
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Pd local structure and size correlations to the activity of Pd/TiO2 for photocatalytic reforming of methanol†
Norli
Abdullah
 abc,
Hasliza
Bahruji
*ad,
Scott M.
Rogers
ab,
Peter P.
Wells
ef,
C. Richard A.
Catlow
abg and
Michael
Bowker
ag
abc,
Hasliza
Bahruji
*ad,
Scott M.
Rogers
ab,
Peter P.
Wells
ef,
C. Richard A.
Catlow
abg and
Michael
Bowker
ag
aThe UK Catalysis Hub, Research Complex at Harwell, Harwell, Oxon, OX11 0FA, UK
bDepartment of Chemistry, University College London, 20 Gordon Street, London WC1H 0AJ, UK
cDepartment of Chemistry, Centre for Foundation Defence Studies, National Defence University of Malaysia, 57000, Malaysia
dCentre of Advance Material and Energy Sciences, Universiti Brunei Darussalam, Jalan Tungku Link, BE 1410, Brunei. E-mail: hasliza.bahruji@ubd.edu.bn
eUniversity of Southampton, University Road, Southampton SO17 1BJ, UK
fDiamond Light Source Ltd Harwell Science and Innovation Campus, Chilton, Didcot OX11 0DE, UK
gCardiff Catalysis Institute, School of Chemistry, Cardiff University, Main Building, Park Place, Cardiff, CF10 AT, UK
First published on 25th June 2019
Abstract
The interaction between Pd and TiO2 for promoting photocatalytic activity was investigated by tailoring the size of Pd nanoparticles and monitoring the photocatalytic activity of methanol photo-reforming reaction for hydrogen gas production. We show that at 0.6 wt% Pd loading, the catalyst with highly dispersed nanoparticles obtained at 1 °C temperature exhibits superior photocatalytic activity for hydrogen gas production. At different weights of Pd loading, tailoring two sets of catalysts with different structural properties provides correlation between the changes in the Pd local structures and the rate of hydrogen production. The impact of controlling the structural properties of metal nanoparticles on influencing H2 production outweighs the effect of metal loading variation. The differences of Pd/TiO2 activity at the different metal loadings were correlated with the changes in the Pd local structure consequently affecting the electronic transfer and photocatalytic efficiency.
Introduction
Photocatalytic hydrogen production offers a green sustainable route for clean and renewable energy.1,2 The performance of TiO2 is greatly enhanced by the deposition of metal nanoparticles; Pt/TiO2, Au/TiO2 and Pd/TiO2 catalysts have been widely investigated and shown to promote hydrogen generation with UV light.3–8 The enhanced activity is associated with the ability of the photo-induced electron in the conduction band of TiO2 to be transferred to the metal co-catalysts.9,10 Such transfer occurs at the metal island and the periphery of the TiO2 interface, which promotes electron–hole separation and consequently extends the lifetime of the energy carriers.11 The work function differences between the metal and TiO2 semiconductor create a Schottky barrier that allows spontaneous injection of photo-generated electrons from the conduction band of TiO2 into the metal.12 The interfacial charge transfer between TiO2 and the metal is a single electron transfer process, although hydrogen generation from water and methanol photo-reforming reaction requires a multi-electron transfer reaction.13 In order to improve the electronic transfer at the metal–TiO2 interface, we must note that the efficiency relies strongly on the interfacial atomic geometry of metal nanoparticles and TiO2.14 Understanding the geometric strain at metal/TiO2 is important for designing an active catalyst. Au nanoparticles deposited on TiO2 with 3–30 nm size produced hydrogen gas from ethanol, but with 3–12 nm size, the effect of size variation was less significant to enhance photocatalytic activity.15 We proposed in our previous studies that the catalytic activity is significantly correlated to Pd loading, with high activity achieved at very low Pd loading.11 Model studies by Bloh et al. found that the optimal metal to TiO2 ratio ∼2.4 doping atoms per nanometer of particle size was required for an ideal catalytic improvement.16 Exceeding this ratio causes the metal to act as a recombination centre. The decrease in activity at high loading is due to the shadowing effect of metal nanoparticles that reduced the light penetration onto TiO2.17 Excess metal nanoparticles on TiO2 are also suggested to become electron and hole trapping sites.18,19Pd is a precious noble metal with a large work function to trap electrons for efficient electron hole separation. We have previously investigated the activity of Pd/TiO2 for photocatalytic reforming of alcohols with the mechanism of hydrogen production involving dehydrogenation of alcohol on the Pd surface and water reduction occurring at the Pd–TiO2 interface.20–23 The efficiency of Pd to promote electron transfer from the conduction band of the semiconductor is not limited to TiO2, and studies on visible light driven photocatalysts such as BiFeO4 and GaN–ZnO showed enhancement in the photocatalytic performance for hydrogen production.24–26 Alteration of the Pd morphology to nanocubes improved the hydrogen production from Pd/2D-C3N4 composites due to the efficient separation of photogenerated energy carriers.27 Understanding the role of the metal nanoparticles in promoting the catalytic activity of TiO2 is often hampered by morphology alterations, particularly at high metal loading.15 The aims of the work are to improve the Pd/TiO2 photocatalytic activity and to gain fundamental understanding of structural changes of Pd upon variation of metal loading. In these studies, tailored Pd metal nanoparticles were prepared using the controlled kinetic growth of Pd colloids via synthesis temperature variations to obtain Pd nanoparticles of different but controlled nanoparticle sizes. Systematic Pd/TiO2 catalyst design managed to differentiate the promotional effect caused by the size of Pd nanoparticles and the amount of metal loading, allowing us to gain an understanding of the effect of the Pd–TiO2 interaction on promoting hydrogen production from photo-reforming of methanol:
| CH3OH + H2O + hν → 3H2 + CO2 |
Results
To enhance the activity of Pd/TiO2 catalysts, the particle size of Pd was tailored by controlling the synthesis temperature of the Pd colloid at a fixed loading of 0.6 wt%. EXAFS is a useful technique that allows the study of nanoparticles and provides local structural information of palladium. The local coordination of Pd was obtained by recording ex situ Pd K-edge EXAFS spectra of catalysts after drying at ambient temperature (Fig. 1). Ex situ Pd K-edge EXAFS spectra were recorded for 0.6 Pd wt% TiO2 catalysts when Pd synthesis was carried out at temperatures of 1 °C, 25 °C, 50 °C and 75 °C. The Pd K-edge XANES of 0.6% Pd/TiO2 prepared at 1 °C shows the Pd environment of the samples corresponding to the Pd metal with a Pd–Pd distance of 2.78 Å.28 The sample also shows the peak associated with the Pd–O first shell with a distance of 2.00 Å.29 The absence of a second shell of Pd–(O)–Pd at 3.3 Å shows that the Pd particles may exist as a subnanometer Pd cluster.30 As the synthesis temperature increased to 25 °C, the Pd–O first shell peak height with a distance of 1.99 Å decreased, accompanied by an increase of the Pd–Pd peak with a radius distance of 2.74 Å, suggesting the growth of Pd0 nanoparticles. The amplitude of the Pd–O first shell decreased with the temperature, which correlates well with the increasing intensity of the Pd–Pd peak at 2.74 Å. This signifies the enlargement of Pd metal nanoparticles as the temperature of the colloidal Pd solution was increased up to 75 °C.31 The EXAFS fitting parameter in Table 1 also revealed the coordination number of the Pd–Pd and Pd–O first shell. It is clear that the Pd–O first shell coordination number reduced from 2.8 for the catalyst obtained at 1 °C to 0.7 as the temperature increased to 75 °C. In contrast, the Pd–Pd coordination numbers increased from 2.8 to 8.4. The variation of the coordination number is associated with the structural and morphological changes, which in this case are due to the enlargement of Pd nanoparticles as the temperature of the colloidal Pd solution increased.32 We can conclude that there are no large PdO crystallites and rather the PdO component originates from the surface oxide, due to the absence of the Pd–Pd scattering at 3.00 Å, which would occur in such crystallites. For the catalyst obtained at 1 °C, the Pd–O and Pd–Pd coordination numbers of 2.8 signify that the Pd nanoparticles for this sample are significantly smaller in comparison to the catalyst obtained at higher temperatures of synthesis. | ||
| Fig. 1 k 3 weighted Fourier transform (magnitude) EXAFS data for the 0.6 wt% Pd/TiO2 catalysts prepared at 1 °C, 25 °C, 50 °C and 75 °C. | ||
| Catalysts | Pd size/nm | Abs. Sc. | N | R/Å | 2/A | E f/eV | R factor | Reference standards (%) | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Pd2+ | Pd0 | R factor | Pd2/Pd0 | ||||||||
| 0.6% Pd/TiO2 | 2.3 ± 0.6 | Pd–Pd | 2.8(3) | 2.78(1) | 0.008 | 4(1) | 0.02 | 69 | 31 | 0.063 | 2.22 |
| 1 °C | Pd–O | 2.8(2) | 2.00(1) | 0.003 | |||||||
| 0.6% Pd/TiO2 | 3.0 ± 0.4 | Pd–Pd | 5.4(3) | 2.738(7) | 0.008 | 7(1) | 0.02 | 46 | 54 | 0.025 | 0.85 |
| 25 °C | Pd–O | 1.8(2) | 1.99(1) | 0.003 | |||||||
| 0.6% Pd/TiO2 | 3.8 ± 0.3 | Pd–Pd | 7.2(2) | 2.743(3) | 0.008 | 8(0) | 0.003 | 30 | 70 | 0.038 | 0.43 |
| 50 °C | Pd–O | 1.1(1) | 1.98(1) | 0.003 | |||||||
| 0.6% Pd/TiO2 | 5.2 ± 0.8 | Pd–Pd | 8.4(2) | 2.750(3) | 0.008 | 8(0) | 0.003 | 22 | 78 | 0.026 | 0.28 |
| 75 °C | Pd–O | 0.7(1) | 1.98(1) | 0.003 | |||||||
| 1% Pd/TiO2 | 2.6 ± 0.4 | Pd–Pd | 4.2(4) | 2.75(1) | 0.008 | 5(1) | 0.019 | 57 | 43 | 0.099 | 1.32 |
| 1 °C | Pd–O | 1.7(4) | 2.00(0) | 0.006 | |||||||
| 1% Pd/TiO2 | 3.3 ± 0.7 | Pd–Pd | 7.7(3) | 2.744(4) | 0.008 | 5(1) | 0.007 | 33 | 67 | 0.048 | 0.49 |
| 50 °C | Pd–O | 1.1(2) | 1.99(1) | 0.006 | |||||||
| 2% Pd/TiO2 | 3.5 ± 0.8 | Pd–Pd | 6.6(3) | 2.739(5) | 0.008 | 5(1) | 0.009 | 35 | 65 | 0.198 | 0.53 |
| 1 °C | Pd–O | 1.7(2) | 2.00(1) | 0.006 | |||||||
| 4% Pd/TiO2 | 3.8 ± 0.8 | Pd–Pd | 8.3(4) | 2.735(5) | 0.008 | 6(1) | 0.013 | 33 | 67 | 0.494 | 0.49 |
| 1 °C | Pd–O | 0.5(3) | 1.97(5) | 0.006 | |||||||
XANES linear combination analysis (LCA) of the 1st derivative, using PdO and Pd foil as the reference standard, was performed to calculate the ratio between Pd2+ and Pd0, as summarised in Table 1. Small Pd nanoparticles form an oxidic surface layer at room temperature. Since smaller particles have a higher surface contribution to XAFS compared with larger particles, the relative size of the Pd nanoparticles can be inferred from the ratio of Pd2+/Pd0. Analysis data given in Table 1 showed that the influence of the synthesis temperature on the Pd2+/Pd0 ratio is shown by a higher percentage of Pd2+ for the catalyst prepared at 1 °C (69% Pd2+), in comparison to that prepared at 75 °C (22% Pd2+). The ratio of Pd2+/Pd0 for the catalyst synthesised at 1 °C is 2.2, significantly higher than the rest of the catalysts. The calculated Pd2+/Pd0 ratio for 0.6% Pd/TiO2 decreases with increasing synthesis temperatures.
The variation of the Pd nanoparticle size distribution from the 0.6 wt% Pd/TiO2 catalyst was further confirmed by TEM analysis, as shown in Fig. 2. The Pd size was measured based on the average diameter of 100 Pd particles observed from TEM images. Controlling the temperature at 1 °C produced Pd nanoparticles with an average particle size of 2.3 nm. Increasing the synthesis temperature to 25 °C shows relatively larger particles with an average diameter of 3.0 nm. The Pd nanoparticle size continues to grow with an average diameter of 3.8 nm for 50 °C synthesis, and 5.2 nm for 75 °C. In general, upon increasing the synthesis temperature for the 0.6% Pd/TiO2 catalyst, the Pd–O (at 2.00 Å) coordination increases, meanwhile the Pd–Pd (∼2.78 Å) coordination decreases, with decreasing average Pd particle size.
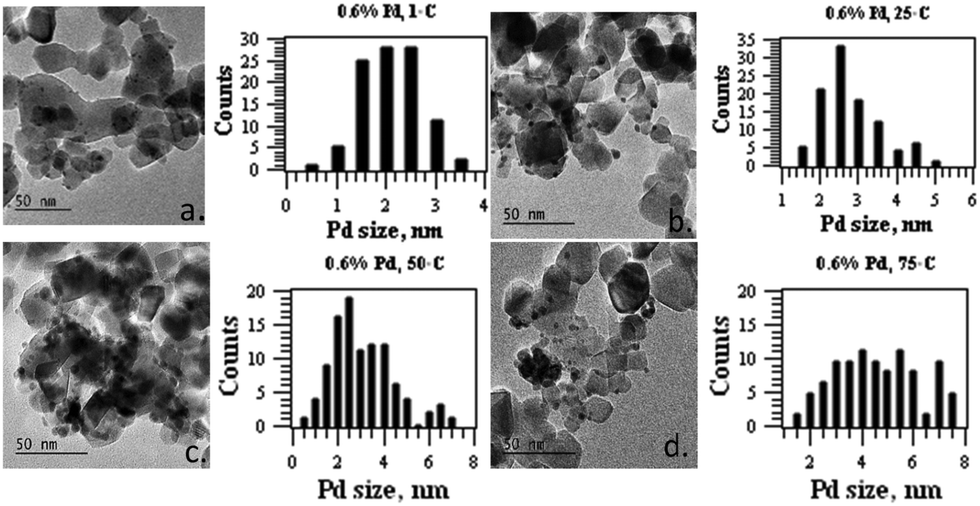 | ||
| Fig. 2 TEM images and Pd size distribution histograms of 0.6 wt% Pd TiO2 synthesised at (a) 1 °C, (b) 25 °C, (c) 50 °C and (d) 75 °C. | ||
The resulting 0.6% Pd/TiO2 catalysts were subsequently used in photocatalytic hydrogen generation by photoreforming of methanol. Fig. 3 shows the plot of hydrogen gas production analysed for every 30 minutes during 3 h reaction. The 0.6% Pd/TiO2 catalyst obtained from the colloidal sol that was prepared at 1 °C, produced ∼676 μmol of H2 in 3 h of reaction. The value is significantly higher than that of the rest of the catalysts at a similar weight loading.
 | ||
| Fig. 3 Photocatalytic hydrogen production from methanol on 0.6 wt% Pd/TiO2 prepared at different synthesis temperatures. | ||
As the temperature of the colloidal sol increases, the results of the 0.6% Pd/TiO2 catalysts showed that the hydrogen generation is significantly affected with the total hydrogen volume reduced to ∼434 μmol for the catalyst produced at 25 °C, 230 μmol for 50 °C and only 186 μmol when the synthesis temperature was raised to 75 °C. The increased activity of 0.6% Pd/TiO2 catalysts produced at 1 °C was compared with our previous finding in which the rate of hydrogen production showed a twofold enhancement in comparison with Pd/TiO2 produced via an impregnation method.10,20
As the impact of controlling the size of metal nanoparticles on increasing H2 productivity is dramatic, we therefore increased the Pd metal loading to study its effect on photocatalytic performance. The Pd loadings were varied at 0.6 wt%, 1 wt%, 2 wt% and 4 wt%. Two sets of catalysts were prepared at different weight loadings, with the first set of catalysts produced at 1 °C to ensure that a narrow Pd nanoparticle distribution was achieved. The second set of catalysts was prepared by varying the synthesis temperature in order to obtain Pd nanoparticles with similar particle sizes. TEM analysis data in Table 1 showed that the particle size of Pd on TiO2 in the first set of catalysts increased with the amount of metal loading, despite the undertaking of the synthesis at 1 °C. Fig. 4 shows the TEM images of 1%, 2% and 4% Pd/TiO2, and the particle size distribution histogram obtained from the sol immobilisation method carried out at 1 °C. Pd at 1% loading shows a Pd diameter of 2.6 nm meanwhile at 2% loading, the average diameter was measured to be 3.5 nm. As the Pd loading increases, the size increases to 3.8 nm at 4% loading.
 | ||
| Fig. 4 TEM images and Pd size distribution histograms of (a) 1% Pd/TiO2 synthesised at 1 °C, (b) 1% Pd/TiO2 synthesised at 50 °C, (c) 2% Pd/TiO2 prepared at 1 °C and (d) 4% Pd/TiO2 prepared at 1 °C. | ||
Table 1 also shows the XAFS analysis data of the Pd/TiO2 catalysts at different weight loadings. The value of Pd–Pd coordination number increased with the amount of Pd loading, implying that larger Pd particles were produced at higher Pd loadings. Similar occurrences for the Pd2+/Pd0 ratios derived from the XANES analysis revealed that the ratio decreases with the amount of Pd loading, from 1.32 at 1% Pd, to 0.53 at 2% Pd and 0.49 at 4% Pd loading. In general, for the first set of catalysts obtained at 1 °C, the amount of Pd loading was varied in the range of 0.6–4 wt%, producing catalysts with an average Pd nanoparticle size of 2–4 nm, with the Pd2+/Pd0 ratios in the range of 2.2–0.49. The catalysts were subsequently used for photocatalytic hydrogen production by photoreforming of methanol. The plot of hydrogen production against reaction time when using the first set of catalysts is shown in Fig. 5a. The catalysts produced a high volume of hydrogen within 3 h of reaction at low Pd loading, with ∼636 μmol of hydrogen for 0.6% Pd loading. The volume was significantly reduced to 512 μmol for 1% Pd/TiO2. As the Pd loading was increased, the photocatalytic activity of the catalysts was significantly reduced with only ∼181 μmol of hydrogen produced at 4% Pd loading.
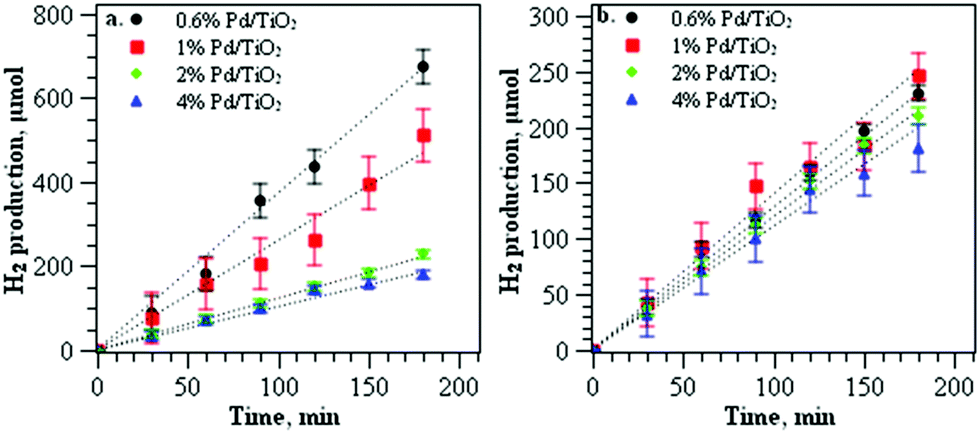 | ||
| Fig. 5 (a) Photocatalytic hydrogen production from methanol on Pd/TiO2 prepared at 1 °C at different weight loadings with an average diameter of 2–4 nm; (b) photocatalytic hydrogen production from methanol on Pd/TiO2 prepared at different weight loadings with an average diameter of 3.5 nm. | ||
The decrease of TiO2 activity at high metal loading is often related to the shadowing effect of the metal that reduces the TiO2 photosensitivity.17 To gain further understanding of the factors influencing the catalytic activity at high metal loading, the second set of catalysts was used for the evaluation of photocatalytic performance. The catalysts were obtained by varying the synthesis temperature for 0.6 wt%. 1 wt%, 2 wt% and 4 wt% of Pd in order to produce catalysts with approximately similar Pd nanoparticle intrinsic structural properties. For 0.6% Pd/TiO2 and 1% Pd/TiO2 catalysts, the synthesis temperature was set at 50 °C, meanwhile for 2% and 4% Pd/TiO2 catalysts, the temperature was set at 1 °C to give a Pd diameter of 3.5 nm. The list of particle sizes of Pd nanoparticles analysed using TEM and the local structure of Pd from XAFS analysis are summarised in Table 1. Apart from similar Pd diameters within 3.3–3.8 nm, the catalysts also consisted of palladium nanoparticles with approximately similar Pd2+/Pd0 ratios ∼0.43–0.53 (Table 1). The photocatalytic performance of the catalysts for hydrogen gas production is shown in Fig. 5b. The catalysts produced similar rates of hydrogen production with the volume of hydrogen production ∼180 to 230 μmol in 3 h of reaction. In comparison to the first set of catalysts that were produced at 1 °C, controlling the size of Pd nanoparticles around 3.5 nm appears to reduce the differences in the catalytic performance of Pd/TiO2 catalysts despite the different weights of metal loading deposited on TiO2.
Discussion
We tailored the size of Pd nanoparticles deposited on TiO2 by controlling the kinetic growth of Pd in the sol solutions. The correlations between the catalytic performance, and the size and the local structure of Pd for 0.6% Pd obtained at different temperatures are shown in Fig. 6. The catalyst obtained from the colloids prepared at 1 °C with an average Pd diameter of 2.3 nm shows superior activity for hydrogen production. The photocatalytic performance is appreciably reduced upon increasing the size of Pd particles. Fig. 6 also shows a clear relationship between the extent of Pd2+ and the Pd particle diameter by TEM. The presence of Pd2+ in the oxidic surface layer of Pd is associated with the surface contribution of Pd to form an interfacial interaction with TiO2. It is important for Pd to be in the Pd0 oxidation state in order to catalyse the hydrogen production steps.8 PdO on TiO2 is reduced to Pd0 in the presence of methanol under UV irradiation prior to sustainable hydrogen production from methanol.10 Despite PdO being reduced to Pd by methanol it is important that some Pd2+ is present at the interface. The susceptibility of Pd2+ to accept an electron increased the efficiency of photogenerated electron trapping that occurs at the Pd–TiO2 interface. The small Pd nanoparticles on TiO2 enhanced the interfacial interaction between TiO2 and palladium, subsequently promoting the efficient electronic transfer from the conduction bands of TiO2 to the Pd metal. Clearly, at a similar metal loading, the catalytic activity in promoting hydrogen generation is significantly influenced by the particle size of Pd, which we relate to the surface Pd–TiO2 interaction. Since all the catalysts were pre-treated under similar conditions, i.e. drying at room temperature prior to the reaction, a strong metal support interaction that often occurs via high temperature annealing is not applicable in this case. However, generating small Pd nanoparticles produced a higher degree of interfacial interaction between TiO2 and palladium. The increase in the oxide–metal interface area promotes the Schottky effect for electron excitation and subsequently enhances the electronic transfer between the conduction bands of TiO2 and the Pd. | ||
| Fig. 6 Comparison of the catalyst performance in hydrogen production plotted against the size of tailored 0.6% Pd prepared by temperature controlled colloidal synthesis. Percentages of Pd oxidation states derived from normalised XANES spectra were also plotted against the particle size of Pd derived using TEM analysis. | ||
Understanding the fundamental aspect of the effect of particle size and metal weight loading is a challenging task due to the agglomeration of nanoparticles at high metal loading. Controlling the particle size of Pd while increasing the weight of the metal deposited on TiO2 allows an understanding of the geometric influence of Pd nanoparticles on photocatalytic performance. Fig. 7a shows the hydrogen production rate dependence on Pd loading and the normalised H2 rate over Pd loading for the first set of catalysts with an average Pd size of 2–4 nm. The rate of hydrogen evolution is significantly reduced as the Pd loading increased to 4%. When the activity is normalised to the amount of Pd loading, the differences in the catalytic activity are even more significant. Varying the amount of metal loading is often associated with the agglomeration of nanoparticles particularly at high metal loading, thus altering the local structure of the metal. This proved that the reduction of Pd/TiO2 activity at high metal loading is due to the differences in the Pd local structure that affected the efficiency of electronic transfer at Pd–TiO2 interfaces.
 | ||
| Fig. 7 Photocatalytic hydrogen generation on Pd/TiO2 at various Pd loadings. (a) Plot of hydrogen generation rate --- and normalised hydrogen rate ---- against Pd weight loading on the catalyst with a variation of Pd diameter in the range of 2–4 nm. (b) Plot of hydrogen generation rate --- and normalised hydrogen rate --- against Pd weight loading on the catalyst with a controlled Pd diameter of 3.5 nm. | ||
On the other hand, the second set of Pd/TiO2 catalysts prepared by varying the synthesis temperature showed a near invariant rate of hydrogen evolution of ∼80 μl h−1 regardless of the amount of Pd loading. When the rate of hydrogen production is normalised to the amount of Pd content, there is only a slight decrease in the hydrogen production rate at higher Pd loading (Fig. 7b). A linear relationship was observed between the rate of hydrogen and the amount of Pd loading implying a similar Pd local structure of the catalysts. We can deduce that the catalytic activity is affected by Pd structural properties, and varies little with the loading amount. The results showed that the impact of controlling the amount of surface palladium interacting with TiO2 in increasing H2 productivity outweighs the effect of metal weight loading.
Experimental
Catalyst preparation
To synthesize the Pd colloidal sol, PVA was used as a stabiliser ligand with NaBH4 as the reducing agent. The size of Pd nanoparticles was controlled by controlling the temperature of Pd nucleation, in which the kinetic growth was altered by variation of the temperature from 1–75 °C. This method was previously reported for Au and found to be reliable for the synthesis of small Au nanoparticles.33 An aqueous solution of K2PdCl4 (Alfa Aesar, 99.9% metal basis) at the desired concentration was prepared. Polyvinylalcohol (PVA) (1 wt% solution, Aldrich, Mw = 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, 80% hydrolysed) was added (PVA/Pd = 0.65 weight ratio). A 0.1 M freshly prepared solution of NaBH4 (>96% Aldrich, NaBH4/Pd = 5 molar ratio) was then added to form a dark-brown sol. After 30 min of sol generation, the colloid was immobilised by adding TiO2 (acidified at pH 1 by sulphuric acid) under vigorous stirring conditions. After 2 h, the slurry was filtered and washed with deionised water before drying in air at ambient temperature for 16 hours. Palladium loading was varied at 0.6 wt%, 1 wt% and 2 wt% and 4 wt% on the TiO2 P25 support.
000, 80% hydrolysed) was added (PVA/Pd = 0.65 weight ratio). A 0.1 M freshly prepared solution of NaBH4 (>96% Aldrich, NaBH4/Pd = 5 molar ratio) was then added to form a dark-brown sol. After 30 min of sol generation, the colloid was immobilised by adding TiO2 (acidified at pH 1 by sulphuric acid) under vigorous stirring conditions. After 2 h, the slurry was filtered and washed with deionised water before drying in air at ambient temperature for 16 hours. Palladium loading was varied at 0.6 wt%, 1 wt% and 2 wt% and 4 wt% on the TiO2 P25 support.
To provide detailed morphological and compositional information about the studied samples at micro and nano-scale, transmission electron microscope (TEM) system JEOL 2100 (LaB6) is employed. For the analysis, the material was ground and mixed with water and 2 mL of materials in water suspension was placed on the TEM grid and dried. The instrument is equipped with a high-resolution Gatan digital camera (2k × 2k) providing maximum resolution of 0.2 Å which makes possible detailed observation of the crystal lattice, obtaining diffraction patterns and accurate measurement of the lattice d spacing with the help of Digital Micrograph software. In scanning transmission electron microscopy (STEM) mode, dark field (HAADF/Z-contrast) detector was used to provide excellent compositional contrast. An energy dispersive X-ray spectrometer (EDS) system (Oxford Instruments) was equipped with a large-area 80 mm2 SDD (Silicon Drift Detector). X-MaxN 80 T was employed for the elemental analysis in line scans, and elemental mapping modes. To analyse the EDS data, the latest version of AZtecTEM software was utilized. The particle diameter of Pd was obtained based on 100 Pd particles observed in TEM images using ImageJ software.
X-ray photoelectron spectra (XPS) were recorded on a Kratos Axis Ultra-DLD XPS spectrometer with a monochromatic Al kα source (75–150 W) and analyser pass energies of 160 eV (for survey scans) or 40 eV (for detailed scans). Samples were mounted using a double-sided adhesive tape and binding energies referenced to the C (1s) binding energy of adventitious carbon contamination which was taken to be 284.7 eV. Data were analyzed using Casa XPS software.
Pd K edge X-ray absorption spectra (XAS) were obtained on the B18 beamline at Diamond Light Source, Didcot, UK. Measurements were performed using a QEXAFS set-up with a fast-scanning Si (311) double crystal monochromator. The time resolution of the spectra reported herein was 1 min per spectrum (kmax = 16, step size 0.5 eV), on average six scans were acquired to improve the signal to noise level of the data for transmission measurements. All samples were diluted with cellulose and pressed into pellets to optimize the effective edge-step of the X-ray absorption fine structure (XAFS) data and measured in transmission mode using ion chamber detectors. Detailed experimental procedures and characterisation data are available in the ESI.†
Photocatalytic measurement
Photocatalytic activity was determined from gas phase photoreforming of methanol under UV irradiation (280–380 nm). 50 mg of the catalyst was mixed with water to form a paste and was dispersed on a glass slide with an irradiated area of the film of ∼7.5 cm2. The catalyst was left to dry under ambient conditions. The glass slide was then placed on a three neck flask containing 15 ml of water and 100 μl of methanol. The system was purged with Ar for 30 minutes and sealed using a rubber stopper. The catalyst was irradiated using an Oriel Xe lamp with 150 Watt from the side of the flask. A gas sample was analysed every 30 min for 3 hours using a PerkinElmer Clarus GC with a TCD detector.Conclusions
Temperature controlled immobilisation of Pd on TiO2 is a reliable method to produce tailored Pd nanoparticles with controlled size and local structures. In correlation with the TEM and XAFS analysis, it is clear that reducing the size of Pd nanoparticles while maintaining the amount of metal weight loading significantly enhanced the photocatalytic activity of Pd/TiO2 for hydrogen gas production. Tailoring two sets of catalysts by increasing the Pd loading allows an understanding of the geometrical effect of metal nanoparticles in enhancing the photocatalytic performance of TiO2 in the UV region. The first set of catalysts revealed that increasing the Pd weight loading also affects the Pd intrinsic structure, which consequently produced large Pd particles and reduced the efficiency of electronic transfer between the Pd and TiO2 interface. However, the second set of catalysts, in which the particle diameter and structural properties of Pd were tailored to be approximately similar, showed negligible differences in the rate of hydrogen generation. The presence of surface Pd2+ to accept an electron from the conduction band of TiO2 increased the efficiency of photogenerated electron trapping that occurs at the Pd–TiO2 interface. This shows the dominant effect of the Pd size and local structure in influencing the photocatalytic activity of the Pd/TiO2 catalyst for hydrogen production from photoreforming of methanol.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge UK Catalysis Hub and EPSRC for research funding to H. Bahruji through grant EP/I038748/1. The Ministry of Education, Malaysia (MOE) is thanked for financial support for a Post-doctoral award to N. Abdullah. The authors wish to thank Diamond Light Source for award of beam time through the BAG access to B18 (allocation number SP10306). The authors also wish to thank the help and assistance of the B18 staff, specifically Dr Diego Gianolio. UK Catalysis Hub is kindly thanked for resources and support provided via our membership of the UK Catalysis Hub Consortium and funded by EPSRC grant: EP/K014706/2, EP/K014668/1, EP/K014854/1, EP/K014714/1 or EP/M013219/1.Notes and references
- C. Acar, I. Dincer and G. F. Naterer, Int. J. Energy Res., 2016, 40, 1449–1473 CrossRef CAS.
- A. A. Ismail and D. W. Bahnemann, Sol. Energy Mater. Sol. Cells, 2014, 128, 85–101 CrossRef CAS.
- A. L. Luna, D. Dragoe, K. Wang, P. Beaunier, E. Kowalska, B. Ohtani, D. Bahena Uribe, M. A. Valenzuela, H. Remita and C. Colbeau-Justin, J. Phys. Chem. C, 2017, 121, 14302–14311 CrossRef CAS.
- Z. H. N. Al-Azri, W.-T. Chen, A. Chan, V. Jovic, T. Ina, H. Idriss and G. I. N. Waterhouse, J. Catal., 2015, 329, 355–367 CrossRef CAS.
- J.-J. Zou, H. He, L. Cui and H.-Y. Du, Int. J. Hydrogen Energy, 2007, 32, 1762–1770 CrossRef CAS.
- G. M. Haselmann and D. Eder, ACS Catal., 2017, 7, 4668–4675 CrossRef CAS.
- J. B. Priebe, J. Radnik, A. J. J. Lennox, M.-M. Pohl, M. Karnahl, D. Hollmann, K. Grabow, U. Bentrup, H. Junge, M. Beller and A. Brückner, ACS Catal., 2015, 5, 2137–2148 CrossRef CAS.
- H. Bahruji, M. Bowker, P. Davies, D. A. Morgan, C. A. Morton, T. Egerton, J. Kennedy and W. Jones, Top. Catal., 2014, 2–3, 70–76 Search PubMed.
- M. Ni, M. K. H. Leung, D. Y. C. Leung and K. Sumathy, Renewable Sustainable Energy Rev., 2007, 11, 401–425 CrossRef CAS.
- H. Bahruji, M. Bowker, P. R. Davies, J. Kennedy and D. J. Morgan, Int. J. Hydrogen Energy, 2015, 40, 1465–1471 CrossRef CAS.
- A. Dickinson, D. James, N. Perkins, T. Cassidy and M. Bowker, J. Mol. Catal. A: Chem., 1999, 146, 211–221 CrossRef CAS.
- M. R. Khan, T. W. Chuan, A. Yousuf, M. N. K. Chowdhury and C. K. Cheng, Catal. Sci. Technol., 2015, 5, 2522–2531 RSC.
- H. Park, H.-i. Kim, G.-h. Moon and W. Choi, Energy Environ. Sci., 2016, 9, 411–433 RSC.
- H. Chen, P. Li, N. Umezawa, H. Abe, J. Ye, K. Shiraishi, A. Ohta and S. Miyazaki, J. Phys. Chem. C, 2016, 120, 5549–5556 CrossRef CAS.
- M. Murdoch, G. I. N. Waterhouse, M. A. Nadeem, J. B. Metson, M. A. Keane, R. F. Howe, J. Llorca and H. Idriss, Nat. Chem., 2011, 3, 489–492 CrossRef CAS PubMed.
- J. Z. Bloh, R. Dillert and D. W. Bahnemann, J. Phys. Chem. C, 2012, 116, 25558–25562 CrossRef CAS.
- J. B. Zhong, et al. , J. Hazard. Mater., 2009, 168, 1632–1635 CrossRef CAS PubMed.
- O. Ola and M. M. Maroto-Valer, Appl. Catal., A, 2015, 502, 114–121 CrossRef CAS.
- K. Bhattacharyya, S. Varma, A. K. Tripathi, S. R. Bharadwaj and A. K. Tyagi, J. Phys. Chem. C, 2008, 112, 19102–19112 CrossRef CAS.
- J. Kennedy, H. Bahruji, M. Bowker, P. R. Davies, E. Bouleghlimat and S. Issarapanacheewin, J. Photochem. Photobiol., A, 2018, 356, 451–456 CrossRef CAS.
- H. Bahruji, M. Bowker, P. R. Davies and F. Pedrono, Appl. Catal., B, 2011, 107, 205–209 CrossRef CAS.
- H. Bahruji, M. Bowker, P. R. Davies, L. S. Al-Mazroai, A. Dickinson, J. Greaves, D. James, L. Millard and F. Pedrono, J. Photochem. Photobiol., A, 2010, 216, 115–118 CrossRef CAS.
- H. Bahruji, M. Bowker and P. R. Davies, J. Chem. Sci., 2019, 131, 33 CrossRef.
- S. Wang, D. Chen, F. Niu, N. Zhang, L. Qin and Y. Huang, Appl. Phys. A: Mater. Sci. Process., 2016, 122, 867 CrossRef.
- S. Wang, D. Chen, F. Niu, N. Zhang, L. Qin and Y. Huang, RSC Adv., 2016, 6, 34574–34587 RSC.
- Z. Li, F. Zhang, J. Han, J. Zhu, M. Li, B. Zhang, W. Fan, J. Lu and C. Li, Catal. Lett., 2018, 148, 933–939 CrossRef CAS.
- Z. Mo, H. Xu, X. She, Y. Song, P. Yan, J. Yi, X. Zhu, Y. Lei, S. Yuan and H. Li, Appl. Surf. Sci., 2019, 467–468, 151–157 CrossRef CAS.
- E. K. Dann, E. K. Gibson, R. A. Catlow, P. Collier, T. E. Eralp and D. Gianolio, et al., Molecular Precursors, Chem. Mater., 2017, 29(17), 177515 CrossRef.
- K. Okumura, R. Yoshimoto, T. Uruga, H. Tanida, K. Kato and S. Yokota, et al. , J. Phys. Chem. B, 2004, 108, 6250–6255 CrossRef CAS PubMed.
- E. J. Peterson, A. T. DeLaRiva, S. Lin, R. S. Johnson, H. Guo, J. T. Miller, J. Hun Kwak, C. H. F. Peden, B. Kiefer, L. F. Allard, F. H. Ribeiro and A. K. Datye, Nat. Commun., 2014, 5, 4885 CrossRef CAS PubMed.
- A. M. Beale and B. M. Weckhuysen, Phys. Chem. Chem. Phys., 2010, 12(21), 5562–5574 RSC.
- M. A. Newton, C. Belver-Coldeira, A. Martinez-Arias and M. Fernandez-Garcia, Nat. Mater., 2007, 6, 528–532 CrossRef CAS PubMed.
- S. M. Rogers, C. R. A. Catlow, C. E. Chan-Thaw, D. Gianolio, E. K. Gibson, A. L. Gould, N. Jian, A. J. Logsdail, R. E. Palmer, L. Prati, N. Dimitratos, A. Villa and P. P. Wells, ACS Catal., 2015, 5, 4377–4384 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cp00826h |
| This journal is © the Owner Societies 2019 |