
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAtmospheric oxidation reactions of imidazole initiated by hydroxyl radicals: kinetics and mechanism of reactions and atmospheric implications†
Zahra
Safaei
 a,
Abolfazl
Shiroudi
*b,
Ehsan
Zahedi
c and
Mika
Sillanpää
a
a,
Abolfazl
Shiroudi
*b,
Ehsan
Zahedi
c and
Mika
Sillanpää
a
aDepartment of Green Chemistry, LUT University, Sammonkatu 12, FI-50130 Mikkeli, Finland
bYoung Researchers and Elite Club, East Tehran Branch, Islamic Azad University, Tehran, Iran. E-mail: abolfazl.shiroudi@iauet.ac.ir
cChemistry Department, Shahrood Branch, Islamic Azad University, Shahrood, Iran
First published on 5th April 2019
Abstract
The atmospheric oxidation mechanism of imidazole initiated by hydroxyl radicals is investigated via OH-addition and H-abstraction pathways by quantum chemistry calculations at the M06-2X/aug-cc-pVTZ level of theory coupled with reaction kinetics calculations using statistical Rice–Ramsperger–Kassel–Marcus (RRKM) theory and transition state theory (TST). It was found that OH addition proceeds more rapidly than H-abstraction by several orders of magnitude. Moreover, H-abstraction reactions with submerged barriers exhibit positive temperature dependence. Effects of reaction temperature and pressure on the reaction between imidazole and OH radicals are studied by means of RRKM calculations. Effective rate coefficients involve two-step mechanisms. According to the experiment, the obtained branching ratios show that the kinetically most efficient process corresponds to OH addition onto a carbon atom which is adjacent to a nitrogen atom having a lower energy barrier. These ratios also reveal that the regioselectivity of the oxidation reaction decreases with increasing temperatures and decreasing pressures. Because of negative activation energies, pressures larger than 100 bar are required to reach the high pressure limit. The atmospheric lifetime of imidazole in the presence of OH radicals is estimated to be ∼4.74 days, based on the calculated overall kinetic rate constant of 1.22 × 10−12 cm3 molecule−1 s−1 at a pressure of 1 bar and nearly ambient temperature. NBO analysis demonstrates that the calculated energy barriers are dictated by charge transfer effects and aromaticity changes because of the delocalization of nitrogen lone pairs to empty π* orbitals.
1. Introduction
The aromatic heterocycle imidazole is widespread in organic compounds. Imidazole plays an important role in biochemical processes because of its special acid–base characteristics. The side chain of histidine contains a weakly basic imidazole group that is a versatile catalyst; it is a strong Lewis base, besides a nucleophile, and it is a Brønsted acid in its protonated form. Accordingly, some histidine residues are essential for the activity of proteins such as hemoglobin, carbonic anhydrase, adenosine deaminase, triose phosphate isomerase, serine proteases, chymotrypsin, etc. Moreover, two nitrogenous bases found in nucleotides, adenine and guanine, are derivatives of purine, a heterocycle consisting of fused pyrimidine and imidazole rings. Nucleotides are building blocks of nucleic acids and coenzymes with diverse functions.1–3 It is well-known that the reactions of the OH radicals with aromatic and heterocyclic compounds are much favored.In the reaction between aromatic compounds and OH radicals, an addition of hydroxyl radicals and the consequent unimolecular decay of the [aromatic-OH]˙ adduct back to the isolated reactants is assumed to be the main reaction path.4 Since only some investigations have been carried out on the reaction between aromatic heterocyclic compounds and OH radicals,5–12 Witte and Zetzsch13 have tried to get further information on the oxidation reaction mechanism of imidazole initiated by OH radicals in the gas phase. In order to consider the temperature dependence, a flash photolysis resonance fluorescence (FP-RF) technique over the temperature range 297–447 K has been used in this study. Biexponential decays of OH radicals are found in the presence of imidazole at a pressure of 133 mbar with argon (Ar) as an inert gas over the temperature range from 353 to 425 K. The reaction between imidazole and OH radicals shows negative activation energy, which indicates an electrophilic addition of OH to the imidazole. The ratio estimation for the amounts of biexponential decay curve yield equilibrium constants, and therefore kinetic rate constants for the unimolecular decay of the [imidazole-OH]˙ adducts leading back to the isolated reactants. Using activation energy, it is possible to derive the bond dissociation energies of the [imidazole-OH]˙ adducts forming back hydroxyl radicals.
An Arrhenius plot of the rate coefficients measured at the temperature range from 297 to 440 K is shown in Fig. 1. The kinetic rate coefficient between imidazole and hydroxyl radicals shows negative temperature dependences over the temperature range 297–440 K, which is equivalent to Arrhenius activation energies of −(1847.91 ± 158.96) cal mol−1.13 Hence, the least-square fit of the experimental rate constants yields as follows:13–15
kforward = (1.7 ± 0.4) × 10−12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp[+(930 ± 80)/T] cm3 molecule−1 s−1 exp[+(930 ± 80)/T] cm3 molecule−1 s−1 |
| kreverse = 5 × 109exp[−(7800 ± 700)/T] s−1 |
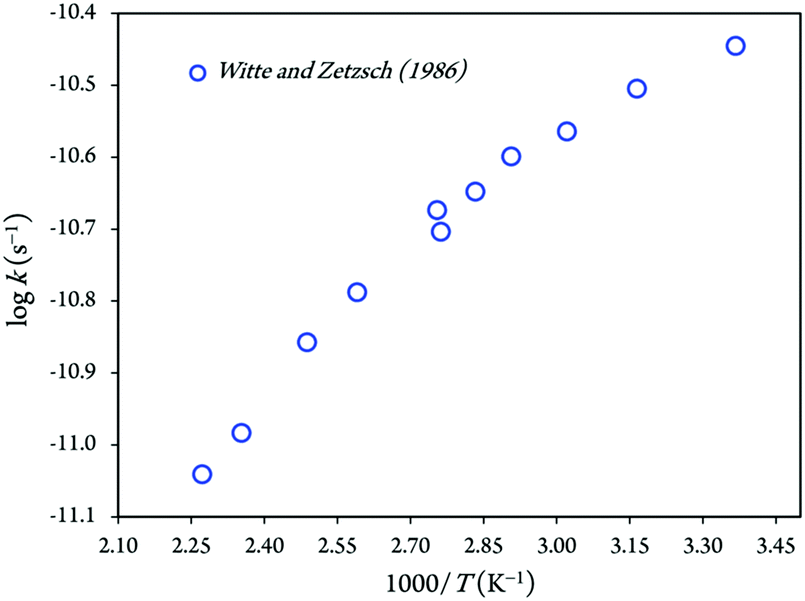 | ||
| Fig. 1 Arrhenius plot for the kinetic rate constant of reaction between imidazole and OH radicals.29 | ||
Experiments show that hydroxyl radicals react with imidazoles by addition at a carbon adjacent to the nitrogen (C2 or C5 positions).17,18 The oxidation reaction of imidazole (I) initiated by hydroxyl radicals might yield three possible adducts, i.e., the 2-hydroxyimidazolyl (I2-OH˙), 4-hydroxyimidazolyl (I4-OH˙), and 5-hydroxyimidazolyl (I5-OH˙) radicals. Nevertheless, electron spin resonance (ESR) measurements and experimental data17 as well as a theoretical study by Llano and Eriksson3 revealed that in neutral and alkaline (pH = 9–10) aqueous solutions, hydroxyl radicals exactly add on the carbon adjacent to the nitrogen (C5 position) and in acidic media (pH = 2), to the same position leading to the 5-adduct.17–19 In spite of the site specificity, I2-OH˙ adducts are energetically more favored by ∼4 kcal mol−1 than I5-OH˙ adducts.
To explain the difference of the kinetic rate constants, six various reactions (1)–(6) were proposed for the oxidation of imidazole by OH radicals in the gas phase (Fig. 2) via H-abstraction and OH-addition reactions. In this study, the main purpose is to investigate theoretically these pathways, upon the assumption of a two-step mechanism. To our knowledge, the present work is the first one, which investigates the oxidation mechanisms between imidazole and hydroxyl radicals under experimental conditions:
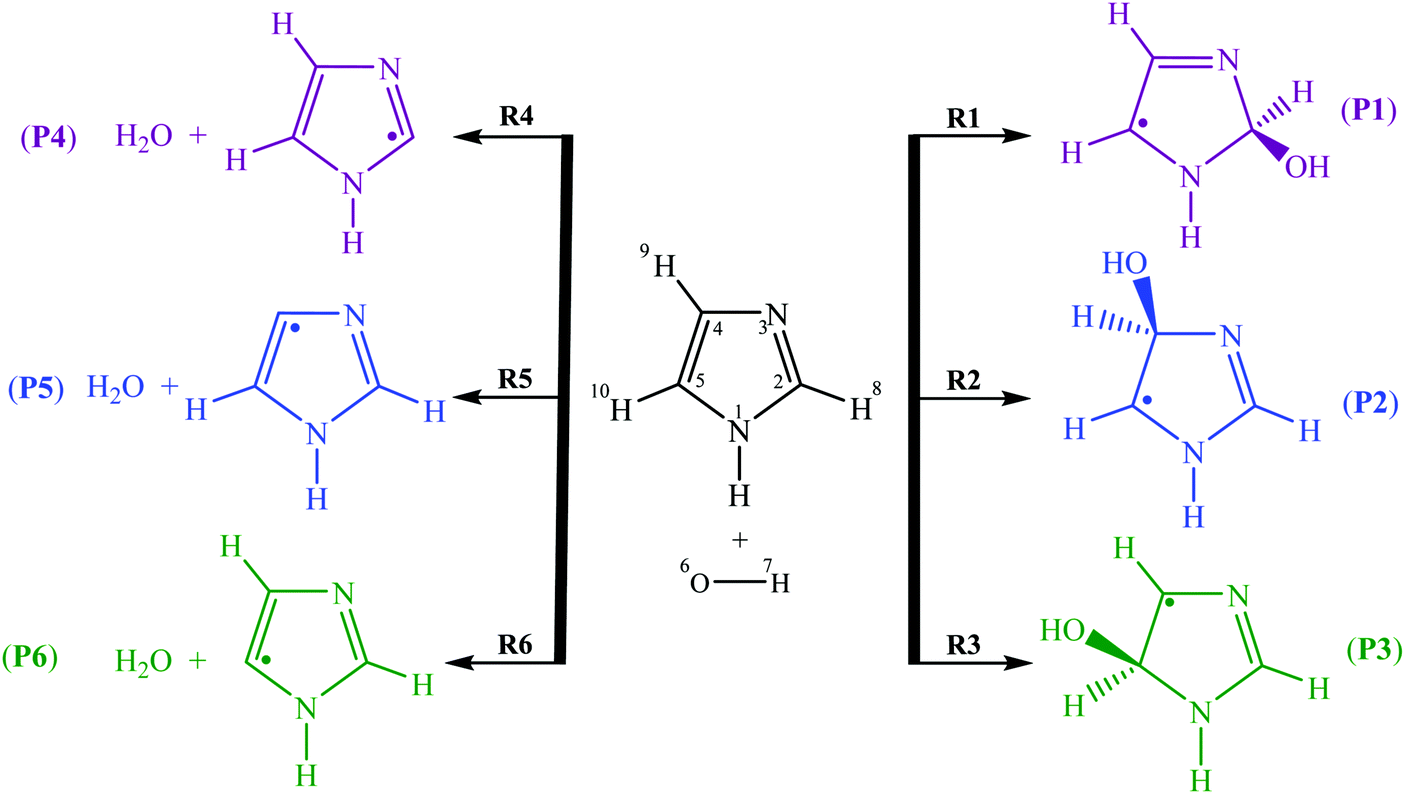 | ||
| Fig. 2 Oxidation reactions between imidazole and hydroxyl radicals. | ||
– OH-addition pathway through attack of hydroxyl radicals onto C2, C4 and C5 atoms yields the products P1–P3, respectively.
– H-abstraction pathway via attack of OH radicals bonded to C2, C4 and C5 atoms gives the products P4–P6, respectively.
In proportion to the hypothesis of the first reversible addition step, the negative energy barrier of this pathway at 298 K points out that the main pathway of the [imidazole-OH]˙ adducts is loss of OH radicals to regenerate the isolated reactants.6 Upon investigating the regioselectivity of OH-addition pathways on imidazole under an inert atmosphere, it was observed that the pathways 1 and 3 associated with the OH addition onto C2 and C5 atoms dominate over hydroxyl radical addition onto the C4 atom (pathway 2).
The obtained M06-2X results will be investigated concerning natural bond orbital (NBO) occupancies,25,26 nucleus independent chemical shift (NICS) indices of aromaticity,20–24 and the nature of electron donor–acceptor interactions for the sake of chemical insights. In this work, we studied the kinetics of oxidation of imidazole. Ab initio calculations of the transition state theory (TST) as well as RRKM theory were used to determine the kinetic rate coefficients via H-abstraction and OH-addition pathways over the temperatures ranging from 297 to 440 K using the M06-2X method to explore the intrinsic insight of the imidazole in the gas-phase which can provide important information for the experimental mechanism investigation. Finally, our obtained theoretical results were compared with experimental data and the results from preceding theoretical studies.
2. Theory and computational details
All the electronic structure calculations were achieved using the Gaussian 09 program.27 The geometrical structures of the stationary points are optimized and vibrational frequencies are calculated at the M06-2X/aug-cc-pVTZ theoretical level28,29 which was found to be a suitable exchange–correlation functional for thermochemical and kinetics calculations.29 Zero-point vibrational energies (ZPVE) were calculated from the M06-2X harmonic frequencies with the scaling factor of 0.971.30,47The intrinsic reaction coordinates (IRC)31 for both directions (forward and reverse) were also performed in order to confirm that the transition state structure properly connects reactants and products.32 IRC calculation uses 30 points in the forward direction and 30 points in the reverse direction, in steps of 0.1 amu1/2 Bohr along the path. The NICS values are achieved by implementation of the gauge-independent atomic orbital (GIAO) method33 to determine the diamagnetic ring current intensity on the optimized geometries of all stationary points along the studied reaction pathways. Schleyer et al.23 state that there is a good linear relationship between the geometric, energetic, and magnetic properties in the organic molecules. The magnetic shielding tensor is calculated for Bq ghost atoms which located at the ring critical point, the point of lowest density in the ring plane to yield NICS(0) values.34–36 The NICS(0) values calculated at the center of the ring were influenced by σ-bonds, whereas the NICS(1) values calculated at 1 Å above the plane were more affected by the π-system which is considered to better reflect the π-electron effects than NICS(0).22,23
The oxidation reaction of imidazole by hydroxyl radicals was analyzed consistent with the scheme37 that is expected; the pathway takes place consistent with the two-step reaction mechanism38 including first the fast pre-equilibrium between the reactants (C3H4N2 + OH˙) and a prereactive complex [C3H4N2⋯OH]˙ (IM) leading to the related products as follows:
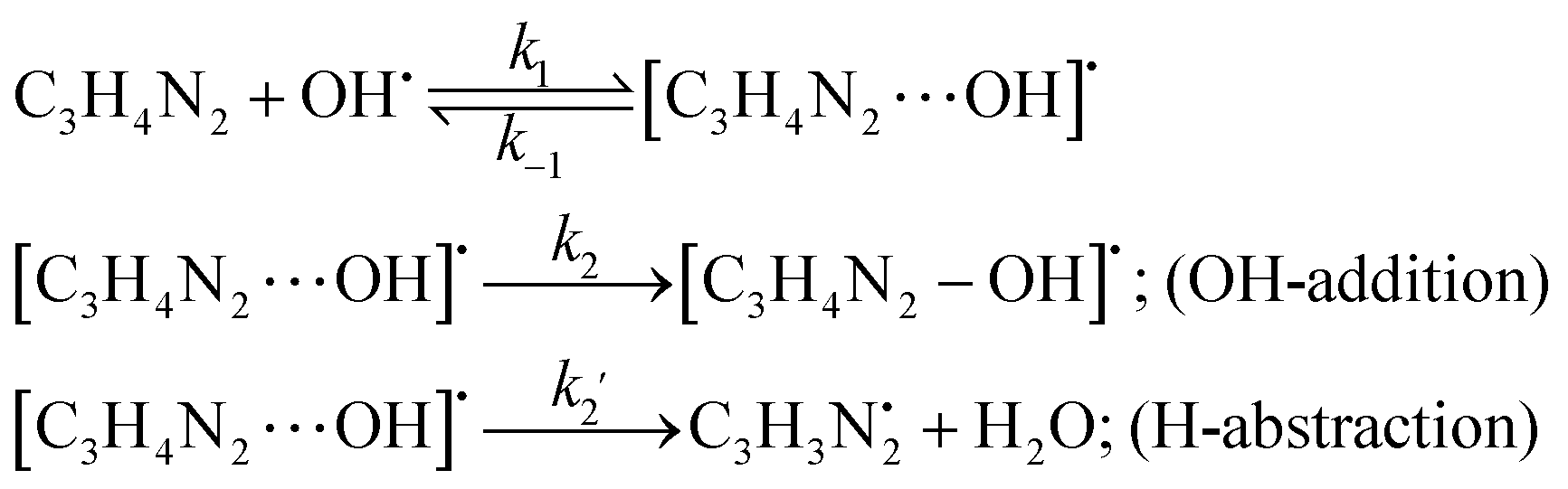
| keff = Kck2 | (1) |
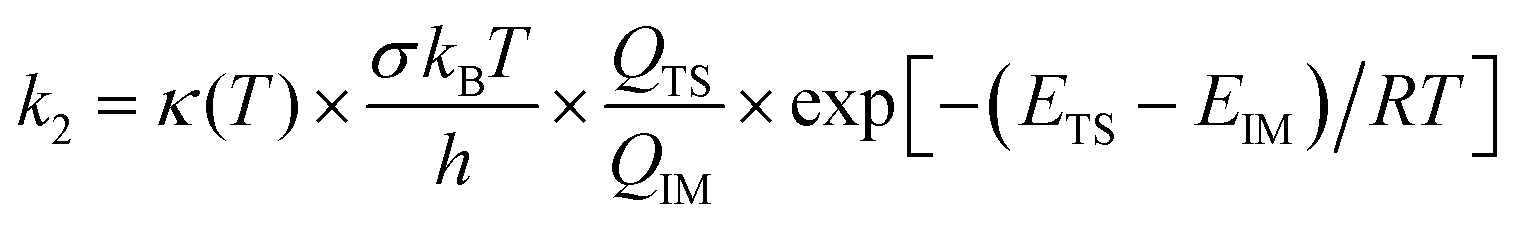 | (2) |
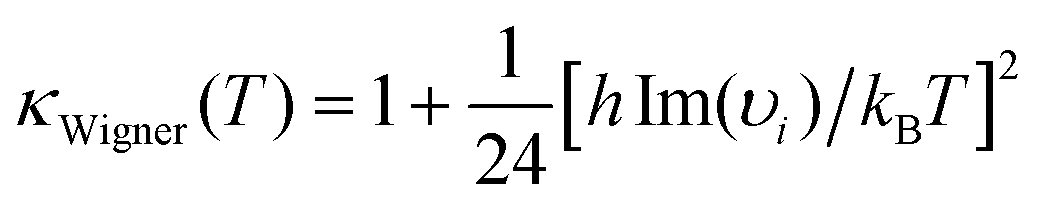 | (3) |
The high-pressure limit kinetic rate coefficients for unimolecular (kuni, in s−1) and bimolecular (kb, in cm3 molecule−1 s−1) reactions using TST are given by:42,43
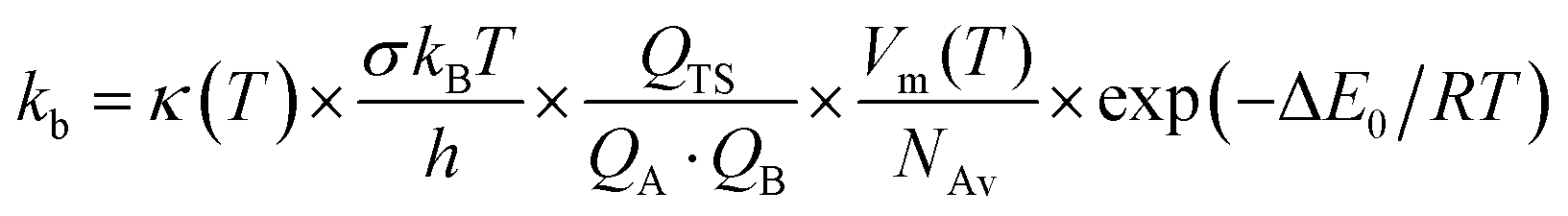 | (4) |
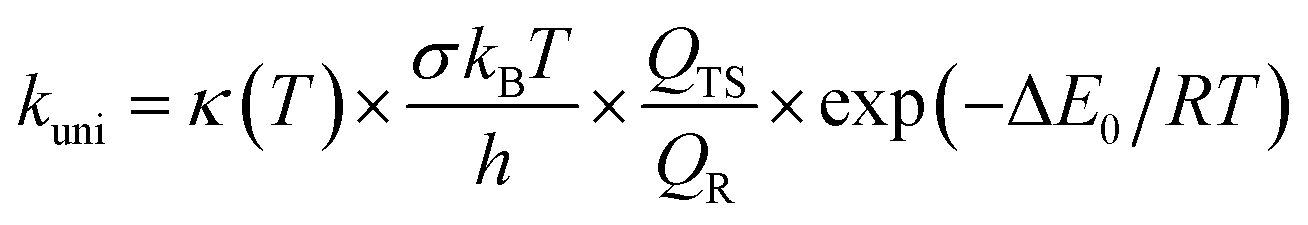 | (5) |
In the oxidation of benzene initiated by OH radicals, there are some unimolecular reactions which might not reach their high-pressure limits under typical atmospheric conditions,44 though the resulting product yields from RRKM theory and high-pressure limit agreed excellently. Therefore to check the validity of the high-pressure limit in imidazole oxidation, RRKM calculations were carried out for the oxidation of imidazole by OH radicals.
The energy-dependent microcanonical rate coefficients, k(E) for the unimolecular reaction according to the RRKM theory is given by:42
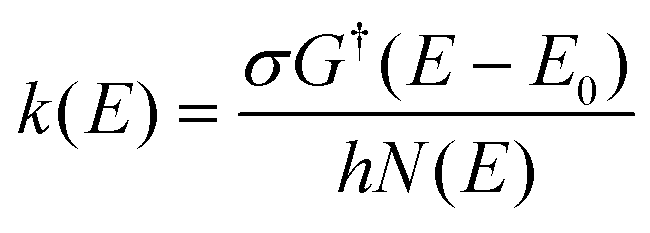 | (6) |
Kinetic rate constants by means of TST and RRKM theories for the studied pathways were obtained using the KiSThelP program.46 It is worth mentioning that every collision deactivates the molecule with ω = βc·ZLJ·[M], which is consistent with the effective collision frequency ω, along with the total gas concentration [M], the collisional efficiency βc, and the Lennard-Jones (LJ) collision frequency ZLJ that was computed from the LJ parameters. The employed Lennard-Jones potential parameters (σ and ε/kB) for [C3H4N2–OH]˙ adducts and argon (as diluent gas) are equal to (σ = 4.8 Å and ε/kB = 492.7 K)48 and (σ = 3.465 Å and ε/kB = 113.5 K),49 respectively.
3. Result and discussion
3.1. The reaction of imidazole with OH radicals
The reactions between imidazole and OH radicals involve two kinds of reaction pathways:(1) Attack of hydroxyl radicals onto the C2, C4, and C5 positions.
(2) Abstraction of hydrogen bonded to the C2, C4, and C5 atoms (H abstraction from C2–H8, C4–H9, and C5–H10 bonds).
In line with a study by Alvarez-Idaboy et al.,50 such a pre-reactive intermediate complex IM is a common feature in reactions between radical species and unsaturated molecules, which finds its origin in long range Coulomb interactions between the reactant molecules.51
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C5 bond of imidazole are not equivalent; thus, three different [C3H4N2–OH]˙ energized adducts (P1–P3) can be formed via addition of hydroxyl radicals onto the C2, C4, and C5 positions, respectively, as depicted in Fig. 2. Under atmospheric conditions, the [C3H4N2–OH]˙ energized adducts could easily further react with oxygen molecules, and form intermediate [C3H4N2–OH–O2]˙ radicals, transition states (TS1–TS3) for the three OH-additions and confirmed by the intrinsic reaction coordinate (IRC) approach. The reaction energies and energy barriers, as well as thermodynamical parameters, are calculated at the M06-2X/aug-cc-pVTZ level of theory for hydroxyl radical attacks onto C2, C4, and C5 positions of imidazole and are shown in Table 1.
C5 bond of imidazole are not equivalent; thus, three different [C3H4N2–OH]˙ energized adducts (P1–P3) can be formed via addition of hydroxyl radicals onto the C2, C4, and C5 positions, respectively, as depicted in Fig. 2. Under atmospheric conditions, the [C3H4N2–OH]˙ energized adducts could easily further react with oxygen molecules, and form intermediate [C3H4N2–OH–O2]˙ radicals, transition states (TS1–TS3) for the three OH-additions and confirmed by the intrinsic reaction coordinate (IRC) approach. The reaction energies and energy barriers, as well as thermodynamical parameters, are calculated at the M06-2X/aug-cc-pVTZ level of theory for hydroxyl radical attacks onto C2, C4, and C5 positions of imidazole and are shown in Table 1.
| Species | Parameters | |||||
|---|---|---|---|---|---|---|
| ΔE0K |
|
|
|
|
|
|
| Imidazole + OH˙ | 0.000 | 0.000 | 0.000 | |||
| IM1 [C2 position] | −4.472 | −4.725 | 2.703 | |||
| IMx (x = 2,3)[C4 & C5 positions] | −3.996 | −4.209 | 2.708 | |||
| OH-Addition pathways: [R–OH]˙ | ||||||
| P1 (2-hydroxyimidazolyl radical) | −27.657 | −28.559 | −19.081 | |||
| P2 (4-hydroxyimidazolyl radical) | −17.607 | −18.445 | −8.993 | |||
| P3 (5-hydroxyimidazolyl radical) | −24.962 | −25.921 | −16.303 | |||
| TS1 [C2 position] | −1.529 | −2.449 | 6.919 | |||
| TS2 [C4 position] | 0.703 | −0.230 | 9.219 | |||
| TS3 [C5 position] | −2.966 | −3.801 | 5.313 | |||
| H-Abstraction pathways: [R]˙ + H 2 O | ||||||
| P4 (2-dehydroimidazolyl + H2O) | −1.814 | −1.522 | −2.621 | |||
| P5 (4-dehydroimidazolyl + H2O) | −1.681 | −1.414 | −2.469 | |||
| P6 (5-dehydroimidazolyl + H2O) | 1.748 | 2.030 | 0.944 | |||
| TS4 [C2 position] | 6.075 | 5.555 | 13.665 | |||
| TS5 [C4 position] | 5.856 | 5.301 | 13.577 | |||
| TS6 [C5 position] | 7.605 | 7.039 | 15.358 | |||
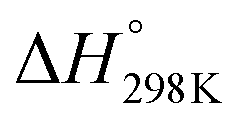
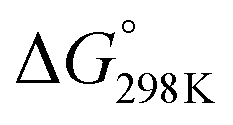
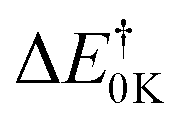
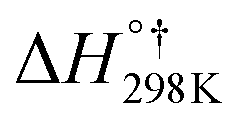
The hydroxyl radicals attack on the imidazole ring to form two prereactive intermediates IM1 and IMx (x = 2,3) for pathways 1–3. In IM1 and IMx (x = 2,3), the lengths of the C2–O6, C4–O6, and C5–O6 bonds are 2.542, 2.555 and 2.847 Å, respectively. The IM1 and IMx (x = 2,3) prereactive complexes are located at 4.47 and 4.0 kcal mol−1 under the total energy of imidazole and OH radicals (as the isolated reactants), respectively. The energy barriers (IM1 → TS1 or IMx → TS2/TS3) encountered along the pathways 1–3 amount to 2.94, 4.70, and 1.03 kcal mol−1, respectively. Two isomerization processes are identified from the IM1 and IMx (x = 2,3): the oxidation process to product P1via the transition state TS1, and the reaction between imidazole and OH radicals to produce pre-reactive intermediate IMx (x = 2,3) via the transition states TS2 and TS3.
In line with experimental data,13–15 all our calculations identify the TS3 structure (as the lowest transition state) on reaction (3), which is located at 2.97 kcal mol−1 under the reactants. The TS1 structure on reaction (1) is found at 1.53 kcal mol−1 under the isolated reactants while the TS2 structure on reaction (2) is located at 0.71 kcal mol−1 above the isolated reactants. The barrier height for pathway 3 is therefore lower by 1.44 and 3.67 kcal mol−1 than that for reactions (1) and (2), respectively (Table 1). This difference in energy barriers for the reactions R + OH˙ → Pi, i = 1–3 shows that the production of product P3 species via OH attack at the C5 position will be kinetically favored over the production of P1 and P2 species (through OH attack onto C2 and C4 positions, respectively).
The obtained results indicate that all considered reactions (1)–(3) are extremely exoergic processes (ΔG < 0), and extremely exothermic processes (ΔH ≈ −28.56, −18.45, and −25.92 kcal mol−1, respectively) at P = 1 bar and T = 298 K. Obviously, it is formation of product P1 (OH attack at the C2 position), which will also be thermodynamically favored (see Fig. 3), because the reaction is strongly exoergic (ΔG = −19.08 kcal mol−1) and strongly exothermic (ΔH = −28.56 kcal mol−1).
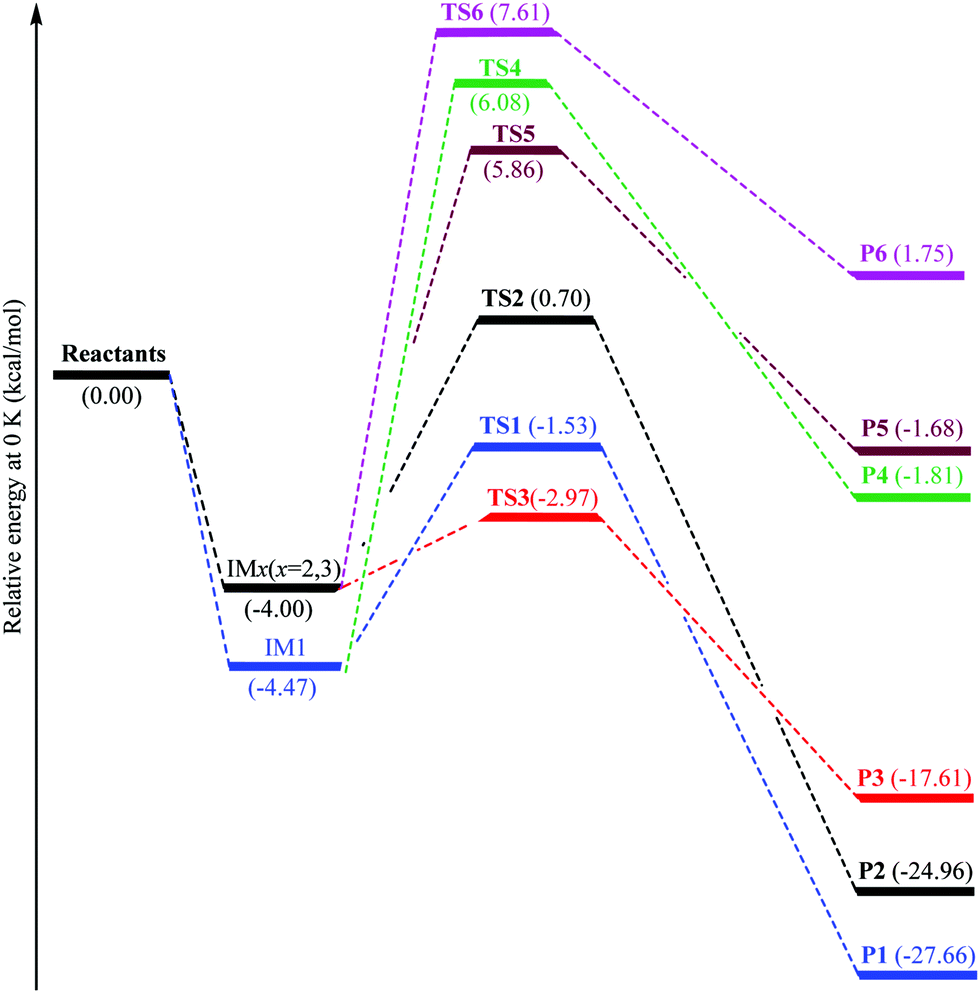 | ||
| Fig. 3 Energy profiles of the H-abstraction and OH-addition reactions. | ||
The transition states TS1–TS3 are considered by nucleus independent chemical shifts equal to −17.41, −9.82 and −19.01, respectively. Obviously, the more noticeable aromatic nature of the TS3 structure describes its higher stability, compared with the TS1 and TS2 ones. Furthermore, the lesser energy of intermediate IM1 compared with IMx (x = 2,3) reveals the more noticeable aromatic nature of the former pre-reactive molecular complex: indeed, the IM1 and IMx intermediates are characterized by NICS indices equal to −12.25 and −11.77, respectively.
According to the M06-2X data, two prereactive intermediates IM1 and IMx (x = 2,3) are characterized by H8–O6, H9–O6, and H10–O6 bond distances equal to 2.639, 2.722, and 3.253 Å, respectively. Proceeding further on reactions (1)–(3), the hydrogen H8, H9 or H10 abstraction via the transition states TS4, TS5, and TS6 requires activation energies of 6.07, 5.86, and 7.61 kcal mol−1 corresponding to the isolated reactants energies (Fig. 3), respectively.
The breaking C–H bond length is elongated by ∼15.7–18.3% (0.17–0.20 Å in absolute value) at the level of the transition states TS4, TS5 and TS6, compared with the equilibrium structure. On the other hand, the forming O–H bond distance is larger than in H2O. The elongation of the O–H bond in the TS4, TS5, and TS6 structures is of the order of ∼24.3–27.5% (0.23–0.27 Å in absolute value) compared with the latter. The optimized structures show that the relative elongation of the C–H bond is less than that of the forming O–H bond, which shows that the TS4, TS5, and TS6 structures are closer to the reactants than the associated products, which is in proportion to Hammond's principle.52
According to the characteristic transition states, the obtained results at ambient temperature and pressure show that the chemical pathways for 4 and 5 are found to be exothermic (ΔH < 0) as well as exoergic (ΔG < 0) processes while the pathway for 6 is found to be an endothermic (ΔH > 0) as well as endoergic (ΔG > 0) process. Because pathway 4 is exoergic (ΔG = −2.62 kcal mol−1) and exothermic (ΔH = −1.52 kcal mol−1), it is clear that the formation of P4 species will be thermodynamically stable. Addition of hydroxyl radicals onto the H atom bonded to the carbon C2 has the lowest Gibbs energy barriers 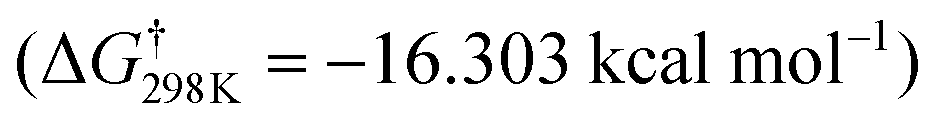 .
.
In proportion to the experimental activation energy of (−840 ± 30) cal mol−1 for oxidation of imidazole by OH radicals, the corresponding activation energy of these reactions (4)–(6) is 5.86 to 7.61 kcal mol−1 above the isolated reactants. The obtained energy barriers for the studied reactions R + OH˙ → Pi (i = 4–6) show that the formation of P5 species can be obtained by removal of a hydrogen atom bonded to the C4 atom leading to a H2O molecule and that 4-dehydroimidazolyl radical formation is kinetically favorable over the production of P4 and P6 species. The transition state TS5 on pathway 5 is the lowest transition state, which is located at 5.86 kcal mol−1 above the isolated reactants. The transition states TS4 and TS6 on reaction pathways 4 and 6 are located at 6.08 and 7.61 kcal mol−1 above the isolated reactants, respectively. The barrier height for reaction (5) is therefore less by 0.22 and 1.75 kcal mol−1 than the activation energy for reactions (4) and (6), respectively (Table 1).
We can see from Table 1 that oxidation processes via OH-addition reactions have negative Gibbs free energy of reaction ΔGr which indicates that OH attacks onto different carbon atoms (C2, C4, and C5) are thermodynamically spontaneous processes. Moreover, the hydroxyl radical addition occurring at the C2 position is thermodynamically the most favorable (ΔE0K = −27.66 kcal mol−1), followed by addition onto the C5 atom, and hydroxyl radical addition bonded to the C5 position leading to a H2O molecule and the related radical is the most unfavorable (ΔE0K = 1.75 kcal mol−1). Instead, from the kinetic viewpoint, addition onto the C5 atom has the lowest Gibbs free activation energy (ΔG† = 5.31 kcal mol−1), followed by addition occurring at the C2 position, while OH addition occurring at the C5 position for the H-abstraction pathways has the highest ΔG† value (ΔG† = 15.36 kcal mol−1).
The energy barriers from the OH-addition processes (1–3) are lower than those from the H-abstraction pathways (4–6) by ∼10.57 kcal mol−1 for reactions between imidazole and OH radicals ( , 6.92 vs. 13.67 kcal mol−1 for C2 position,
, 6.92 vs. 13.67 kcal mol−1 for C2 position, 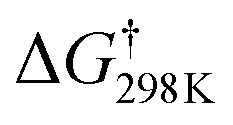 , 9.22 vs. 13.58 kcal mol−1 for C4 atom and 5.31 vs. 15.36 kcal mol−1 for C5 position). Accordingly, the hydroxyl addition pathways are more important than the hydrogen atom abstraction processes. Evidently, the process reactions via OH-addition pathways are faster than the consistently H-abstraction pathways, as they involve noticeably lower activation energies (Table 1). Thus, from a thermodynamic viewpoint, OH addition pathways 1–3 are favored for the oxidation reaction. The obtained results show that P1–P3 species can be obtained from pathways 1–3 and are important intermediates produced in the processes. The products P1–P3 are activated radicals and can further react with the global oxygen molecules to form the related organic peroxy radicals in the atmosphere.
, 9.22 vs. 13.58 kcal mol−1 for C4 atom and 5.31 vs. 15.36 kcal mol−1 for C5 position). Accordingly, the hydroxyl addition pathways are more important than the hydrogen atom abstraction processes. Evidently, the process reactions via OH-addition pathways are faster than the consistently H-abstraction pathways, as they involve noticeably lower activation energies (Table 1). Thus, from a thermodynamic viewpoint, OH addition pathways 1–3 are favored for the oxidation reaction. The obtained results show that P1–P3 species can be obtained from pathways 1–3 and are important intermediates produced in the processes. The products P1–P3 are activated radicals and can further react with the global oxygen molecules to form the related organic peroxy radicals in the atmosphere.
Furthermore, as can be seen in Table 1, the difference in activation energies shows that the production of P1–P3 species related to the OH addition onto C2, C4, and C5 positions will be kinetically the most favorable compared to the other products [P4–P6]. Among the reactions (1)–(3) leading to products P1–P3, the supplied data demonstrate that the most favorable process is OH˙ attack onto the C5 position from a kinetic viewpoint. The predicted Gibbs free activation energies are 6.92, 9.22, 5.31, 13.67, 13.58, and 15.36 kcal mol−1 for the reactions (1)–(6), respectively. Obviously, formation reactions of (1)–(3) related to the OH attack onto different positions on the ring (C2, C4, and C5) dominate. Therefore in the kinetic section, we only focus on the OH-addition pathways.
Addition of OH radicals to the hydrogen bonded C2 and C4 atoms is exothermic by −1.52 and −1.41 kcal mol−1, whereas for the C5 position it is endothermic by 2.03 kcal mol−1. The corresponding Gibbs free activation barriers are 13.67, 13.58, and 15.36 kcal mol−1, respectively. Due to the significantly higher activation barriers, these three H-abstraction reactions will not be further discussed. The Gibbs free activation barriers of the hydroxyl radical attacks onto the C2, C4, and C5 atoms via pathways 1–3 are 5.31–9.22 kcal mol−1, and the Gibbs reaction energies are from −8.99 to −19.08 kcal mol−1. Particularly, the hydroxyl radical attack onto the C2 position is the most exothermic (ΔH = −28.56 kcal mol−1), while OH attacks onto the C5 position have the lowest activation barrier (ΔH = −2.97 kcal mol−1). Evidently, pathways (1–3) have lower barrier heights and are more exothermic compared with pathways (4–6).
3.2. Kinetic analysis
RRKM and TST bimolecular kinetic rate coefficients of the atmospheric reactions between imidazole and OH radicals are listed in Tables 2 and 3 at the pressures of 1 bar and 133 mbar and over the considered temperatures. Further RRKM data computed at higher and lower pressures are shown for the same temperature regimes in Tables S1a–j of the ESI.† Effective rate coefficients [keff] of the reactions (1)–(3) involve a two-step mechanism: at first, a fast and reversible pre-equilibrium between the reactants (R) and the pre-reactive complex IM is established, followed by a further irreversible step leading to the related products:Thus, the effective rate coefficients for the reactions (1)–(3) are given by:
 | (7) |
| T (K) | Rate constants | Branching ratio (%) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| IM1 → P1 | IMx → P2 | IMx → P3 | R → P1 | R → P2 | R → P3 | R (1) | R (2) | R (3) | |
| k 2 (1) (s−1) | k 2 (2) (s−1) | k 2 (3) (s−1) | k eff (1) (cm3 mol−1 s−1) | k eff (2) (cm3 mol−1 s−1) | k eff (3) (cm3 mol−1 s−1) | ||||
| The kinetic parameters in parentheses were calculated at the M06-2X/aug-cc-pVTZ level by means of RRKM theory. | |||||||||
| 297 | 1.75 × 109 | 3.80 × 107 | 4.04 × 1010 | 6.61 × 10−14 | 1.15 × 10−15 | 1.22 × 10−12 | 5.12 | 0.09 | 94.79 |
| (4.49 × 108) | (4.33 × 107) | (1.57 × 109) | (1.70 × 10−14) | (1.31 × 10−15) | (4.77 × 10−14) | (25.72) | (1.99) | (72.29) | |
| 316 | 2.37 × 109 | 6.11 × 107 | 4.43 × 1010 | 6.88 × 10−14 | 1.52 × 10−15 | 1.10 × 10−12 | 5.87 | 0.13 | 94.00 |
| (5.38 × 108) | (6.57 × 107) | (1.55 × 109) | (1.56 × 10−14) | (1.64 × 10−15) | (3.86 × 10−14) | (27.99) | (2.93) | (69.08) | |
| 331 | 2.93 × 109 | 8.54 × 107 | 4.73 × 1010 | 7.11 × 10−14 | 1.85 × 10−15 | 1.03 × 10−12 | 6.46 | 0.17 | 93.37 |
| (6.05 × 108) | (8.76 × 107) | (1.53 × 109) | (1.47 × 10−14) | (1.90 × 10−15) | (3.32 × 10−14) | (29.47) | (3.83) | (66.71) | |
| 344 | 3.48 × 109 | 1.12 × 108 | 4.98 × 1010 | 7.32 × 10−14 | 2.18 × 10−15 | 9.75 × 10−13 | 6.97 | 0.21 | 92.82 |
| (6.59 × 108) | (1.09 × 108) | (1.50 × 109) | (1.39 × 10−14) | (2.14 × 10−15) | (2.95 × 10−14) | (30.52) | (4.71) | (64.77) | |
| 353 | 3.89 × 109 | 1.33 × 108 | 5.15 × 1010 | 7.49 × 10−14 | 2.42 × 10−15 | 9.42 × 10−13 | 7.34 | 0.24 | 92.42 |
| (6.95 × 108) | (1.26 × 108) | (1.49 × 109) | (1.34 × 10−14) | (2.30 × 10−15) | (2.72 × 10−14) | (31.20) | (5.37) | (63.43) | |
| 362 | 4.32 × 109 | 1.56 × 108 | 5.32 × 1010 | 7.64 × 10−14 | 2.69 × 10−15 | 9.15 × 10−13 | 7.69 | 0.27 | 92.04 |
| (7.28 × 108) | (1.44 × 108) | (1.47 × 109) | (1.29 × 10−14) | (2.47 × 10−15) | (2.53 × 10−14) | (31.71) | (6.08) | (62.21) | |
| 386 | 5.59 × 109 | 2.34 × 108 | 5.76 × 1010 | 8.40 × 10−14 | 4.08 × 10−15 | 8.28 × 10−13 | 8.61 | 0.37 | 91.02 |
| (8.07 × 108) | (1.95 × 108) | (1.42 × 109) | (1.17 × 10−14) | (2.90 × 10−15) | (2.11 × 10−14) | (32.73) | (8.12) | (59.15) | |
| 402 | 6.52 × 109 | 2.98 × 108 | 6.04 × 1010 | 8.40 × 10−14 | 4.08 × 10−15 | 8.28 × 10−13 | 9.16 | 0.45 | 90.39 |
| (8.50 × 108) | (2.32 × 108) | (1.39 × 109) | (1.09 × 10−14) | (3.19 × 10−15) | (1.90 × 10−14) | (33.01) | (9.61) | (57.38) | |
| 425 | 7.98 × 109 | 4.08 × 108 | 6.43 × 1010 | 8.89 × 10−14 | 5.05 × 10−15 | 7.96 × 10−13 | 10.00 | 0.57 | 89.44 |
| (9.00 × 108) | (2.88 × 108) | (1.34 × 109) | (1.00 × 10−14) | (3.57 × 10−15) | (1.66 × 10−14) | (33.23) | (11.82) | (54.96) | |
| 440 | 8.99 × 109 | 4.92 × 108 | 6.67 × 1010 | 9.24 × 10−14 | 5.74 × 10−15 | 7.78 × 10−13 | 10.54 | 0.66 | 88.80 |
| (9.25 × 108) | (3.25 × 108) | (1.31 × 109) | (9.50 × 10−15) | (3.79 × 10−15) | (1.53 × 10−14) | (33.24) | (13.28) | (53.48) | |
| T (K) | Rate constants | Branching ratio (%) |
k
exp × 101213–15 (cm3 mol−1 s−1) |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
| IM1 → P1 | IMx → P2 | IMx → P3 | R → P1 | R → P2 | R → P3 | R (1) | R (2) | R (3) | ||
| k 2 (1) (s−1) | k 2 (2) (s−1) | k 2 (3) (s−1) | k eff (1) (cm3 mol−1 s−1) | k eff (2) (cm3 mol−1 s−1) | k eff (3) (cm3 mol−1 s−1) | |||||
| 297 | 7.54 × 107 | 1.59 × 107 | 2.14 × 108 | 2.85 × 10−15 | 4.82 × 10−16 | 6.49 × 10−15 | 29.02 | 4.91 | 66.08 | (35.9 ± 3.3) |
| 316 | 8.80 × 107 | 2.26 × 107 | 2.11 × 108 | 2.40 × 10−15 | 5.28 × 10−16 | 4.93 × 10−15 | 30.54 | 6.72 | 62.74 | (31.1 ± 0.1) |
| 331 | 9.71 × 107 | 2.86 × 107 | 2.07 × 108 | 2.11 × 10−15 | 5.57 × 10−16 | 4.03 × 10−15 | 31.51 | 8.32 | 60.18 | (27.3 ± 2.5) |
| 344 | 1.04 × 108 | 3.42 × 107 | 2.04 × 108 | 1.89 × 10−15 | 5.78 × 10−16 | 3.45 × 10−15 | 31.94 | 9.77 | 58.30 | (25.2 ± 0.3) |
| 353 | 1.09 × 108 | 3.83 × 107 | 2.01 × 108 | 1.77 × 10−15 | 5.89 × 10−16 | 3.09 × 10−15 | 32.48 | 10.81 | 56.71 | (22.5 ± 1.0) |
| 362 | 1.13 × 108 | 4.24 × 107 | 1.99 × 108 | 1.64 × 10−15 | 5.98 × 10−16 | 2.81 × 10−15 | 32.49 | 11.85 | 55.67 | (19.8 ± 1.0) |
| 386 | 1.23 × 108 | 5.37 × 107 | 1.92 × 108 | 1.37 × 10−15 | 6.15 × 10−16 | 2.20 × 10−15 | 32.74 | 14.70 | 52.57 | (16.3 ± 0.3) |
| 402 | 1.28 × 108 | 6.11 × 107 | 1.87 × 108 | 1.22 × 10−15 | 6.19 × 10−16 | 1.89 × 10−15 | 32.72 | 16.60 | 50.68 | (13.9 ± 0.5) |
| 425 | 1.33 × 108 | 7.12 × 107 | 1.80 × 108 | 1.04 × 10−15 | 6.16 × 10−16 | 1.56 × 10−15 | 32.34 | 19.15 | 48.51 | (10.4 ± 0.7) |
| 440 | 1.35 × 108 | 7.74 × 107 | 1.76 × 108 | 9.36 × 10−16 | 6.10 × 10−16 | 1.39 × 10−15 | 31.88 | 20.78 | 47.34 | (9.1 ± 0.1) |
A more quantitative insight into the evolution of the regioselectivity for the effective rate coefficients (1)–(3), which is obtained by means of TST and RRKM theories at the pressures of 1.0 bar and 133 mbar and temperatures ranging from 297 to 440 K, is given by:
 | (8) |
The comparison of the kinetic rate coefficients and branching ratios that have been achieved at the studied temperatures and at a pressure of 1 bar are summarized in Table 2. The main difference is found for the kinetic rate coefficient of the second step in reaction (3) [k2(3)]. Because the pressure issues have to be taken into consideration for a reliable interpretation of the experimental kinetic data, it seems preferable to consider the RRKM approach for evaluating the kinetic rate constant. According to the calculated M06-2X energy profiles and related vibrational frequencies, Wigner tunneling correction κ(T) values equal to 1.18, 1.17 and 1.08 were found with TST calculation for the second reaction step [IM1 → P1; IMx → P2,P3] of the pathways 1–3. These results show that tunneling effects are practically negligible.
As can be seen in Table S2 of the ESI,† because the effective rate coefficient kr cannot be compared directly with the corresponding kf [R + OH˙ → Pi; i = 1–3], we can treat the 2nd order oxidation reaction as a pseudo 1st order reaction. Using an average hydroxyl radical concentration of 2 × 106 molecule cm−3 in the atmosphere,53 apparent kinetic rate constants for forward OH-addition processes, kf[OH] are obtained. All reactions (1)–(3) occurring on the C2, C4, and C5 positions indicate that they are reversible processes. Because the kf[OH] values are small, their contribution to atmospheric oxidation of imidazole by OH radicals is negligible. The values of kr are consistently greater than that of kf[OH] (see Table S2, ESI†). The obtained results in Table S2 of the ESI,† show that the backward processes of the reactions (1)–(3) are much faster than the forward chemical pathways. Hence, to calculate the possibility of the reactions studied here [reactions (1)–(3)], the subsequent reactions of the corresponding adducts in these reactions should be taken into account.
RRKM estimates for unimolecular (k2) and effective bimolecular (keff) kinetic rate coefficients were performed over a temperature range 297–440 K and pressure of 133 mbar and the obtained effective rate coefficients compared with the available experimental data (Table 3).13–15 More kinetic data were calculated by means of RRKM theory at higher and lower pressures for the same temperature regimes (summarized in Tables S1a–j of the ESI†).
The Arrhenius plot of the effective rate coefficients for the reaction channels 1–3 using RRKM theory clearly confirms that the formation of P3 species via OH radical attacks onto the C5 position will consequently dominate over the formation of the other species (P1 and P2) over a temperature range of 297–440 K and pressure of 133 mbar (see Fig. 4).
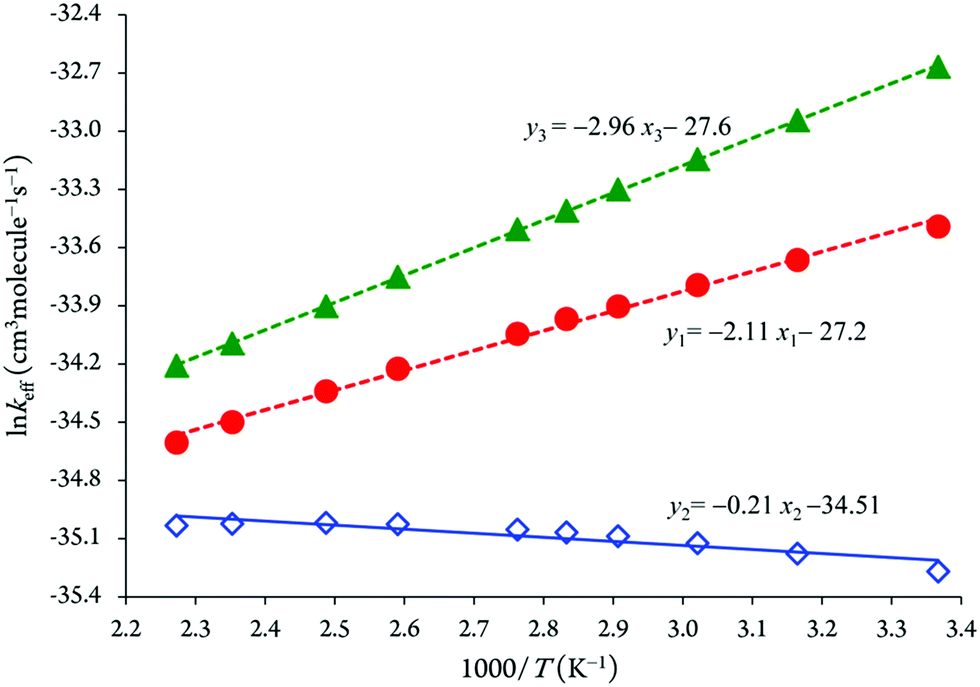 | ||
| Fig. 4 The Arrhenius plot of the obtained RRKM effective rate coefficients (P = 133 mbar). Legend: (●) effective kinetic rates for reaction (1); (◇) effective kinetic rates for reaction (2); (▲) effective kinetic rates for reaction (3). | ||
The obtained results in Table 3 indicate that RRKM effective rate constants keff(3) for reaction (3) are larger by factors 1.49 to 2.28 and 2.28 to 13.46, respectively, than the effective rate constants keff(1) and keff(2) for reaction pathways 1 and 2. One can find a similar trend for pressures ranging from 10−10 to 108 bars in Tables S3a–j of the ESI.† Because of the involved negative energy barriers for the reactions (1) and (3), the RRKM effective rate coefficients for OH addition onto the C2 and C5 positions will decrease gradually with increasing temperatures, while for reaction (2) (OH radical onto the C4 atom), it is in the opposite manner (see also Tables S1a–j of the ESI†). Thus, the reaction channel 3 is a favorable process compared with the reactions (1) and (2) from a kinetic point of view.
It appears that the branching ratios for OH addition onto the C2, C4 and C5 positions leading to the related energized adducts are predicted as 29.02–32.74%, 4.91–20.78%, and 47.34–66.08%, respectively (see Fig. 5 and Table 3). Branching ratios of the chemical channels 1 and 3 increase gradually with increasing temperatures. In contrast, for the reaction (2) a decrease in the branching ratio is observed. Under atmospheric conditions, the OH addition onto the C5 position accounts for ∼66% of the branching ratio, whereas the remaining ∼29% and ∼5% for OH addition onto the C2 and C4 positions, respectively, are based on the evaluated preceding theoretical results. We remind that the kinetically less efficient reaction is precisely pathway 2, with branching ratios equal to 4.91–20.78% over the temperature range from 297 to 440 K.
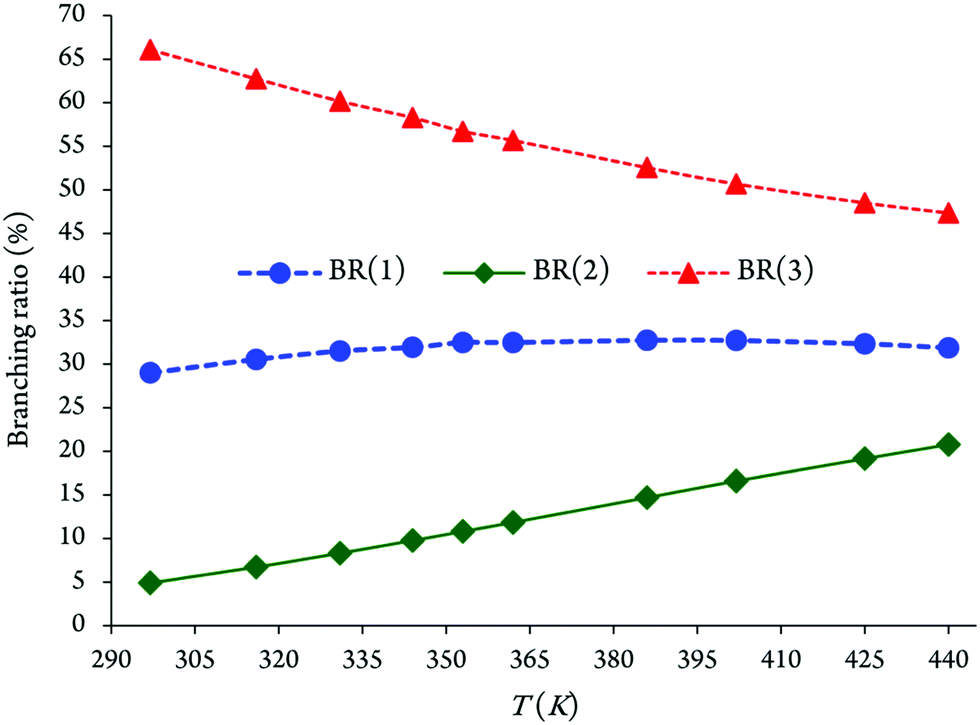 | ||
| Fig. 5 Evolution of branching ratios as a function of the increasing temperatures for chemical reactions (1)–(3). | ||
We display in Fig. 5 the evolution of branching ratios for the addition of OH attack onto the C2, C4 and C5 atoms as a function of the temperature and pressure, respectively (see also Table 3 and Tables S3a–j of the ESI,† for detailed numerical values at various temperatures ranging from 297 to 440 K, and pressures ranging from 10−10 to 108 bar). In line with the computed energy profiles and kinetic rate constants (RRKM data), the production of the P3 species (via channel 3) clearly dominates over the formation of the P1 and P2 species (via channels 1 and 2) at the studied temperatures, down to extremely low pressures, P > 10−10 bar. Nevertheless, from these data, and the correspondingly computed regioselectivity indices {RSI = R(3) − [R(1) + R(2)]/[R(1) + R(2) + R(3)]}, the regioselectivity of the oxidation process decreases with increasing temperatures and decreasing pressures (see Fig. 6).
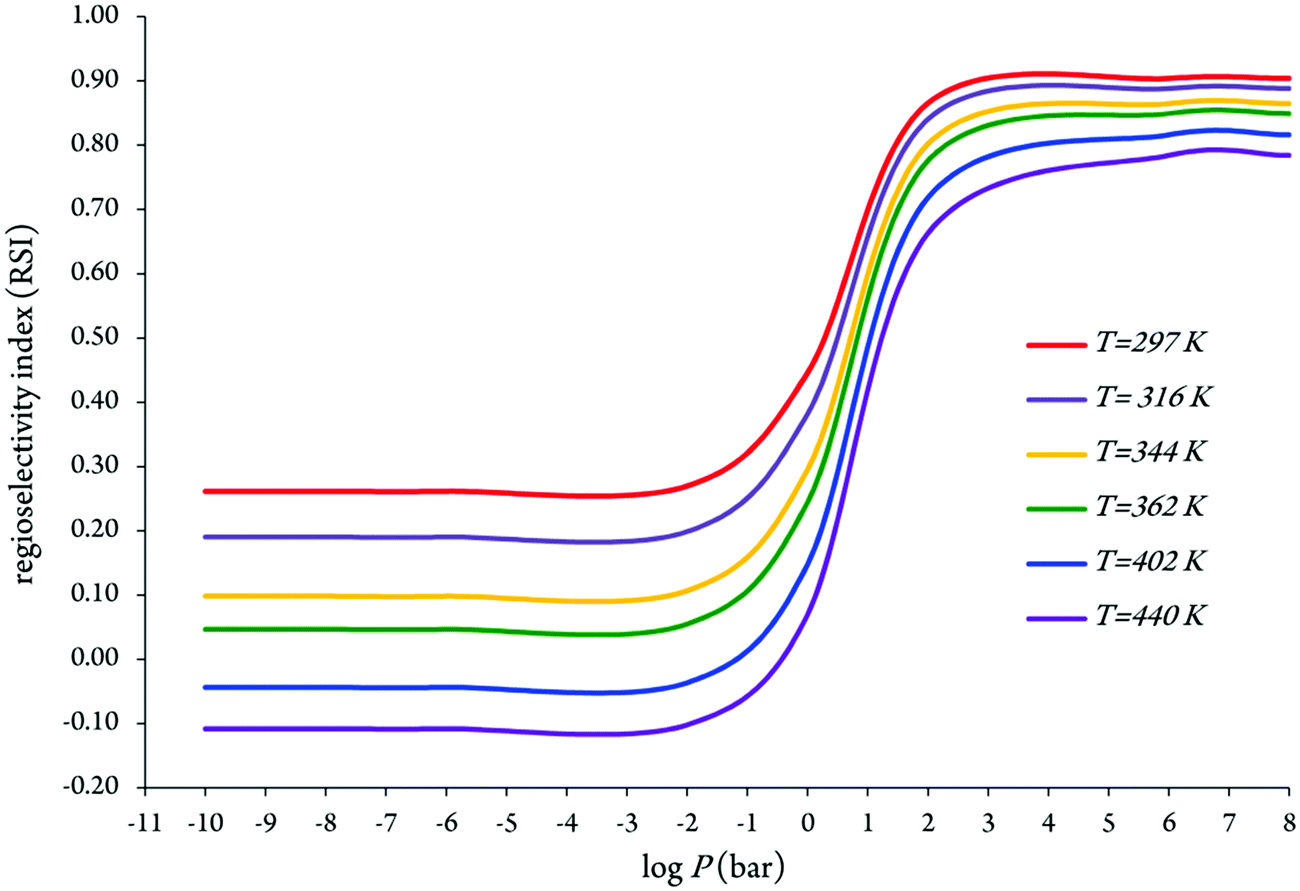 | ||
| Fig. 6 Temperature and pressure dependence of the regioselectivity in the oxidation of imidazole by OH radicals based on the obtained effective rate constants [keff(1), keff(2), and keff(3)] by means of RRKM theory. | ||
The pressure dependence of the calculated effective rate coefficients at 297, 344, 386 and 440 K is shown in Fig. 7. As can be seen in Fig. 7, for ensuring their saturation to the high-pressure limit, pressures P > 102 bar are required. This observation shows that the calculation of the effective rate constants by means of transition state theory is not valid at atmospheric pressure (1 bar). Hence, pressure effects need to be taken into account on the kinetic study using RRKM theory for consistent insight into the experiment,29,33 which was obtained at a pressure of 133 mbar. The obtained RRKM effective rate coefficients related to the reaction (3) are about 2–3 orders of magnitude underestimated. Both the theoretical and experimental rate coefficients for this reaction with increasing temperatures are decreased, which confirms that there is a negative activation energy for this reaction.
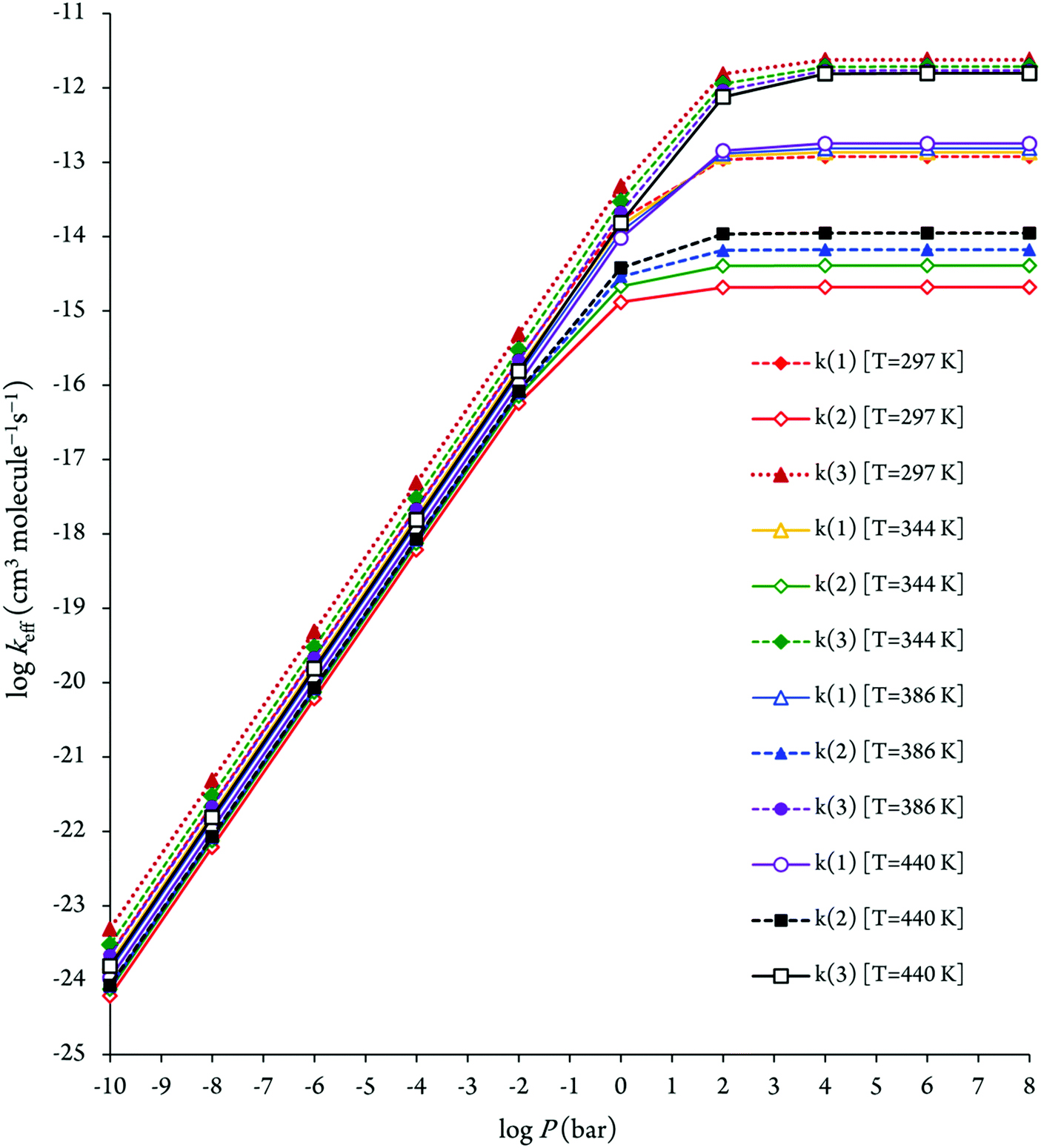 | ||
| Fig. 7 Pressure dependence of the RRKM bimolecular rate constants for the pathways 1–3. | ||
3.3. Atmospheric implications
In this study, we used DFT calculations for the chemical reaction (1)–(3) to consider the atmospheric oxidation reaction of imidazole initiated by hydroxyl radicals. The overall TST rate constant for reactions between imidazole with OH radicals at P = 1 bar and T = 297 K was 1.22 × 10−12 cm3 molecule−1 s−1. The obtained kinetic rate coefficient can be applied to measure the lifetime for imidazole under the atmospheric conditions. Assuming typical tropospheric 12 h daytime average concentrations of OH radicals as 2 × 106 molecule cm−3,53 the lifetime of imidazole relative to reaction with hydroxyl radicals is calculated to be ∼4.74 days which suggests imidazole can contribute to the formation of various secondary pollutants like ozone, peroxyacyl nitrates (PANs), and nitric acid following emission into the atmosphere.3.4. Natural bond orbital analysis
Natural bond orbital analysis was initially established as a way of quantifying resonance structure contributions to molecular systems. The energetic stabilizations are taken into account for all possible interactions between “filled” (donor) Lewis-type NBOs and “empty” (acceptor) non-Lewis NBOs and then their energies are estimated by 2nd-order perturbation theory.54 Delocalization energy (E2) for each donor NBO(i) and acceptor NBO(j) is given by:55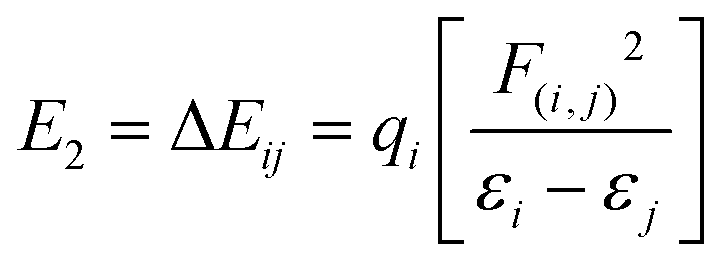 | (9) |
Based on the optimized geometries of the 2-hydroxyimidazolyl radical (P1) and 5-hydroxyimidazolyl (P3) radical, the natural bond orbital analysis demonstrates that the delocalization energies related to the electron delocalization from the non-bonding nitrogen lone pair orbital LP(1)N1 to σ*(C4–C5) antibonding orbitals are equal to 4.58 and 4.61 kcal mol−1, respectively. Furthermore, NBO analysis shows that the σ(C4–C5) bonding orbital occupancies for energized adducts P1 and P3 are both equal to ∼0.994, whereas the σ*(C4–C5) antibonding orbital occupancies in the energized adducts P1 and P3 are equal to ∼0.040 and ∼0.022, respectively.
Table 4 shows that for the transition states TS1 and TS3, strong interaction prevails between one nitrogen lone pair of imidazole and the unoccupied π*(C4–C5) orbital, resulting for the latter orbital in occupancies around 0.157 and 0.167, respectively. The delocalization energies for TS1 and TS3 are equal to 20.31 and 27.12 kcal mol−1, respectively. Moreover, charge transfer delocalization donates the larger stability of the TS3 (OH attack onto C5 atom) compared with the TS1 (OH attack onto C2 atom). NBO analysis demonstrates that, compared with imidazole, the LP(1)N1 → π*(C4–C5) delocalization energies have a strong effect on the energy barriers which was computed for pathway 1 (E2 = 20.31 kcal mol−1) and pathway 3 (E2 = 27.12 kcal mol−1). The occupancies of the π(C4–C5) bonding orbital in the transition states TS1 and TS3 amount to 0.948 and 0.911, respectively, whereas the occupancies of π*(C4–C5) antibonding orbitals in these structures are equal to 0.157 and 0.167, respectively. Furthermore, the NBO data for the transition states TS1 and TS3 show that a slight interaction prevails between one of the nitrogen lone pairs of imidazole and the unoccupied σ*(C4–C5) orbital, resulting for the latter orbital in an occupancy around 0.011. The corresponding stabilization energies for TS1 and TS3 are equal to 3.68 and 3.88 kcal mol−1, respectively.
| Imidazole | TS1 | TS3 | P1 | P3 | |
|---|---|---|---|---|---|
| Occupancies | |||||
| σ(C4–C5) | 0.99369 | 0.99388 | 0.99363 | 0.99300 | 0.99361 |
| π(C4–C5) | 0.92975 | 0.94807 | 0.91073 | 0.98827 | 0.82432 |
| LP(1)N1 | 0.79424 | 0.80502 | 0.78782 | 0.93083 | 0.90059 |
| LP(1)N3 | 0.96186 | 0.96280 | 0.96233 | 0.96419 | 0.96009 |
| σ*(C4–C5) | 0.01014 | 0.01078 | 0.01091 | 0.03945 | 0.02207 |
| π*(C4–C5) | 0.14731 | 0.15739 | 0.16649 | 0.16680 | 0.19802 |
| Stabilization energies (E2) | |||||
| LP(1)N1 → π*(C4–C5) | 20.68 | 20.31 | 27.12 | 9.01 | 4.45 |
| LP(1)N3 → σ*(C4–C5) | 3.50 | 3.88 | 3.68 | 4.58 | 4.61 |
4. Conclusions
The atmospheric oxidation mechanisms of imidazole initiated by hydroxyl radicals via OH-addition and H-abstraction processes were studied for the first time at the M06-2X/aug-cc-pVTZ level of theory. The difference in reaction energies shows that the production of species related to the OH-addition process will be thermodynamically favorable, and these reactions seem to be strongly exothermic reactions. This first reaction step for OH addition onto the different carbon positions was found to be an exoergic process (ΔG < 0) at ambient temperature and pressure. In line with the experiment, due to the formation of a prereactive complex [C3H4N2⋯OH]˙, the corresponding transition state lies below the isolated reactant; therefore, effective negative activation energies around −1.53 and −2.97 kcal mol−1 are related to the OH radical attacks onto the C2 and C5 positions, respectively.Kinetic rate coefficients for unimolecular and bimolecular reaction steps were estimated by means of transition state theory and Rice–Ramsperger–Kassel–Marcus theory. Effective rate coefficients involve a two-step mechanism: at first, a fast and reversible pre-equilibrium between the isolated reactants (imidazole and OH radicals) and the pre-reactive complex [C3H4N2⋯OH]˙ is established, followed by a further irreversible step leading to the related products. In proportion to the experiment, the obtained branching ratios show that the kinetically most efficient process over the temperature range 297–440 K correspond to OH addition onto a carbon atom which is adjacent to the nitrogen atom having a lower energy barrier. These ratios also reveal that the regioselectivity of the oxidation reaction decreases with increasing temperatures and decreasing pressures.
A comparison with TST results seems to validate RRKM theory for all OH additions onto different carbon atoms of imidazole (C2, C4, and C5 positions). The obtained RRKM effective rate coefficients for the favorable reaction appear to be sufficient for achieving semi-quantitative insights into the available experimental kinetic data with discrepancies, which are about 2–3 orders of magnitude underestimated. The theoretical rate coefficient for the favorable reaction shows that it decreases with increasing temperatures, which confirms that there is a negative activation energy.
In line with negative activation energies, it was found that the standard transition-state-approximation breaks down at ambient pressure for the first bimolecular reaction steps. RRKM calculations reveal that overwhelmingly high pressures, where P > 102 bar, are required to ensure the validity of the transition state theory approximation for the OH addition onto all carbon position pathways.
NICS indices and natural bond orbital analysis show that the computed activation energies are dictated by charge transfer effects and aromaticity changes because of the delocalization of nitrogen lone pairs to neighboring empty π* orbitals.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding by Maa-ja vesitekniikan tuki ry (MVTT) is acknowledged. The authors would also like to acknowledge the contributions of Environics OY.References
- A. L. Lehninger, D. L. Nelson and M. M. Cox, Principles of Biochemistry, Worth Publishers, New York, 1993 Search PubMed.
- L. Stryer, Biochemistry, Freeman & Company, New York, 2nd edn, 1981 Search PubMed.
- J. Llano and L. A. Eriksson, Mechanism of hydroxyl radical addition to imidazole and subsequent water elimination, J. Phys. Chem. B, 1999, 103, 5598–5607 CrossRef CAS.
- R. A. Perry, R. Atkinson and J. N. Pitts Jr, Kinetics and mechanism of the gas phase reaction of hydroxyl radicals with aromatic hydrocarbons over the temperature range 296–473 K, J. Phys. Chem., 1977, 81, 296–304 CrossRef CAS.
- D. Martin, J. L. Jourdain and G. Le Bras, Kinetic study for the reactions of OH radicals with dimethylsulfide, diethylsulfide, tetrahydrothiophene, and thiophene, Int. J. Chem. Kinet., 1985, 17, 1247–1261 CrossRef CAS.
- P. H. Wine and R. J. Thompson, Kinetics of OH reactions with furan, thiophene, and tetrahydrothiophene, Int. J. Chem. Kinet., 1984, 16, 867–878 CrossRef CAS.
- R. Atkinson, S. M. Aschmann and W. P. L. Carter, Kinetics of the reactions of O3 and OH radicals with furan and thiophene at 298 ± 2 K, Int. J. Chem. Kinet., 1983, 15, 51–61 CrossRef CAS.
- H. MacLeod, J. L. Jourdain and G. Le Bras, Absolute rate constant for the reaction of OH with thiophene between 293 and 473 K, Chem. Phys. Lett., 1983, 98, 381–385 CrossRef CAS.
- J. H. Lee and I. N. Tang, Absolute rate constants for the hydroxyl radical reactions with ethane, furan, and thiophene at room temperature, J. Chem. Phys., 1982, 77, 4459–4463 CrossRef CAS.
- T. J. Wallington, Kinetics of the gas phase reaction of OH radicals with pyrrole and thiophene, Int. J. Chem. Kinet., 1986, 18, 487–496 CrossRef CAS.
- E. C. Tuazon, R. Atkinson, A. M. Winer and J. N. Pitts Jr., A study of the atmospheric reactions of 1,3-dichloropropene and other selected organochlorine compounds, Arch. Environ. Contam. Toxicol., 1984, 13, 691–700 CrossRef CAS.
- R. Atkinson, S. M. Aschmann, A. M. Winer and W. P. L. Carter, Rate constants for the gas phase reactions of OH radicals and O3 with pyrrole at 295 ± 1 K and atmospheric pressure, Atmos. Environ., 1984, 18, 2105–2107 CrossRef CAS.
- F. Witte and C. Zetzsch, Examination of the temperature dependence for the reaction of OH radicals with heterocyclic aromatics (imidazole, furan, pyrrole and thiophene) and the unimolecular decay of the adducts imidazole-OH and thiophene-OH, 9th International Symposium on Gas Kinetics, University of Bordeaux, Bordeaux, France, 1986.
- R. Atkinson, Kinetic and mechanism of the gas-phase reactions of the hydroxyl radical with organic compounds under atmospheric conditions, Chem. Rev., 1985, 85, 69–201 Search PubMed.
- R. Atkinson, Kinetics and mechanisms of the gas-phase reactions of the hydroxyl with organic compounds, J. Phys. Chem. Ref. Data, Monogr., 1989, 1, 1–246 Search PubMed.
- D. Lucas and N. J. Brown, The influence of thiophene on the selective reduction of NO by NH3, Combust. Flame, 1983, 49, 283–288 CrossRef CAS.
- A. Samuni and P. Neta, Electron spin resonance study of the reaction of hydroxyl radicals with pyrrole, imidazole, and related compounds, J. Phys. Chem., 1973, 77, 1629–1635 CrossRef.
- G. Lassmann, L. A. Eriksson, F. Himo, F. Lendzian and W. Lubitz, Electronic structure of a transient histidine radical in liquid aqueous solution: EPR continuous-flow studies and density functional calculations, J. Phys. Chem. A, 1999, 103, 1283–1290 CrossRef CAS.
- L. A. Eriksson and F. Himo, Radicals in biophysical systems-A theoretical perspective, Trends Phys. Chem., 1997, 6, 153–170 CAS.
- P. Cysewski, An ab initio study on nucleic acid bases aromaticities, THEOCHEM, 2005, 714, 29–34 CrossRef CAS.
- P. v. R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao and N. J. R. van Eikema Hommes, Nucleus-independent chemical shifts: a simple and efficient aromaticity probe, J. Am. Chem. Soc., 1996, 118, 6317–6318 CrossRef CAS PubMed.
- S. Nigam, C. Majumder and S. K. Kulshreshtha, Theoretical study of aromaticity in inorganic tetramer clusters, J. Chem. Sci., 2006, 118, 575–578 CrossRef CAS.
- P. v. R. Schleyer, M. Manoharan, Z. X. Wang, B. Kiran, H. J. Jiao, R. Puchta and N. Hommes, Dissected nucleus-independent chemical shift analysis of π-aromaticity and antiaromaticity, Org. Lett., 2001, 3, 2465–2468 CrossRef CAS PubMed.
- P. v. R. Schleyer, H. Jiao, B. Goldfuss and P. K. Freeman, Aromaticity and antiaromaticity in five-membered C4H4X ring systems: “classical” and “magnetic” concepts may not be “orthogonal”, Angew. Chem., Int. Ed. Engl., 1995, 34, 337–340 CrossRef CAS.
- A. E. Reed, R. B. Weinstock and F. Weinhold, Natural population analysis, J. Chem. Phys., 1985, 83, 735–746 CrossRef CAS.
- J. K. Badenhoop and F. Weinhold, Natural steric analysis of internal rotation barriers, Int. J. Quantum Chem., 1999, 72, 269–280 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, et al., Gaussian 09, Revision B.01, Gaussian, Wallingford, CT, 2009 Search PubMed.
- Y. Zhao and D. G. Truhlar, Density functionals with broad applicability in chemistry, Acc. Chem. Res., 2008, 41, 157–167 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- J. P. Merrick, D. Moran and L. Radom, An evaluation of harmonic vibrational frequency scale factors, J. Phys. Chem. A, 2007, 111, 11683–11700 CrossRef CAS PubMed.
- F. Fukui, A formulation of the reaction coordinate, J. Phys. Chem., 1970, 74, 4161–4163 CrossRef.
- Z. Safaei, A. Shiroudi, R. Padash, M. Sillanpää and E. Zahedi, Reaction mechanisms and kinetics of the β-elimination processes of compounds CHF2CH2SiFnMe3−n (n = 0–3): DFT and CBS-QB3 methods using Rice–Ramsperger–Kassel–Marcus and transition state theories, J. Fluorine Chem., 2018, 216, 71–80 CrossRef CAS.
- K. Wolinski, J. F. Hilton and P. Pulay, Efficient implementation of the gauge-independent atomic orbital method for NMR chemical shift calculations, J. Am. Chem. Soc., 1990, 112, 8251–8260 CrossRef CAS.
- R. F. W. Bader, Atoms in Molecules: A Quantum Theory, Clarendon, Oxford, 1990 Search PubMed.
- F. P. Cossío, I. Morao, H. J. Jiao and P. v. R. Schleyer, In-plane aromaticity in 1,3-dipolar cycloadditions. Solvent effects, selectivity, and nucleus-independent chemical shifts, J. Am. Chem. Soc., 1999, 121, 6737–6746 CrossRef.
- I. Morao, B. Lecea and F. P. Cossío, In-plane aromaticity in 1,3-dipolar cycloadditions, J. Org. Chem., 1997, 62, 7033–7036 CrossRef.
- D. L. Singleton and R. J. Cvetanovic, Temperature dependence of the reaction of oxygen atoms with olefins, J. Am. Chem. Soc., 1976, 98, 6812–6819 CrossRef CAS.
- J. R. Alvarez-Idaboy, N. M. Diez and A. Vivier-Bunge, A quantum chemical and classical transition state theory explanation of negative activation energies in OH addition to substituted ethenes, J. Am. Chem. Soc., 2000, 122, 3715–3720 CrossRef CAS.
- V. H. Uc, J. R. Alvarez-Idaboy, A. Galano and A. Vivier-Bunge, Theoretical explanation of nonexponential OH decay in reactions with benzene and toluene under pseudo-first-order conditions, J. Phys. Chem. A, 2008, 112, 7608–7615 CrossRef CAS PubMed.
- E. Wigner, Calculation of the rate of elementary association reactions, J. Chem. Phys., 1937, 5, 720–725 CrossRef CAS.
- E. P. Wigner, Uber das uberschreiten von potentialschwellen bei chemischen reaktionen, Z. Phys. Chem., Abt. B, 1932, 19, 203–216 Search PubMed.
- K. A. Holbrook, M. J. Pilling, S. H. Robertson and P. J. Robinson, Unimolecular Reactions, Wiley, New York, 2nd edn, 1996 Search PubMed.
- M. J. Pilling and P. W. Seakins, Reaction Kinetics, Oxford University Press Inc., New York, 1999 Search PubMed.
- D. R. Glowacki, L. Wang and M. J. Pilling, Evidence of Formation of Bicyclic Species in the Early Stages of Atmospheric Benzene Oxidation, J. Phys. Chem. A, 2009, 113, 5385–5396 CrossRef CAS PubMed.
- R. G. Gilbert and S. C. Smith, Theory of Unimolecular and Recombination Reactions, Blackwell Scientific Publications, Boston, MA, 1990 Search PubMed.
- S. Canneaux, F. Bohr and E. Henon, KiSThelP: a program to predict thermodynamic properties and rate constants from quantum chemistry results, J. Comput. Chem., 2013, 35, 82–93 CrossRef PubMed.
- Computational chemistry comparison and benchmark database, precomputed vibrational scaling factors. (http://cccbdb.nist.gov/vibscalejust.asp).
- R. J. Kee, F. M. Rupley, J. A. Miller, M. E. Coltrin, J. F. Grcar, E. Meeks, H. K. Moffat, A. E. Lutz, G. Dixon-Lewis, M. D. Smooke, J. Warnatz, G. H. Evans, R. S. Larson, R. E. Mitchell, L. R. Petzold, W. C. Reynolds, M. Caracotsios, W. E. Stewart, P. Glarborg, C. Wang, C. L. McLellan, O. Adigun, W. G. Houf, C. P. Chou, S. F. Miller, P. Ho, P. D. Young, D. J. Young, D. W. Hodgson, M. V. Petrova and K. V. Puduppakkam, Chemkin, Reaction design, San Diego, California, 2010 Search PubMed.
- F. M. Mourits and H. A. Rummens, A critical evaluation of Lennard-Jones and Stockmayer potential parameters and of some correlation methods, Can. J. Chem., 1977, 55, 3007–3020 CrossRef CAS.
- J. R. Alvarez-Idaboy, N. Mora-Diez, R. J. Boyd and A. Viver-Bunge, On the importance of prereactive complexes in molecule-radical reactions: hydrogen abstraction from aldehydes by OH, J. Am. Chem. Soc., 2001, 123, 2018–2024 CrossRef CAS PubMed.
- C. W. Zhou, A. M. Mebel and X. Y. Li, An ab initio/Rice–Ramsperger–Kassel–Marcus study of the reactions of propenols with OH. Mechanism and kinetics of H abstraction channels, J. Phys. Chem. A, 2009, 113, 10667–10677 CrossRef CAS PubMed.
- G. S. Hammond, A correlation of reaction rates, J. Am. Chem. Soc., 1953, 77, 334–338 CrossRef.
- R. G. Prinn, R. F. Weiss, B. R. Miller, J. Huang, F. N. Alyea, D. M. Cunnold, P. J. Fraser, D. E. Hartley and P. G. Simmonds, Atmospheric trends and lifetime of CH3CCl3 and global OH concerns, Science, 1995, 269, 187–192 CrossRef CAS PubMed.
- J. E. Carpenter and F. Weinhold, Analysis of the geometry of the hydroxymethyl radical by the different hybrids for different spins natural bond orbital procedure, THEOCHEM, 1988, 169, 41–62 CrossRef.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Intermolecular interactions from a natural bond orbital, donor–acceptor viewpoint, Chem. Rev., 1988, 88, 899–926 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary data (Tables S1–S3) associated with this article can be found, in the online version. Table S1: kinetic rate constants for the reactions involved in the reaction pathways 1–3 by means of RRKM theory at different pressures and temperatures, according to the computed M06-2X energy profiles; Table S2: kinetic rate constants (k) for OH attack onto C2, C4, and C5 positions of imidazole for different addition products by means of TST (P = 1 bar) (x = 2, 3); Table S3: effective bimolecular rate constants (cm3 molecule−1 s−1), branching ratios (%) and regioselectivity for the pathways 1–3 (x = 1, 2) at different pressures and temperatures, using RRKM theory, and according to the computed M06-2X energy profiles. See DOI: 10.1039/c9cp00632j |
| This journal is © the Owner Societies 2019 |