
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Insight into the internal structure of amyloid-β oligomers by isotope-edited Fourier transform infrared spectroscopy†
Cesare M.
Baronio
 ,
Maurizio
Baldassarre
and
Andreas
Barth
*
,
Maurizio
Baldassarre
and
Andreas
Barth
*
Department of Biochemistry and Biophysics, Stockholm University, Sweden. E-mail: barth@dbb.su.se
First published on 2nd April 2019
Abstract
The internal structure of amyloid-β (Aβ) oligomers was investigated with isotope-edited Fourier transform infrared spectroscopy. Homo-oligomers of Aβ40 and Aβ42 were prepared from unlabeled and 13C, 15N-labeled monomeric Aβ and from mixtures of these. For the unlabeled peptides, two main bands were observed in 2H2O at 1685 and 1622 cm−1 for Aβ40 and at 1685 and 1626 cm−1 for Aβ42. These band positions indicate that the number of strands per sheet is at least four. The obtained experimental amide I spectra were simulated using a number of structural models (antiparallel β-sheets, β-barrels and a dodecamer structure). According to experiments and calculations, the main 13C-band shifts down at increasing molar ratio of labeled peptides. This shift occurs when vibrational coupling becomes possible between 13C-amide groups in close-by strands. It is small, when intervening 12C-strands increase the distance between 13C-strands; it is large, when many neighboring strands are labeled. The shift depends on the internal structure of the peptides within the oligomers, i.e. on the building block that each peptide molecule contributes to the β-sheets of the oligomers. The shift is largest, when individual peptides contribute just a single strand surrounded by strands from other peptide molecules. It is smaller when each molecule forms two or three adjacent strands. As indicated by a comparison between experiment and computation, the number of adjacent β-strands per peptide molecule is two for Aβ40 oligomers and two or more for Aβ42 oligomers. Our results are well explained by regular, antiparallel β-sheets or β-barrels.
Introduction
Alzheimer's disease is the most frequent neurodegenerative disease. It is associated with the formation of amyloid plaques in the brain which mainly consist of aggregated amyloid-β peptide (Aβ). Aβ is formed by proteolytic cleavage of the amyloid precursor protein in two main variants which are 40 (Aβ40) and 42 (Aβ42) residues long. Both have a propensity to aggregate, which is stronger for Aβ42 due to two additional hydrophobic residues (isoleucine, alanine) at the C-terminus.1–9 The aggregation process from monomers to fibrils is complex and involves a large number of oligomeric species, which are thought to contribute considerably to the toxicity of Aβ.4,10–15 These oligomers are difficult to investigate because they have not yet been crystallized without modification, they are too small for cryo-electron microscopy and aggregates larger than tetramers are too large for solution NMR.16 Nevertheless, several structural models have been proposed, either for a dimer unit from solution NMR,16 from solid state NMR studies17–21 or from X-ray crystallography of Aβ stabilized by a protein scaffold22 or by modifying the sequence.23 Further information has been obtained from Fourier transform infrared (FTIR) spectroscopy of dried Aβ42 oligomers, the spectrum of which resembled most closely that of a bacterial porin with 16 antiparallel β-strands.24 The proposed oligomer structures differ considerably, those that have identified β-sheet structures – relevant to this work – agree on the existence of antiparallel β-sheets in the oligomers. Whereas some studies detect them in the C-terminal half17,22,23 or in the C-terminal quarter,19 a further study identifies them in the central portion but not in the C-terminal tail.16 The discrepancies between the oligomer models call for further investigation, in particular for studies in aqueous solution, considering that most of the present models are based on results in non-aqueous environments.Infrared spectroscopy is widely used in amyloid research25–28 focusing mostly on the absorption of the amide I vibrations. These vibrations consist mainly of the stretching vibration of the carbonyl groups of the peptide backbone. The study of the amide I band is particularly powerful in combination with isotope labeling, which can be used to reveal mixing of two different peptides29,30 or to study the structure of aggregates.31–36
The interpretation of the amide I band profile is enhanced by spectrum calculations. Density function theory can only be applied to small sections of proteins so most approaches focus on a computer-time-efficient description of the amide I vibrations.37–40 Each amide I oscillator is assigned an intrinsic frequency, which depends on the electrostatic environment.41–44 The individual amide I oscillators couple electrostatically with other amide I oscillators, which is often described by transition dipole coupling.45–47 These approximations are insufficient for nearest neighbor interactions which are therefore modeled from density functional theory calculations of small peptides.48–53 Conformational dynamics can be accounted for either by statistical variation of the parameters,54–56 by averaging spectra of snapshot structures from molecular dynamics simulations57–59 or by direct time-domain approaches which also take into account motional narrowing.60–63
The relevant model structure for this work are antiparallel β-sheets. Their amide I vibrations are characterized by a strong coupling between amide groups in adjacent strands. This coupling explains the large splitting of the absorption band into a high and a low wavenumber component.45,64 Coupling is less effective when the frequencies of the coupled oscillators are different, for example because the amide groups contain different carbon isotopes. Thus, mixing of different carbon isotopes in a β-sheet leads to a downshift of the high wavenumber band and an upshift of the low wavenumber band which decreases the splitting between the two bands.65
Using a combination of isotope-edited FTIR spectroscopy and spectrum calculation, we have found that mixtures of monomeric Aβ40 and Aβ42 form mixed hetero-oligomers.65 This work is here extended to homo-oligomers of either Aβ40 or Aβ42. In particular, we describe calculations that aimed to achieve a quantitative agreement with the experiments. Our results show that the internal structure of each peptide in homo-oligomers is such that it contributes at least two adjacent strands to the β-sheets.
Experimental
Recombinant, unlabeled and 13C, 15N uniformly labeled Aβ40 and Aβ42 were obtained from AlexoTech (Umeå, Sweden). 15N-Labeling has a very minor effect on the spectrum and will therefore not be mentioned hereafter. Experimental procedures were as described previously.65 Briefly, hexafluoroisopropanol- (HFIP) treated Aβ was dissolved in 20 mM NaO2H in order to obtain a monomeric peptide solution at a concentration of 200 μM. Unlabeled and labeled peptides were mixed and 4 μL of this solution added to a dry film produced from 4 μL of 100 mM sodium phosphate to bring the solution to p2H 7.4 and initiate aggregation. This preparation was chosen to prevent dilution of the peptide when the pH was lowered. It produced small-sized oligomers for Aβ40 and Aβ42 and additional larger aggregates for Aβ42 as shown by gel electrophoresis of photo-crosslinked samples.65The peptide solution was placed between two CaF2 windows that were separated by a 50 μm plastic spacer. FTIR spectra were recorded approximately 20 min after initiation of aggregation with a Tensor 37 FTIR spectrometer (Bruker, Germany) that was equipped with a sample shutter. The reference position of the sample shuttle contained a buffer sample that was prepared in the same way as the Aβ samples but without peptide. The spectral resolution was 2 cm−1 and a zero-filling factor of 2 was used. The second derivative spectra were smoothed over 13 data points (approx. 13 cm−1) in order to determine the band positions and for the figures in this article. The band position of the 13C-band at 0.1 molar ratio of 13C-peptides was determined by simultaneous fitting of the absorbance and the second derivative spectrum as described in our previous work.65 The band positions at all other 13C![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :12C ratios were determined directly from the second derivative spectra.
:12C ratios were determined directly from the second derivative spectra.
For an estimation of the secondary structure content, buffer contributions to the absorbance spectrum that were not accounted for by the buffer sample in the reference position of the sample shuttle were subtracted from the sample spectrum as described previously.65 Then a straight line between the absorbance values at 1700 and 1600 cm−1 was subtracted from the absorbance spectrum. The data points in the intervals 1710–1700 cm−1 and 1600–1590 cm−1 were set to zero and data points above 1710 cm−1 and below 1590 cm−1 removed. The absorbance spectrum and its second derivative were simultaneously fitted66 in the spectral range between 1700 and 1600 cm−1 with the program Kinetics written by Erik Goormaghtigh (Université Libre de Bruxelles) using a scaling factor (weight) of 50 and Savitzky–Golay windows of 9, 13, and 17 points for the second derivative. Six bands were fitted with initial positions at 1683, 1678, 1664, 1645, 1632, and 1624 cm−1 without baseline. These fits produced two bands in the region of the main β-sheet absorption near 1625 cm−1 at very similar band positions but with widths that differed by a factor of two. The wider band was placed 2 cm−1 higher than the narrower band for both Aβ40 and Aβ42. The areas obtained were averaged for all three Savitzky–Golay windows used and for three independent experiments for each peptide. They are listed in Table S1 (ESI†).
Fits with only five bands were also tested for one Aβ40 and one Aβ42 spectrum. The standard deviation between fit and experimental spectra increased by a factor of more than 2 compared to the 6-band fit. In particular, there was less agreement below 1615 cm−1 for both absorbance spectrum and second derivative spectrum. The band placed initially in the random coil region at 1645 cm−1 moved during the fit into the 1635–1630 cm−1 region, assigned to β-sheets. This increased the total area assigned to β-sheets by up to 60% relative to the area obtained in the 6-band fits. For the 5-band fit to the Aβ42 spectrum, the total band area assigned to β-sheets decreased from 65% to 50% when the Savitzky–Golay window increased from 9 to 17 data points. In contrast, the 6-band fits to this spectrum produced a consistent β-sheet area of 41%, 37%, and 42% for windows of 9, 13, and 17 data points, respectively. Because of the missing but expected band for random coils in the 5-band fits, the more consistent results with the 6-band fits and the better agreement between experimental data and 6-band fits, we regard the 6-band fits as a better representation of our experimental data and discuss only these fits hereafter.
Computational methods
The amide I spectrum was simulated using a Matlab program65,67 and model structures with antiparallel β-sheets. Parallel β-sheets cannot be used because our approach is based on a matching between experimental and computational spectra (see below). This requires the presence of a distinct high wavenumber β-sheet band that is not observed in spectra calculated for parallel β-sheets.Antiparallel β-sheets of different sizes were created according to the atomic coordinates suggested by Fraser and MacRae.68 Most of the sheets consisted of strands with 10 residues (9 complete amide groups) corresponding to the number of residues in the β-strands of Aβ4069,70 and Aβ4271 fibrils. Antiparallel β-barrels of different sizes were also created, selecting barrel-residing residues from available structures in the protein data bank. The complete list of PDB files and the selected residues for each of these files are listed in the ESI.† Additionally, we considered a complete atomic structure for Aβ oligomers.23 The structure is a dodecamer, consisting of four triangular trimers that are tetrahedrically arranged around a central cavity. The structures used are shown in Fig. 1.
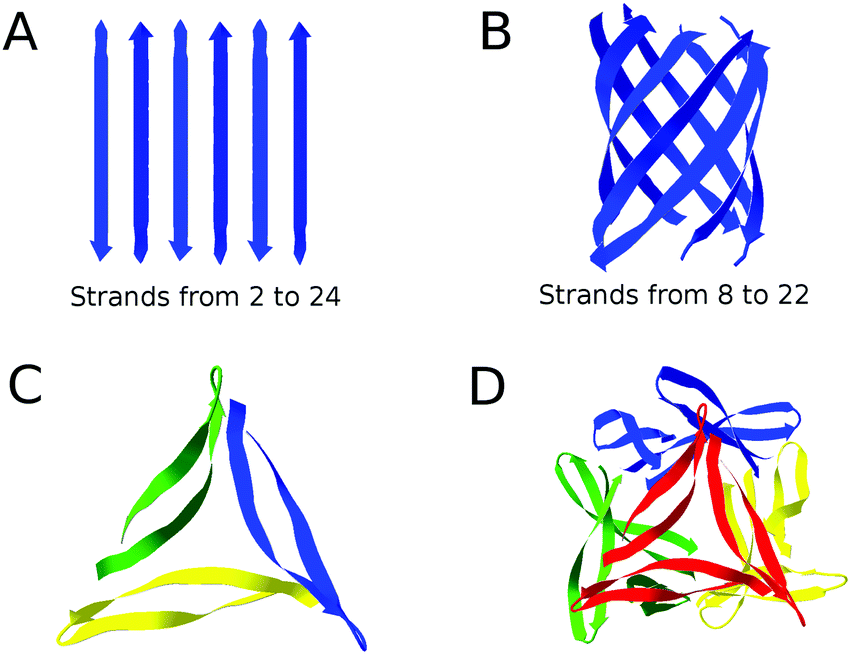 | ||
| Fig. 1 Representation of the structures used in our calculations. (A) Antiparallel β-sheets, composed of 2 to 24 strands, (B) β-barrels, composed of 8 to 22 strands with antiparallel orientation, (C) a trimer unit of the dodecamer structure, (D) the entire dodecamer structure of pdb entry 5HOW.23 The structure of each monomer in the dodecamer is a β-hairpin where two β-strands are connected by a short loop. In (C) different monomers have different colors. In (D) different trimer units have different colors. The figure was created using Swiss PDB Viewer.72 The complete list of models can be found in the ESI.† | ||
The amide I spectrum of our model structures was calculated according to the floating oscillator model37 where a protein is regarded as an assembly of individual amide I oscillators that couple via transition dipole coupling. The rest of the structure is not considered. Each oscillator is associated with an intrinsic frequency or wavenumber and a transition dipole moment. The latter is proportional to the dipole derivative, which is the change of dipole moment associated with the amide I vibration when the oscillator passes through the equilibrium position. Further effects on the infrared spectrum can be considered as described below. The frequencies and the intensities of the normal modes of vibration are then obtained by a diagonalization of the mass normalized force constant matrix as in our previous work.65,67
The diagonal elements of the force constant matrix were calculated from the intrinsic wavenumber, which was assumed to be the same for each unlabeled amide group. The intrinsic wavenumber includes the effects of hydrogen bonding. A constant intrinsic wavenumber for our model structures is therefore equivalent to the assumption of the same hydrogen bonding strength to all amide groups either due to hydrogen bonding to other amide groups or to water. In case of the dodecamer structure we explicitly considered hydrogen-bonding and nearest neighbor effects as in our original version of the program.67
The non-diagonal elements of the matrix were obtained using coupling constants from density functional theory calculations for nearest neighbor interactions48,49 and transition dipole coupling for all other interactions37 with parameters adjusted to match the experimental spectra as described below. The position of the dipole derivative was as suggested by Moore and Krimm.37,73 We assumed that the transition dipole moment was the same for each amide group. The angle of the dipole derivative was fixed to 10°, 20° or 30° degrees with respect to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond, which made the dipole derivative pointing towards the N atom.
O bond, which made the dipole derivative pointing towards the N atom.
The composition of the force constant matrices and, consequently, the simulation of the absorption spectra depended on the values of the dipole derivative and of the intrinsic wavenumber of the unperturbed amide group. Panel A of Fig. 2 shows that the maximum absorbance and the splitting between high and low wavenumber bands increase when the magnitude of the dipole derivative is increased. Panel B shows that the whole spectrum is shifted towards higher wavenumbers when the intrinsic wavenumber is increased.
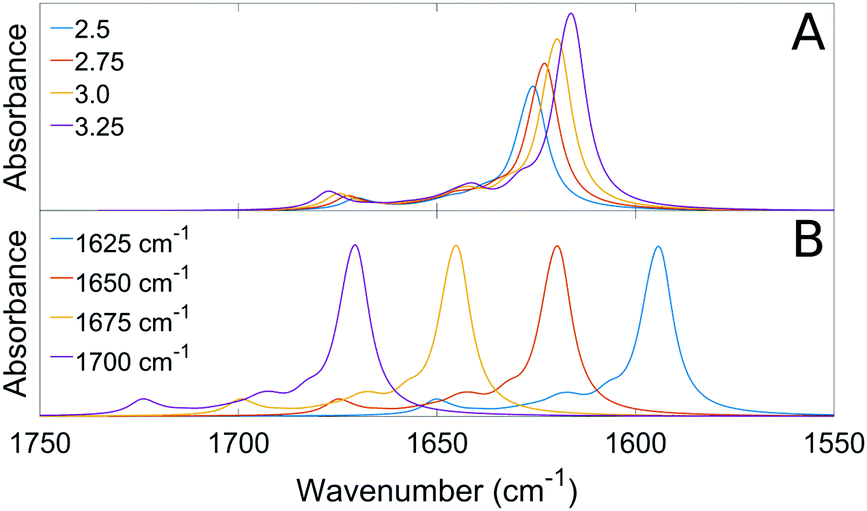 | ||
| Fig. 2 The calculated absorption spectrum depends on the magnitude of the dipole derivative (panel A) and on the intrinsic wavenumber (panel B). The magnitude of the dipole derivative is given in D Å−1 amu−1/2 in panel A and the intrinsic wavenumber in panel B. | ||
The magnitude of the dipole derivative and the intrinsic wavenumber were adjusted for each model structure so that the low and high wavenumber band positions of the calculated spectrum matched those in the experimental spectrum of the all-unlabeled sample. The matching between experimental and simulated band positions was performed separately for the Aβ40 and the Aβ42 spectra. Once the calculation parameters were adjusted for one particular model structure, we used these parameters for the calculation of the spectra for all other 13C:12C ratios.
We simulated the presence of 13C strands according to the 13C:12C molar ratios used in the experiments. The strands were labeled according to five different patterns, as shown in Fig. 3, that model the contribution of individual peptides to the structure of the oligomers. This contribution is further on termed building block. All amide groups within one building block are thought to belong to the same peptide molecule and contain the same carbon isotope (with the exception of a 10% impurity of 12C-atoms in 13C-peptides, see below). A building block does not comprise the entire peptide molecule and the rest of the peptide is assumed not to participate in β-sheets. Three building blocks had a different number of adjacent strands from the same peptide. These were:
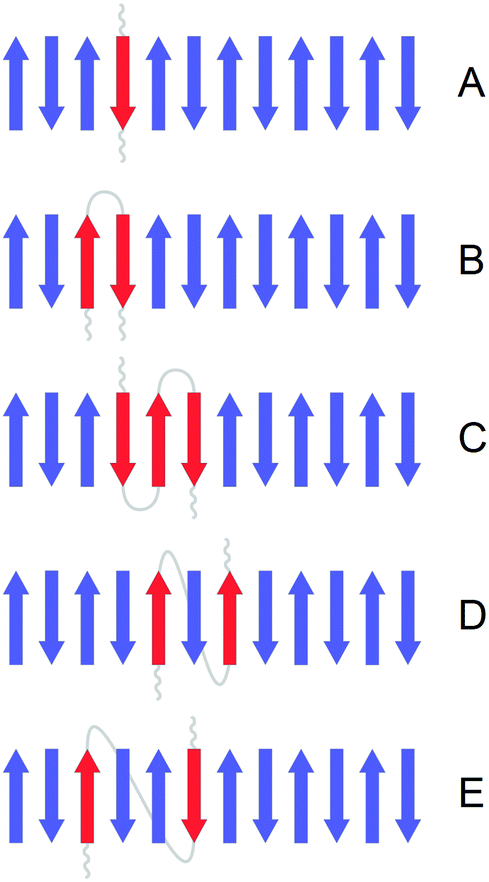 | ||
| Fig. 3 Illustration of the considered building blocks. A building block is the contribution of an individual peptide (red) to our β-sheet and β-barrel model structures. The figure shows the example of an Aβ oligomer with a 12-stranded antiparallel β-sheet. Strands from other peptides are shown in blue. (A) Single strand, (B) 2-strand block, (C) 3-strand block, (D) interlaced model 1, (E) interlaced model 2. See text for a detailed description of the building blocks. The non-β-sheet sections of the peptide with the red β-strands are schematically shown in grey. The respective sections of other peptides are not shown. | ||
(A) Single strand: each strand has neighboring strands from other peptides meaning that a given strand might have two adjacent strands with a different carbon isotope.
(B) 2-strand block: each peptide molecule contributes two adjacent strands. Therefore, each strand has one neighboring strand from the same peptide with the same carbon isotope. An example of such a structure is a β-hairpin where the two antiparallel strands are connected by a loop.
(C) 3-strand block: each peptide molecule contributes three adjacent strands. The middle strand of a 3-strand block has two adjacent strands with the same carbon isotope. An example of such a structure is a 3-stranded β-sheet where the three strands are connected by two loops. This building block can only be applied to a subset of our model structures where the number of strands is a multiple of 3: β-sheets with 6, 12 and 24 strands; β-barrels with 12 and 18 strands.
In addition, we considered two types of non-adjacent arrangements:
(D) Interlaced model 1: each peptide molecule contributes two strands which are separated by one strand from a different peptide. This arrangement requires that the number of strands is a multiple of 4, we used it for our β-sheets with 4, 12 and 24 strands and our β-barrels with 8, 12 and 16 strands.
(E) Interlaced model 2: each peptide molecule contributes two strands that are separated by two strands from two different peptides. For this model the number of strands needs to be a multiple of 6: β-sheets with 6, 12 and 24 strands; β-barrels with 12 and 18 strands.
For labeled strands, the diagonal elements of the mass-normalized force constant matrix were multiplied by 0.94725. This mass factor was obtained from Aβ40 experiments by first matching the simulated and the experimental high and low wavenumber band positions for the 12C-Aβ40 spectrum, as described above, and then simulating the 13C-spectrum (considering 10% 12C-impurities in 13C-strands, see below) with different mass factors until agreement was obtained between the experimental and simulated band positions of the main β-sheet band. Our mass factor is close to the ratio of the reduced masses for labeled and unlabeled CO oscillators (0.95604, resulting in a 37 cm−1 down shift from 1650 cm−1 upon 13C-labeling). Our mass factor is also close to the ratio of the reduced masses inferred from DFT calculations of trans unlabeled and 13C N-methylacetamide (0.94771, resulting in a 45 cm−1 down shift from 1735 cm−1 upon 13C-labeling).74
Three situations could occur for the non-diagonal elements. If the non-diagonal element represented an interaction between two unlabeled groups, the force constant remained unchanged. If the non-diagonal element represented an interaction between two labeled groups, the force constant was multiplied by the mass factor (as for the diagonal elements of the labeled groups). If the non-diagonal element represented an interaction between a labeled and an unlabeled group, the force constant was multiplied by the square root of the mass factor.
In order to account for deviations from the used structures (and consequently from the calculated force constants), statistical variations were added to the elements of the force constant matrix. The diagonal elements were subjected to a random variation between −1% and 1% of their original value and the off-diagonal elements between −10% and 10%. Furthermore, we considered the 10% impurity of the labeled peptides used in the experiments: in other words, there is a 10% chance in our calculations to find unlabeled amide groups inside labeled strands. The presence of the unlabeled amide groups inside labeled strands weakens the coupling within and between labeled strands: in the absorbance spectrum the band position of the main band is upshifted and the splitting between high and low wavenumber bands decreases. For example, the splitting for a 6-stranded 13C-sheet reduces from 61.0 cm−1 to 57.4 cm−1. As a result of including isotopic impurity, the average splitting for sheets with 4–24 strands and 10 residues/strand and all barrels was 57.5 cm−1, which is very similar to the experimental splitting for 13C-Aβ40, which was 57.7 cm−1 (using parameters adjusted to the 12C-spectrum of Aβ40 and a dipole derivative angle of 20°).
From the diagonalization of the force constant matrix, we obtained the wavenumbers and the intensities of the normal modes in the amide I region. With this information, the absorbance spectrum was calculated using Gaussian band shapes with a full width at half maximum of 8 cm−1. For a comprehensive statistical sampling of the variations in the force constants, of the isotope composition of the sheets at a given 13C:12C ratio and of different positions of unlabeled amide groups within labeled strands, 3000 calculations for each ratio were performed and the spectra averaged. The number of 3000 calculations was chosen because it provided a good reproducibility of the spectrum. The calculations were repeated 20 times in order to assess the deviation in the band positions between different simulation runs.
Results
Experiments
We measured the infrared spectra of Aβ oligomers that consisted of mixtures of 12C- and 13C-isotopomers of either Aβ40 or Aβ42. These homo-oligomers are denoted Aβ40 oligomers or Aβ42 oligomers in the following. The peptides were mixed as monomers in 2H2O at alkaline p2H, then brought to p2H 7.4 and measured after 20 min, corresponding to the minimal waiting time to accurately purge the spectrometer sample chamber after sample insertion. Fig. 4 shows the second derivatives of the infrared absorption spectra of Aβ40 and Aβ42 for different 13C:12C-ratios. The minima in second derivatives correspond to the band positions of the component bands in the absorbance spectra. The two major bands are found in spectral regions of β-sheet absorption. Their band positions are listed in Table 1. The band at 1685 cm−1 is an indicator of oligomers and often considered a marker band for antiparallel β-sheets as discussed previously.27,65 The absence of distinct bands in the non-β-sheet spectral regions indicate a large content of β-sheets. Indeed, a fit to the absorbance spectra revealed a contribution of β-sheet bands to the total absorbance in the amide I region of 28% for Aβ40 and of 39% for Aβ42. Since β-sheets absorb stronger in 2H2O than unordered structures by a factor of ∼1.5,75,76 the β-sheet content is estimated to ∼20% for Aβ40 and to ∼25% for Aβ42. The β-sheet content in structured Aβ oligomers might be higher than these values because of the presence of unstructured monomers and aggregates.
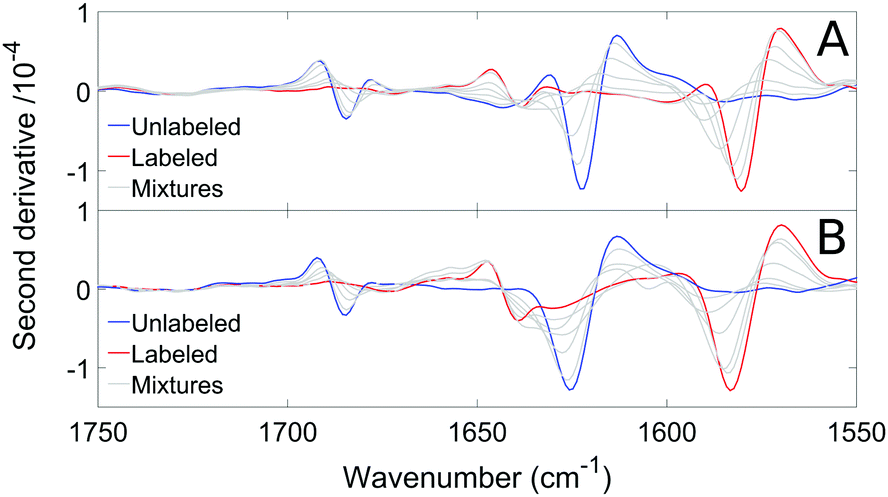 | ||
| Fig. 4 Spectra of the second derivative of IR absorbance of (A) Aβ40 oligomers and (B) Aβ42 oligomers at different 13C:12C-ratios. The spectrum of the all-unlabeled sample is blue and that of the all-labeled sample red. Mixtures of unlabeled and labeled Aβ (9:1, 3:1, 1:1, 1:3, 1:9) are grey. | ||
| Band position ± standard deviation in cm−1 | Number of experiments | |||
|---|---|---|---|---|
| 12C | 13C | 12C | 13C | |
| Aβ40 | 1622.3 ± 0.1 | 1580.6 ± 0.2 | 3 | 5 |
| 1684.6 ± 0.1 | 1638.3 ± 0.3 | |||
| Aβ42 | 1625.5 ± 0.4 | 1582.9 ± 0.4 | 6 | 3 |
| 1684.6 ± 0.2 | 1639.3 ± 0.2 | |||
The spectra of Aβ40 and Aβ42 are similar in shape but the band positions of the main band for unlabeled and labeled Aβ42 are found at 2–3 cm−1 higher wavenumbers than for Aβ40, indicating different β-sheet structures.65 The spectra of the oligomers are similar to those of Aβ42:Aβ40 hetero-oligomers that we have published previously.65
The main band of the unlabeled oligomers at 1622–1625 cm−1 and that of the labeled oligomers at 1580–1583 cm−1 shift towards higher wavenumber upon admixture of peptides with a different isotope. In the following, we will focus on the 13C-band because the 12C-band is not clearly seen at low 12C abundance and because it is overlapped by the high wavenumber band of the 13C-spectrum. The shift of the 13C-band position for both oligomers is shown in Fig. 5.
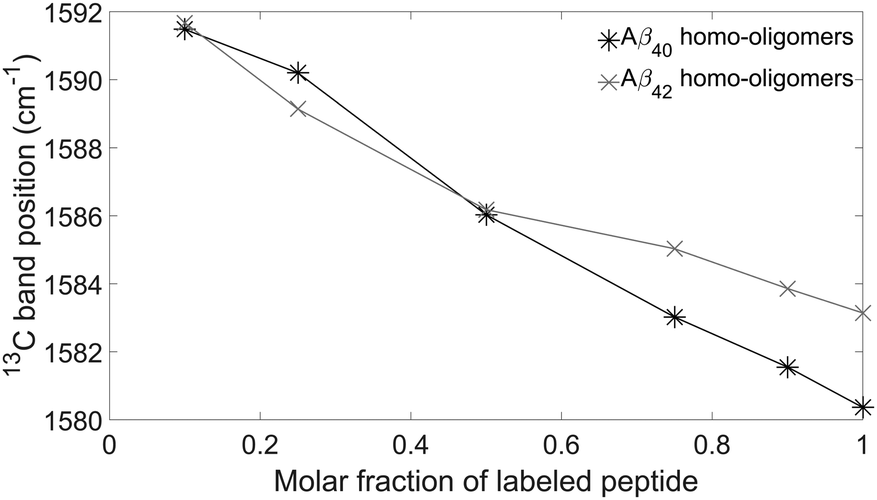 | ||
| Fig. 5 Experimental 13C-band positions for Aβ40 (black) and Aβ42 (grey) oligomers. | ||
Calculation strategy
In order to get information on the internal structure of individual peptide molecules within the oligomers, we calculated the 13C-band shift for a variety of oligomer model structures. The aim of these calculations was a quantitative agreement between calculated and experimental spectra. Here, the calculations faced two challenges: (i) the atomic structure of Aβ oligomers is unknown and (ii) the values for the parameters that enter the spectrum calculations are not known to the necessary precision. Regarding (i) we used a number of model structures including a published atomic model. Regarding (ii) we adjusted the calculation parameters to fit the experimental spectra of the entirely unlabeled and the entirely labeled samples as described in Computational methods. With these constraints, the spectra of the isotopic mixtures were calculated without further adjustments.In our calculations, we considered different internal structures of the peptide molecules within the oligomers giving rise to different building blocks for the β-sheets (see Fig. 3). A building block in our calculations is the contribution of a single peptide molecule to the β-sheets of the oligomers. Within a given building block the carbon isotope is the same (with the exception of 12C impurities in 13C peptides, see Computational methods) and the vibrational coupling represents intramolecular coupling. Coupling between amide groups in different building blocks models the intermolecular coupling. We considered the following building blocks: a single strand, where the strand contributed by one peptide has neighboring strands from other peptides; a 2-strand block, where two adjacent strands contributed by one peptide molecule have 2-strand blocks from other peptides as neighbors; a 3-strand sheet, where each peptide monomer contributes three adjacent strands to the β-sheet of the oligomer. In addition, we considered two types of two non-adjacent strand arrangements: in interlaced model 1, the two strands from one peptide molecule are separated by a strand from another peptide, while in interlaced model 2 the two strands are separated by two strands from two different peptides.
General features of the calculated spectra
Fig. 6 shows an example of an absorbance spectrum and its second derivative calculated by our program, using an ideal antiparallel sheet with 12 strands and 10 residues per strand (9 complete amide groups) and the single strand building block. Each line in Fig. 6 is the average of 20 repetitions of 3000 statistical compositions for each 13C:12C ratio. The blue line and the red line represent the two isotopically pure sheets. Each grey line represents a mixture of unlabeled and labeled species, using the same 13C:12C ratios as in the experiments.
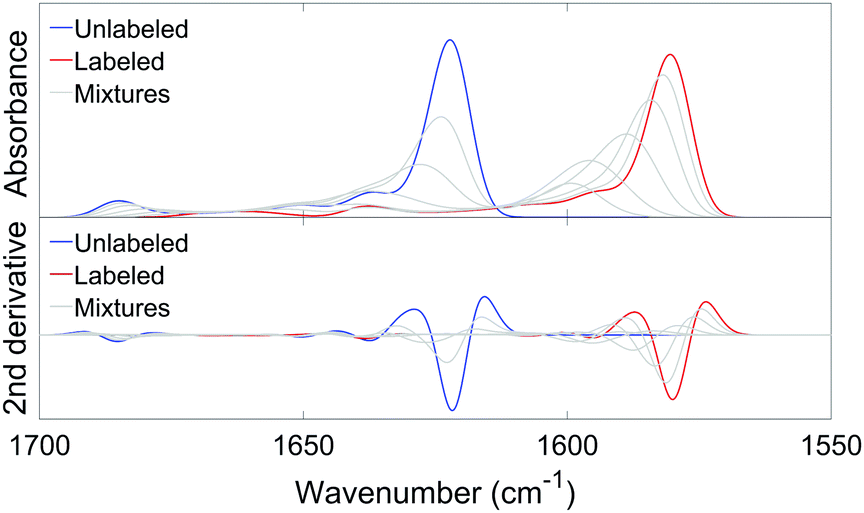 | ||
| Fig. 6 Calculated absorbance and second derivative spectra for an antiparallel sheet with 12 strands and 10 residues per strand (9 complete amide groups), using the single strand building block. In the upper panel, the absorbance spectra are shown for the unlabeled structure (blue), the labeled structure (red) and mixtures (9:1, 3:1, 1:1, 1:3, 1:9) of unlabeled and labeled strands (grey). In the lower panel, the second derivatives of the absorbance spectra in the upper panel are shown, using the same color code. | ||
As in the experimental spectra, the main 12C- and 13C-bands shift down when the β-sheets become isotopically purer and the high wavenumber β-sheet band shifts up. We will focus on the shift of the main 13C-band in the 1600–1580 cm−1 range and use the term 0.1 → 1 13C-band shift for a downshift of the 13C-band position upon changing the 13C:12C ratio from 0.1 to 1.
The role of interstrand coupling
The shift of the 13C-band upon 13C-enrichment is due to vibrational coupling between different strands of a β-sheet. Without interstrand coupling, an isolated (but hydrogen bonded) strand would absorb at the same wavenumber as an entire sheet. This is also true for a single 13C-strand surrounded by 12C-strands, which would absorb at the same wavenumber as an entirely labeled sheet. Therefore, in the absence of interstrand coupling the 13C-band shift would be zero.Even in the presence of interstrand coupling, a 13C-strand that is surrounded by 12C-strands is rather uncoupled from the rest of the sheet. In test calculations of such a system with a β-sheet composed of 12 strands with 10 residues (9 complete amide groups) each and the labeled strand close to the middle of the sheet, only ∼15% of the vibrational energy of the two vibrations with highest intensity in the 13C spectral region was contributed by 12C-amide groups. Accordingly, a single 13C-strand in an otherwise 12C-sheet absorbs similarly (1600 cm−1 in our calculation) to a 13C-strand that is entirely decoupled from the 12C-strands (1605 cm−1). The situation is similar at low 13C:12C ratios because the large distance between the 13C-strands prevents an effective coupling. When more 13C-strands are incorporated into the sheet, 13C-interstrand-coupling becomes noticeable because the 13C-strands get closer and this shifts the main β-sheet band to lower wavenumbers.45 In summary, the 13C-band shift is different from zero when 13C-strands are sufficiently close to enable effective coupling between amide I vibrations in different 13C-strands.
Influence of β-sheet structure
In the following, we discuss the calculations for Aβ40. We used a set of model structures consisting of 7 β-sheets (4–24 strands) and 11 β-barrels (8–22 strands). For each of the model structures, the parameters were adjusted so that the calculated spectra matched the experimental spectrum of the unlabeled sample. Then the spectra of the isotope mixtures were calculated and the 13C-band shifts at a certain molar ratio of labeled peptides averaged for all structures.The shifts calculated for β-barrels of various sizes are rather independent of the number of strands in the barrel, whereas those calculated for flat antiparallel β-sheets increase with the number of strands in the sheet, in particular for small sheets (see Table S4, ESI†). When the building block is a single strand, the steepest dependency is between two and six strands, whereas for building blocks of two and three strands, considerable sensitivity is observed up to 12 strands.
In contrast, the number of residues per strand has a minor role: reducing it from 10 residues (9 complete amide groups) to 6 residues or 4 residues, decreases the 13C-band shift by at most 0.5 cm−1 using the strand building block and sheets with 6 and 12 strands (see Table S4, ESI†).
A further influence that we considered is that of intra-chain coupling where the dominant contribution is the interaction between the nearest neighbors. This interaction depends on the backbone dihedral angles and can be smaller or larger than for ideal antiparallel β-sheets when the dihedral angles deviate from the ideal values.48–51 In the context of the evaluation of our dodecamer results (see below), we tested the effect of reducing the intra-chain nearest neighbor coupling to 1/3 of the original value. This required us to increase the magnitude of the dipole derivative in order to match the splitting of the β-sheet bands of experiment and calculation. This led to stronger interstrand coupling and thus to a larger 13C-band shift (4 cm−1 larger for a 12-stranded sheet and the single strand building block).
Influence of the dipole derivative angle
The angle of the dipole derivative with respect to the CO bond is a parameter that could not be adjusted from a comparison with experiment. Instead, we used fixed values of 10°, 20°, and 30° for a sub-set of four sheets (4, 6, 12 and 24 strands) and four barrels (8, 12, 18 and 22 strands). Similar 13C-band shifts were found for these angles, which are shown in Fig. 7 and tabulated in Table 2. They demonstrate that the calculation results are robust against a change in dipole derivative angle. Therefore, we will discuss only the results for an angle of 20° in the following.
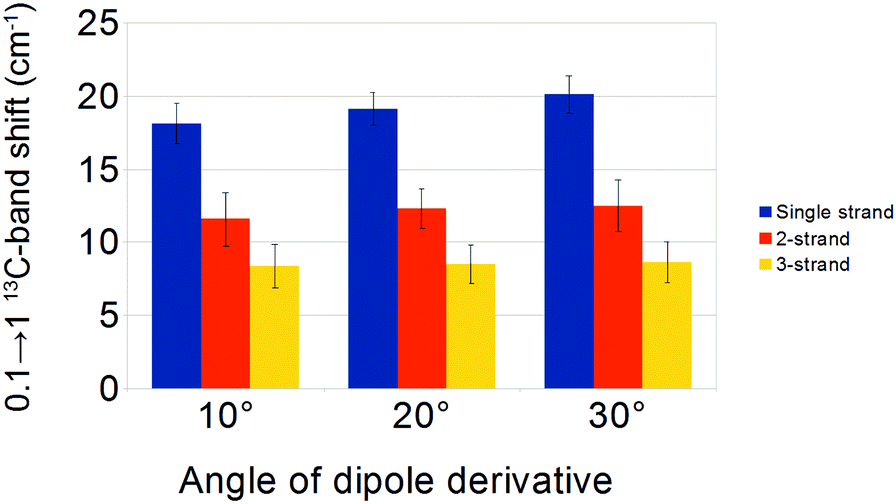 | ||
| Fig. 7 0.1 → 1 13C-band shifts, averaged from sets of four β-sheets and four β-barrels, using different angles of the dipole derivative. The calculation parameters were adjusted to match the all-unlabeled experimental spectrum for Aβ40. Because of the adjustment, the magnitude of the dipole derivative had to be increased when we increased the angle of the dipole derivative. A higher magnitude led to a slightly stronger coupling between the strands, so the shifts of the 13C-band position for the three building blocks are somewhat larger for larger angles. | ||
:12C ratio from 0.1 to 1
| 0.1 → 1 13C-band shift (in cm−1) ± standard deviation | ||||||
|---|---|---|---|---|---|---|
| Single strand | 2-strand block | 3-strand block | Interlaced model 1 | Interlaced model 2 | ||
| Aβ40 | Experiment | 11.1 | ||||
| Angle: 10° | 18.2 ± 1.4 | 11.6 ± 1.8 | 8.4 ± 1.5 | |||
| Angle: 20° | 19.2 ± 1.1 | 12.3 ± 1.3 | 8.5 ± 1.3 | 15.5 ± 1.8 | 17.3 ± 1.3 | |
| Angle: 30° | 20.1 ± 1.3 | 12.5 ± 1.8 | 8.6 ± 1.4 | |||
| Angle: 20° – dodecamer | 12.5 | |||||
| Aβ42 | Experiment | 8.5 | ||||
| Angle: 20° – sheet | 17.1 | 11.5 | 8.2 | |||
| Angle: 20° – barrel | 18.6 | 11.7 | 7.7 | |||
Aβ40 oligomers
Fig. 8 shows the average 13C-band shifts obtained for the Aβ40 calculations and Table 2 collates the 0.1 → 1 13C-band shifts.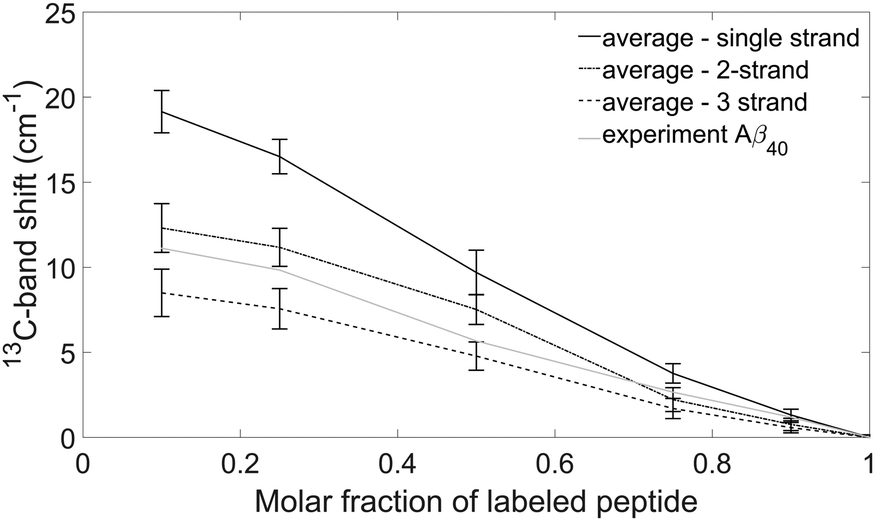 | ||
| Fig. 8 Experimental and simulated 13C-band shifts for Aβ40 oligomers. The calculations were done for 18 model structures (see text) with a dipole derivative angle of 20° and the obtained band shifts averaged. The black solid line is the average 13C-band shift using the single strand building block. The dash-dot line is the average 13C-band shift using the 2-strand building block. The dashed line is the average 13C-band shift using the 3-strand building block. The grey solid line shows the experimental 13C-band shift. The error bars indicate the standard deviations of the band positions calculated for the different model structures. | ||
The figure shows that the 13C-band shift depends on the building block that each peptide monomer contributes to the oligomer structure. The shifts at 10% and 25% 13C-peptide content are largest for the strand building block, smaller for the 2-strand building block and even smaller for the 3-strand building block.
For a particular building block, the standard deviation for all model structures is smaller than the difference between different building blocks (see Fig. 8). Accordingly, the 13C-band shift is more sensitive to the nature of the building block than to the particular structure used for the calculation. This demonstrates that the 13C-band shift can be used to study the internal structure of individual peptides in Aβ oligomers.
The 13C-band shifts for the interlaced building blocks are in between the shifts for the single strand and the 2-strand building blocks. They are listed in Table 2, but not shown in Fig. 8. The results can be explained at the limiting case of highly diluted labeled peptide. Then the neighboring peptides will be unlabeled and the insertion of one or two unlabeled strands between the two strands of the labeled peptide weakens the coupling between the labeled strands, which leads to a higher 13C-band position than in the case of two adjacent strands.
The experimental 13C-band shift is shown as grey line in Fig. 8. The 0.1 → 1 13C-band shift is 11.1 cm−1 which is very close to the average shift calculated for our model structures and the 2-strand building block (12.3 cm−1). This shift depends to some degree on the structure of the β-sheets (see section Influence of β-sheet structure), which is reflected in the error bars in Fig. 8. For example, when a sheet consists of only two strands, the calculated 0.1 → 1 13C-band shift for the single strand building block is 12.6 cm−1, which is close to the shift observed experimentally for Aβ40. However, such a small sheet with only two strands can be excluded from the experimental position of the main β-sheet band as further discussed below (see section Structural constraints from experiments and calculations for Aβ oligomers). In conclusion, our results indicate that each Aβ40 molecule contributes two adjacent strands to the β-sheets of the oligomers.
In addition to our model structures, we investigated a structure obtained by X-ray crystallography of a modified Aβ17–36 peptide. The structure of this peptide has been stabilized by introducing a disulfide bridge between residues 24 and 29 and by linking the N- and C-terminus via an ornithine molecule. In the X-ray structure (pdb entry 5HOW23), each peptide forms a β-hairpin and assembles with two other peptides in a triangular unit that consists of distorted 4-stranded β-sheets at both ends of the hairpins. Four triangular units assemble to a dodecamer, which was used in our calculations. Because of the β-hairpin structure of individual peptides, this structure corresponds to our 2-strand building block. In our calculation the 0.1 → 1 13C-band shift is 12.5 cm−1. It is close to the average shift calculated with the 2-strand building block for our model structures (12.3 cm−1). This shift seems to be caused by two opposing effects. (i) The small number of strands in the sheet of the triangular unit is expected to cause a smaller 13C-band shift. An even smaller shift of only 8.3 cm−1 was calculated for a model sheet with four strands and the 2-strand building block (Table S4, ESI†). (ii) The deviation of the dihedral angles from those of an ideal antiparallel β-sheet is expected to increase the 13C-band shift. An inspection of the Ramachandran plot of the structure revealed that many dihedral backbone angles deviate from those of an ideal antiparallel sheet. The average angles were Φ = −98.5° and Ψ = 113.4° (the dihedral angles of our ideal antiparallel β-sheets were −138.6° and 134.5°, respectively). These angles lead to a weaker nearest neighbor coupling48 which generates a larger 13C-band shift with our approach as discussed above in section Influence of β-sheet structure. Taken together, the 13C-band shift calculated for the dodecamer structure can be explained by the small number of strands in the sheets and a smaller nearest neighbor coupling than in ideal sheets.
We note that the calculated all-unlabeled spectrum of the dodecamer contains two bands at 1622 and 1639 cm−1 in the main region of β-sheet absorption. The higher wavenumber band is not observed in our experiments.
Summarizing the results for the 0.1 → 1 13C-band shift of Aβ40 oligomers, the experimental shift is close to that calculated for the dodecamer and to the average shift of β-sheets and β-barrels when the building block consists of two adjacent strands. We conclude that our experimental isotope shift for Aβ40 oligomers is well explained by β-sheet structures in which individual Aβ40 peptides adopt a β-hairpin structure.
Aβ42 oligomers
In Aβ42 oligomers, the experimental 0.1 → 1 13C-band shift is 8.5 cm−1, which is less than for Aβ40 (11.1 cm−1). For our Aβ42 calculations, we considered only one β-sheet and one β-barrel, both with 12 strands. These particular structures were chosen because they generate 13C-band shifts close to the average shifts in the Aβ40 calculations shown in Fig. 8. Their calculated 12C-spectra were matched to the experimental spectrum and the 13C-band positions of isotope mixtures are shown in Fig. 9 for the three building blocks. Table 2 lists the 13C-band shifts obtained. As shown in Fig. 9, the experimental 13C-band shift matches closely the calculated shift for the 3-strand model. This might indicate that individual Aβ42 peptides contribute more than two adjacent strands to the β-sheets of the oligomers. We consider this conclusion to be valid for sheets of moderate size (12 and more strands). However, when the number of strands per sheet is smaller, the 13C-band shift is also smaller and is similar to the experimental shift for a 4-stranded sheet calculated with the 2-strand building block. Therefore, we cannot exclude that the building block consists of only two strands. We conclude that the β-sheets of Aβ42 either contain a small number of strands, where each peptide molecule contributes a β-hairpin, or contain a larger number of strands, where each peptide contributes more than two adjacent strands to the β-sheets.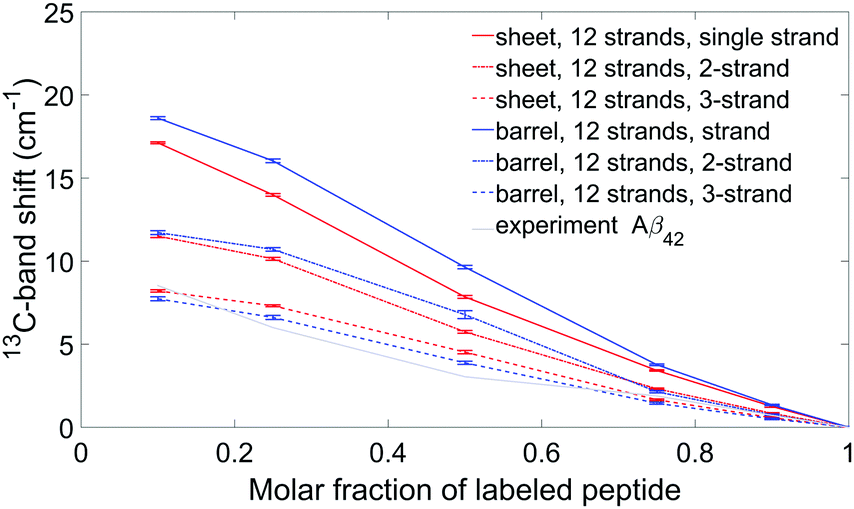 | ||
| Fig. 9 Experimental and simulated 13C-band shifts for Aβ42 oligomers for two specific models: a sheet and a barrel, each composed of 12 strands. The dipole derivative angle was 20°. Each model is represented by a different color, while the three building blocks are shown using three different line style (as in Fig. 8). Experimental band positions are shown using a grey solid line. | ||
Discussion
Comparison with previous infrared studies
We studied the effects of the isotope composition of β-sheets on the amide I infrared spectrum. We focused on the 13C-band shift in order to elucidate the internal structure of Aβ peptides in Aβ42 and Aβ40 oligomers. For all model structures investigated, the shift depends on the structural unit (building block) that each individual peptide molecule contributes to the structure of the oligomers. The differences between the shifts of different building blocks were larger than the standard deviations calculated from the results for different model structures and therefore are useful for structural studies. This is particularly true for the most diluted cases (0.1 and 0.25 13C:12C-ratio) which highlights their importance for the analysis.56
Related to this work, Matos et al.30 investigated mixtures of Aβ42 and pyroglutamylated Aβ3–42. Upon admixture of 50% pyroglutamylated 12C-Aβ3–42 to Aβ42, they observed an upshift of the 13C-Aβ42 band from 1585 cm−1 to 1595 cm−1. This is much larger than the shift of 3 cm−1 for the corresponding isotope dilution step for our Aβ42 oligomers, and of 6 cm−1 for our Aβ40 oligomers. Therefore, the structure of an Aβ42 peptide in the aggregates with pyroglutamylated Aβ3–42 seems to be different from that in our Aβ40 and Aβ42 homo-oligomers as well as in our Aβ40:Aβ42 hetero-oligomers. The 10 cm−1 shift observed by Matos et al. is slightly larger than the average shifts for the single strand building block calculated for our model structures (Fig. 8 and 9). Therefore, individual Aβ42 molecules likely contribute only a single strand to the aggregates with pyroglutamylated Aβ3–42.
Moran et al.56 investigated amyloid fibrils formed by the eye lens protein γD-crystallin in 2H2O. Their 0.1 → 1 13C-band shift of 22 cm−1 was judged to be consistent with contributions of either one or two strands per protein.56 An even larger shift of nearly 30 cm−1 was observed for thermally induced aggregates, which is consistent with a contribution of one strand per protein to the β-sheets of the aggregates.77 We note that both the unlabeled and labeled γD-crystallin aggregates exhibit lower band positions than our Aβ samples, which seems to indicate stronger interstrand coupling and thus explains that Moran et al. observe larger shifts than we calculate.
From a similar isotope dilution experiment, Buchanan et al.59 concluded that each monomer within their antiparallel polyQ amyloid fibrils contributes two adjacent strands. Their experimental 0.1 → 1 13C-band shift of 12 cm−1 is close to our average shifts for the 2-strand block (11.6 cm−1, 12.3 cm−1 and 12.5 cm−1 for the Aβ40 calculations and dipole derivative angles of 10°, 20°, and 30°, respectively) demonstrating agreement between the two studies.
Structural constraints from experiments and calculations for Aβ oligomers
In the present study, we analyzed the 13C-band shift of Aβ oligomers that contained a mixture of labeled and unlabeled peptides. Because the structure of Aβ oligomers is debated, we used a large number of model structures for our analysis. We also matched the calculations to the experimental spectra. This approach removed two biases – those of selecting a particular structure and particular calculation parameters. This made possible a direct quantitative comparison between experiment and calculations. It turned out that the results were robust in the sense that different internal structures of Aβ produce quantitatively different shifts irrespective of the particular overall β-sheet structure used for the calculations. For further improvement of the calculations, more information about the structure of Aβ oligomers is needed.According to the results of our study, Aβ molecules contribute more than just a single strand to the structure of the oligomers. Also the two interlaced building blocks, the 13C-band shifts of which are located between those of the single strand block and of the 2-strand block, failed to reproduce the experimentally observed shifts. Instead, our experiments with Aβ40 are best described by a contribution of two adjacent strands from each peptide molecule. For Aβ42, contributions of two or more adjacent strands are possible or a combination of these. These structural constraints are fulfilled by (successive) β-hairpins, which are also in line with the antiparallel orientation of the β-strands inferred from the observation of a distinct high wavenumber band.
A further structural constraint can be derived from the band position of the main β-sheet band and the splitting between high and low wavenumber band, which are sensitive to the number of strands in a sheet.45,78 For the all-unlabeled samples it is found below 1626 cm−1 for both peptides and the splitting is ∼60 cm−1. Designed β-hairpins and 3-stranded antiparallel sheets absorb at a higher wavenumber, near ∼1635 cm−1 in 2H2O, and the splitting is smaller: ∼40 cm−1.79–81 From this comparison we deduce that the β-sheets of our Aβ oligomers are formed by at least four strands.
We conclude that our experimental spectra and 13C-band shifts can be well explained by flat, antiparallel β-sheets or by β-barrels with at least four strands in which each peptide molecule contributes two adjacent strands to Aβ40 oligomers and two or more adjacent strands to Aβ42 oligomers. Less uniform structures than flat sheets and barrels, like that of the dodecamer,23 are likely to produce more distinct bands than the two bands that we observed.
In our previous study,65 we studied Aβ40:Aβ42 hetero-oligomers in which one of the peptides was labeled. A comparison with homo-oligomers (see Fig. 2 of that publication) shows very similar shifts of the 13C-band upon isotope enrichment which we regard identical within experimental error. This indicates that the structure of each peptide is similar in homo- and hetero-oligomers.
Our structural constraints are consistent with oligomer models that propose a hairpin structure for individual peptide molecules.17,23,24,82 They are also in line with the suggestion that Aβ42 may fold into units with more strands per molecule.21 Models in which a considerable section of the peptide contributes only one strand to the oligomers16,18 do not well explain the isotope effect seen for our oligomers. The hairpins should be organized in a β-sheet with at least four strands or in a β-barrel. The latter has been suggested previously24 on the basis of the infrared absorption spectrum. Sheets and barrels with little structural distortions account well for our data. The trends seen in our results can be generalized also to other aggregating peptides. Those aggregates with amide I band positions similar to ours will produce 13C-band shifts that can be compared quantitatively to our calculations. This enables to deduce the internal structure of individual peptide molecules in these aggregates.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to Eeva-Liisa Karjalainen for the original version of the Matlab program for spectrum calculations and to Erik Goormaghtigh (Université Libre de Bruxelles) for providing the program Kinetics. We acknowledge support from Stockholms läns landsting, Wenner-Gren Stiftelserna, Knut och Alice Wallenbergs Stiftelse, Magnus Bergvalls Stiftelse and Stiftelsen Lars Hiertas Minne.References
- D. R. Thal, J. Walter, T. C. Saido and M. Fändrich, Acta Neuropathol., 2015, 129, 167–182 CrossRef CAS PubMed.
- R. Aleksis, F. Oleskovs, K. Jaudzems, J. Pahnke and H. Biverstål, Biochimie, 2017, 140, 176–192 CrossRef CAS PubMed.
- F. Chiti and C. M. Dobson, Annu. Rev. Biochem., 2017, 86, 27–68 CrossRef CAS PubMed.
- I. Benilova, E. Karran and B. De Strooper, Nat. Neurosci., 2012, 15, 349–357 CrossRef CAS PubMed.
- J. A. Hardy and G. A. Higgins, Science, 1992, 256, 184–185 CrossRef CAS PubMed.
- J. Hardy and D. Allsop, Trends Pharmacol. Sci., 1991, 12, 383–388 CrossRef CAS PubMed.
- D. J. Selkoe, Neuron, 1991, 6, 487–498 CrossRef CAS PubMed.
- K. Beyreuther and C. L. Masters, Brain Pathol., 1991, 1, 241–251 CrossRef CAS.
- D. J. Selkoe and J. Hardy, EMBO Mol. Med., 2016, 8, 595–608 CrossRef CAS PubMed.
- B. Caughey and P. T. Lansbury, Annu. Rev. Neurosci., 2003, 26, 267–298 CrossRef CAS PubMed.
- K. L. Viola and W. L. Klein, Acta Neuropathol., 2015, 129, 183–206 CrossRef CAS PubMed.
- C. Haass and D. J. Selkoe, Nat. Rev. Mol. Cell Biol., 2007, 8, 101–112 CrossRef CAS PubMed.
- E. Y. Hayden and D. B. Teplow, Alzheimer's Res. Ther., 2013, 5, 60 CrossRef PubMed.
- S. Lesné, Swiss Med. Wkly., 2014, 144, w14021 Search PubMed.
- M. Fändrich, J. Mol. Biol., 2012, 421, 427–440 CrossRef PubMed.
- L. Yu, R. Edalji, J. E. Harlan, T. F. Holzman, A. P. Lopez, B. Labkovsky, H. Hillen, S. Barghorn, U. Ebert, P. L. Richardson, L. Miesbauer, L. Solomon, D. Bartley, K. Walter, R. W. Johnson, P. J. Hajduk and E. T. Olejniczak, Biochemistry, 2009, 48, 1870–1877 CrossRef CAS PubMed.
- C. Lendel, M. Bjerring, A. Dubnovitsky, R. T. Kelly, A. Filippov, O. N. Antzutkin, N. C. Nielsen and T. Härd, Angew. Chem., Int. Ed., 2014, 53, 12756–12760 CrossRef CAS PubMed.
- W. M. Tay, D. Huang, T. L. Rosenberry and A. K. Paravastu, J. Mol. Biol., 2013, 425, 2494–2508 CrossRef CAS PubMed.
- D. Huang, M. I. Zimmerman, P. K. Martin, A. J. Nix, T. L. Rosenberry and A. K. Paravastu, J. Mol. Biol., 2015, 427, 2319–2328 CrossRef CAS PubMed.
- S. Chimon, M. A. Shaibat, C. R. Jones, D. C. Calero, B. Aizezi and Y. Ishii, Nat. Struct. Mol. Biol., 2007, 14, 1157–1164 CrossRef CAS PubMed.
- M. Ahmed, J. Davis, D. Aucoin, T. Sato, S. Ahuja, S. Aimoto, J. I. Elliott, W. E. Van Nostrand and S. O. Smith, Nat. Struct. Mol. Biol., 2010, 17, 561–567 CrossRef CAS PubMed.
- V. A. Streltsov, J. N. Varghese, C. L. Masters and S. D. Nuttall, J. Neurosci., 2011, 31, 1419–1426 CrossRef CAS PubMed.
- A. G. Kreutzer, I. L. Hamza, R. K. Spencer and J. S. Nowick, J. Am. Chem. Soc., 2016, 138, 4634–4642 CrossRef CAS PubMed.
- E. Cerf, R. Sarroukh, S. Tamamizu-Kato, L. Breydo, S. Derclaye, Y. F. Dufrêne, V. Narayanaswami, E. Goormaghtigh, J.-M. Ruysschaert and V. Raussens, Biochem. J., 2009, 421, 415–423 CrossRef CAS PubMed.
- B. Martial, T. Lefèvre and M. Auger, Biophys. Rev., 2018, 10, 1133–1149 CrossRef CAS PubMed.
- H. Komatsu, L. Liu, I. V. J. Murray and P. H. Axelsen, Biochim. Biophys. Acta, Biomembr., 2007, 1768, 1913–1922 CrossRef CAS PubMed.
- R. Sarroukh, E. Goormaghtigh, J.-M. Ruysschaert and V. Raussens, Biochim. Biophys. Acta, Biomembr., 2013, 1828, 2328–2338 CrossRef CAS PubMed.
- K. Nunez, J. Kay, A. Krotow, M. Tong, A. R. Agarwal, E. Cadenas and S. M. de la Monte, J. Alzheimer's Dis., 2016, 51, 151–163 CAS.
- J. Nguyen, M. A. Baldwin, F. E. Cohen and S. B. Prusiner, Biochemistry, 1995, 34, 4186–4192 CrossRef CAS PubMed.
- J. O. Matos, G. Goldblatt, J. Jeon, B. Chen and S. A. Tatulian, J. Phys. Chem. B, 2014, 118, 5637–5643 CrossRef CAS PubMed.
- S. D. Moran and M. T. Zanni, J. Phys. Chem. Lett., 2014, 5, 1984–1993 CrossRef CAS PubMed.
- S. M. Decatur, Acc. Chem. Res., 2006, 39, 169–175 CrossRef CAS PubMed.
- Z. Ganim, H. S. Chung, A. W. Smith, L. P. DeFlores, K. C. Jones and A. Tokmakoff, Acc. Chem. Res., 2008, 41, 432–441 CrossRef CAS PubMed.
- Y. S. Kim and R. M. Hochstrasser, J. Phys. Chem. B, 2009, 113, 8231–8251 CrossRef CAS PubMed.
- J. Manor and I. T. Arkin, Biochim. Biophys. Acta, Biomembr., 2013, 1828, 2256–2264 CrossRef CAS PubMed.
- J. Ma, I. M. Pazos, W. Zhang, R. M. Culik and F. Gai, Annu. Rev. Phys. Chem., 2015, 66, 357–377 CrossRef CAS PubMed.
- H. Torii and M. Tasumi, J. Chem. Phys., 1992, 96, 3379–3387 CrossRef CAS.
- H. Torii and M. Tasumi, in Infrared spectroscopy of biomolecules, ed. H. H. Mantsch and D. Chapman, Wiley-Liss, New York, 1996, pp. 1–18 Search PubMed.
- A. Amadei, I. Daidone, A. Di Nola and M. Aschi, Curr. Opin. Struct. Biol., 2010, 20, 155–161 CrossRef CAS PubMed.
- J. Jeon, S. Yang, J.-H. Choi and M. Cho, Acc. Chem. Res., 2009, 42, 1280–1289 CrossRef CAS PubMed.
- T. la Cour Jansen, J. Knoester and C. Jansen, J. Chem. Phys., 2006, 124, 44502 CrossRef PubMed.
- H. Torii, T. Tatsumi and M. Tasumi, J. Raman Spectrosc., 1998, 29, 537–546 CrossRef CAS.
- H. Torii, T. Tatsumi, T. Kanazawa and M. Tasumi, J. Phys. Chem. B, 1998, 102, 309–314 CrossRef CAS.
- S. Ham, J.-H. Kim, H. Lee and M. Cho, J. Chem. Phys., 2003, 118, 3491–3498 CrossRef CAS.
- Y. N. Chirgadze and N. A. Nevskaya, Biopolymers, 1976, 15, 607–625 CrossRef CAS PubMed.
- S. Krimm and Y. Abe, Proc. Natl. Acad. Sci. U. S. A., 1972, 69, 2788–2792 CrossRef CAS.
- T. C. Cheam and S. Krimm, Chem. Phys. Lett., 1984, 107, 613–616 CrossRef CAS.
- T. la Cour Jansen, A. G. Dijkstra, T. M. Watson, J. D. Hirst and J. Knoester, J. Chem. Phys., 2006, 125, 44312 CrossRef PubMed.
- T. la Cour Jansen, A. G. Dijkstra, T. M. Watson, J. D. Hirst and J. Knoester, J. Chem. Phys., 2012, 136, 209901 CrossRef.
- H. Torii and M. Tasumi, J. Raman Spectrosc., 1998, 229, 81–86 CrossRef.
- S. Ham and M. Cho, J. Chem. Phys., 2003, 118, 6915–6922 CrossRef CAS.
- J.-H. Choi and M. Cho, J. Chem. Phys., 2004, 120, 4383–4392 CrossRef CAS PubMed.
- R. D. Gorbunov, D. S. Kosov and G. Stock, J. Chem. Phys., 2005, 122, 224904 CrossRef PubMed.
- J.-H. Choi, S. Ham and M. Cho, J. Chem. Phys., 2002, 117, 6821 CrossRef CAS.
- J. P. Lomont, K. L. Rich, M. Maj, J.-J. Ho, J. S. Ostrander and M. T. Zanni, J. Phys. Chem. B, 2018, 122, 144–153 CrossRef CAS PubMed.
- S. D. Moran, A. M. Woys, L. E. Buchanan, E. Bixby, S. M. Decatur and M. T. Zanni, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 3329–3334 CrossRef CAS PubMed.
- Z. Ganim and A. Tokmakoff, Biophys. J., 2006, 91, 2636–2646 CrossRef CAS PubMed.
- T. M. Watson and J. D. Hirst, Phys. Chem. Chem. Phys., 2004, 6, 998–1005 RSC.
- L. E. Buchanan, J. K. Carr, A. M. Fluitt, A. J. Hoganson, S. D. Moran, J. J. de Pablo, J. L. Skinner and M. T. Zanni, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 5796–5801 CrossRef CAS PubMed.
- S. Hahn, S. Ham and M. Cho, J. Phys. Chem. B, 2005, 109, 11789–11801 CrossRef CAS PubMed.
- L. Wang, C. T. Middleton, M. T. Zanni and J. L. Skinner, Methods, 2011, 3713–3724 CAS.
- E. Małolepsza and J. E. Straub, J. Phys. Chem. B, 2014, 118, 7848–7855 CrossRef PubMed.
- H. Torii, J. Phys. Chem. B, 2007, 111, 5434–5444 CrossRef CAS PubMed.
- Y. Abe and S. Krimm, Biopolymers, 1972, 11, 1817–1839 CrossRef CAS PubMed.
- M. Baldassarre, C. M. Baronio, L. A. Morozova-Roche and A. Barth, Chem. Sci., 2017, 8, 8247–8254 RSC.
- M. Baldassarre, C. Li, N. Eremina, E. Goormaghtigh and A. Barth, Molecules, 2015, 20, 12599–12622 CrossRef CAS PubMed.
- E.-L. Karjalainen, T. Ersmark and A. Barth, J. Phys. Chem. B, 2012, 116, 4831–4842 CrossRef CAS PubMed.
- R. Fraser and T. MacRae, in Conformation in fibrous proteins and related synthetic polypeptides, ed. R. Fraser, Academic Press, New York, 1973, pp. 218–246 Search PubMed.
- I. Bertini, L. Gonnelli, C. Luchinat, J. Mao and A. Nesi, J. Am. Chem. Soc., 2011, 133, 16013–16022 CrossRef CAS PubMed.
- A. T. Petkova, Y. Ishii, J. J. Balbach, O. N. Antzutkin, R. D. Leapman, F. Delaglio and R. Tycko, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 16742–16747 CrossRef CAS PubMed.
- T. Lührs, C. Ritter, M. Adrian, D. Riek-Loher, B. Bohrmann, H. Döbeli, D. Schubert and R. Riek, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 17342–17347 CrossRef PubMed.
- N. Guex and M. C. Peitsch, Electrophoresis, 1997, 18, 2714–2723 CrossRef CAS PubMed.
- W. H. Moore and S. Krimm, Biopolymers, 1976, 15, 2439–2464 CrossRef CAS PubMed.
- G. Cuevas, V. Renugopalakrishnan, G. Madrid and A. T. Hagler, Phys. Chem. Chem. Phys., 2002, 4, 1490–1499 RSC.
- Y. N. Chirgadze, B. V. Shestopalov and S. Y. Venyaminov, Biopolymers, 1973, 12, 1337–1351 CrossRef CAS PubMed.
- M. Jackson, P. I. Haris and D. Chapman, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 1989, 998, 75–79 CrossRef CAS.
- S. D. Moran, T. O. Zhang and M. T. Zanni, Protein Sci., 2014, 23, 321–331 CrossRef CAS PubMed.
- J. Kubelka and T. A. Keiderling, J. Am. Chem. Soc., 2001, 123, 12048–12058 CrossRef CAS PubMed.
- J. Hilario, J. Kubelka and T. A. Keiderling, J. Am. Chem. Soc., 2003, 125, 7562–7574 CrossRef CAS PubMed.
- D. Scheerer, H. Chi, D. McElheny, T. A. Keiderling and K. Hauser, J. Phys. Chem. B, 2018, 122, 10445–10454 CrossRef CAS PubMed.
- Y. Xu, P. Purkayastha and F. Gai, J. Am. Chem. Soc., 2006, 128, 15836–15842 CrossRef CAS PubMed.
- A. Sandberg, L. M. Luheshi, S. Söllvander, T. Pereira de Barros, B. Macao, T. P. J. Knowles, H. Biverstål, C. Lendel, F. Ekholm-Petterson, A. Dubnovitsky, L. Lannfelt, C. M. Dobson and T. Härd, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 15595–15600 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cp00717b |
| This journal is © the Owner Societies 2019 |