhan
Loganathan
a,
Geoffrey M.
Bowers
b,
Brice F.
Ngouana Wakou
c,
Andrey G.
Kalinichev
 c,
R. James
Kirkpatrick
ad and
A. Ozgur
Yazaydin
*ae
c,
R. James
Kirkpatrick
ad and
A. Ozgur
Yazaydin
*ae
aDepartment of Chemistry, Michigan State University, East Lansing, Michigan 48824, USA
bDepartment of Chemistry and Biochemistry, St. Mary's College of Maryland, St. Mary's City, Maryland 20686, USA
cLaboratoire SUBATECH (UMR 6457 – Institut Mines-Télécom Atlantique, Université de Nantes, CNRS/IN2P3), 44307, Nantes, France
dDepartment of Earth and Environmental Sciences, Michigan State University, East Lansing, Michigan 48824, USA
eDepartment of Chemical Engineering, University College London, London, WC1E 7JE, UK. E-mail: ozgur.yazaydin@ucl.ac.uk
First published on 7th March 2019
Abstract
The interactions among fluid species such as H2O, CO2, and CH4 confined in nano- and meso-pores in shales and other rocks is of central concern to understanding the chemical behavior and transport properties of these species in the earth's subsurface and is of special concern to geological C-sequestration and enhanced production of oil and natural gas. The behavior of CO2, and CH4 is less well understood than that of H2O. This paper presents the results of a computational modeling study of the partitioning of CO2 and CH4 between bulk fluid and nano- and meso-pores bounded by the common clay mineral montmorillonite. The calculations were done at 323 K and a total fluid pressure of 124 bars using a novel approach (constant reservoir composition molecular dynamics, CRC-MD) that uses bias forces to maintain a constant composition in the fluid external to the pore. This purely MD approach overcomes the difficulties in making stochastic particle insertion–deletion moves in dense fluids encountered in grand canonical Monte Carlo and related hybrid approaches. The results show that both the basal siloxane surfaces and protonated broken edge surfaces of montmorillonite both prefer CO2 relative to CH4 suggesting that methods of enhanced oil and gas production using CO2 will readily displace CH4 from such pores. This preference for CO2 is due to its preferred interaction with the surfaces and extends to approximately 20 Å from them.
Introduction
The interactions among fluid species such as water, carbon dioxide and methane confined in nano- and meso-pores in shales and other rocks is of central concern to understanding the chemical behavior and transport properties of these species in the earth's subsurface and is of special concern to geological C-sequestration and enhanced production of oil and natural gas.1–6 The behavior of water and cations at mineral surfaces and in the interlayer galleries of expandable clays (smectites) has been studied experimentally and computationally for many years,7–19 and more recently there has been increased interest in carbon dioxide and hydrocarbons.20–44 The mutual interactions between CO2 and CH4 in nano- and meso-scale confinement, however, remain poorly understood. Most simulation studies have focused on single component adsorption in the interlayers of clay minerals. These studies show that the CO2 is more strongly adsorbed to clay minerals than CH4.45–47 Recent Grand Canonical Monte Carlo (GCMC) calculations, for instance, have shown preferential adsorption of CO2 over CH4 in the interlayers of dry Na-montmorillonite and other inorganic shale components as functions of temperature (313–373 K) and pressure (0–20 MPa).47–52 In shales and other sedimentary rocks, pores have a wide range of sizes, and the effects of pore size on CO2/CH4 partitioning are poorly understood. This paper presents the results of a computational molecular dynamics (MD) modeling study of the partitioning of CO2 and CH4 between bulk fluid and slit-like nano- and meso pores from 4 to 73 Å thick and the structure of the fluid in those pores. In contrast to most computational studies of clays, the montmorillonite substrate model here is of finite size (Fig. 1), allowing study of the fluid interaction with the siloxane basal surfaces and the protonated sites on broken edges of the clay layers. The results show that the partitioning of CO2 and CH4 between the bulk fluid (representing fluid in large pores in shales and other rocks) and the nano- and meso-pores is substantially influenced by the pore thickness, with CO2 being largely preferred in narrow confined pores. CO2 is also greatly preferred at the protonated edges of broken clay layers. | ||
| Fig. 1 Schematic representations of the simulation cells used in the constant reservoir composition molecular dynamics, CRC-MD calculations of CO2/CH4 partitioning into pores bounded by montmorillonite basal surfaces. (a) The full simulation cell showing the different regions. Pink arrows represent bias forces. (b) Enlarged image of the silt-like pore and montmorillonite T–O–T layers. Color code: Si – yellow; O – red; Mg – green; Al – pink; H – white; Na – blue; CCO2 – cyan; CCH4 – black. Substrate atoms are represented using sticks, and the CO2/CH4 fluids and Na+ ions are represented in balls. The directions of the X and Z axis are indicated by the arrows on the lower left. | ||
The MD simulations were done using a novel approach that uses self-adjusting bias forces to maintain a constant composition in the fluid external to the pore (constant reservoir composition molecular dynamics, CRC-MD). Such self-adjusting bias forces have been used previously to maintain a constant chemical potential of the fluid species in modelling the growth of urea crystals from solution (constant chemical potential MD)53,54 and to create a concentration gradient across a membrane for modelling the gas transport and separation of mixtures in membranes (concentration gradient driven MD).55 The advantages of using a purely MD approach to maintain the composition of dense fluids rather than using the stochastic particle insertion–deletion moves used in grand canonical Monte Carlo and related hybrid approaches, such as dual control volume grand canonical molecular dynamics,56 have been discussed in detail in ref. 55. Inspired by these studies,53–55 we have implemented the CRC-MD method, which has proven to be an efficient way to evaluate partitioning of fluid species between bulk fluid and nano- and meso-pores and also onto the surfaces of finite size solid substrates.
Methods
In the CRC-MD method, each simulation cell consists of a bulk fluid reservoir, two bias force regions, two composition control regions, two transition regions, and the substrate (clay) + pore assemblage in the center (Fig. 1a). The concentrations of the fluid species in the control regions are maintained at constant values by forces in the bias force regions. These bias forces act in such a way that if the concentration of a given species in a control region is different than the target concentration molecules of that species are moved into or out of the control region from or to the reservoir. A detailed explanation of the functional forms of the bias forces and how they work can be found elsewhere.55The substrate used in the simulations was the expandable smectite clay mineral, montmorillonite, which develops a permanent negative structural charge by the isomorphic substitution of Al3+ for Si4+ in the tetrahedral sheet and Mg2+ for Al3+ in the octahedral sheet. The model here has a structural formula of M+0.75(Si7.75Al0.25)(Al3.5Mg0.5)O20(OH)4.12 The distribution of the isomorphic substitutions has a quasi-disordered pattern and was prepared using the “Supercell” program,57 which creates substitutions in accordance with an extension of Lowenstein's rule,58 which forbids tetrahedral Al–O–Al and octahedral Mg–O–Mg linkages. The simulated montmorillonite particles have lateral dimensions of ∼73.0 Å × 41.4 Å along the a and b crystallographic axes, respectively, with an orthorhombic unit cell. To create a finite size particle, the montmorillonite structure was cleaved along (010), creating broken edge sites on that surface. On the broken edges, the dangling tetrahedral Al3+ and Si4+ sites are saturated by single OH− groups, and most of the dangling octahedral Al3+ sites are saturated with 1 OH− and 1 H2O molecule.59 Meanwhile, according to ab initio calculations,60 when the octahedral Mg2+ sites are saturated with 1 H2O molecule and 1 OH− group, proton transfer reactions occur with the neighboring octahedral Al3+ sites and tetrahedral Si4+ sites to generate octahedral Mg2+ sites saturated with 2 H2O molecules (as predicted by the crystal growth theory),61 and a neighboring Al3+ site with 2 OH− groups. These coordination environments were used in the clay substrate. Furthermore, the oxygen atoms of the OH− groups of the broken edge sites were assigned a slightly higher negative charge (−0.9659 |e|) compared to the original CLAYFF62 value (−0.95 |e|). This was done not only to ensure that the broken edges of the montmorillonite particle are electrostatically neutral, but also because the OH− groups of the edge sites have fewer bond connections compared to those in the interior of the clay structure. Importantly, the two (010) surfaces of this model have different compositions. One contains only unsubstituted tetrahedral Si4+ and octahedral Al3+ sites (left side of the particle in Fig. 1b), whereas the other contains two substituted tetrahedral Al3+ and two octahedral Mg2+ sites (right side of Fig. 1b).
The modeled montmorillonite particle consists of three T–O–T layers which encompass two anhydrous interlayers and expose two external basal surfaces to the pore (Fig. 1a). Because of the 3-dimensional periodic boundary conditions used, only the broken (010) surfaces and the basal surfaces bounding the pore are exposed to the fluid phase. The lateral dimensions of the simulation cell are 300 Å × 41.4 Å (Fig. 1a). The thickness of the slit-like pore between the external basal surfaces (Fig. 1b) were varied from 4 to 73 Å. For pore thicknesses less than 10 Å, at the start of each simulation the charge compensating Na+ ions were placed at the midplane of the pore far from the substitution sites on the external surfaces, thereby allowing the Na+ ions to choose their preferred adsorption sites during the course of the simulation. For models with pore thicknesses >10 Å, the Na+ ions were initially placed 5 Å from the external basal surface. The CO2 and CH4 molecules were initially placed external to the clay particle at distances >∼30 Å from the two broken (010) surfaces. The number of CO2 and CH4 molecules in the full simulation cell varies from 3456 to 8063 and increases with increasing pore thickness in order to maintain the desired target densities in the control region. We analyze the CO2/CH4 partitioning and density profiles near the clay particle in three different dimensions: (i) normal to the basal surfaces bounding the pore (z direction); (ii) parallel to the broken edges in the transition region (z direction), and (iii) normal to the broken edges in the transition region (x direction). We define the pore thickness as the distance between the centers of the oxygen atoms of the basal surfaces bounding the pore. Similarly, we define the surfaces of the broken edges as the plane containing the centers of the protonated oxygen atoms on the surface. Further details about the analysis methods are described elsewhere.10,11,14,15,31–33
We performed the molecular dynamics simulations in the canonical NVT ensemble at 323 K using the LAMMPS simulation package.63 A Nosé–Hoover thermostat was used to control the temperature.64 The interatomic interactions for the montmorillonite were obtained from the CLAYFF62 force field, and the parameters for the broken edge sites were obtained from newly developed metal–O–H bending potentials consistent with ClayFF.65,66 The CO2 and CH4 molecules were represented by the EPM267 and TraPPE-UA68 models, respectively. Three-dimensional periodic boundary conditions were employed with a cutoff of 14.0 Å for short-range non-electrostatic interactions, and the long-range electrostatic interactions were computed using the particle–particle particle-mesh (PPPM) summation algorithm69 with an accuracy of 10−6. A time step of 1 fs was employed to integrate the equations of motion. Each system was equilibrated for 10 ns followed by another 2 ns of data production with the atomic coordinates recorded every 10 fs. Importantly, we fixed the positions of 18 octahedral atoms (Al3+ or Mg2+) in the simulated montmorillonite structure (2 left: 2 middle: 2 right per TOT layer) in order to prevent the clay layers from moving with respect to each other.
A modified version of the PLUMED 2.3.0 plugin70 was used to apply the bias forces to maintain the fluid composition in the control regions. The target CO2 and CH4 densities in the control regions were set to 2 CO2 and 2 CH4 molecules per nm3. At 323 K, these densities yield a total fluid pressure of 124 bars, as calculated by using the Peng–Robinson equation of state.71 These temperature and pressure conditions are relevant to the upper part of the Earth's crust and petroleum reservoirs. The width of the transition regions, control regions and the bias force regions were set to 25 Å, 25 Å and 2.5 Å, respectively. The target composition of the fluid species, i, in the control regions was maintained using the force constants kLi = kRi = 5000 kJ nm3 mol−1 which are placed at the center of the bias force regions located on the left, L, and right, R, side of the clay particle. The compositions in the control regions were monitored at intervals of 0.5 ps during the entire simulation.
Results and discussion
The mole fractions and number densities of CO2 and CH4 molecules in the pores vary greatly depending on the pore thickness (Fig. 2a and b). At the smallest thickness studied (4.0 Å), neither CO2 nor CH4 enters the pore. At a thickness of 6.0 Å, CO2 fills the pore and there is only a negligible fraction of CH4 molecules near the entry points. These results are in good agreement with recent experimental and simulation studies that show that the basal spacing required for CO2 intercalation in the interlayer galleries of smectite clays is less than for CH4 due to steric effects.6,31,32,38,39 At a pore thicknesses of 7.5 Å some CH4 enters the pore, and the CO2/CH4 ratio decreases rapidly with increasing pore thickness up to 33.0 Å (Fig. 2a). At larger pore thickness, the CO2/CH4 ratio decreases less rapidly and slowly approaches the composition in the control regions (2 molecules per nm3), although for the thicknesses studied here it never reaches the control region composition. The number density of CO2 molecules in the pores is always larger than in the control region, with the highest value of ∼8.2 molecules per nm3 at 9.0 Å and progressively smaller values at larger thicknesses. In contrast, the CH4 density increases with increasing thickness to 23.0 Å and then decreases with increasing thickness. However, it is never much greater than the value in the control region of 2 molecules per nm3. (Fig. S1, ESI,† shows that the fluid compositions in the control regions were kept at the target values with great accuracy throughout the production runs.) These changes highlight the important conclusion that pores with dimensions of 1 to a few nm bounded by basal clay surfaces preferentially incorporate CO2 relative to CH4, with the smallest pores showing the greatest preference (Fig. 2b). These results are in agreement with recent GCMC simulation studies of CO2/CH4 adsorption in the interlayers of Na-montmorillonite at 318 K and Pfluid = 0 to 300 bars.48 Importantly, the CO2/CH4 ratios at small pore sizes (<23.0 Å) are in excellent agreement with the selectivity parameters obtained from recent GCMC simulation studies at similar thermodynamic conditions (323 K and Pfluid = 100 bar).49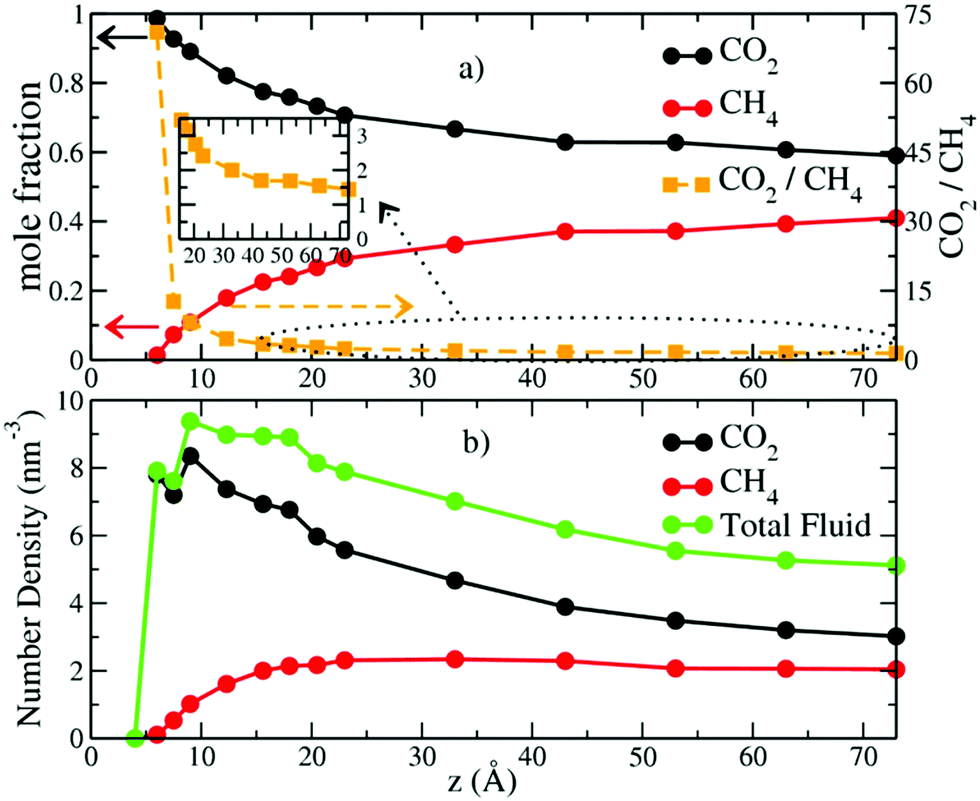 | ||
| Fig. 2 (a) Computed mole fractions of CO2 (black) and CH4 (red) molecules and the CO2/CH4 ratio (gold) as functions of pore thickness. Solid and dashed lines correspond to left and right y axis, respectively. (b) Number density of the fluid species within the pore region bounded by the oxygens of the montmorillonite basal surfaces. | ||
The probability density profiles (PDPs) of CO2 and CH4 in the pores perpendicular to their surfaces show that the changes in their concentrations with pore thickness are due to preferential sorption of CO2 and structuring of the fluid near the pore surfaces (Fig. 3). Significant structuring of the fluid into three discernable layers extends to ∼15.0 Å from each pore surface, with CO2/CH4 ratios larger than in the composition control regions extending to ∼20 Å from the surfaces. At a pore thickness of 6.0 Å, the CO2 molecules are located at the mid-plane of the pore (3.0 Å from each surface), as expected since this thickness corresponds to a clay interlayer with 1 layer of intercalated molecules.32 At a pore thickness of 7.5 Å, the CO2 begins to form two layers, and the small amount of CH4 is at the middle of the pore. At 9.0 Å there are two well developed layers of CO2 and CH4. At 12.3 Å a third layer of CO2 and CH4 begins to develop, and the layer of CO2 nearest the surfaces develops a shoulder. At 15.6 Å the central layer begins to split into two, and at 20.5 Å a fifth layer begins to develop. By 43.0 Å the fluid structuring near the surfaces is fully developed, and the CO2 and CH4 concentrations in the middle of the pore are essentially equal. At 73.0 Å the concentrations in the central region of the pore are close to those in the control volume, 2 molecules per nm3. For all pore thicknesses greater than 33.0 Å the fluid at each surface is structured into three layers of CO2 and CH4 with maxima near 3.0, 6.8, and 10.4 Å and peak densities that decrease with increasing distance from the surface. The PDP's are in good agreement with those from GCMC simulation studies of single component adsorption in slit nanopores in Na-montmorillonite at 323 K and Pfluid = 100 bars with a thickness of ∼21.0 Å49 and also in interlayers of Na-montmorillonite at 298 K and Pfluid = 40 bars.45,46 Such three-layer structures commonly occur near surfaces for many fluids, including H2O.72
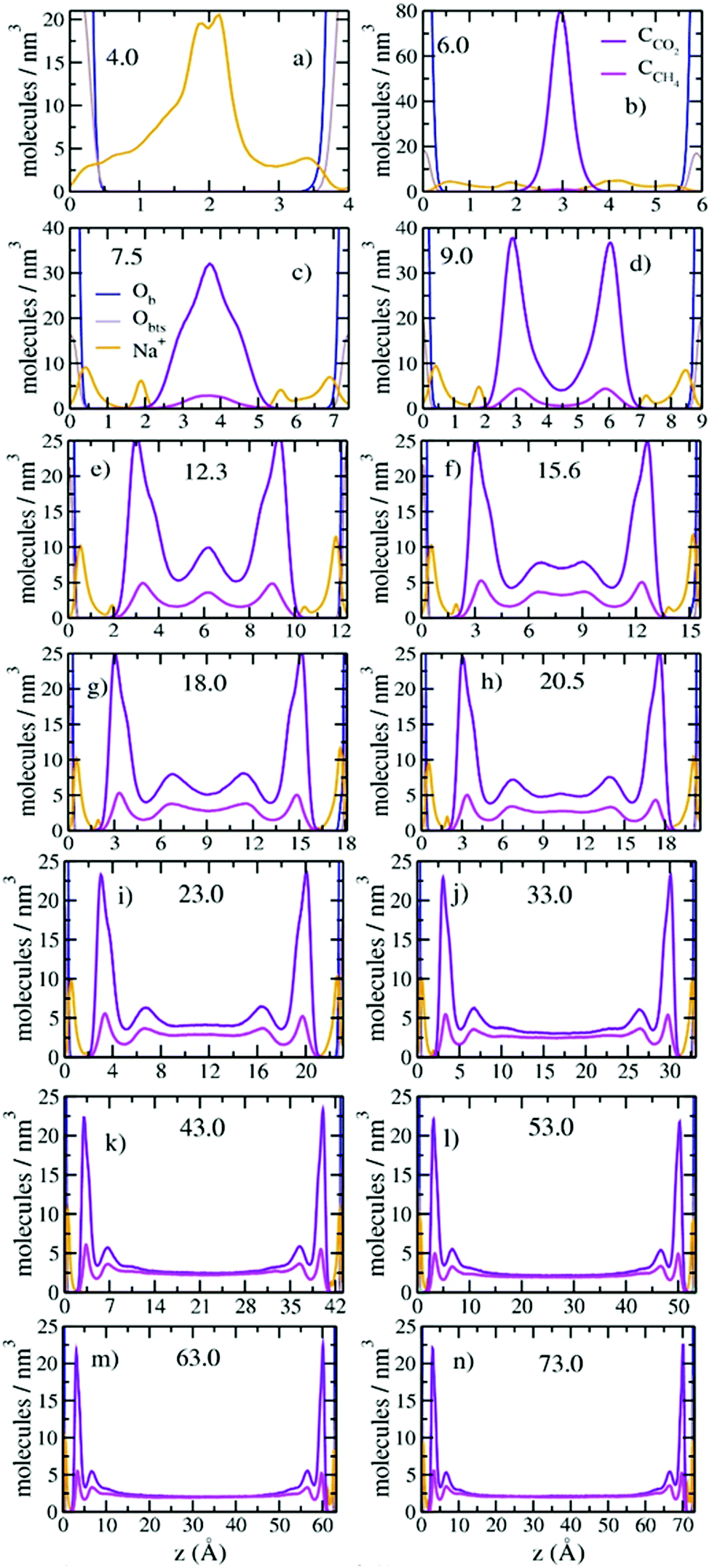 | ||
| Fig. 3 Computed probability density profiles (PDPs) of Na+ (orange), CCO2 (violet) and CCH4 (magenta) as functions of distance normal to the pore surfaces (montmorillonite basal surface) with varying pore thicknesses. The vertical brown and blue lines represent the positions of oxygen atoms of the montmorillonite basal surface coordinated to tetrahedral Si sites (Ob – blue) and tetrahedral Al sites (Obts – brown). | ||
The differences in the structure of the CO2 and CH4 layers near the pore surfaces are due to the differences in their interaction with the basal surface of the clay. For the CO2 layer nearest to the surface, the peak at 3.0 Å is due to CO2 molecules adsorbed with one OCO2 located above the center of a ditrigonal cavity on the basal surface and the other OCO2 located above a Si tetrahedron, as observed in earlier simulation studies of smectite interlayers.25,31,32 The CO2 molecules at 3.9 Å are located with their CCO2 closer to a tetrahedral site. This structuring is consistent with the well-known ability of CO2 to enter smectite clay interlayers.6,20–28,31–34,37,38,40 In contrast, the lack of structuring in the CH4 peak nearest the surface and the equal distances between the three layers suggest that these molecules are interacting much more weakly with the surfaces and behave like hard spheres with the layers packed on one top of each other. This conclusion is consistent with the incorporation of CH4 into the interlayer galleries of expandable clays occurring by a passive, space filling mechanism with basal spacings similar to 7.5 Å.39
Increasing CO2 and CH4 incorporation with increasing pore thickness also greatly effects the coordination of the Na+ ions to the pore surfaces (Fig. 3). At 4.0 Å, most of the Na+ ions are located at the mid-plane of the pore (near 2.0 Å from each surface) with broad shoulders at 0.5 Å from each surface. Based on previous simulation studies by Greathouse et al.,11 the Na+ ions at 0.5 Å are located above the centers of ditrigonal cavities, and those at 2.0 Å are adsorbed near the Si/Al tetrahedra. At 6.0 Å, where CO2 begins to fill the pore, more Na+ occur above the ditrigonal cavities, and as the pore thickness increases progressively more occur on these sites. At larger pore thicknesses (>7.5 Å), the Na+ ions located at 2.0 Å from the surfaces are adsorbed only near tetrahedral Al3+ sites.
As near the basal surfaces bounding the pores, the concentrations of CO2 and CH4 on the surfaces of the broken edges of the clay layers are greater than in the control volumes, and CO2 is preferentially associated with the surface (Fig. 4). The layered structure of the fluid near the broken edge surfaces is also generally similar to that on the basal surfaces, and concentrations greater than in the control regions extending to ∼20 Å from the surface. The concentrations of CO2 and CH4 in the well-defined layers near the broken surfaces are, however, significantly lower than above the basal surfaces (compare values in Fig. 4b with those in Fig. 3), suggesting that both are attracted less strongly to the protonated edge sites than to basal siloxane surfaces. The PDP's show three peaks for CO2 (1.7 Å, 2.8 Å and 6.3 Å) and two peaks for CH4 (3.4 Å and 6.7 Å). The positions of the two peaks for CH4 are the same as above the basal surfaces, again illustrating the weak, non-specific interaction of CH4 with silicate surfaces. In contrast, the positions of the two peaks for CO2 are significantly closer to the broken edge surfaces. Recent GCMC simulation studies showed a greater preference for CO2 relative to CH4 in hydroxylated pores in quartz, which the authors attributed to stronger interaction of CO2 with the protonates sites.49 The origin of this difference will require substantial additional work, but we note that in the results here the differences between the two broken (010) surfaces with differently substituted octahedral and tetrahedral sites are not significant. Note that in these simulations the structuring of the fluid above the broken surfaces does not depend on pore size, since the broken surfaces face the transition region, for which the thickness is independent of pore size. The PDP's of the fluid species parallel to the broken edges (sum of all molecules within 15.0 Å of the surface) further illustrate the preferential association of CO2 with the broken edges (Fig. 4a). These plots also demonstrate that the effects of the surface extend only ∼10 Å into the fluid in the transition region above the opening of the pore.
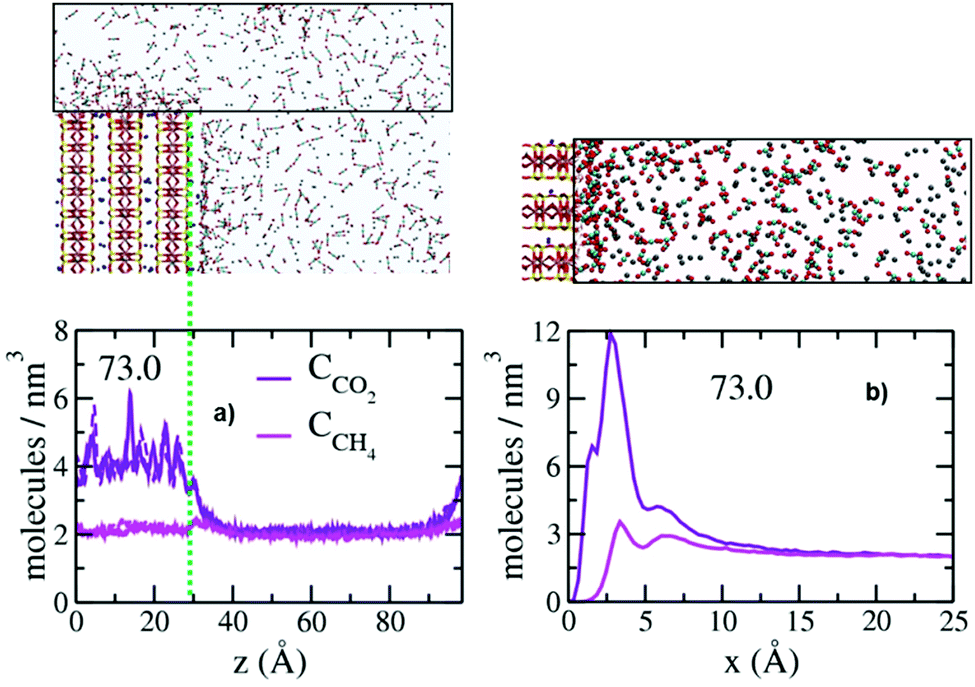 | ||
| Fig. 4 Computed PDP's of CCO2 (violet) and CCH4 (magenta) near the broken and protonated (010) surfaces. (a) Average concentration within 15.0 Å of the surfaces and its extension above the pore (box on the top of the figure) plotted parallel to the surface. (b) PDP normal to the broken edges (box to the right of the surface shown). The solid and dashed lines represent the distributions on the left and right sides of the montmorillonite model which expose differently substituted sites. The differences between these surfaces is not significant. Green dotted lines in (a) represent the plane of basal surface oxygen atoms. | ||
Conclusions
Computational molecular modeling of the partitioning of supercritical CO2 and CH4 between bulk fluid and nano- and meso-pores 4 to 73 Å thick bounded by the basal surfaces of the expandable clay mineral montmorillonite show that both species are preferentially incorporated into the pores with the preference for CO2 much greater than for CH4. This behavior is due to the association of the fluid species with the clay surface and with a much greater preference of the surface for CO2 relative to CH4. Structuring of the fluid extends to ∼15 Å from the surface, and the CO2/CH4 ratio is greater than in the control region to ∼20 Å from the surface. The total CO2/CH4 ratio in the pores is greatest at a pore thickness of 6 Å, decreases rapidly to 33 Å, and then gradually approaches the bulk value at larger thickness. Even at a pore thickness of 73 Å, however, it has not reached this value. The behavior of CO2 and CH4 on protonated, charge-neutral broken (010) edges of the clay layers is broadly similar, but the concentrations of the fluid species near these surfaces are lower than near the basal surfaces, suggesting that they have weaker interactions with protonated edge sites than with siloxane basal surfaces.The calculations were done at 323 K and a total fluid pressure of 124 bars using a novel approach (constant reservoir composition molecular dynamics, CRC-MD) that maintains a constant composition in the fluid external to the pore. In this method, bias forces are employed to maintain the target composition of the external fluid, here 2 molecules per nm3.49–51 This purely MD approach overcomes the difficulties in making stochastic particle insertion–deletion moves in dense fluids encountered in grand canonical Monte Carlo and related hybrid approaches. The simulation cell contains a finite size solid substrate, and the approach can be readily applied to many kinds of porous materials such as zeolites, MOFs and C-based materials. It can also be readily applied to pores bounded by non-porous materials, such as silicates and carbonates.
The results here clearly show that nano- and meso-pores in shales bounded by clay minerals have a preference for CO2 relative to CH4 and thus methods of enhanced petroleum production such as CO2 floods or the use of CO2-based fracking fluids should readily displace CH4 from such pores. The results are in good agreement with previous experimental and computational, high T and P studies that show that there can be an energetic driving force for CO2 incorporation in the interlayer galleries of expandable clay minerals, whereas CH4 enters these spaces by a passive, space filling mechanism.6,31,32,38,39,49 The thickness of the surface layer in which the fluid is structured (∼15 Å) is similar to that in which H2O is structured on oxide and hydroxide surfaces,72,73 suggesting that computational modeling of geochemically relevant fluids in nano- and meso-pores bounded by such materials need only include pores with thicknesses less than a few nm to capture the essential features of their behavior. The results parallel the preference of other inorganic shale components (e.g., calcite and quartz) for CO2 relative to CH4, suggesting that for oxide and oxysalt materials, CH4 adsorption is relatively weak and its filling of pores bounded by them occurs by a passive, space filling mechanism.48–52 The organic component of shale known as kerogen is commonly thought to contain much of the CH4, but recent GCMC studies support the conclusion that although it prefers CO2 relative to CH4, the behavior of these species is strongly dependent on the chemical composition, functionality, moisture content and pore structure of the kerogen.74,75 Overall, the structure, dynamics and energetics of the adsorption environments of CO2, CH4, other hydrocarbons, and H2O in kerogen have not been well explored, and the CRC-MD methods used here can be readily applied to help address these important questions.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge computational resources from the UK Materials and Molecular Modelling Hub, which is partially funded by EPSRC (EP/P020194/1), the National Energy Research Scientific Computing Center, which is supported by the Office of Science of the U.S. Department of Energy under ECARP No. m1649 and the iCER computational facility at Michigan State University. The work in this manuscript was supported by the United States Department of Energy, Office of Science, Office of Basic Energy Science, Chemical Science, Biosciences, and Geosciences division through the sister grants DE-FG02-08ER15929 (Kirkpatrick, P.I. and Yazaydin, co-PI) and DE-FG02-10ER16128 (Bowers, P.I.). A. G. K. and B. F. N. W. acknowledge financial support of the industrial chair “Storage and Disposal of Radioactive Waste” at the IMT-Atlantique, funded by ANDRA, Orano, and EDF, and of the European Union's Horizon 2020 research and innovation program under grant agreements No. 640979 and 764810. We also acknowledge support from the Michigan State University Foundation.References
- A. Busch, S. Alles, Y. Gensterblum, D. Prinz, D. N. Dewhurst, D. M. Raven, H. Stanjek and B. M. Krooss, Int. J. Greenhouse Gas Control, 2008, 2, 297–308 CrossRef CAS.
- B. Cantucci, G. Montegrossi, O. Vaselli, F. Tassi, F. Quattrocchi and H. E. Perkins, Chem. Geol., 2009, 265, 181–197 CrossRef CAS.
- S. J. Altman, B. Aminzadeh, T. M. Balhoff, P. C. Bennett, S. L. Bryant, M. B. Cardenas, K. Chaudhary, R. T. Cygan, W. Deng, T. Dewers, D. A. DiCarlo, P. Eichhubl, M. A. Hesse, C. Huh, E. N. Matteo, Y. Mehmani, C. M. Tenney and H. Yoon, J. Phys. Chem. C, 2014, 118, 15103–15113 CrossRef CAS.
- P. Pei, K. G. Ling, J. He and Z. Z. Liu, J. Nat. Gas Sci. Eng., 2015, 26, 1595–1606 CrossRef CAS.
- A. G. Ilgen, J. E. Heath, I. Y. Akkutlu, L. T. Bryndzia, D. R. Cole, T. J. Kneafsey, K. L. Milliken, L. J. Pyrak-Nolte and R. Suarez-Rivera, Earth-Sci. Rev., 2017, 166, 132–152 CrossRef CAS.
- H. T. Schaef, N. Loganathan, G. M. Bowers, R. J. Kirkpatrick, A. O. Yazaydin, S. D. Burton, D. W. Hoyt, E. S. Ilton, K. S. Thanthiriwatte, D. A. Dixon, B. P. McGrail, K. M. Rosso, E. S. Ilton and J. S. Loring, ACS Appl. Mater. Interfaces, 2017, 9, 36783–36791 CrossRef CAS PubMed.
- G. M. Bowers, J. W. Singer, D. L. Bish and R. J. Kirkpatrick, J. Phys. Chem. C, 2011, 115, 23395–23407 CrossRef CAS.
- C. A. Weiss, R. J. Kirkpatrick and S. P. Altaner, Am. Mineral., 1990, 75, 970–982 CAS.
- C. A. Weiss, R. J. Kirkpatrick and S. P. Altaner, Geochim. Cosmochim. Acta, 1990, 54, 1655–1669 CrossRef CAS.
- C. P. Morrow, A. O. Yazaydin, M. Krishnan, G. M. Bowers, A. G. Kalinichev and R. J. Kirkpatrick, J. Phys. Chem. C, 2013, 117, 5172–5187 CrossRef CAS.
- J. A. Greathouse, D. B. Hart, G. M. Bowers, R. J. Kirkpatrick and R. T. Cygan, J. Phys. Chem. C, 2015, 119, 17126–17136 CrossRef CAS.
- B. F. Ngouana Wakou and A. G. Kalinichev, J. Phys. Chem. C, 2014, 118, 12758–12773 CrossRef.
- R. J. Kirkpatrick, A. G. Kalinichev, G. M. Bowers, A. O. Yazaydin, K. Marimuthu, M. Saharay and C. P. Morrow, Am. Mineral., 2015, 100, 1341–1354 CrossRef.
- N. Loganathan, A. O. Yazaydin, G. M. Bowers, A. G. Kalinichev and R. J. Kirkpatrick, J. Phys. Chem. C, 2016, 120, 10298–10310 CrossRef CAS.
- N. Loganathan, A. O. Yazaydin, G. M. Bowers, A. G. Kalinichev and R. J. Kirkpatrick, J. Phys. Chem. C, 2016, 120, 12429–12439 CrossRef CAS.
- B. Rotenberg, V. Marry, R. Vuilleumier, N. Malikova, C. Simon and P. Turq, Geochim. Cosmochim. Acta, 2007, 71, 5089–5101 CrossRef.
- S. L. Teich-McGoldrick, J. A. Greathouse, C. F. Colon and R. T. Cygan, J. Phys. Chem. C, 2015, 119, 20880–20891 CrossRef CAS.
- G. M. Bowers, J. W. Singer, D. L. Bish and R. J. Kirkpatrick, Am. Mineral., 2014, 99, 318–331 CrossRef CAS.
- U. V. Reddy, G. M. Bowers, N. Loganathan, M. Bowden, A. O. Yazaydin and R. J. Kirkpatrick, J. Phys. Chem. C, 2016, 120, 8863–8876 CrossRef CAS.
- H. T. Schaef, J. S. Loring, V. A. Glezakou, Q. R. S. Miller, J. Chen, A. T. Owen, M. S. Lee, E. S. Ilton, A. R. Felmy, B. P. McGrail and C. J. Thompson, Geochim. Cosmochim. Acta, 2015, 161, 248–257 CrossRef CAS.
- J. S. Loring, H. T. Schaef, R. V. F. Turcu, C. J. Thompson, Q. R. Miller, P. F. Martin, J. Hu, D. W. Hoyt, O. Qafoku, E. S. Ilton, A. R. Felmy and K. M. Rosso, Langmuir, 2012, 28, 7125–7128 CrossRef CAS PubMed.
- J. S. Loring, E. S. Ilton, J. Chen, C. J. Thompson, P. F. Martin, P. Benezeth, K. M. Rosso, A. R. Felmy and H. T. Schaef, Langmuir, 2014, 30, 6120–6128 CrossRef CAS PubMed.
- J. S. Loring, H. T. Schaef, C. J. Thompson, R. V. F. Turcu, Q. R. Miller, J. Chen, J. Hu, D. W. Hoyt, P. F. Martin, E. S. Ilton, A. R. Felmy and K. M. Rosso, Energy Procedia, 2013, 37, 5443–5448 CrossRef CAS.
- E. G. Krukowski, A. Goodman, G. Rother, E. S. Ilton, G. Guthrie and R. J. Bodnar, Appl. Clay Sci., 2015, 114, 61–68 CrossRef CAS.
- A. Botan, B. Rotenberg, V. Marry, P. Turq and B. Noetinger, J. Phys. Chem. C, 2010, 114, 14962–14969 CrossRef CAS.
- A. Kadoura, A. K. N. Nair and S. Sun, J. Phys. Chem. C, 2017, 121, 6199–6208 CrossRef CAS.
- M. Makaremi, K. D. Jordan, G. D. Guthrie and E. M. Myshakin, J. Phys. Chem. C, 2015, 119, 15112–15124 CrossRef CAS.
- A. Rao and Y. Leng, Langmuir, 2016, 32, 11366–11374 CrossRef PubMed.
- M. M. Sena, C. P. Morrow, R. J. Kirkpatrick and M. Krishnan, Chem. Mater., 2015, 27, 6946–6959 CrossRef CAS.
- A. O. Yazaydin, G. M. Bowers and R. J. Kirkpatrick, Phys. Chem. Chem. Phys., 2015, 17, 23356–23367 RSC.
- N. Loganathan, A. O. Yazaydin, G. M. Bowers, A. G. Kalinichev and R. J. Kirkpatrick, J. Phys. Chem. C, 2017, 121, 24527–24540 CrossRef CAS.
- N. Loganathan, G. M. Bowers, A. O. Yazaydin, H. T. Schaef, J. S. Loring, A. G. Kalinichev and R. J. Kirkpatrick, J. Phys. Chem. C, 2018, 122, 4391–4402 CrossRef CAS.
- N. Loganathan, G. M. Bowers, A. O. Yazaydin, A. G. Kalinichev and R. J. Kirkpatrick, J. Phys. Chem. C, 2018, 122, 23460–23469 CrossRef CAS.
- D. R. Cole, A. A. Chialvo, G. Rother, L. Vlcek and P. T. Cummings, Philos. Mag., 2010, 90, 2339–2363 CrossRef CAS.
- D. R. Cole, S. Ok, A. Striolo and A. Phan, Rev. Mineral. Geochem., 2013, 75, 495–545 CrossRef CAS.
- G. Rother, E. S. Ilton, D. Wallacher, T. Hauss, H. T. Schaef, O. Qafoku, K. M. Rosso, A. R. Felmy, E. G. Krukowski, A. G. Stack, N. Grimm and R. J. Bodnar, Environ. Sci. Technol., 2013, 47, 205–211 CrossRef CAS PubMed.
- E. S. Ilton, H. T. Schaef, O. Qafoku, K. M. Rosso and A. R. Felmy, Environ. Sci. Technol., 2012, 46, 4241–4248 CrossRef CAS PubMed.
- G. M. Bowers, H. T. Schaef, J. S. Loring, D. W. Hoyt, S. D. Burton, E. D. Walter and R. J. Kirkpatrick, J. Phys. Chem. C, 2017, 121, 577–592 CrossRef CAS.
- G. M. Bowers, J. S. Loring, H. T. Schaef, E. D. Walter, S. D. Burton, D. W. Hoyt, S. S. Cunniff, N. Loganathan and R. J. Kirkpatrick, ACS Earth Space Chem., 2018, 2, 640–652 CrossRef CAS.
- P. Giesting, S. Guggenheim, A. F. K. van Groos and A. Busch, Int. J. Greenhouse Gas Control, 2012, 8, 73–81 CrossRef CAS.
- A. Phan, D. R. Cole and A. Striolo, J. Phys. Chem. C, 2014, 118, 4860–4868 CrossRef CAS.
- A. Phan, D. R. Cole and A. Striolo, Langmuir, 2014, 27, 8066–8077 CrossRef PubMed.
- A. Phan, D. R. Cole, R. G. Weiss, J. Dzubiella and A. Striolo, ACS Nano, 2016, 8, 7646–7656 CrossRef PubMed.
- A. Phan, D. R. Cole and A. Striolo, Philos. Trans. R. Soc., A, 2016, 374, 20150019 CrossRef PubMed.
- Z. Jin and A. Firoozabadi, Fluid Phase Equilib., 2013, 360, 456–465 CrossRef CAS.
- Z. Jin and A. Firoozabadi, Fluid Phase Equilib., 2014, 382, 10–20 CrossRef CAS.
- N. Yang, S. Liu and X. Yang, Appl. Surf. Sci., 2015, 356, 1262–1271 CrossRef CAS.
- Q. Wang and L. Huang, Fuel, 2019, 239, 32–43 CrossRef CAS.
- H. Sun, H. Zhao, N. Qi, X. Qi, K. Zhang and Y. Li, Mol. Simul., 2017, 43, 1004–1011 CrossRef CAS.
- H. Sun, H. Zhao, N. Qi, X. Qi, K. Zhang, W. Sun and Y. Li, RSC Adv., 2016, 6, 104456 RSC.
- H. Sun, W. Sun, H. Zhao, Y. Sun, D. Zhang, X. Qi and Y. Li, RSC Adv., 2016, 6, 32770–32778 RSC.
- H. Sun, H. Zhao, N. Qi and Y. Li, Energy Technol., 2017, 5, 2065–2071 CrossRef CAS.
- C. Perego, M. Salvalaglioa and M. Parrinello, J. Chem. Phys., 2015, 142, 144113 CrossRef CAS PubMed.
- T. Karmakar, P. M. Piaggi, C. Perego and M. Parrinello, J. Chem. Theory Comput., 2018, 14, 2678–2683 CrossRef CAS PubMed.
- A. Ozcan, C. Perego, M. Salvalaglio, M. Parrinello and A. O. Yazaydin, Chem. Sci., 2017, 8, 3858–3865 RSC.
- G. S. Heffelfinger and F. van Swol, J. Chem. Phys., 1994, 100, 7548 CrossRef CAS.
- K. Okhotnikov, T. Charpentier and S. Cadars, J. Cheminf., 2016, 8, 17 Search PubMed.
- W. Lowenstein, Am. Mineral., 1954, 39, 92–96 Search PubMed.
- S. V. Churakov, J. Phys. Chem. B, 2006, 110, 4135–4146 CrossRef CAS PubMed.
- X. Liu, X. Lu, E. J. Meijer, R. Wang and H. Zhou, Geochim. Cosmochim. Acta, 2012, 81, 56–68 CrossRef CAS.
- G. N. White and L. W. Zelazny, Clays Clay Miner., 1988, 36, 141–146 CrossRef CAS.
- R. T. Cygan, J. J. Liang and A. G. Kalinichev, J. Phys. Chem. B, 2004, 108, 1255–1266 CrossRef CAS.
- S. Plimpton, J. Comp. Physiol., 1995, 117(1), 1–19 CrossRef CAS.
- W. Shinoda, M. Shiga and M. Mikami, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 134103 CrossRef.
- M. Pouvreau, J. A. Greathouse, R. T. Cygan and A. G. Kalinichev, J. Phys. Chem. C, 2017, 121, 14757–14771 CrossRef CAS.
- M. Pouvreau, J. A. Greathouse, R. T. Cygan and A. G. Kalinichev, J. Phys. Chem. C, 2019 DOI:10.1021/acs.jpcc.9b00514.
- R. T. Cygan, V. N. Romanov and E. M. Myshakin, J. Phys. Chem. C, 2012, 116, 13079–13091 CrossRef CAS.
- M. G. Martin and J. I. Siepmann, J. Phys. Chem. B, 1998, 102, 2569–2577 CrossRef CAS.
- S. J. Plimpton, R. Pollock and M. J. Stevens, Proceedings of the Eighth SIAM Conference on Parallel Processing for Scientific Computing, Minneapolis, Minnesota, 1997.
- G. A. Tribello, M. Bonomi, D. Branduardi, C. Camilloni and G. Bussi, Comput. Phys. Commun., 2014, 185, 604–613 CrossRef CAS.
- D. Y. Peng and D. B. Robinson, Ind. Eng. Chem. Fundam., 1976, 15, 59–64 CrossRef CAS.
- J. Wang, A. G. Kalinichev and R. J. Kirkpatrick, Geochim. Cosmochim. Acta, 2006, 70, 562–582 CrossRef CAS.
- S. Gruener, T. Hofmann, D. Wallacher, A. V. Kitky and P. Huber, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2009, 79, 067301 CrossRef PubMed.
- H. Sun, H. Zhao, N. Qi and Y. Li, J. Phys. Chem. C, 2017, 121, 10233–10241 CrossRef CAS.
- L. Huang, Z. Ning, Q. Wang, W. Zhang, Z. Cheng, X. Wu and H. Qin, Appl. Energy, 2018, 210, 28–43 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cp00851a |
| This journal is © the Owner Societies 2019 |