
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structural determinants of coiled coil mechanics†
Patricia
López-García
 a,
Melis
Goktas
a,
Ana E.
Bergues-Pupo
b,
Beate
Koksch
c,
Daniel
Varón Silva
d and
Kerstin G.
Blank
*a
a,
Melis
Goktas
a,
Ana E.
Bergues-Pupo
b,
Beate
Koksch
c,
Daniel
Varón Silva
d and
Kerstin G.
Blank
*a
aMax Planck Institute of Colloids and Interfaces, Mechano(bio)chemistry, Science-Park Potsdam Golm, 14424 Potsdam, Germany. E-mail: kerstin.blank@mpikg.mpg.de
bMax Planck Institute of Colloids and Interfaces, Department of Theory and Bio-Systems, Science-Park Potsdam Golm, 14424 Potsdam, Germany
cFreie Universität Berlin, Institute of Chemistry and Biochemistry – Organic Chemistry, Takustrasse 3, 14195 Berlin, Germany
dMax Planck Institute of Colloids and Interfaces, Department of Biomolecular Systems, Science-Park Potsdam Golm, 14424 Potsdam, Germany
First published on 16th April 2019
Abstract
The natural abundance of coiled coil (CC) motifs in the cytoskeleton and the extracellular matrix suggests that CCs play a crucial role in the bidirectional mechanobiochemical signaling between cells and the matrix. Their functional importance and structural simplicity has allowed the development of numerous applications, such as protein-origami structures, drug delivery systems and biomaterials. With the goal of establishing CCs as nanomechanical building blocks, we investigated the importance of helix propensity and hydrophobic core packing on the mechanical stability of 4-heptad CC heterodimers. Using single-molecule force spectroscopy, we show that both parameters determine the force-induced dissociation in shear loading geometry; however, with different effects on the energy landscape. Decreasing the helix propensity lowers the transition barrier height, leading to a concomitant decrease in the distance to the transition state. In contrast, a less tightly packed hydrophobic core increases the distance to the transition state. We propose that this originates from a larger side chain dynamics, possible water intrusion at the interface as well as differences in solvation of the hydrophobic amino acids at the transition state. In conclusion, the different contributions of helix propensity and hydrophobic core packing need to be considered when tuning the mechanical properties of CCs for applications.
Coiled coils (CCs) are self-assembled, superhelical motifs that are naturally found in numerous proteins in the cytoskeleton and the extracellular matrix.1 CCs consist of two (or more) α-helices, each characterized by a repetitive pattern of seven amino acids, called heptad (abcdefg)n (Fig. 1a). Positions a and d form the hydrophobic core of the superhelical structure; e and g are mostly charged amino acids, which participate in interhelical salt bridges; b, c and f are solvent-exposed, often polar amino acids, which contribute to the helix propensity of the individual helices.2,3
 | ||
| Fig. 1 Experimental design. (a) CC heptad pattern. (b) SMFS setup showing mechanical loading of a CC heterodimer in the shear geometry. (c) Sequences of the CCs used in this study. The terminal cysteines define the shear pulling geometry. The helix propensity of the CC-forming peptides was calculated using AGADIR.4 | ||
Utilizing this simple design, CCs serve as model systems for studying protein folding and stability. As a result, they are increasingly used as templates for protein design and sequences with a pre-determined thermodynamic stability can now be synthesized de novo.5–8 Such sequences find application in peptide-based hydrogels9–11 and protein origami structures12,13 as well as in biosensors14 and drug delivery systems.15 Considering their natural role as a mechanical scaffold, surprisingly little information is available about the sequence–structure–mechanics relationship of CCs. With the goal of introducing CCs as nanomechanical building blocks, we have characterized three different CC heterodimers with atomic force microscope (AFM)-based single-molecule force spectroscopy (SMFS; Fig. 1b). We show that hydrophobic core packing and helix propensity affect the thermodynamic stability in similar ways; however, the underlying changes to the energy landscape are different.
Using the thermodynamically and mechanically well-characterized CC A4B4 as the starting point (Fig. 1c),7,16 we used the standard rules of CC design2,5 to obtain one sequence with a reduced helix propensity and a second sequence with a different hydrophobic core packing. Throughout the manuscript, we define hydrophobic core packing as the combination of hydrophobicity and side chain packing of the amino acids in positions a and d. To reduce the helix propensity, Ala in position b was substituted with Ser in all heptads17 (A4SB4S; Fig. 1c). Hydrophobic core packing was altered using another β-branched amino acid in position a (Val instead of Ile; A4VB4V). The Asn in the third heptad was not replaced to maintain heterospecificity.18,19 For A4VB4V, it needs to be considered that this substitution also decreases the helix propensity, which is lower for Val than for Ile (Fig. 1c). To define the points of force application, Cys was introduced at the desired termini for coupling the CC to the surface and the AFM cantilever. For A-peptides, Cys was located at the N-terminus, while it was placed at the C-terminus of B-peptides, thus establishing a shear pulling geometry (Fig. 1b).
To validate the design and to compare mechanical and thermodynamic stability, the CCs were first investigated with circular dichroism (CD) spectroscopy. Wavelength scans, showing an α-helical signature with minima at 208 nm and 222 nm and thermal denaturation experiments are displayed in Fig. S1 (ESI†). As expected, A4B4 shows the highest melting temperature Tm, while A4SB4S and A4VB4V were significantly destabilized (Table 1). The corresponding free energy difference between the folded (F) and the unfolded (U) state ( ) was obtained from van’t Hoff plots (Fig. S2, ESI†). In comparison to A4B4, both modified CCs also show a lower
) was obtained from van’t Hoff plots (Fig. S2, ESI†). In comparison to A4B4, both modified CCs also show a lower  , which follows a similar trend compared to other CCs reported in the literature.5,20
, which follows a similar trend compared to other CCs reported in the literature.5,20
| T m (°C) |
|
F (pN) | ΔxF–TS (nm) | k off (s−1) | ΔGF–TS (kBT) | |
|---|---|---|---|---|---|---|
| a Most probable rupture force F determined at a retract speed of 400 nm s−1. All values are depicted as mean ± standard error of the mean (SEM). | ||||||
| A4B4 | 77.0 ± 0.3 | 14.2 ± 0.3 | 43.1 ± 0.2 | 1.3 ± 0.2 | (3.2 ± 2.1) × 10−4 | 29.2 ± 1.4 |
| A4SB4S | 54.3 ± 0.3 | 5.3 ± 0.2 | 23.6 ± 4.8 | 0.9 ± 0.1 | (2.8 ± 1.1) × 10−1 | 21.3 ± 0.2 |
| A4VB4V | 59.0 ± 0.6 | 7.1 ± 0.6 | 28.2 ± 0.1 | 1.7 ± 0.0 | (2.4 ± 1.7) × 10−3 | 26.6 ± 0.7 |
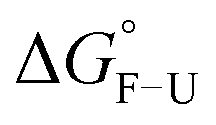
To address the question whether the thermodynamic and mechanical stabilities are correlated and how the respective substitutions affect the energy landscape, SMFS was carried out. The B-peptide was immobilized to the cantilever, while the corresponding A-peptide was immobilized to the surface (Fig. S3, ESI†). The CC forming peptides cooperatively fold and associate when the cantilever is in contact with the surface. When retracting the cantilever, the CC experiences a steadily increasing force until it ruptures, yielding two unfolded monomers (Fig. 1b). At a retract speed of 400 nm s−1, both A4SB4S and A4VB4V are mechanically less stable than A4B4 (Fig. 2a). At this retract speed, the most probable rupture forces decrease 20 pN and 15 pN for A4SB4S and A4VB4V, respectively. This provides a first indication that the substitutions also lower the mechanical stability of the CCs.
 | ||
| Fig. 2 Single-molecule force spectroscopy. (a) Representative rupture force histograms obtained at a retract speed of 400 nm s−1, with n = 284 (A4B4), n = 243 (A4SB4S) and n = 420 (A4VB4V). The dashed lines show Gaussian fits, applied to extract the most probable rupture forces. The inset shows representative force–extension curves for each CC. (b) Dynamic SMFS plot. Each CC was measured in triplicate using a different cantilever and surface (different shades of the same colour). The solid lines represent fits to the Bell–Evans model. | ||
Subsequent dynamic SMFS, performed over a range of loading rates (r = dF/dt)21 from approximately 15 pN s−1 to 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 pN s−1, revealed that both modified CCs possess a lower mechanical stability over the complete range of loading rates (Fig. S4–S6, ESI†); however, their dependence on the loading rate is different. Fitting the data to the Bell–Evans model21 (Fig. 2b) allows for obtaining more detailed information about the energy landscape of the CC two-state system (folded CC vs. random coil peptides): koff, the force-free dissociation rate, and ΔxF–TS, the distance from the folded to the transition state (TS). Using the Arrhenius equation, also the barrier height between the folded and the transition state (ΔGF–TS) can be calculated, provided that the Arrhenius pre-factor is known. Here, we use an Arrhenius pre-factor of 5 × 108 s−1, which was estimated for the dimeric GCN4 leucine zipper.22Table 1 summarizes the obtained fit values, as well as the calculated ΔGF–TS values. In addition to the results shown in Table 1, where the mean ± standard error of the mean (SEM) was calculated from three independent experiments, a Peacock test23 was performed to determine if the modified CCs A4SB4S and A4VB4V are significantly different from the reference sequence A4B4. This test shows p-values smaller than 0.01 (see ESI† for details).
000 pN s−1, revealed that both modified CCs possess a lower mechanical stability over the complete range of loading rates (Fig. S4–S6, ESI†); however, their dependence on the loading rate is different. Fitting the data to the Bell–Evans model21 (Fig. 2b) allows for obtaining more detailed information about the energy landscape of the CC two-state system (folded CC vs. random coil peptides): koff, the force-free dissociation rate, and ΔxF–TS, the distance from the folded to the transition state (TS). Using the Arrhenius equation, also the barrier height between the folded and the transition state (ΔGF–TS) can be calculated, provided that the Arrhenius pre-factor is known. Here, we use an Arrhenius pre-factor of 5 × 108 s−1, which was estimated for the dimeric GCN4 leucine zipper.22Table 1 summarizes the obtained fit values, as well as the calculated ΔGF–TS values. In addition to the results shown in Table 1, where the mean ± standard error of the mean (SEM) was calculated from three independent experiments, a Peacock test23 was performed to determine if the modified CCs A4SB4S and A4VB4V are significantly different from the reference sequence A4B4. This test shows p-values smaller than 0.01 (see ESI† for details).
Comparing the koff values presented in Table 1 shows that both modifications (A4SB4S and A4VB4V) lower the height of the transition state barrier with respect to the reference sequence A4B4 (higher koff and lower ΔGF–TS). This lower barrier height is correlated with the different thermodynamic stabilities (Table 1) and helix propensities of the three CCs (Fig. 1c). This suggests that a reduced helix propensity lowers the barrier height, thereby affecting both the thermodynamic and mechanical stability of the CCs. Interestingly, the ΔxF–TS values do not correlate with the thermodynamic stabilities. The Ala-Ser modification (A4SB4S) reduces ΔxF–TS, whereas the Ile-Val modification (A4VB4V) increases ΔxF–TS. This suggests that modifications in the solvent-exposed residues affect the energy landscape of the CC interaction differently when compared to hydrophobic core modifications.
In contrast to cooperative dissociation and unfolding upon increasing temperature, CCs respond to an applied axial stretching force in three phases.16,24–26 Initially, the force increases linearly with extension and the helices remain intact (phase I). At a strain of 10–20%, the individual helices start uncoiling at an almost constant force (phase II). In long CCs, the force increases sharply after the helices are uncoiled and the resulting structure is extended further (phase III). For CCs with a length of ≤4 heptads loaded in the shear geometry, the CC chains separate in phase I or just at the transition to phase II.16 This is a direct result of the force-induced chain separation mechanism. At loading rates typically used in SMFS, the applied force causes the uncoiling of helical structure at the points of force application. This, in turn, destabilizes the CC thermodynamically and facilitates the subsequent dissociation of the CC chains (uncoiling-assisted dissociation). This mechanism allows for explaining the effects of helix propensity and hydrophobic core packing on CC shearing.
When mechanically loaded in shear geometry, the hydrogen bonds stabilizing the individual helices are aligned parallel to the force vector, whereas the hydrophobic side chains are arranged almost perpendicularly. The torsional angles and helical propensities of the individual amino acids, which are responsible for maintaining stable hydrogen bonds in the helices,17,27,28 are thus critically determining the resistance of CCs to shear forces. For CCs with a lower overall helix propensity less force is required to uncoil the individual helices. In addition, a lower helix propensity is correlated with a lower thermodynamic stability. Uncoiling of only small parts of helical structure thus destabilizes an already less stable CC further, an effect that was observed earlier when decreasing CC length.16 Assuming that the hydrophobic core is not altered, this suggests that chain separation occurs at smaller extensions. In the case of A4SB4S, a higher koff value is thus accompanied by a shorter ΔxF–TS. This result is in line with the observation that artificial constraints, which stabilize the helices against uncoiling, lead to an increase in the forces required for chain separation.3,11,29
Following this line of argumentation, ΔxF–TS for A4VB4V is expected to lie in between the values obtained for A4SB4S and A4B4; however, A4VB4V shows an increase in ΔxF–TS. This suggests that substituting Ile with Val does not only affect the helix propensity. We propose that the increased ΔxF–TS originates from the combined effect of side chain hydrophobicity and packing. A reduced packing is expected to increase side chain dynamics at the hydrophobic interface, thus facilitating the rearrangement of the Val side chains in response to the applied force. In other words, a less densely packed hydrophobic core may allow the relative displacement of the helices prior to or during helix uncoiling. At the same time, changes in side chain hydrophobicity affect the interaction of the CC structure with water. Shear deformation may lead to water intrusion at the interface (wetting), followed by force-induced helix uncoiling. Alternatively, the helices may unfold without significant wetting. Independent of the presence of wetting, helix unfolding exposes hydrophobic amino acids to solvent, which need to be hydrated (solvation).20,30–33 While both wetting and solvation are primarily determined by side chain hydrophobicity, also the local environment31,32,34,35 and protein flexibility36 are expected to play important roles. As the two-state energy landscape model condenses the contribution of all these factors onto a single reaction coordinate (Fig. 3), it remains an open question which factor(s) determine the observed change in ΔxF–TS.
 | ||
Fig. 3 Energy landscape of the CCs. The horizontal line represents the distance from the folded (F) to the transition state (TS) (ΔxF–TS), while the vertical solid arrow represents the transition barrier height (ΔGF–TS). The dotted arrow shows the energy difference between the folded and the unfolded state (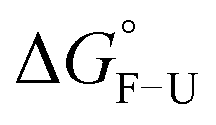 ). ). | ||
An increase in ΔxF–TS has also been observed when introducing destabilizing substitutions in the hydrophobic core of the globular proteins GB1 and protein L.37,38 This effect, termed mechanical softening, was absent for the titin I27 domain, however, where mechanical unfolding is determined by a number of key hydrogen bonds.39,40 This suggests that substitutions in the hydrophobic core only affect the energy landscape parameters when positioned at a mechanically loaded interface. Clearly, CCs represent an attractive model system to dissect the different contributions of hydrophobicity and side chain packing to the mechanics of hydrophobic interfaces, e.g. via temperature-dependent SMFS experiments.
In summary, the combination of helix propensity and hydrophobic core packing determines the mechanical stability of CCs; however, with different effects on the energy landscape (Fig. 3). Whereas a reduced helix propensity decreases both the barrier height and the distance to the transition state, an increase in the transition state distance is obtained when decreasing the hydrophobic core packing. Clearly, both parameters can be utilized when designing CCs with controlled mechanical properties for future applications. Most interestingly, these parameters can be used to obtain CCs with similar thermodynamic stabilities that possess a different dynamic response to an externally applied force. Such systems will find application as molecular force sensors41 or as physical hydrogel crosslinks, which show a pre-defined response to mechanical deformation.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Isabell Tunn, Alberto Sanz de Leon and Ana Vila Verde for inspiring discussions. This work was funded by the Max Planck Society and the International Max Planck Research School (IMPRS) on Multiscale Bio-Systems. Open Access funding provided by the Max Planck Society.References
- A. N. Lupas and J. Bassler, Trends Biochem. Sci., 2017, 42, 130–140 CrossRef CAS PubMed.
- D. N. Woolfson, Subcell. Biochem., 2017, 82, 35–61 CAS.
- I. Drobnak, H. Gradisar, A. Ljubetic, E. Merljak and R. Jerala, J. Am. Chem. Soc., 2017, 139, 8229–8236 CrossRef CAS PubMed.
- E. Lacroix, A. R. Viguera and L. Serrano, J. Mol. Biol., 1998, 284, 173–191 CrossRef CAS PubMed.
- J. R. Litowski and R. S. Hodges, J. Biol. Chem., 2002, 277, 37272–37279 CrossRef CAS PubMed.
- H. Gradišar and R. Jerala, J. Pept. Sci., 2011, 17, 100–106 CrossRef PubMed.
- F. Thomas, A. L. Boyle, A. J. Burton and D. N. Woolfson, J. Am. Chem. Soc., 2013, 135, 5161–5166 CrossRef CAS PubMed.
- R. O. Crooks, A. Lathbridge, A. S. Panek and J. M. Mason, Biochemistry, 2017, 56, 1573–1584 CrossRef CAS PubMed.
- J. Yang, C. Xu, C. Wang and J. Kopeček, Biomacromolecules, 2006, 7, 1187–1195 CrossRef CAS PubMed.
- C. Aronsson, S. Danmark, F. Zhou, P. Oberg, K. Enander, H. Su and D. Aili, Sci. Rep., 2015, 5, 14063 CrossRef CAS PubMed.
- I. Tunn, A. S. de Léon, K. G. Blank and M. J. Harrington, Nanoscale, 2018, 10, 22725–22729 RSC.
- A. L. Boyle, E. H. C. Bromley, G. J. Bartlett, R. B. Sessions, T. H. Sharp, C. L. Williams, P. M. G. Curmi, N. R. Forde, H. Linke and D. N. Woolfson, J. Am. Chem. Soc., 2012, 134, 15457–15467 CrossRef CAS PubMed.
- A. Ljubetic, F. Lapenta, H. Gradisar, I. Drobnak, J. Aupic, Z. Strmsek, D. Lainscek, I. Hafner-Bratkovic, A. Majerle, N. Krivec, M. Bencina, T. Pisanski, T. C. Velickovic, A. Round, J. M. Carazo, R. Melero and R. Jerala, Nat. Biotechnol., 2017, 35, 1094–1101 CAS.
- H. Chao, D. L. Bautista, J. R. Litowski, R. T. Irvin and R. S. Hodges, J. Chromatogr. B: Biomed. Sci. Appl., 1998, 715, 307–329 CrossRef CAS.
- F. Thomas, W. M. Dawson, E. J. M. Lang, A. J. Burton, G. J. Bartlett, G. G. Rhys, A. J. Mulholland and D. N. Woolfson, ACS Synth. Biol., 2018, 7, 1808–1816 CrossRef CAS PubMed.
- M. Goktas, C. Luo, R. M. A. Sullan, A. E. Bergues-Pupo, R. Lipowsky, A. Vila Verde and K. G. Blank, Chem. Sci., 2018, 9, 4610–4621 RSC.
- C. N. Pace and J. M. Scholtz, Biophys. J., 1998, 75, 422–427 CrossRef CAS PubMed.
- E. O'Shea, R. Rutkowski and P. Kim, Science, 1989, 243, 538–542 CrossRef.
- J. M. Fletcher, G. J. Bartlett, A. L. Boyle, J. J. Danon, L. E. Rush, A. N. Lupas and D. N. Woolfson, ACS Chem. Biol., 2017, 12, 528–538 CrossRef CAS PubMed.
- A. Akmal and V. Muñoz, Proteins: Struct., Funct., Bioinf., 2004, 57, 142–152 CrossRef CAS PubMed.
- E. Evans and K. Ritchie, Biophys. J., 1997, 72, 1541–1555 CrossRef CAS PubMed.
- B. Ibarra-Molero, G. I. Makhatadze and C. R. Matthews, Biochemistry, 2001, 40, 719–731 CrossRef CAS PubMed.
- A. E. Bergues-Pupo, M. Goktas, I. Tunn, P. Lopez-Garcia, A. V. Verde, K. G. Blank and A. Valleriani, J. Chem. Phys., 2018, 149, 244120 CrossRef PubMed.
- D. D. Root, V. K. Yadavalli, J. G. Forbes and K. Wang, Biophys. J., 2006, 90, 2852–2866 CrossRef CAS PubMed.
- S. G. Falkovich, I. M. Neelov and A. A. Darinskii, Polym. Sci., Ser. A, 2010, 52, 662–670 CrossRef.
- Z. Qin and M. J. Buehler, Phys. Rev. Lett., 2010, 104, 198304 CrossRef PubMed.
- P. Chakrabarti and D. Pal, Prog. Biophys. Mol. Biol., 2001, 76, 100–102 CrossRef.
- A. Chakrabartty, T. Kortemme and R. L. Baldwin, Protein Sci., 1994, 3, 843–852 CrossRef CAS PubMed.
- A. E. Bergues-Pupo, K. G. Blank, R. Lipowsky and A. Vila Verde, Phys. Chem. Chem. Phys., 2018, 20, 29105–29115 RSC.
- M. S. Cheung, A. E. García and J. N. Onuchic, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 685–690 CrossRef CAS PubMed.
- R. Zhou, X. Huang, C. J. Margulis and B. J. Berne, Science, 2004, 305, 1605–1609 CrossRef CAS PubMed.
- P. Liu, X. Huang, R. Zhou and B. J. Berne, Nature, 2005, 437, 159–162 CrossRef CAS PubMed.
- I. Popa, J. M. Fernandez and S. Garcia-Manyes, J. Biol. Chem., 2011, 286, 31072–31079 CrossRef CAS PubMed.
- A. K. Jana, J. C. Jose and N. Sengupta, Phys. Chem. Chem. Phys., 2013, 15, 837–844 RSC.
- A. K. Jana and N. Sengupta, Biophys. J., 2012, 102, 1889–1896 CrossRef CAS PubMed.
- R. C. Remsing, E. Xi and A. J. Patel, J. Phys. Chem. B, 2018, 122, 3635–3646 CrossRef CAS PubMed.
- D. P. Sadler, E. Petrik, Y. Taniguchi, J. R. Pullen, M. Kawakami, S. E. Radford and D. J. Brockwell, J. Mol. Biol., 2009, 393, 237–248 CrossRef CAS PubMed.
- T. Bu, H. C. Wang and H. Li, Langmuir, 2012, 28, 12319–12325 CrossRef CAS PubMed.
- D. J. Brockwell, G. S. Beddard, J. Clarkson, R. C. Zinober, A. W. Blake, J. Trinick, P. D. Olmsted, D. A. Smith and S. E. Radford, Biophys. J., 2002, 83, 458–472 CrossRef CAS PubMed.
- R. B. Best, S. B. Fowler, J. L. T. Herrera, A. Steward, E. Paci and J. Clarke, J. Mol. Biol., 2003, 330, 867–877 CrossRef CAS PubMed.
- M. Goktas and K. G. Blank, Adv. Mater. Interfaces, 2017, 4, 1600441 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cp00665f |
| This journal is © the Owner Societies 2019 |