
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructural dynamics of an excited donor–acceptor complex from ultrafast polarized infrared spectroscopy, molecular dynamics simulations, and quantum chemical calculations†‡
Christopher A.
Rumble
 * and
Eric
Vauthey
*
* and
Eric
Vauthey
*
Department of Physical Chemistry, University of Geneva, 30 Quai Ernest-Ansermet, CH-1211 Geneva 4, Switzerland. E-mail: christopher.rumble@unige.ch; eric.vauthey@unige.ch
First published on 15th May 2019
Abstract
The structural dynamics of an electron donor/acceptor complex (DAC) consisting of benzene and tetracyanoethylene (Bz/TCNE) solvated in CH2Cl2 have been investigated using ultrafast spectroscopy and mixed quantum/classical computer simulations. Population dynamics from visible and infrared transient absorption (TRIR) spectroscopy point to complex sub-10 ps dynamics followed by charge recombination on a 55–60 ps timescale. Structural dynamics involving large-scale reorganization of Bz˙+/TCNE˙− radical ion pairs are revealed using TRIR anisotropy measurements. A computational study combining quantum chemical calculations and classical molecular dynamics simulations was able to reproduce the experimental electronic absorption lineshape and TRIR anisotropy dynamics, allowing for a detailed investigation of the pair conformational dynamics. Contrary to the static single structure typically assumed in descriptions of DACs, we find that neither the ground nor excited state can be described using a single, well-defined species. Instead, the pair undergoes a rearrangement from disordered π-stacks to edge-to-face T-shaped structures following excitation. Translational diffusion of the radical ion pairs following excitation was found to be heterogeneous and dependent on both pair separation and orientation coordinates. Given the sensitivity of charge-transfer reactions to the arrangement of donor/acceptor pairs, the structural heterogeneity and corresponding dynamics demonstrated herein must be taken into account in future modeling of charge recombination processes in DACs.
1 Introduction
The electronic absorption spectrum of electron donor (D) and acceptor (A) mixtures in solution is often characterized by the presence of a low-energy electronic absorption band not present in the spectra of the individual constituents. This band is due to a charge-transfer (CT) transition of a donor–acceptor complex (DAC),1 and its excitation leads to the population of an excited state that is essentially a radical ion pair (IP).2,3 Such direct optical generation of ion pairs has made DACs attractive targets for testing electron-transfer theories, as it allows one to monitor the charge-recombination (CR) dynamics of ion pairs without the complications associated with their formation upon bimolecular photoinduced electron transfer.3–6 In this respect, significant advances in our understanding of electron-transfer processes have been achieved thanks to studies of DACs, including the role of intramolecular modes7–9 and the effect of non-equilibrium dynamics.10–12Surprisingly, little is known about the structure of DACs in liquids, although such information is crucial for a quantitative analysis of their electron-transfer dynamics. DACs are often assumed to have a well-defined structure consisting of D and A moieties in a π-stacked face-to-face arrangement. This picture is based primarily on X-ray diffraction studies of crystalline DACs,13,14 spectroscopic measurements in supersonic jets,15,16 and quantum-chemical calculations.17–21 However, a significant number of experimental observations point toward an influence of DA pair structure on DAC photophysics in liquids. Complexes with substituted benzene donors exhibit two CT absorption bands ascribed to different mutual donor/acceptor orientations favoring the overlap of the acceptor LUMO with either the HOMO or HOMO−1 of the donor.22,23 Excitation wavelength-dependent transient absorption studies of CR in DACs have been rationalized in terms of non-equilibrium CR in a mixture of tightly and loosely bound complexes.24,25 Braun and Sutin explained variations in free-ion yields following excitation at different wavelengths by invoking an equilibrium between well-defined DACs and ‘random’ solvent-separated pairs with a distribution of DA separations.26,27 Vibrational wavepackets observed in ultrafast studies of DACs in liquids were originally ascribed to intermolecular modes of the complexes,28,29 similar to those measured below ∼100 cm−1 in crystalline DACs.30 However, subsequent investigations revealed that these wavepackets originated from vibrational modes localized on either D or A subunits.31–33 It was concluded that the absence of intermolecular vibrational wavepackets most probably reflects a distribution of DAC geometries in the ground state. Finally, polarized transient electronic absorption measurements revealed significantly lower anisotropy values than those expected for perfectly formed π stacked ‘sandwich’ structures.25,29,34,35 These low values were interpreted in terms of either multiple distinct geometries or structural disorder, but no detailed modeling of the anisotropies was carried out.
The present work attempts to address three primary questions regarding the structure and dynamics of DACs:
1. What structures do DACs adopt in the ground state? Are they predominately face-to-face π-stacks? How much heterogeneity is present?
2. Of these ground-state structures, which can undergo CT excitation? These excitable neutral pairs will determine the structure of the ion pairs at the onset of the CR process.
3. How does the excited-state population evolve after excitation? Do the IPs keep the same structure from which they were excited, or do they adopt a different conformation or set of conformations than in the ground state?
To this end, we have combined transient visible absorption (visTA) and polarized time-resolved infrared absorption (TRIR) spectroscopies with mixed quantum/classical computer simulations. We have chosen to focus on the well-known pair of benzene (Bz, donor) and tetracyanoethylene (TCNE, acceptor) solvated in CH2Cl2, which has been the subject of recent experimental36,37 and computational investigations.38,39 The Bz/TCNE pair has a single CT absorption band peaked near 400 nm, allowing for easy excitation using the doubled fundamental of a Ti:Sapphire laser system, and exhibits sufficiently slow (∼50 ps) CR to enable observation of the complete excited state structural dynamics. Additionally, the TCNE radical anion, TCNE˙−, has strong vibrational markers associated with the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N stretching modes.40 These vibrational transition are located around 2150 cm−1, a relatively uncongested region of the IR spectrum, and have proven useful in previous infrared studies of DACs and bimolecular electron transfer using TCNE as an acceptor.41,42
N stretching modes.40 These vibrational transition are located around 2150 cm−1, a relatively uncongested region of the IR spectrum, and have proven useful in previous infrared studies of DACs and bimolecular electron transfer using TCNE as an acceptor.41,42
We employ both visTA and TRIR measurements in order to provide an overview of the excited-state population dynamics. Polarized TRIR is then used to measure time-resolved vibrational anisotropy and obtain unprecedented insight into the geometry of excited DACs and their structural dynamics. To support the experiments, we have undertaken a systematic computational study of the Bz/TCNE/CH2Cl2 system combining quantum chemical calculations at the density-functional level of theory (DFT) and classical molecular dynamics (MD) simulations. Using umbrella sampling techniques and time-dependent density functional theory (TD-DFT) calculations we are able to characterize the ground- and excited-state distribution of Bz/TCNE structures as well as identify a distribution of ground-state geometries that allow for CT excitation. We then perform non-equilibrium MD in order to model the experimental TRIR anisotropy dynamics with help from the TD-DFT calculations. After validation by comparison to the experimental visible absorption lineshape and TRIR anisotropy, the results of the non-equilibrium MD simulations are used to examine translational diffusion of the ion pairs. The results of this study reveal that the Bz/TCNE DAC cannot be thought of as a well-defined ground-state complex that undergoes CT excitation to a static ion-pair structure. The anisotropy experiments and computations point to an initial population of structurally disordered ion pairs that relax to a predominantly T-shaped arrangement and not a π-stack as is usually assumed. Given the demonstrated sensitivity of charge-transfer processes to donor/acceptor orientation,43,44 theoretical descriptions of charge recombination dynamics in DACs must take into account DA pair structural diversity and the evolution of the excited state in order to properly model the charge recombination reaction.
2 Methods
2.1 Materials
HPLC grade benzene, toluene, p-xylene, and CH2Cl2 were purchased from Sigma-Aldrich and used as received. Tetracyanoethylene was purchased from Sigma-Aldrich and sublimed before use. Samples were prepared by adding donor to 0.04 M TCNE in CH2Cl2 to reach an absorbance at 400 nm of 0.1 in the 1 mm visTA cell and 0.2–0.3 in the 0.3 mm TRIR cell. Given the small association constant of the binary complexes45 and the low donor and acceptor concentrations we have employed, we expect negligible amounts of the ternary complex to be present. The steady-state UV-vis absorption spectra of the visTA and TRIR samples were identical despite the differences in benzene concentration.2.2 Spectroscopy
Steady-state UV-vis absorption measurements were performed using a Cary 50 spectrophotometer. Previously described instruments were used for the visible transient absorption (visTA),46,47 and infrared transient absorption (TRIR) measurements.48,49 Instrumental and data processing details are provided in Sections S1 and S2 of the ESI.‡2.3 Computations
All quantum chemical calculations were carried out using the Gaussian09 Rev. D.01 package50 with the CAM-B3LYP functional51 and a 6-31G(d,p) basis set. Electrostatic potentials generated from these calculations were fit using the CHELPG method52 to assign atomic charges to Bz, Bz˙+, TCNE, and TCNE˙− for use as solutes in the MD simulations. Linear response TD-DFT calculations using the same functional and basis set were performed on select Bz/TCNE conformations in order to calculate the 5 lowest energy electronic transitions of the pairs. Molecular orbitals were visualized using the Avogadro 1.2.0 software package.53All molecular dynamics (MD) simulations were performed using the GROMACS 2018.1 package,54 and the OPLS-AA force field.55 Simulation parameters and procedural details for equilibration, umbrella sampling, potential of mean force, and simulated TRIR anisotropy calculations are provided in Sections S3–S5 of the ESI.‡ The MD trajectories were visualized using the VMD 1.9.3 package56 and the Tachyon ray tracing library.57
3 Results and discussion
3.1 Electronic transient absorption
The steady-state electronic absorption spectrum of Bz/TCNE in CH2Cl2 consists of a single broad, featureless, CT band with a maximum at 388 nm (Fig. S1, ESI‡). Neither Bz nor TCNE absorb in this region, meaning that the excited-state population upon 400 nm excitation originates solely from CT transitions of Bz/TCNE. Representative visTA spectra of Bz/TCNE in CH2Cl2 are presented in the top panel of Fig. 1. Although these data are generally poor in structural information, they provide a detailed view of the excited-state population dynamics. VisTA spectra collected at early times consist of a weak negative band below 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 cm−1 (606 nm), and a broad positive band peaking at 22000 cm−1 (455 nm). During the first 10 ps this broad feature undergoes a narrowing on its low-energy side, whereas the negative band decays to zero. At later times, the relaxed positive band decays entirely within ∼200 ps.
500 cm−1 (606 nm), and a broad positive band peaking at 22000 cm−1 (455 nm). During the first 10 ps this broad feature undergoes a narrowing on its low-energy side, whereas the negative band decays to zero. At later times, the relaxed positive band decays entirely within ∼200 ps.
 | ||
| Fig. 1 Top: VisTA spectra of benzene/TCNE in CH2Cl2 following 400 nm excitation. Bottom: DADS from a four exponential global analysis of the visTA data in the top panel. | ||
Decay-associated difference spectra (DADS) and time constants resulting from a global multiexponential analysis of the visTA spectra are presented in the bottom panel of Fig. 1. Corresponding evolution-associated difference spectra (EADS), obtained by assuming successive exponential steps in the global analysis, are presented in Fig. S2 of the ESI.‡ A sum of four exponential functions is required to reproduce the spectra consisting of three components with sub-10 ps time constants that appear in the DADS as both rises and decays with a complicated wavelength dependence. A fourth component is present with a 55 ps time constant that is always a decay, and its DADS is nearly identical to the long-time visTA spectrum. The negative band coincides well with the steady-state fluorescence spectrum of this DAC reported by Chiu et al.36 and can be assigned to stimulated emission. Its sub-10 ps decay together with the concomitant narrowing on the low-energy side of the positive band are interpreted as relaxation from the emissive Franck–Condon state to the equilibrium, IP-like, excited-state of the DAC. This is consistent with the electronic absorption spectra of TCNE˙− and Bz˙+ reported in literature.58–60 Equilibration of the Franck–Condon state should involve vibrational, structural and solvent relaxation processes which cannot be disentangled, as they occur on similar timescales. Structural relaxation results in a decrease of the electronic coupling between the D and A subunits, hence the rapid decay of the stimulated emission. The complexity of the early dynamics is reflected by the need for three exponential functions to reproduce it. On the other hand, the 55 ps component can be attributed to the CR of the Bz˙+/TCNE˙− ion pairs. The visTA spectra do not substantially decrease in intensity before 10 ps, suggesting that no significant CR occurs before equilibration of the excited DAC. Our recombination time of 55 ps is somewhat slower than the 29 ps reported by Chiu et al.36 using time-resolved fluorescence. This experiment is only able to examine the recombination of fluorescent pairs, whereas our visTA measurements include contributions from all pairs, possibly explaining the discrepancy in the recombination times. The 55 ps time constant is consistent with values reported for other DACs with a similar energy gap for recombination in medium polarity solvents.25,61,62
3.2 Time-resolved IR absorption and anisotropy
N stretching modes of TCNE˙− centered near 2150 cm−1 and 2185 cm−1.40 Such spectra of Bz/TCNE in CH2Cl2 are presented the top panel of Fig. 2. These data show that both CN stretching bands undergo a frequency upshift of ∼4 cm−1 and a slight (∼10%) increase in intensity before decaying. Similar dynamics have been explained in previous TRIR studies of bimolecular ET processes involving TCNE as indicative of tight (lower frequency) and loose (higher frequency) ion pairs, but were not observed in direct CT excitation due to prohibitively fast recombination.42 Blue shifts and narrowing in TRIR spectra are also observed in vibrational cooling and energy redistribution processes.63,64 Given the relatively large amount of excess energy deposited into the pair following 400 nm excitation, these processes are also expected to contribute to the spectral dynamics.
 | ||
| Fig. 2 Top: TRIR spectra of benzene/TCNE in CH2Cl2 in the nitrile stretch region following 400 nm excitation. Bottom: DADS from a three exponential global analysis of TRIR data in the top panel. | ||
Three exponential components were sufficient for reproducing the TRIR dynamics using a global analysis. The resulting time constants and DADS are presented in the bottom panel of Fig. 2 and EADS in Fig. S3 (ESI‡). The initial rise and frequency up-shift are associated with the 4.3 and 13 ps time constants whereas the decay occurs with a 63 ps time constant. These data are consistent with the visTA and draw a similar picture of the excited state population dynamics. The DAC Franck–Condon state equilibrates on a ∼10 ps time scale through vibrational, solvent, and conformational relaxation, and the ensuing ion pair recombines with a ∼60 ps time constant. Unfortunately, the TRIR population dynamics themselves do not yield significant structural information about the pairs.
![[M with combining circumflex]](https://www.rsc.org/images/entities/i_char_004d_0302.gif) ex, relative to the probed IR transition moment i (outlined in Fig. 3), according to65
ex, relative to the probed IR transition moment i (outlined in Fig. 3), according to65 | (1) |
ex and i, providing a direct link to the excited-state structure and dynamics.
 | ||
| Fig. 3 Schematic representation of the Bz/TCNE DAC with experimentally relevant visible and vibrational transition dipole moments labeled. The vector ex represents the pumped CT transition dipole moment, and 1 and 2 represent the nitrile vibrations probed in TCNE˙−. | ||
Before presenting the anisotropy data, we must first describe the transition dipole moments involved in the TRIR measurements and the anticipated geometry of the Bz/TCNE pair. Fig. 3 illustrates a canonical π-stacked ‘sandwich’ DAC with the orientations of the electronic and vibrational transition dipole moments labeled. The electronic CT transition dipole of DACs is known to lie perpendicular to the molecular planes of the donor and acceptor, whereas the probed CN vibrational transition dipole moments both lie in the molecular plane of TCNE and are mutually perpendicular.42 We define 1 as the transition dipole moment of the 2150 cm−1 antisymmetric stretch which lies along the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond and 2 as the transition dipole moment of the symmetric stretch near 2185 cm−1 which lies in the TCNE molecular plane orthogonal to 1.
C bond and 2 as the transition dipole moment of the symmetric stretch near 2185 cm−1 which lies in the TCNE molecular plane orthogonal to 1.
The TRIR anisotropy of Bz/TCNE measured at 2147 and 2188 cm−1 is presented in Fig. 4 and best-fit parameters in Table 1. Population and anisotropy time constants from fits at different frequencies of the TRIR spectrum are provided in Table S1 of the ESI.‡ We observe a clearly bimodal anisotropy at 2147 cm−1 with a 5.9 ps rise and a 47 ps decay, whereas the 2188 cm−1 anisotropy begins negative and simply relaxes to zero on the order of 10 ps. The decay of the negative anisotropy of both bands occurs on a similar timescale as the spectral dynamics in the visTA and TRIR spectra, possibly reflecting a shared physical origin. The best-fit parameters in Table S1 (ESI‡) do not exhibit a significant frequency dependence within a band and the variation most likely reflects uncertainties in the data.
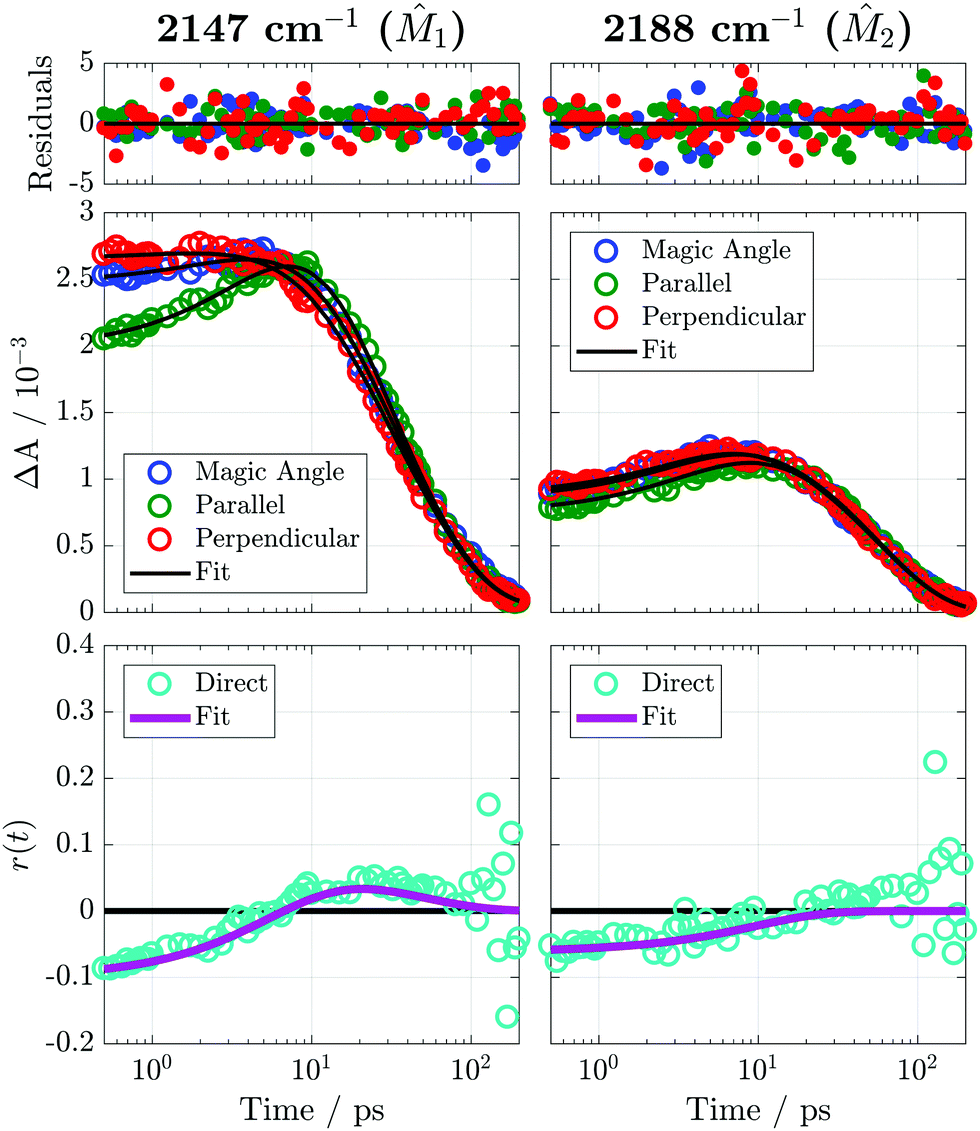 | ||
| Fig. 4 Polarized TRIR time traces and anisotropy measured with Bz/TCNE/CH2Cl2 at 2147 cm−1 and 2188 cm−1 following 400 nm excitation. Residuals were weighted using the standard error of the 8 individual measurements as uncertainties. These uncertainties would be approximately the size of the symbols if plotted as error bars. Fitting of the TRIR traces and the anisotropy was performed using the model described in Section S2 of the ESI.‡ The points labeled ‘direct’ are the anisotropy calculated using the definition r(t) = [ΔA‖(t) − ΔA⊥(t)]/[ΔA‖(t) + 2ΔA⊥(t)]. This representation of the anisotropy was not analyzed directly but is intended to serve as a check on the fitting procedure. | ||
| Population dynamics | ||||||||
|---|---|---|---|---|---|---|---|---|
![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) (cm−1) (cm−1) |
h | bkg | a 1 | τ 1 (ps) | a 2 | τ 2 (ps) | a 3 | τ 3 (ps) |
| 2147 | 4.77 | 0.04 | −0.25 | 4.9 | 0.26 | 15 | 0.50 | 50 |
| 2188 | 2.29 | 0.02 | −0.14 | 2.1 | −0.18 | 8.6 | 0.68 | 51 |
| Anisotropy dynamics | ||||||
|---|---|---|---|---|---|---|
|
(cm−1) |
G | r 0 | r 1 | τ r,1 (ps) | r 2 | τ r,2 (ps) |
| 2147 | 0.976 | −0.10 | −0.16 | 5.9 | 0.06 | 47 |
| 2188 | 0.979 | −0.06 | −0.06 | 9.8 | — | — |
A number of processes could be responsible for the temporal evolution of the anisotropy including: (1) changes in the mutual orientation of the D and A subunits, (2) reorientation of the whole excited complex, and (3) decay of subpopulations of excited DACs with a well-defined geometry and faster CR dynamics. As discussed earlier, no significant population decay occurs during the first 10 ps and, consequently, process (3) should not be important here. The Stokes–Einstein–Debye (SED) prediction for the reorientation time of the whole complex is 19.4 ps using a van der Waals volume of 194 Å3,66 a CH2Cl2 viscosity of 0.411 mPa s at 298 K,67 and stick boundary conditions. This is significantly longer than the τr,1 values measured here. Time constants on the order of SED predictions were observed in earlier TRIR anisotropy studies of exciplexes of 9-cyanoanthracene and phthalic anhydride in acetonitrile and tetrahydrofuran.48 Therefore, the decay of the negative anisotropy can be attributed to structural relaxation of the DAC after excitation and the slow decay of the positive anisotropy found with 1 to the reorientation of the complex and randomization of Bz/TCNE orientations.
A qualitative interpretation of the initial anisotropies can be attempted using the schematic in Fig. 3 as a guide. If all excited DACs were sandwich structures as depicted in Fig. 3, we would expect an initial anisotropy of r(i)0 = −0.2 for both probed vibrations and not the values of r(1)0 = −0.10 and r(2)0 = −0.06 seen in the experiment. Other sandwich structures obtained by rotating TCNE about the ex axis would have the same initial anisotropy of −0.2. Therefore, the reduced initial anisotropies must result from rotation of the Bz and TCNE molecular planes with respect to each other. The observed values of r(i)0 could be the result of (1) significant orientational disorder, (2) multiple well-defined structures with different anisotropies, or (3) a single structure with an angle of 65° between ex and 1 and 61° between ex and 2. Because the anisotropy experiment reports only on ensemble averaged orientations, one cannot discriminate between these three cases without resorting to detailed modeling of the system. The sign change from negative to positive in the 2147 cm−1 anisotropy indicates that 1 initially rotates into the same direction as ex, whereas 2 either randomizes or adopts a magic angle (54.7°) orientation with respect to ex. These dynamics are consistent with the formation of edge-to-face (T-shaped) or edge-to-edge structures, but we do not have the benzene-specific anisotropies required to discriminate between the two, as the TRIR bands of Bz˙+ modes were too weak to reliably extract anisotropies.
It should be noted that these anisotropies are not unique to the Bz/TCNE complex. TRIR anisotropy measurements of toluene/TCNE and p-xylene/TCNE complexes in CH2Cl2, presented in Fig. S4, S5 and Tables S2, S3 (ESI‡), exhibit somewhat stronger initial anisotropies with r0 ranging from −0.14 to −0.17 with single exponential relaxation that appears slower than in Bz/TCNE. Unfortunately, charge recombination in these pairs is 5–10 times faster than in Bz/TCNE due to different driving forces for these reactions, quickly depopulating the excited state and obscuring the long-time (>10 ps) anisotropy. Therefore, we cannot be confident in the anisotropy time constants and cannot determine if the 1 anisotropy changes sign, an unambiguous indicator of structural rearrangement. Regardless of the lack of long time data, the larger initial anisotropies and clearly slower anisotropy dynamics may be due to the formation or preferential excitation of tighter face-to-face complexes. Further interpretation of the experimental anisotropies is difficult, as there is no unique distribution of structures that can reproduce the observed anisotropy; the experiments only report on the ensemble average of structures and their corresponding anisotropies. In order to learn more about the structures and dynamics reported on by the anisotropy we must turn to computer simulations validated by comparison to the experiments.
3.3 Mixed quantum/classical computations
A combined quantum/classical computational study of the Bz/TCNE system was undertaken in order to aid interpretation of the TRIR anisotropies. There are a number of key physical processes which must be modeled in order to simulate the experimental anisotropy in a sufficiently rigorous manner. First, we must determine which Bz/TCNE structures are present in the ground state and which of these are able to undergo CT excitation. Then we must simulate the dynamics of the ion pairs resulting from the CT excitation and calculate anisotropies according to eqn (1). To accomplish this, we combine classical MD simulations with TD-DFT calculations using the following procedure:1. Characterize the ground-state Bz/TCNE structures using the potential of mean force calculated from umbrella sampling MD simulations.
2. Select a representative set of ground-state Bz/TCNE conformations from the umbrella sampling simulations and use TD-DFT to calculate CT transition energies, transition dipole moments, and oscillator strengths.
3. Using the same set of configurations, run non-equilibrium MD trajectories after substituting Bz/TCNE charges with those of Bz˙+/TCNE˙−. This serves to model the CT transition as an instantaneous transfer of a full electron from Bz to TCNE.
4. Calculate an anisotropy for each non-equilibrium trajectory according to eqn (1) using CT transition dipole moments from the TD-DFT calculations for ex(0) and TCNE˙− coordinates from the simulations to calculate 1(t) and 2(t). The final simulated anisotropy is calculated by averaging the individual trajectory anisotropies weighted by the CT transition oscillator strength at t = 0.
 | ||
| Fig. 5 Potentials of mean force (PMFs) for neutral Bz/TCNE and ionic Bz˙+/TCNE˙− pairs. The black dashed lines are intended to guide the eye toward the minima of the neutral and ion pair PMFs. Inset: Neutral Bz/TCNE pair distribution function used for selection of conformations for the TD-DFT calculations and non-equilibrium MD simulations. | ||
The neutral pair w(r) has a minimum of −0.3kBT at r = 0.37 nm (−0.66 kJ mol−1 at T = 293.15 K). Because this free energy is significantly smaller than kBT, we conclude there is little attraction between neutral Bz and TCNE in room temperature CH2Cl2. Rather than as ground state complexes, it is better to consider Bz/TCNE pairs in this solvent as dilute cosolvents. The shallow w(r) reported here is consistent with the weak association constant of 0.1 L mol−1 measured for Bz/TCNE complexes using steady-state UV-vis absorption.45 In contrast to the neutrals, the ion pair w(r) has a significantly deeper minimum of −22.5kBT and is found at a longer separation, r = 0.54 nm. The difference in the location of the minimum and the large change in interaction strength point to significant structural rearrangement following excitation. The initially disordered (in r) ground-state population will be funneled into the deep and narrow minimum of the ionic w(r) after excitation, effecting a significant conformational change. The depth of this minimum also suggests that no free ions will form on an experimentally significant timescale. We expect that the high degree of structural diversity demonstrated in the Bz/TCNE DAC studied here to be present in other DACs as well. Preliminary PMF calculations for pairs of dimethylaniline and aromatic acceptors in acetonitrile (Fig. S6, ESI‡) exhibit similarly shallow minima and would be expected to have structural disorder similar to that of Bz/TCNE.
We turn to orientational distribution functions, G(θ), to characterize mutual Bz/TCNE orientations of the neutral and ionic pairs. After selection of appropriate molecular vectors, these distributions are calculated from Bz/TCNE conformations generated by the PMF calculations according to:
| G(θ) = 〈δ(θ − ϕ)〉 | (2) |
1 in Fig. 3) and for Bz originates at its center-of-mass and points between two Bz H atoms. The out-of-plane vectors are simply normal to the Bz and TCNE molecular planes. These vectors are illustrated by the inset images in Fig. 6. Note that these distributions are by definition weighted by sin(θ), therefore, an isotropic distribution of orientations corresponds to a sin function as illustrated by the gray lines in Fig. 6. A similar approach was recently used to predict the relative orientation of the individual constituents in perylene diimide aggregates.69
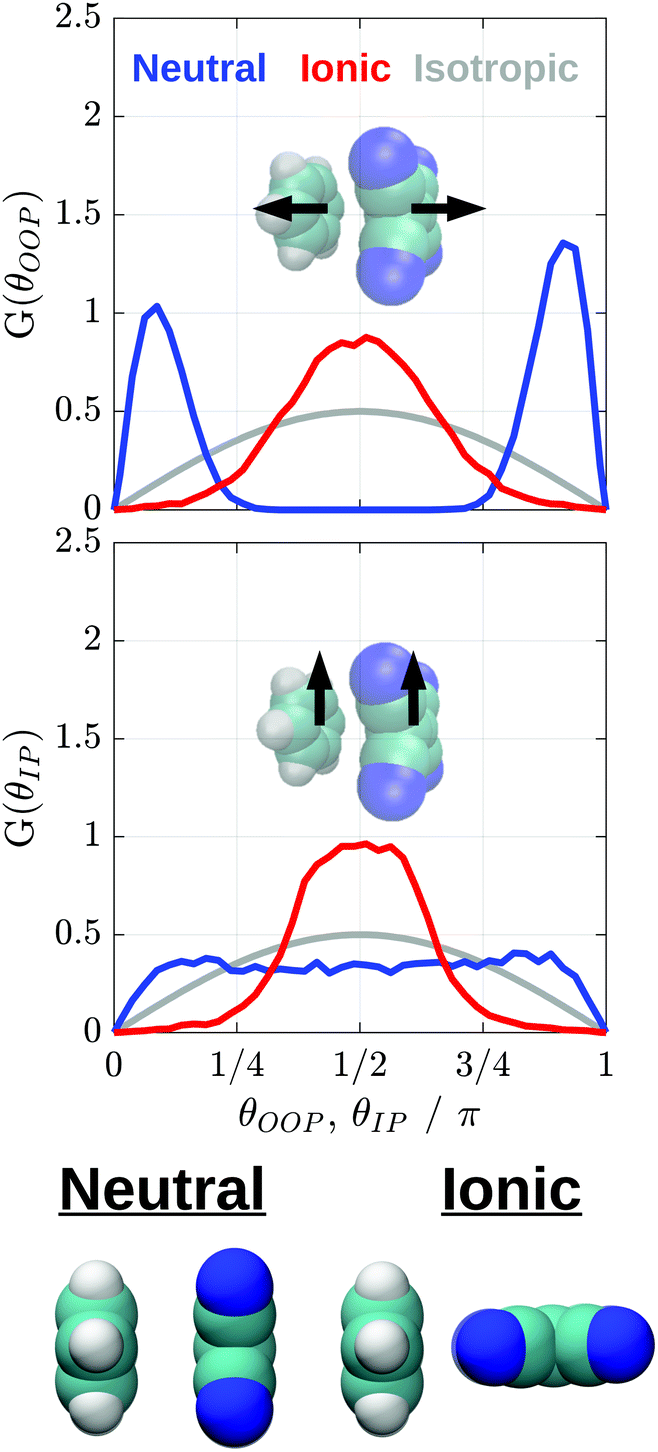 | ||
| Fig. 6 Orientational distributions for Bz and TCNE pairs within 0.025 nm of the minimum of w(r) for the neutral (blue) and ionic (red) species. The angles θOOP, and θIP represent the angles between the out-of-plane and in-plane molecular vectors indicated on each plot. These distributions are normalized for unit area. The gray lines represent the expectation for an isotropic distribution of orientations. Conformations depicted beneath the plots represent the most probable Bz/TCNE structures predicted by the orientational distributions. | ||
The out-of-plane vectors of neutral Bz/TCNE show a strong preference for mutual orientations of 0 and π, as would be expected for a face-to-face sandwich structure. There are much weaker preferences for orientations of 0 and π for the in-plane angle. Together, these distributions allow us to conclude that the neutral pair w(r) minimum does indeed correspond to a sandwich structure, but this geometry is not strongly preferred due to the weak attraction between neutral Bz and TCNE exemplified by the shallow PMF. On the other hand, the ion pairs assume a significantly different structure than their neutral counter parts. Both out-of-plane and in-plane vectors now show strong preferences for orientations of π/2. These distributions suggest that the dominant ion pair structure is edge-to-face or T-shaped, with the long-edge of TCNE˙− rotated toward the benzene molecular plane. By taking the difference of the ion pair PMF at the neutral and ion pair PMF minima, we estimate a driving force of ∼–5kBT for the formation of the T-shaped pair. Illustrations of the conformations predicted for the neutral and ion pair are depicted at the bottom of Fig. 6. These structures are roughly consistent with our experimentally observed anisotropies. The sandwich structures predicted by the neutral w(r) and expected to be most easily excited are associated with a negative anisotropy for both 1 and 2 at short times, and diffusion into T-shaped structures is consistent with the change of sign observed in the 1 anisotropy and the lack of sign change for 2.
We begin by modeling the experimental steady-state absorption lineshape, A()/, using the results of TD-DFT calculations on the representative Bz/TCNE conformations selected from the PMF simulations. Bz/TCNE coordinates are extracted from each selected conformation, all solvent molecules stripped away, and a TD-DFT calculation is performed on the remaining Bz/TCNE atoms to determine the electronic transition energies, transition dipole moments, and oscillator strengths for the 5 lowest energy transitions. We find that the energy of the first two transitions ranges from 18000 to nearly 32000 cm−1 and corresponds primarily to one-electron excitations from the HOMO and HOMO−1 of Bz to the LUMO of TCNE. The higher transitions, which are more than 2500 cm−1 higher in energy, correspond primarily to local excitations of Bz or TCNE, and lie far outside the CT band observed in experiment. Oscillator strengths and transition frequencies for all transitions are presented in Fig. S7 and an example Bz/TCNE conformation and frontier molecular orbitals involved in the first two transitions are given in Fig. S8 (ESI‡). All further analyses will only concern the two lowest energy transitions which will be referred to hereafter as the ‘CT transitions’.
The oscillator strengths for the CT transitions are plotted against their transition energy in the top panel of Fig. 7. Despite the two-fold degeneracy of the isolated benzene HOMO, the first two transitions form two slightly separated bands. This is a consequence of the benzene molecular orbital symmetry and the mutual orientation of benzene and TCNE. Although, in isolation, the benzene HOMO is two-fold degenerate, the two HOMOs differ spatially, primarily by rotation of 90° about the Bz C6 symmetry axis (Fig. S9, ESI‡). Consequently, benzene HOMO energies (and therefore CT transition frequencies) will split depending on the relative orientation of benzene and TCNE. This phenomenon is analogous to the split bands observed in the absorption spectra of substituted benzene DACs.22,23 In these DACs, the degeneracy of the HOMO is broken due to asymmetrical substitution and two distinct absorption bands are observed, corresponding to CT from either the HOMO or HOMO−1 of the donor to the LUMO of TCNE. In the case of benzene, the difference in transition energy is due to differences in donor and acceptor MO overlap and not differences in the isolated donor HOMO energies. This splitting is much weaker than that induced by substitution, and therefore the absorption spectrum appears to be a single broad band.
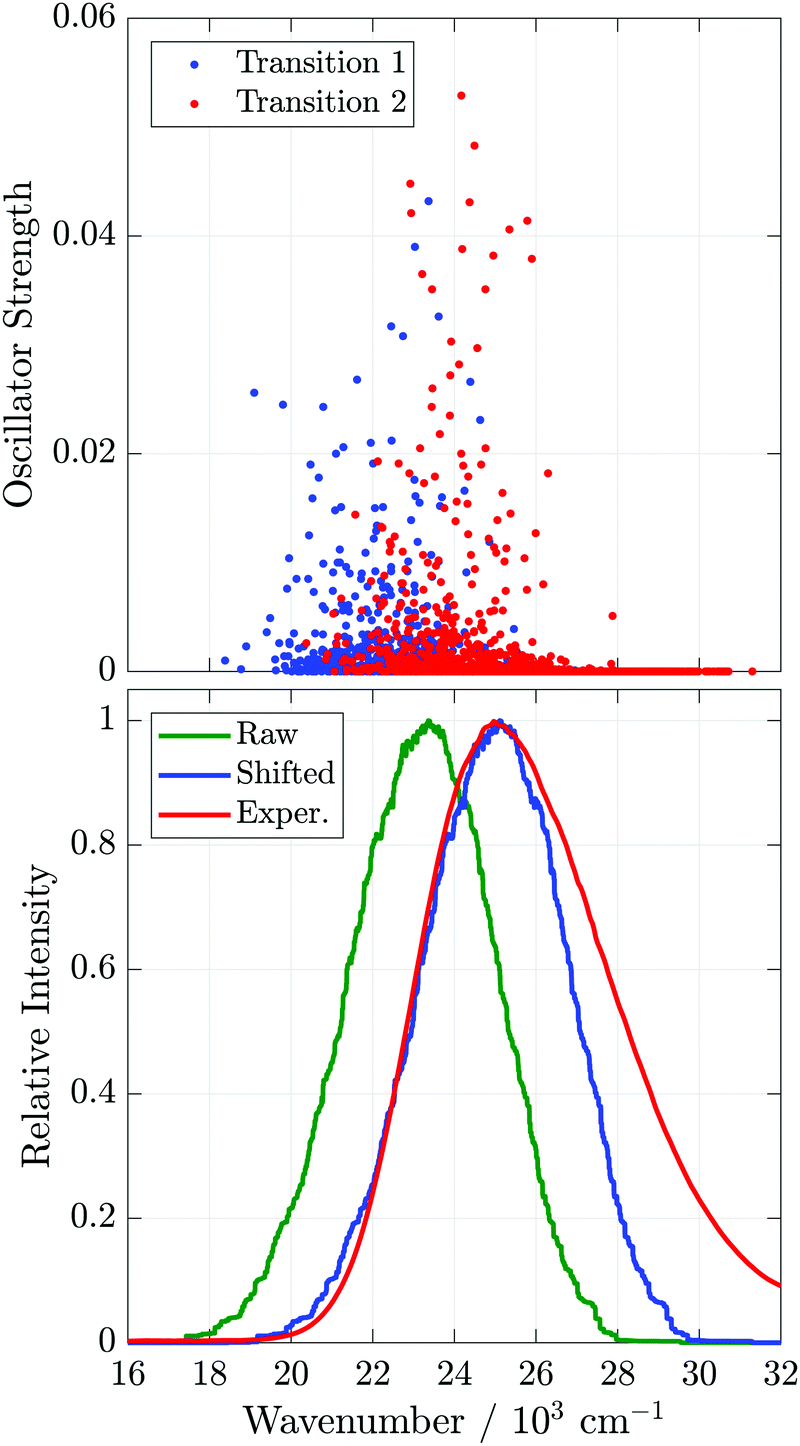 | ||
| Fig. 7 Top: Oscillator strengths of the two Bz/TCNE CT transitions vs. transition energy. Bottom: Experimental and simulated Bz/TCNE/CH2Cl2 absorption lineshapes. The green curve labeled ‘Raw’ is the result of binning and smoothing the oscillator strengths in the top panel, the blue curve labeled ‘Shifted’ is the ‘Raw’ curve up-shifted by 1750 cm−1, and the red curve labeled ‘Exper.’ is the experimental absorption lineshape, A()/. | ||
We approximate the measured absorption lineshape by binning the CT transition oscillator strengths by transition energy and smooth the resulting histogram using a moving-average filter. The results of such calculations and the experimental absorption lineshape are presented in the bottom plot of Fig. 7. The raw simulated lineshape must be up-shifted by 1750 cm−1 (0.22 eV) in order to match the maximum of the experimental lineshape. This is not unexpected, as the TD-DFT calculations are performed in vacuum and do not include the solvent environment, either explicitly or through continuum models. After shifting, the calculated spectra appear to reproduce the red-edge of the absorption spectrum, but do not properly capture the asymmetry of the band at high frequencies, possibly due to the lack of vibronic progression7,9 and solvent environment in the TD-DFT calculations. Despite these omissions, the width of the experimental absorption lineshape (FWHM = 5500 cm−1) is captured reasonably well by the simulation (FWHM = 4200 cm−1), suggesting that the broadening of the experimental lineshape can be well accounted for by Bz/TCNE structural heterogeneity. In previous simulations, which included vibronic transitions but not inhomogeneous broadening due to structural heterogeneity, unexpectedly large reorganization energies had to be assumed to properly reproduce the CT lineshape of DACs in apolar solvents.28,70 The good agreement obtained here suggests the MD generated Bz/TCNE pairs are a reasonable representation of the Bz/TCNE ground state.
Before moving on to the non-equilibrium simulations, we pause to consider the oscillator strength and transition energy dependence on separation distance as depicted in Fig. 8. Although pairs separated by less than 0.40 nm (approximately the first minimum in the neutral pair g(r)) exhibit the largest CT oscillator strengths, a significant number of these short-range pairs have weak or no oscillator strength. Concomitantly, there are pairs at separations up to 0.75 nm with significant oscillator strength, both supporting the r cutoff used in the conformation selection procedure and confirming that long-distance pairs will contribute to the excited-state population at t = 0. Calculated transition energies, (i)CT, are plotted as a function of separation in the bottom panel of Fig. 8. We see that both CT transition energies vary greatly even for the same r, with a spread of ∼4000 cm−1, due to the fact that the same r may be associated with a number of distinct geometries. Consequently, r alone is clearly not a sufficient descriptor of the DAC structure. Smoothing these data with a moving-average filter (green and yellow curves, bottom panel Fig. 8) allow us to more easily follow how the mean excitation energy changes with r. The two transition energies are approximately constant at 22000 and 23600 cm−1 for r < 0.55 nm and increase monotonically to 26200 and 28500 cm−1 at r = 0.91 nm. Although these smoothed curves might appear to suggest the magnitude of transition energy splitting is constant, the splitting can range from 400 to 5000 cm−1 and has no correlation with r (Fig. S10, ESI‡). Therefore, the splitting is a sensitive function of Bz/TCNE mutual orientation and not separation distance.
 | ||
| Fig. 8 Distance dependence of the CT oscillator strengths (top) and transition energies (bottom) obtained from TD-DFT calculations, where r is the center-of-mass separation of the Bz/TCNE pair. The smoothed transition energies in the bottom panel were calculated using a moving average filter. A frequency shift was not applied to these data. | ||
1 anisotropy begins negative and changes sign within a few picoseconds, whereas 2 starts slightly less negative than 1 and relaxes to 0 on a timescale similar to the experiment. Initial anisotropies are reproduced nearly exactly and further demonstrate the realism of the ground state previously described by the PMF and TD-DFT calculations.
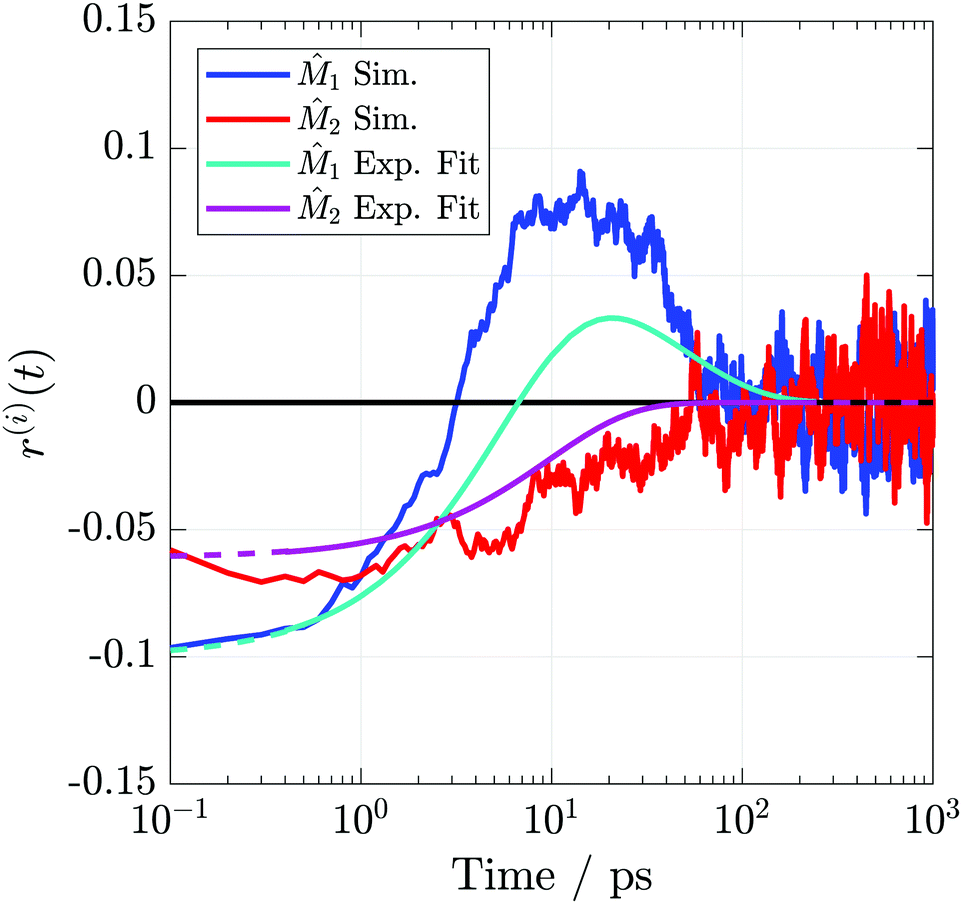 | ||
| Fig. 9 TRIR anisotropies from the simulations and fits to the experimental data. Axes definitions are as outlined in Fig. 3. Dashed portions of the experimental curves indicate extrapolations outside the collected temporal region. | ||
| Vector | r 0 | r 1 | τ r,1 (ps) | r 2 | τ r,2 (ps) |
|---|---|---|---|---|---|
|
1
|
−0.11 | −0.28 | 5.2 | 0.17 | 28 |
|
2
|
−0.07 | −0.07 | 20 | — | — |
The apparent success in reproducing the experimental anisotropy allows us to be confident in using the non-equilibrium simulations to study the excited state structural dynamics in greater detail. Oscillator strength weighted orientational distributions calculated at t = 0 and t = 1 ns (Fig. S11, ESI,‡ analogous to those in Fig. 6 but with orientations weighted by the t = 0 oscillator strength), are consistent with those predicted by the PMF calculations, but with more disorder. The disorder in these distributions compared to those calculated from the PMFs comes from considering all selected initial conformations, ranging from 0.31 to 0.95 nm, instead of only focusing on the minimum in the PMF as was done in Fig. 6. These data confirm that the structural rearrangement predicted by the equilibrium PMF calculations does in fact take place.
To attempt to visualize the Bz/TCNE ‘chromophore’, we have created composite images by overlaying Bz/TCNE conformations at t = 0 and 1 ns from each non-equilibrium MD trajectory. The transparency (alpha channel) of each individual image was weighted by the conformation's t = 0 oscillator strength in order to approximate its contribution to the absorption lineshape/excited state population. The resulting images are presented in Fig. 10, and TCNE carbon atoms have been removed for clarity. The t = 0 image shows that although excitation of Bz/TCNE comes predominately from face-to-face structures, other, less ordered, conformations contribute significantly to the excitation. This disorder is the source of the reduced initial anisotropy seen in the TRIR anisotropy experiments. At t = 1 ns the ion pairs have reached a conformational equilibrium where a majority of the Bz˙+/TCNE˙− pairs are oriented in an edge-to-face configuration. Despite the strong Coulombic attraction between the ions, the pairs are still considerably disordered. These images clearly demonstrate the stark structural differences between neutral and ion pair DACs and their accompanying heterogeneity; this DAC cannot be described as a single static species.
 | ||
| Fig. 10 An approximation of the Bz/TCNE ‘chromophore’ created by superimposing an image of each Bz/TCNE conformation from non-equilibrium MD simulations at t = 0 and 1 ns. The blue spheres represent TCNE nitrogen atoms and the TCNE carbon atoms have been omitted for clarity. The transparency of each individual image is weighted by the conformation's t = 0 oscillator strength in order to approximate its contribution to the excited state population. | ||
In order to follow translational diffusion of the ion pairs, we examine the time-dependent distributions of center-of-mass separation, p(r;t), as presented in Fig. 11. These calculations are related to the time-dependent pair distribution function, g(r;t), but are normalized simply for unit area instead of the bulk density. Therefore, p(r;t) represents the evolution of the ion-pair center-of-mass distribution from t = 0, determined by the neutral pair w(r) to their equilibrium distribution, determined by the ion pair w(r) (blue and red curves, Fig. 5). We note that these distributions will not be weighted by oscillator strength in order to highlight the pure ion pair dynamics.
 | ||
| Fig. 11 Time-dependent distributions of center-of-mass separation from the non-equilibrium MD simulations. | ||
The top panel of Fig. 11 illustrates the short-time evolution of p(r;t). At t = 0, we see a peak near 0.37 nm that corresponds to the minimum in w(r) of the neutral pairs. This peak shifts in 0.2 ps to 0.34 nm before relaxing towards the ion pair w(r) minimum of 0.54 nm with a small side peak at r ≈ 0.40 nm. From the long side, r > 0.70 nm, the distribution contracts towards the ion-pair minimum, but a secondary shoulder also appears at r ≈ 0.7 nm. The long-time evolution of p(r;t) in the bottom panel of Fig. 11 shows that the two side peaks seen in the short time distributions eventually collapse into the ion-pair minimum. The short side of the distribution collapses faster as evidenced by the faster rise on the short side than the long side.
These dynamics suggest that w(r) is not sufficient by itself to properly describe the translational dynamics of the photogenerated ions. There is only a single minimum in w(r), and the derivative of w(r) is always negative for r < rmin and positive for r > rmin (Fig. S12, ESI‡) indicating that there are no local minima in w(r) and therefore no metastable states for the ion pairs to sample (e.g. the short time contraction feature or side peaks in the long-time p(r;t)). We can conclude that the structural relaxation in this system must happen on a multidimensional potential surface that includes both separation and orientation coordinates. This is also supported by the starkly different orientational distributions for the neutral and ionic species shown in Fig. 6. The path a particular ion pair follows to reach the ion pair equilibrium position will then depend on its initial position on this multidimensional surface. Multidimensional diffusion involving separation and solvent coordinates has been used to describe the ion pairing mechanism of monovalent ion pairs in water,71,72 and the diffusion seen in the molecular ions here can be seen as an extension of these dynamics involving additional orientational coordinates.
4 Conclusions
The experiments and simulations herein paint a significantly different portrait of the DAC ground and excited-state in liquids than is typically assumed by the single structure π-stacked ‘sandwich’ model. The excited-state dynamics of the Bz/TCNE DAC begin with a ∼10 ps equilibration involving vibrational, solvent and structural relaxation from the initial Franck–Condon state to an ion-pair-like state, as evidenced by the complicated spectral dynamics observed in both visTA and TRIR measurements. Spectral relaxation is followed by the charge recombination of the ion pairs with a time-constant of 55–60 ps. TRIR anisotropy experiments reveal starkly different dynamics for two different TCNE˙− vibrations and indicate structural reorganization of the ion pairs following excitation.Using a combination of TD-DFT calculations and molecular dynamics simulations, we were able to satisfactorily reproduce the experimental steady-state visible absorption spectrum and TRIR anisotropy. The results of these calculations demonstrate that even though a π-stack is the most probable ground state structure, the attraction between neutral Bz/TCNE in CH2Cl2 is less than kBT and a high degree of disorder is present. On the other hand, Bz˙+/TCNE˙− ion pairs preferentially adopt edge-to-face T-shaped structures and not the π-stack as usually assumed. This result suggests that the ground-state structure of the DAC is primarily determined by dispersion interactions, whereas that of the ion-pair like excited state is governed by Coulombic interactions. The TD-DFT calculations on representative MD generated Bz/TCNE structures revealed a strong sensitivity of the CT transition oscillator strength and transition energy to the mutual Bz/TCNE orientation and a weak dependence on separation. Therefore, optical excitation in the CT band results in a photoselection of geometries with a significant oscillator strength. However, these excitable pairs still form a highly disordered ensemble with disparate center-to-center distances and D/A orientations illustrated in the composite images of Fig. 10. The apparent success in reproducing the visible absorption lineshape using vacuum phase TD-DFT calculations suggests that broadening in experimental spectrum is dominated by structural inhomogeneity. Non-equilibrium MD simulations of the Bz˙+/TCNE˙− pair exhibit dynamics inconsistent with diffusion on a one dimensional potential energy surface and indicates the presence of coupled rotational–translational diffusion and heterogeneous structural dynamics. Further simulations and quantum-chemical calculations on this system and other pairs are needed in order to understand the interplay between pair structure and solvent properties on the DAC absorption spectrum, excited-state orientational and translational dynamics, and charge recombination.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Joseph S. Beckwith for assistance in collecting the visTA data and Alexander Zech for providing the CCParser Python module used for extracting data from the TD-DFT calculations. The quantum chemical and molecular dynamics computations were performed at University of Geneva on the Baobab cluster. We are also grateful for the financial support provided by the Swiss National Science Foundation (project no. 200020-165890) and the University of Geneva.References
- R. S. Mulliken, J. Am. Chem. Soc., 1952, 74, 811–824 CrossRef CAS.
- E. F. Hilinski, J. M. Masnovi, J. K. Kochi and P. M. Rentzepis, J. Am. Chem. Soc., 1984, 106, 8071–8077 CrossRef CAS.
- N. Mataga and H. Miyasaka, Adv. Chem. Phys., 1999, 107, 431–496 CAS.
- J. M. Masnovi and J. K. Kochi, J. Am. Chem. Soc., 1985, 107, 7880–7893 CrossRef CAS.
- T. Asahi and N. Mataga, J. Phys. Chem., 1989, 93, 6575–6578 CrossRef CAS.
- T. Kumpulainen, B. Lang, A. Rosspeintner and E. Vauthey, Chem. Rev., 2017, 117, 10826–10939 CrossRef CAS PubMed.
- F. Markel, N. S. Ferris, I. R. Gould and A. B. Myers, J. Am. Chem. Soc., 1992, 114, 6208–6219 CrossRef CAS.
- A. B. Myers, Chem. Phys., 1994, 180, 215 CrossRef CAS.
- M. Lilichenko, D. Tittelbach-Helmrich, J. W. Verhoeven, I. R. Gould and A. B. Myers, J. Chem. Phys., 1998, 109, 10958–10969 CrossRef CAS.
- O. Nicolet and E. Vauthey, J. Phys. Chem. A, 2002, 106, 5553–5562 CrossRef CAS.
- R. G. Fedunov, S. V. Feskov, A. I. Ivanov, O. Nicolet, S. Pagès and E. Vauthey, J. Chem. Phys., 2004, 121, 3643–3656 CrossRef CAS.
- S. V. Feskov, V. A. Mikhailova and A. I. Ivanov, J. Photochem. Photobiol., C, 2016, 29, 48–72 CrossRef CAS.
- E. Adman, M. Rosenblum, S. Sullivan and T. N. Margulis, J. Am. Chem. Soc., 1967, 89, 4540–4542 CrossRef CAS.
- F. K. Larsen, R. G. Little and P. Coppens, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1975, 31, 430–440 CrossRef.
- F. A. Balmer, P. Ottiger, C. Pfaffen and S. Leutwyler, J. Phys. Chem. A, 2013, 117, 10702–10713 CrossRef CAS PubMed.
- F. A. Balmer, P. Ottiger and S. Leutwyler, J. Phys. Chem. A, 2015, 119, 10462–10474 CrossRef CAS.
- M. Hayashi, T.-S. Yang, J. Yu, A. Mebel and S. H. Lin, J. Phys. Chem. A, 1997, 101, 4156–4162 CrossRef CAS.
- M.-S. Liao, Y. Lu, V. D. Parker and S. Scheiner, J. Phys. Chem. A, 2003, 107, 8939–8948 CrossRef CAS.
- J. A. Ciezak, J. B. Leão and B. S. Hudson, THEOCHEM, 2006, 767, 23–28 CrossRef CAS.
- D. Kuchenbecker and G. Jansen, ChemPhysChem, 2012, 13, 2769–2776 CrossRef CAS.
- T. S. Kolesnikova, M. S. Chernov'yants, M. E. Kletskii, O. N. Burov, G. I. Bondarenko and P. A. Knyazev, J. Phys. Chem. A, 2017, 121, 7000–7008 CrossRef CAS.
- E.-M. Voigt, J. Am. Chem. Soc., 1964, 86, 3611–3617 CrossRef CAS.
- M. Rossi, U. Buser and E. Haselbach, Helv. Chim. Acta, 1976, 59, 1039–1053 CrossRef CAS.
- O. Nicolet, N. Banerji, S. Pagès and E. Vauthey, J. Phys. Chem. A, 2005, 109, 8236–8245 CrossRef CAS PubMed.
- O. F. Mohammed and E. Vauthey, J. Phys. Chem. A, 2008, 112, 5804–5809 CrossRef CAS.
- J. Zhou, B. R. Findley, A. Teslja, C. L. Braun and N. Sutin, J. Phys. Chem. A, 2000, 104, 11512–11521 CrossRef CAS.
- J. Zhou, B. R. Findley, C. L. Braun and N. Sutin, J. Chem. Phys., 2001, 114, 10448–10456 CrossRef CAS.
- K. Wynne, C. Galli and R. M. Hochstrasser, J. Chem. Phys., 1994, 100, 4797–4810 CrossRef CAS.
- K. Wynne, G. D. Reid and R. M. Hochstrasser, J. Chem. Phys., 1996, 105, 2287–2297 CrossRef CAS.
- M. Rossi and E. Haselbach, Helv. Chim. Acta, 1979, 62, 140–151 CrossRef CAS.
- I. V. Rubtsov and K. Yoshihara, J. Phys. Chem. A, 1997, 101, 6138–6140 CrossRef CAS.
- M. Hayashi, T.-S. Yang, J. Yu, A. Mebel, R. Chang, S. H. Lin, I. V. Rubtsov and K. Yoshihara, J. Phys. Chem. A, 1998, 102, 4256–4265 CrossRef CAS.
- I. V. Rubtsov and K. Yoshihara, J. Phys. Chem. A, 1999, 103, 10202–10212 CrossRef CAS.
- B. R. Arnold, A. W. Schill and P. V. Poliakov, J. Phys. Chem. A, 2001, 105, 537–543 CrossRef CAS.
- B. R. Arnold, A. Euler, P. V. Poliakov and A. W. Schill, J. Phys. Chem. A, 2001, 105, 10404–10412 CrossRef CAS.
- C.-C. Chiu, C.-C. Hung, C.-L. Chen and P.-Y. Cheng, J. Phys. Chem. B, 2013, 117, 9734–9756 CrossRef CAS.
- C.-C. Chiu, C.-C. Hung and P.-Y. Cheng, J. Phys. Chem. B, 2016, 120, 12390–12403 CrossRef CAS.
- G. Piacente, A. Amadei, M. D'Abramo, I. Daidone and M. Aschi, Phys. Chem. Chem. Phys., 2014, 16, 20624–20638 RSC.
- A. A. J. Aquino, I. Borges, R. Nieman, A. Köhn and H. Lischka, Phys. Chem. Chem. Phys., 2014, 16, 20586–20597 RSC.
- J. S. Miller, Angew. Chem., Int. Ed., 2006, 45, 2508–2525 CrossRef CAS.
- O. F. Mohammed, N. Banerji, B. Lang, E. T. J. Nibbering and E. Vauthey, J. Phys. Chem. A, 2006, 110, 13676–13680 CrossRef CAS.
- O. F. Mohammed, K. Adamczyk, N. Banerji, J. Dreyer, B. Lang, E. T. J. Nibbering and E. Vauthey, Angew. Chem., Int. Ed., 2008, 47, 9044–9048 CrossRef CAS PubMed.
- G. Angulo, A. Cuetos, A. Rosspeintner and E. Vauthey, J. Phys. Chem. A, 2013, 117, 8814–8825 CrossRef CAS.
- B. Wu, M. Maroncelli and E. W. Castner, J. Am. Chem. Soc., 2017, 139, 14568–14585 CrossRef CAS.
- J. E. Frey, A. M. Andrews, D. G. Ankoviac, D. N. Beaman, L. E. Du Pont, T. E. Elsner, S. R. Lang, M. A. O. Zwart, R. E. Seagle and L. A. Torreano, J. Org. Chem., 1990, 55, 606–624 CrossRef CAS.
- N. Banerji, G. Duvanel, A. Perez-Velasco, S. Maity, N. Sakai, S. Matile and E. Vauthey, J. Phys. Chem. A, 2009, 113, 8202–8212 CrossRef CAS PubMed.
- G. Duvanel, N. Banerji and E. Vauthey, J. Phys. Chem. A, 2007, 111, 5361–5369 CrossRef CAS.
- M. Koch, R. Letrun and E. Vauthey, J. Am. Chem. Soc., 2014, 136, 4066–4074 CrossRef CAS.
- B. Dereka, D. Svechkarev, A. Rosspeintner, M. Tromayer, R. Liska, A. M. Mohs and E. Vauthey, J. Am. Chem. Soc., 2017, 139, 16885–16893 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford, CT, 2013 Search PubMed.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- L. E. Chirlian and M. M. Francl, J. Comput. Chem., 1987, 8, 894–905 CrossRef CAS.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, J. Cheminform., 2012, 4, 17 CrossRef CAS.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1–2, 19–25 CrossRef.
- W. L. Jorgensen, D. S. Maxwell and J. Tirado-Rives, J. Am. Chem. Soc., 1996, 118, 11225–11236 CrossRef CAS.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS.
- J. Stone, MSc thesis, Computer Science Department, University of Missouri-Rolla, 1998.
- T. Shida, Electronic Absorption Spectra of Radical Ions, Elsevier, Amsterdam, 1988, vol. 34 Search PubMed.
- Y. Kodera and T. Hikida, Spectrosc. Lett., 1989, 22, 1229–1239 CrossRef CAS.
- T. M. Bockman, Z. J. Karpinski, S. Sankararaman and J. K. Kochi, J. Am. Chem. Soc., 1992, 114, 1970 CrossRef CAS.
- T. Asahi, M. Ohkohchi and N. Mataga, J. Phys. Chem., 1993, 97, 13132–13137 CrossRef CAS.
- W. Jarzeba, S. Murata and M. Tachiya, Chem. Phys. Lett., 1999, 301, 347–355 CrossRef CAS.
- P. Hamm, S. M. Ohline and W. Zinth, J. Chem. Phys., 1997, 106, 519–529 CrossRef CAS.
- M. Koch, A. Rosspeintner, K. Adamczyk, B. Lang, J. Dreyer, E. T. J. Nibbering and E. Vauthey, J. Am. Chem. Soc., 2013, 135, 9843–9848 CrossRef CAS PubMed.
- G. R. Fleming, Chemical Applications of Ultrafast Spectroscopy, Oxford University Press, 1986 Search PubMed.
- J. T. Edward, J. Chem. Educ., 1970, 47, 261 CrossRef CAS.
- Y. Marcus, The Properties of Solvents, Wiley, New York, 2005 Search PubMed.
- D. Chandler, Introduction to Modern Statistical Mechanics, Oxford University Press, New York, 1987 Search PubMed.
- M. A. Mattson, T. D. Green, P. T. Lake, M. McCullagh and A. T. Krummel, J. Phys. Chem. B, 2018, 122, 4891–4900 CrossRef CAS PubMed.
- K. Kulinowski, I. R. Gould and A. B. Myers, J. Phys. Chem., 1995, 99, 9017–9026 CrossRef CAS.
- S. Roy, M. D. Baer, C. J. Mundy and G. K. Schenter, J. Phys. Chem. C, 2016, 120, 7597–7605 CrossRef CAS.
- S. Roy, M. D. Baer, C. J. Mundy and G. K. Schenter, J. Chem. Theory Comput., 2017, 13, 3470–3477 CrossRef CAS PubMed.
Footnotes |
| † All data are available for download at http://doi.org/10.5281/zenodo.2872150. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cp00795d |
| This journal is © the Owner Societies 2019 |