
Effects of sulfation and the environment on the structure of chondroitin sulfate studied via Raman optical activity†
Václav
Profant
 *a,
Christian
Johannessen
b,
Ewan W.
Blanch
c,
Petr
Bouř
*d and
Vladimír
Baumruk
a
*a,
Christian
Johannessen
b,
Ewan W.
Blanch
c,
Petr
Bouř
*d and
Vladimír
Baumruk
a
aInstitute of Physics, Faculty of Mathematics and Physics, Charles University, Ke Karlovu 5, 121 16 Prague 2, Czech Republic. E-mail: profant@karlov.mff.cuni.cz; Tel: +420 221911346
bDepartment of Chemistry, University of Antwerp, Groenenborgerlaan 171, 2020 Antwerp, Belgium
cSchool of Science, RMIT University, GPO Box 2476, Melbourne, VIC 3001, Australia
dInstitute of Organic Chemistry and Biochemistry, Academy of Sciences, Flemingovo náměstí 2, 166 10 Prague 6, Czech Republic. E-mail: bour@uochb.cas.cz; Tel: +420 220183348
First published on 14th March 2019
Abstract
Glycosaminoglycans are linear carbohydrate polymers with essential roles in many biological processes. Chondroitin sulfate (CS) is one of them, omnipresent in living organisms as an important structural component of cartilage. It provides much of its resistance to compression. Despite its biological importance, little is still known about the relation of the CS structure to chemical composition and interaction with the environment. We therefore measured Raman and Raman optical activity (ROA) spectra of five CS samples of different biological origin and variously sulfated CS building blocks (GlcA, GalNAc, and basic disaccharide units) in a wide frequency range between 200 cm−1 and 1800 cm−1 and analyzed them with respect to specific structure marker bands. We show that ROA spectroscopy is sensitive to the conformational stability and rigidity of pyranose rings of saccharides, the orientation of sugar hydroxyl groups and the secondary structure of the CS's backbone. The CS secondary structure has been found to be quite stable, with a minor variation as a reaction to physicochemical parameters (concentration, pH, temperature, and the presence of cations). Larger changes were observed under chemical changes (sulfation) of the CS chain. ROA spectroscopy thus exhibited useful potential to study the structure of similar biopolymers.
1 Introduction
Chondroitin sulfate1,2 (CS) is one of the most common carbohydrate polymers in the human body. It is an important component of cartilage, providing a resistance to compression. It is also found in other connective tissues, such as ligaments and tendons. It is usually attached to proteins as a part of proteoglycan.3 CS also interacts with a variety of important molecules, such as growth factors, cytokines, adhesion compounds and lipoproteins, and thus takes part in various physiological processes including wound healing, neurite outgrowth and growth factor signaling.4–6 Posttranslational modifications and sulfation of the CS chain were observed for pancreatic,7 gastric,8 rectal,9,10 and ovarian11 cancers along with colon adenocarcinoma.12CS belongs to the class of glycosaminoglycans (GAGs), long unbranched heteropolymers composed of repeating disaccharide units. CS is a sulfated GAG composed of alternating monosaccharides, D-glucuronic acid (GlcA) and N-acetyl-D-galactosamine (GalNAc), joined together by β(1 → 4) and β(1 → 3) glycosidic links (see Fig. 1).13 A single CS chain usually composed of more than 100 sugar residues, each of which can be sulfated in variable positions and quantities. Owing to the sulfate and carboxylic groups, CS occurs as a polyanion. Its charge is usually compensated by metal cations (Na+ and Ca2+, etc.). The CS disaccharide unit (GlcA–GalNAc) is most often sulfated at the C-4 or/and C-6 positions of GalNAc. The terms CS-A and CS-C (or C4S and C6S) are usually used to describe CS chains rich in GlcA–GalNAc-4S and GlcA–GalNAc-6S, respectively, where 4S and 6S stand for 4-O-sulfate and 6-O-sulfate.13,14 In small proportions, CS basic units may also be disulfated or unsulfated, and sporadic sulfation may occur at the C(2) position of GlcA. The structure and the fine chemical pattern of CS strongly depend on the biological source.15
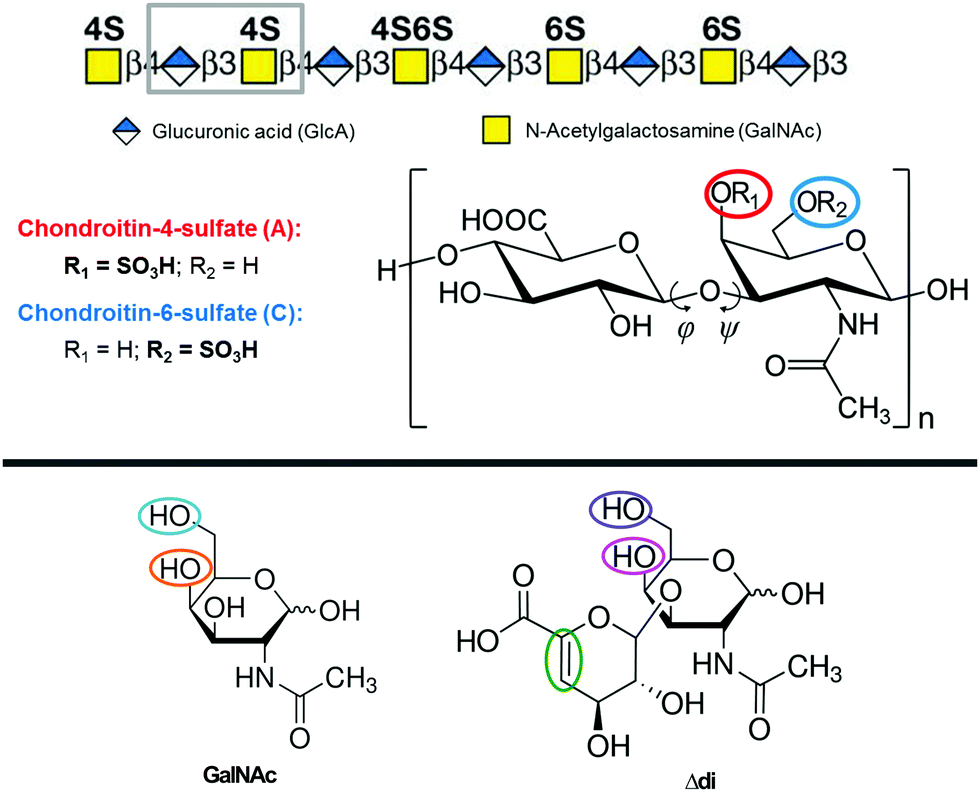 | ||
| Fig. 1 (top) CS polymer chain with the basic disaccharide units (grey rectangle), and positions for possible sulfations (R1 and R2). Adapted from ref. 16. (bottom) The GalNAc and Δdi unit models. OH groups that may be sulfated are indicated. The other Δdi modification (the double bond) is indicated by the green ellipse. | ||
The first crystallographic CS data were reported in the 1970s.17 The C4S polymer chain was found to prefer three- and two-fold helical conformations depending on the humidity, with sulfate groups located on the surface of the molecule.18–21 C6S exhibited similar properties with the sulfate groups even further from the helical axis.22,23 In addition, an eight-fold helix was reported for C6S.24 CS related NMR studies were mainly focused on identification of conformational preferences, identification of intramolecular H-bonding, and determination of characteristic values of the glycosidic link dihedral angles within short fragments of the CS chain (i.e. di-,25 tetra-,26 penta-,27 hexa-,28 and octasaccharides29). The NMR studies were often complemented by molecular modelling.30–32
The structure studies were nicely summarized in a review by Pomin,33 concluding that (i) the CS's solution secondary structure is helical, favoring the left-handed three-fold (32) or eight-fold (85) helix. Other possible conformations are the 21 helix or the right-handed 83 helix. (ii) The values of dihedral angles φ and ψ characterizing the geometry of glycosidic links (Fig. 1) were determined as (−75° ± 10°) and (100° ± 10°) for the β(1 → 3) link, together with (−75° ± 5°) and (−115° ± 10°) for the β(1 → 4) link, respectively. These values correspond to both 32 and 85 helices. (iii) The C-4 sulfation pattern has only a small influence on the overall conformation of the CS polymer.
However, X-ray crystallography and NMR spectroscopy are often limited in their flexibility to study biological processes, typically modelled by CS in different solutions.34 The high-resolution characterization can then be completed via optical spectroscopies, such as electronic circular dichroism (ECD),35,36 infrared (IR) absorption,37,38 and Raman scattering.38–40 These techniques provided a fast and affordable characterization of various GAGs including differently sulfated CS samples,41 although they may be limited by various factors as well. For example, saccharides possess a limited number of chromophores (too few electronic transitions) suitable for ECD, and IR and Raman spectroscopies are rather insensitive to the secondary structure (conformation).
Below, we explore possibilities of the Raman optical activity (ROA)42–44 that can be applied more universally. Previous ROA carbohydrate studies include monosaccharides,45 disaccharides,46 simple polysaccharides47,48 and even complex glycoproteins.49–51 ROA spectra can be measured in aqueous solutions in a wide (∼200–2000 cm−1) spectral region and can appear to be sensitive to both sugar ring puckering and a longer-range order of the glycosidic chain.52 This sensitivity was also confirmed in a recent study of agarose.53 However, there are only a limited number of ROA studies dedicated to GAG polymers. One deals with the structural details of hyaluronic acid,52 and another one documents the possibility of recording decent ROA spectra of other GAGs.54
In the present study we focused on acquiring high-quality ROA spectra of two most common forms of CS differing in sulfation (C4S and C6S) and analyzing possible structural indicators caused by the environmental factors. Vibrational assignment is performed to characterize features arising from the higher order structure. To find features related to sulfation we compared the data to spectra of the CS building blocks – GlcA, differently sulfated variants of GalNAc, and the basic disaccharide unit (GlcA-β(1 → 3)–GalNAc). In addition, we investigated the effect of variations of several important physicochemical properties – concentration, pH, temperature, and the presence of cations – on the CS structure. It appears that chemical modification, i.e. different sulfation of the basic monosaccharide units, causes changes in the CS secondary structure, while changes of physical conditions do not have any significant effect.
2 Methods
2.1 Samples
Chondroitin sulfate (CS) polymers as sodium salts were purchased from Sigma, and their biological sources and product numbers are summarized in Table 1. Most samples provided yellow color with a fluorescence background after dissolution. Nevertheless, the content of impurities was probably very low because neither HPLC nor lyophilization-based purification led to any change.| Chondroitin sample | Sigma-Aldrich product # | C4S contents [%] | |||
|---|---|---|---|---|---|
| Ref. 39 | Ramanb | ROAb | Raman & ROAb | ||
| a EPR = European pharmacopoeia reference. b For this study, see the text. Standard deviation of fitted values is 5%, and ratios of C4S and C6S add up to 100%. | |||||
| (1) Bovine trachea | C9819 | 60 | 63 | 60 | 65 |
| (2) Shark cartilage | C4384 | 10 | 15 | 20 | 14 |
| (3) Bovine cartilage | C6737 | — | 52 | 49 | 49 |
| (4) EPRa standard (marine) | Y0000593 | — | 5 | 6 | 5 |
| (5) EPR standard | Y0000280 | — | 50 | 52 | 52 |
Chemicals containing CS building blocks comprised (i) D-glucuronic acid (GlcA) and N-acetyl-D-galactosamine (GalNAc) obtained from Sigma, and (ii) GalNAc-4S, GalNAc-6S, and modified disaccharide units with various sulfation patterns (Δdi-0S, Δdi-4S, and Δdi-6S) obtained from Dextra. These samples were of high purity (>99%), did not exhibit measurable fluorescence, and were used without further purification.
2.2 Experiment
The samples were dissolved in deionized water to a concentration of 50 mg ml−1 (100 mg ml−1 for GlcA and GalNAc) and filtered through a 0.22 μm Millipore filter. Each solution was subsequently put into a quartz cell (Starna Scientific Ltd, 4 × 3 mm, and an internal volume of ∼100 μl) and measured at room temperature (20 °C). If needed, the samples were left in the laser beam for 30 minutes to quench the fluorescence from residual impurities prior to signal accumulation. Raman and ROA spectra were recorded using a μ-ChiralRaman-2X™ instrument (BioTools, Inc.) based on the concept of W. Hug55 adopting the scattered circular polarization (SCP) modulation scheme and backscattering geometry. Other experimental conditions were set as follows: a spectral range of 200–2000 cm−1, 532 nm excitation wavelength, 800 mW power at the sample, 1.029 s accumulation time, 10 minutes per frame (480 accumulations), and 8 cm−1 spectral resolution. The total acquisition time for each sample was ∼80 hours.2.3 Data treatment
The wavenumber scale was calibrated using a standard Raman spectrum of toluene and then linearly interpolated in regular 1 cm−1 intervals. The post-processing of the Raman data typically involved solvent signal subtraction and subsequent baseline correction by a third order polynomial. This was performed manually using the GRAMS/AI software (Thermo Electron Corporation). It should be noted that the baseline correction has to be performed very carefully, especially in the case of a high fluorescence background, as it may alter the spectral shape and relative band intensities. Spectral series originating from measurements of property dependencies (pH, temperature, and concentration, etc.) were therefore treated using the procedure developed by Palacký et al.56 Based on the factor analysis, the algorithm provides a uniform baseline correction for the whole spectral set. For the ROA spectra only occasional minor baseline adjustment was done. Both the Raman and ROA spectra comprise correction to the instrument response affecting relative band intensities.572.4 Factor analysis
As usual, each spectrum Yi(λ) in a studied series was decomposed into a set of orthonormal basis spectra Sj(λ) as | (1) |
Assuming that the recorded spectra are superpositions of M pure forms Zn(λ)
 | (2) |
 | (3) |
 | (4) |
3 Results and discussion
3.1 Determination of pure C4S and C6S ROA spectra
As mentioned above, natural CS polymers are not sulfated uniformly. Most of them are mixtures of C4S and C6S forms, with minor contributions from disulfated and unsulfated species. To obtain Raman and ROA spectra of pure C4S and C6S, we measured five commercially available CS samples of different origin (Table 1). In particular, the lower wavenumber region (below 600 cm−1) is supposed to contain bands related to delocalized vibrational motions highly specific to the secondary structure.59,60Raman and ROA spectra of all five samples measured at 50 mg ml−1 concentration in water at neutral pH are shown in Fig. 2. It is worth mentioning that all samples suffered from high fluorescence probably arising from some impurities, which made the Raman data treatment more challenging and also led to a lower signal/noise ratio in the ROA spectra. The spectral shapes of all samples are clearly quite similar. Visually, mostly based on the Raman patterns around 400 and 1000 cm−1, we can perhaps distinguish two types of spectral profiles. The first one comprises samples (1), (3) and (5); and the second one includes samples (2) and (4).
 | ||
| Fig. 2 Raman (left) and ROA (right) spectra of CS samples of different origin. Spectral bands arising from the vibration of free sulfate (983 cm−1) are marked by circles; spectral bands corresponding to the impurities are marked by asterisks. | ||
Raman spectra of samples (1) and (2) are consistent with published data;39 small differences can be explained by the presence of impurities. The impurities appear not to be chiral as the corresponding ROA spectra were not affected. The band at 983 cm−1 (Fig. 2, black (1) and red (2) lines) can be assigned to the free sulfate group. The origin of the others (such as 787, 1488, 1530, and 1579 cm−1) is presently unclear.
To obtain spectra of the C4S and C6S pure forms, we used factor analysis.58 Based on this, we found that all spectra in the set (5× Raman and 5× ROA, Fig. 2) may be reconstructed as linear combinations of two subspectra, S1 and S2 (Fig. S1, ESI†). The remaining signal/subspectra represent impurities, variations in baseline correction, noise, and also contributions from differently sulfated CS forms which might occur in samples in a small amount.
Relative ratios of C4S and C6S were determined by a fit according to eqn (4), where initial values for samples 1 and 2 were set according to the literature (the C4S![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :C6S ratio should be 60:40 and 10:90 for CS from bovine trachea and CS from shark cartilage, respectively).39 The fit was performed independently for three sets of spectra: Raman only, ROA only, and Raman and ROA together. The results of the fits (Table 1) are quite consistent, with a 5% standard deviation, and also well correspond to values found previously.39 The spectra of pure C4S and C6S forms are plotted as shown in Fig. 3. In spite of the similarity, there are clearly a number of distinct bands, both present in the Raman and ROA spectra. For the Raman spectra, the differences are mostly found within 700 to 1200 cm−1. Most probably, they arise from vibrations of the SO3− group and the C–O–S link. In ROA, two distinct intervals – the upper one (950–1150 cm−1) covering the sulfate group vibrations, and the lower one (200–500 cm−1), encompassing the more delocalized modes and polymer backbone motions – comprise the most notable differences discussed in the following section.
:C6S ratio should be 60:40 and 10:90 for CS from bovine trachea and CS from shark cartilage, respectively).39 The fit was performed independently for three sets of spectra: Raman only, ROA only, and Raman and ROA together. The results of the fits (Table 1) are quite consistent, with a 5% standard deviation, and also well correspond to values found previously.39 The spectra of pure C4S and C6S forms are plotted as shown in Fig. 3. In spite of the similarity, there are clearly a number of distinct bands, both present in the Raman and ROA spectra. For the Raman spectra, the differences are mostly found within 700 to 1200 cm−1. Most probably, they arise from vibrations of the SO3− group and the C–O–S link. In ROA, two distinct intervals – the upper one (950–1150 cm−1) covering the sulfate group vibrations, and the lower one (200–500 cm−1), encompassing the more delocalized modes and polymer backbone motions – comprise the most notable differences discussed in the following section.
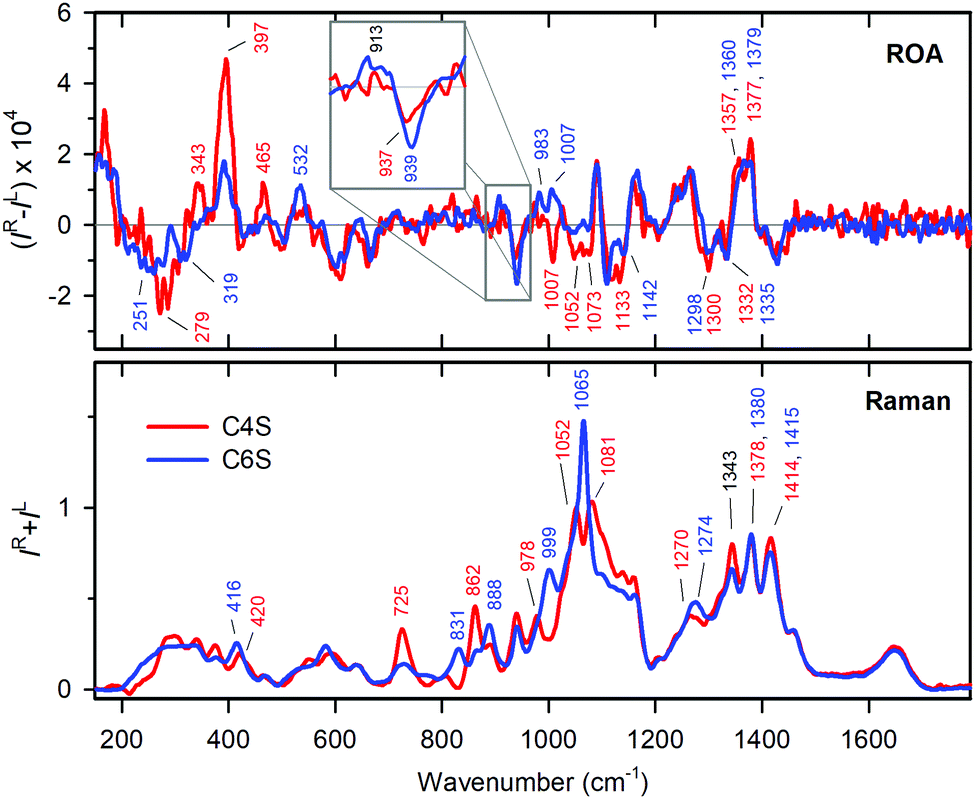 | ||
| Fig. 3 Raman (bottom) and ROA (top) spectra of pure C4S (red lines) and C6S (blue lines) determined by the fit. Vibrational bands characteristic for each CS form are labelled and highlighted by the corresponding color. ROA features corresponding to glycosidic link vibrations are magnified in the inset. | ||
3.2 Analysis of the C4S and C6S spectra and band assignment
Vibrational analysis of the C4S and C6S Raman and ROA spectra was performed on an empirical basis, using the Raman spectra of GlcA (Sigma G8645), GalNAc-4S (Dextra G1054), and GalNAc-6S (Sigma 51947), as well as published data.39,40 We were thus able to assign most spectral features above 700 cm−1, tracking their origin back to component monosaccharide units (see Table S1, ESI† and Fig. 4).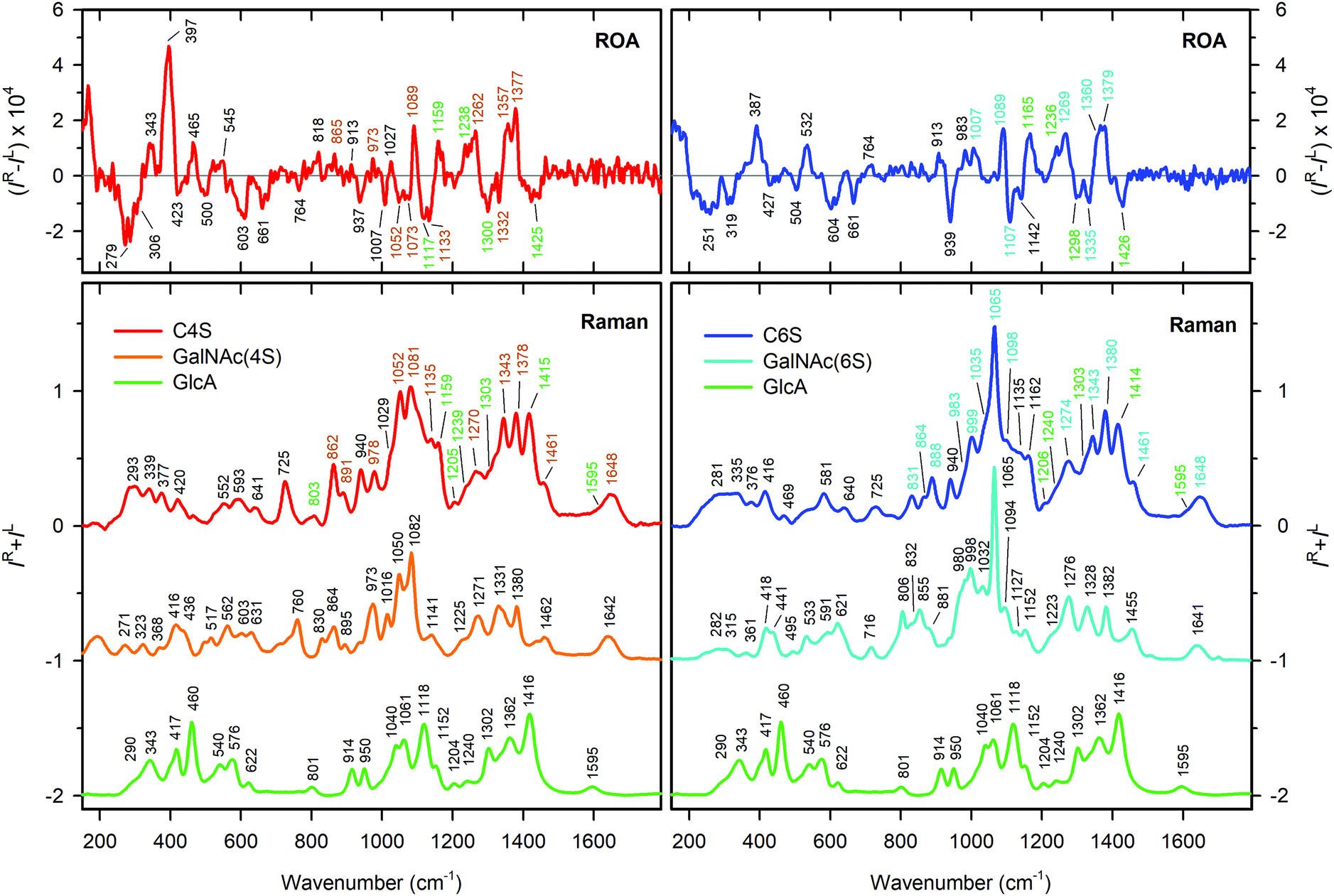 | ||
| Fig. 4 Assignment of Raman and ROA bands in C4S (red, left panels) and C6S (blue, right panels) based on the Raman spectra of GlcA (green), GalNAc-4S (orange) and GalNAc-6S (cyan). CS bands corresponding to the vibrations of the particular building unit are highlighted by corresponding color. The detailed assignment based on ref. 40 is shown in Table S1 (ESI†). | ||
The most distinct differences between Raman spectra of C4S and C6S are located in the region from 700 to 1100 cm−1, dominated by vibrations of the sulfate group and its linkage to GalNAc. The diverse character of these vibrations is directly derived from the position of the sulfate group at the pyranose ring of GalNAc. For the most probable GalNAc 4C1 chair puckering28,33,61 the sulfate group is oriented in the axial plane at the C(4) position, and in the equatorial plane at the C(6) position. In the latter case the sulfate group will be further from the ring. C(4) sulfation therefore may influence more the puckering of the GalNAc ring and/or conformation preferences of the β(1 → 3) glycosidic link.62 When compared to spectra of differently sulfated GalNAc monosaccharides (Fig. 4), it is evident that sulfation manifests itself in the same way as in the spectra of C4S and C6S. The most intense bands at 1081 cm−1 (C4S) and at 1065 cm−1 (C6S) correspond to the symmetric OSO3− group stretching vibration. The asymmetric OSO3− stretch, which is weak in the Raman spectra, appears to be part of a strong band at 1270 cm−1 for C4S and at 1274 cm−1 for C6S.40 Modes at lower wavenumbers (1052, 978, 862, and 725 cm−1 for C4S and 999, 888, and 831 cm−1 for C6S, respectively) correspond mostly to the stretching and deformation vibrations of the C–O–S linkage. The bands corresponding to the C(1)–O–C glycosidic link stretching vibrations are located at 940 cm−1 for both C4S and C6S. This value is in good agreement with previous assignments.63
The upper spectral region (1100–1700 cm−1) is dominated by the lateral groups of GalNAc and GlcA. For GalNAc, we can distinguish amide I (1648 cm−1), amide III (1343 cm−1), and CH3 deformation vibration of the acetylamide moiety (∼1379 cm−1), and the CH2 bending vibration at C(6) (1460 cm−1), from GlcA, we can find asymmetric (1595 cm−1) and symmetric (∼1415 cm−1) deformations of the COO− groups, and the C(5)H bending deformation (∼1310 cm−1). Both CS forms contribute a large number of CH and COH vibrations (the latter under ∼1200 cm−1), underlying the whole region. The shift of the band at 1270 cm−1 (C4S) to 1274 cm−1 (C6S) may be caused by a combination of two effects: (i) a frequency shift of the OSO3− asymmetric stretch due to the different environment of the sulfate groups in C4S and C6S, and (ii) changes in the C(4)H bending frequency of GalNAc due to the presence of sulfate at C(4). Changes in the low frequency region (below 700 cm−1) are smaller but still observable, and cannot be interpreted on the basis of empirical/localized vibration.
Visually, the ROA spectra of C4S and C6S are perhaps more similar than the Raman spectra. This similarity may be caused by the flexibility of lateral functional groups of CS (N-acetyl and COO−, SO3−, etc.), leading to signal averaging. In addition, these groups are achiral. Thus, for example, there is no visible ROA signal in the amide I region. Down to 1200 cm−1, the spectra of C4S and C6S are nearly identical with the exception of bands at ∼1300 and ∼1332 cm−1 differing in relative intensity. This may be explained by lower intensity of the 1300 cm−1 band in C6S (see Fig. 3), corresponding to C(5)H bending in GlcA, which indicates differences in the COO− group environment in C4S and C6S. Within 950–1200 cm−1, however, we observe characteristic spectral patterns for both CS forms. C4S bands at 1052, 1073 and 1133 cm−1 are either not present or are much lower in intensity in C6S. The bands at 1007 cm−1 with approximately the same intensity have opposite signs (see also Fig. 3).
The glycosidic linkage is identified by unbalanced positive/negative couplets at 913/937 and 913/939 cm−1 for C4S and C6S, respectively. Their positions above 900 cm−1 are consistent with previous observations of β type linkages.63 The positive/negative sign of the couplet correlates with the sign observed for the β(1 → 4) linkages. In CS, there are also β(1 → 3) linkages; however, as far as we know their ROA signature is not known.
The region from 400 to 900 cm−1 mostly contains bands of low intensity, for both chondroitin forms, but there are again very strong ROA bands below 400 cm−1. For C4S, there is a broad negative band at 279 cm−1, and three positive bands at 465, 343 and 397 cm−1. The 397 cm−1 band is the most intense one in the whole spectrum. For C6S, the low-frequency pattern is formed by two negative bands at 251 and 319 cm−1, and two positive bands of similar intensity at 397 and 532 cm−1. Vibrational modes of such a low frequency may originate in skeletal motions of larger molecular parts. Therefore, these differences most likely reflect differences in the secondary structure of C4S and C6S.
3.3 Effect of sulfation patterns
As mentioned above, the sulfation of GalNAc residues, either at C(4) or at C(6), may significantly affect the conformation of the CS polymer chain. This may happen via changes in the GalNAc ring puckering, changes in the β(1 → 3) and β(1 → 4) link geometries and less-specific influence on molecular flexibility and conformational freedom. To obtain a deeper insight, we have studied unsulfated, and4-O- and 6-O-sulfated forms of GalNAc and modified the basic disaccharide unit Δdi with a β(1 → 3) link: GalNAc, GalNAc-4S, GalNAc-6S, Δdi-0S, Δdi-4S, and Δdi-6S (Fig. 1). The disaccharide unit with a β(1 → 4) link was not available.Raman and ROA spectra of GalNAc and Δdi samples are shown in Fig. 5. Generally, the changes induced by sulfation are more prominent in the Raman spectra than in the ROA spectra. The major differences occur in bands at 980–1120 cm−1 (colored in gray), corresponding to the OSO3− symmetric stretch and C–O–S stretches, and below 550 cm−1, corresponding to skeletal motions. The ROA/Raman intensity ratio (circular intensity difference, CID) is almost two times higher for Δdi than for GalNAc, which indicates a considerable reduction in flexibility for the disaccharides when compared to the free monosaccharides.
 | ||
Fig. 5 Raman and ROA spectra of the differently sulfated GalNAc and Δdi molecules. GalNAc: on the left, GalNAc (black) and GalNAc-4S (orange). On the right, GalNAc (black) and GalNAc-4S (cyan). Δdi: on the left, Δdi-0S (black) and Δdi-4S (pink). On the right, Δdi-0S (black) and Δdi-6S (purple). Vibration corresponding to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretch is marked by an asterisk. The distinctive vibrational bands are labelled. The spectral region mostly affected by the sulfate group vibration is colored in gray. The most significant difference between the spectra is highlighted by the red rectangle. C stretch is marked by an asterisk. The distinctive vibrational bands are labelled. The spectral region mostly affected by the sulfate group vibration is colored in gray. The most significant difference between the spectra is highlighted by the red rectangle. | ||
Sulfation of GalNAc at C(4) leads to six distinct Raman bands (1082, 1050, 1016, 864, 760, and 562 cm−1, see Table S1 (ESI†) for their assignments). Changes in the ROA spectrum are mostly subtle, including an intensity variation of several bands, small shifts in position of others, the appearance of a negative band at 1104 cm−1, a weak positive band at 760 cm−1, and a shoulder at 562 cm−1. Clearly visible is the replacement of the 878 cm−1 band by the 864 cm−1 band by sulfation (most probably C–O–S deformation). Also the 933 cm−1 band gets smaller. Sulfation at C(6) of GalNAc is accompanied by several new Raman bands (1065, 1032, 998, 855, and 803 cm−1). Again, corresponding changes in ROA are limited to the region of OSO3− and C–O–S stretches and to vibrations below 550 cm−1, while the rest remains almost identical.
For Δdi, sulfation at C(6) also causes limited changes in the ROA spectrum only, mostly within 950–1150 cm−1. Sulfation at C(4) has a much greater impact. Outside of the OSO3− and C–O–S stretching region, we also observe a change around 900 cm−1. The positive 892 cm−1 band vanishes (its shoulder remains as a weak band at 905 cm−1), the negative 964 cm−1 band shifts to 974 cm−1, and most importantly the negative 935 cm−1 band loses most of its intensity. It probably corresponds to a β(1 → 3) glycosidic link vibration. The intensity of several other bands changes as well (1157, 1315, and 1381 cm−1). We can thus deduce that the C(4) Δdi sulfation affects the structure more than for GalNAc. This is consistent with NMR data.62,64
In summary, we have observed that 6-O-sulfation affects neither the conformation of GalNAc itself nor the conformation of the β(1 → 3) glycosidic linkage, while 4-O-sulfation appears to have a measurable effect on the GalNAc conformation and even more noticeable effect on the conformation of the β(1 → 3) glycosidic linkage. The linkage appears to be characterized by the positive/negative ROA couplet at ∼910/935 cm−1.
3.4 CS secondary structure
We saw that above 1200 cm−1 both C4S and C6S have similar spectra. Within 600 and 1200 cm−1 the Raman spectra seem to be selective as compared to ROA, and below 600 cm−1 the ROA spectra are more selective than the Raman spectra. However, the Raman and ROA spectra provide different information about the secondary structure.Raman spectra seem to reflect primarily the primary structure of the CS chain. As shown in Fig. 4, the majority of characteristic Raman features can be directly assigned to vibrational bands of the monosaccharide units, GlcA and sulfated GalNAc. Indeed, the Raman spectrum looks like a sum of these monosaccharides, at least above 800 cm−1.
On the other hand, ROA spectra of C4S and C6S do not resemble the spectra of GlcA, sulfated GalNAc, or disaccharide units so much. ROA spectra of the disaccharide unit share a number of similar features with GalNAc in bands at 550–1300 cm−1 (Fig. 6, seven and nine bands in the case of 4S and 6S, respectively). There are some similarities for bands corresponding to the glycosidic linkage (around 900 cm−1, highlighted by grey lines) and possibly also for positive bands at approximately 1235 and 1265 cm−1. This suggests that the ROA spectra of CS reflect the secondary structure, rather than chemical composition, similarly as for other biopolymers.53,59,60 Intense characteristic ROA bands in the low wavenumber region of the C4S and C6S spectra (Fig. 6) also most probably emerge from the coupling of the CS backbone vibrations, and therefore demonstrate the differences in the CS secondary structure caused by sulfation.
 | ||
| Fig. 6 CS ROA spectra compared to simpler models. On the left C4S (red) versus GlcA (green), and GalNAc-4S (orange) monosaccharides, and Δdi-4S (pink) disaccharide. On the right, C6S (blue) and spectra of monosaccharide (GlcA, green; GalNAc-6S, cyan) and disaccharide (Δdi-6S, purple) units sulfated at C(6). Corresponding GalNAc and Δdi bands are labelled. The most distinct CS bands are indicated by the grey lines. | ||
The difference in the secondary structure of C4S and C6S is also indicated by ROA signals of the C–O–C linkage, both for the β(1 → 3) and β(1 → 4) links. The corresponding couplet (913/937 cm−1) is similar in both CS forms, but there is a difference in intensity, with the positive component of the couplet being significantly weaker for C4S (Fig. 3, inset). Such a variance might be caused by a conformational difference. As shown above, the conformation of the β(1 → 3) link is slightly altered by the adjacent 4-O sulfate group (Fig. 5 left, Fig. 6, ref. 62 and 64). However, C(6) sulfation does not affect the geometry of the β(1 → 3) link, but may change the conformation of the β(1 → 4) link.62
In the future, more detailed characterization of the C4S and C6S secondary structure might be done on the basis of quantum chemical simulations of measured spectra. These would have to deal with conformational flexibility of the monomeric units, interaction with the solvent, and large size of the system. A fragment based approach might overcome some limitations,65,66 but accurate simulations are currently available for smaller systems only.67–69
3.5 Concentration, pH, and temperature dependence
For the concentration dependence an upper limit (100 mg ml−1) was given by the maximum solubility of the sample, and the lower one (12.5 mg ml−1) was given as a minimum sufficient for ROA acquisition. The spectra of sample (1) are plotted in Fig. S2 (ESI†). The upper panel shows spectra normalized to an identical accumulation time, and the lower one displays the same spectra after additional intensity normalization. Within the experimental inaccuracy, the spectra are rather concentration-independent. The concentration 12.5 mg ml−1 may seem to be still relatively high (especially when compared to concentrations used in common Raman or UV measurements); however, CS usually occurs in connective tissues in high concentrations, which makes the concentrations used for ROA measurements closer to the physiological ones.The pH dependence brought only somewhat larger variations, as documented in Fig. 7 (the whole spectral region is shown in Fig. S3, ESI†) for three pH values (2, 6 and 11). Measurements on CS samples (1) (bovine trachea; the highest C4S content) and (4) (EPR marine, the highest C6S content) provided similar results. Fine differences in Raman spectra under pH 6 → 11 change occur at 1068 (1065 in sample (4)), 1300, and 1342 cm−1, which can be assigned to different solvation of the sulfate, carboxyl, and N-acetyl group, respectively. The differences in the ROA spectra at 1046 and 1300 cm−1 can be explained similarly.
 | ||
| Fig. 7 Raman and ROA spectra of CS obtained from bovine trachea (left) and CS EPR marine (right) in a neutral (pH 6, green), basic (pH 11, blue), and acidic (pH 2, red) environment. The major changes in spectra are labelled. | ||
The measurement at pH = 2 was complicated by a reduction in CS solubility and partial precipitation (supernatant spectra are shown). Also at pH = 2 most changes are limited to the Raman spectrum: an intensity drop of the 1300 and 1415 cm−1 bands, corresponding to protonation of the carboxyl group to COOH (pKa ∼ 2.7),70 a tiny decrease of the intensity of the 1342 cm−1 band assigned to amide III vibration, and changes in band characteristics for OSO3− and C–O–S stretching (1026, 1068, and 1099 cm−1 in sample (1), and 1065 cm−1 in sample (4)). A change in the 1300 cm−1 band is also seen in ROA. The small scale of the changes indicates the conformational stability of the CS backbone under pH variation, rather unusual for a biopolymer.
The temperature was varied from 10 to 90 °C for CS samples (1) (bovine trachea; the highest C4S content; 10 °C step) and (4) (EPR marine, the highest C6S content; 5 °C step). Because of the long time of the ROA measurements, detailed dependence could be obtained only for the Raman spectra. In addition, the samples suffered from a relatively high fluorescence that increased with temperature, which made the background subtraction process quite difficult and laborious. Final Raman and difference Raman spectra of samples (4) and (1) are shown in Fig. 8 and Fig. S4 (ESI†), respectively. (The difference spectra are related to that recorded at 10 °C.) As for the pH and concentration factors, the changes induced by the temperature are rather subtle, reduced to slight intensity changes and band shifts. The changes are gradual and smaller than 10% of the parent signals.
 | ||
| Fig. 8 Raman and difference (vs. 10 °C) Raman spectra of CS EPR marine measured at various temperatures. | ||
To uncover any possible structural transitions, we performed the factor analysis of both spectral sets (Fig. S5 and S6, ESI† for samples (4) and (1), respectively). The results were rather similar. The factor dimension was about four, which indicates a rather complex change, perhaps consisting of different interaction with the environment and minor changes in the flexibility of the CS chain. The course of coefficients corresponding to the main spectral change (subspectra S2, almost two magnitudes weaker than the average signal) was almost linear with temperature. The courses of the coefficients corresponding to S3 and S4 were more interesting displaying the linear progress with temperature in combination with some kind of structural transition. However, statistical weights of the third and fourth subspectrum were only a few thousandths of the original signal. For a one-step transition model (Fig. S5 and S6, red lines, ESI†), we obtained a transition temperature of (45 ± 5) °C and (47 ± 4) °C for samples (1) and (4), respectively, connected with ΔH = −(16 ± 4) kcal mol−1 and ΔS = −(50 ± 10) cal mol−1 K−1 per whole CS polymer composed of ∼100 monosaccharide units. ROA spectra were obtained at lower temperatures only (Fig. S7, ESI†), but also do not indicate any significant change in the CS structure.
3.6 Cation effects
The CS polymer, similar to DNA and RNA chains, is a polyanion bearing two negative charges per basic disaccharide unit (the sulfate and carboxyl groups). Therefore, we investigated whether different counter-ions in the form of metal cations cause any changes in the CS structure. Four different cations were investigated – Na+ (medium – 1.16 Å,71 univalent), K+ (large – 1.52 Å,71 univalent), Ca2+ (medium – 1.14 Å,71 divalent), and Cu2+ (small – 0.87 Å,71 divalent). The first three are omnipresent in living organisms, while copper is an essential trace element. Also this factor did not lead to dramatic changes (Fig. 9), but minor spectra variations may indicate some specific interactions of CS with the environment. The sodium/potassium exchange leaves both Raman and ROA spectra virtually unaltered. The sodium/calcium exchange already caused a slight intensity change in the 1098 cm−1 Raman band and the 1047 cm−1 ROA band, both corresponding to the sulfate group vibrations. This may be related to the divalent character of the calcium ion. Finally, the sodium/copper exchange caused most variations. In the Raman spectra, there is an intensity decrease in the 1415 cm−1 band (COO− symmetric deformation). Other changes (shift of the 1274 cm−1 band, an intensity decrease in the 1098 cm−1 band, and sharpening of the 1068 cm−1 band) relate to sulfate group vibrations. Changes in the ROA spectra have similar character – change in the 1300 cm−1 band reflects a weak binding to the COO− group, while changes in the 1000–1150 cm−1 interval correspond to the vibrations of sulfate moieties. These changes can be explained by the different character of copper, containing the d electrons and preferring octahedral coordination geometry. | ||
| Fig. 9 Raman and ROA spectra of CS obtained from bovine trachea sodium (red), potassium (green), calcium (blue), and copper (orange) salt. Significant changes in spectra are indicated by the band positions. | ||
4 Conclusions
We used ROA spectroscopy to obtain insight into the CS structure, stability and interactions with the environment. Based on the five CS samples of different origin we identified Raman and ROA spectra of pure C4S and C6S sulfated forms. As for other biopolymers, a complementarity was observed for the Raman and ROA spectra, the first ones reflecting the primary structure, whereas the second ones were found more sensitive to the secondary structure.Based on a comparison with simpler molecules, a number of characteristic ROA features could be related to the structure of the polysaccharide chain. The spectra of CS building blocks also revealed that the 4-O-sulfation affects the conformation of the β(1 → 3) glycosidic linkage, while the 6-O-sulfation does not have any measurable effect.
Also changes in physico-chemical properties (concentration, pH, temperature, and the presence of cations) could conveniently be studied via the Raman and ROA spectroscopies. These factors, however, caused only marginal changes in the CS structure, typically limited to the polar groups exposed to the solvent. Unlike other biopolymers (proteins and nucleic acids) the conformation of the CS backbone is thus remarkably stable and remains unaffected by the environmental variations.
In the future, theoretical simulations of Raman and ROA spectra may reveal even more detailed characteristics of the CS polymer; these are, however, currently very challenging because of the complexity of the system.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Czech Science Foundation (16-00270S and 18-05770S), the Ministry of Education (LTC17012 and CZ.02.1.01/0.0/0.0/16_019/0000729) and Charles University (project UNCE 010).References
- P. A. Levene and F. B. La Forge, J. Biol. Chem., 1913, 15, 69–79 CAS.
- E. A. Davidson and K. Meyer, J. Biol. Chem., 1954, 211, 605–611 CAS.
- L. Rodén, in The Biochemistry of Glycoproteins and Proteoglycans, ed. W. J. Lennarz, Plenum Press, New York, 1980, pp. 267–371 Search PubMed.
- F. N. Lamari, A. D. Theocharis, A. P. Asimakopoulou, C. J. Malavaki and N. K. Karamanos, Biomed. Chromatogr., 2006, 20, 539–550 CrossRef CAS PubMed.
- C. M. Willis, R. D. Prinz and M. Klüppel, in Signaling, Gene Regulation and Cancer, ed. S. R. Singh and M. K. M. Mishra, Nova Science Publishers, Inc., New York, 2012, pp. 19–38 Search PubMed.
- C. Malavaki, S. Mizumoto, N. Karamanos and K. Sugahara, Connect. Tissue Res., 2008, 49, 133–139 CrossRef CAS PubMed.
- A. D. Theocharis, M. E. Tsara, N. Papageorgacopoulou, D. D. Karavias and D. A. Theocharis, Biochim. Biophys. Acta, Mol. Basis Dis., 2000, 1502, 201–206 CrossRef CAS.
- A. D. Theocharis, D. H. Vynios, N. Papageorgakopoulou, S. S. Skandalis and D. A. Theocharis, Int. J. Biochem. Cell Biol., 2003, 35, 376–390 CrossRef CAS PubMed.
- A. D. Theocharis and D. A. Theocharis, Biomed. Chromatogr., 2002, 16, 157–161 CrossRef CAS PubMed.
- M. E. Tsara, A. D. Theocharis and D. A. Theocharis, Anticancer Res., 2002, 22, 2893–2898 CAS.
- M. J. E. Vallen, S. Schmidt, A. Oosterhof, J. Bulten, L. F. A. G. Massuger and T. H. van Kuppevelt, PLoS One, 2014, 9, e111806 CrossRef PubMed.
- A. D. Theocharis, Biochim. Biophys. Acta, Mol. Basis Dis., 2002, 1588, 165–172 CrossRef CAS.
- F. N. Lamari and N. K. Karamanos, in Chondroitin sulfate: structure, role and pharmcological activity, ed. N. Volpi, Advances in Pharmacology, London, UK, 2006, vol. 53, pp. 33–48 Search PubMed.
- K. Meyer and M. M. Rapport, Science, 1951, 113, 596–599 CrossRef CAS PubMed.
- C. D. Nandini and K. Sugahara, in Chondroitin sulfate: structure, role and pharmcological activity, ed. N. Volpi, Advances in Pharmacology, London UK, 2006, vol. 53, pp. 253–279 Search PubMed.
- J. Salbach, T. D. Rachner, M. Rauner, U. Hempel, U. Anderegg, S. Franz, J.-C. Simon and L. C. Hofbauer, J. Mol. Med., 2012, 90, 625–635 CrossRef PubMed.
- E. D. T. Atkins, Pure Appl. Chem., 1977, 49, 1135–1149 CAS.
- D. H. Isaac and E. D. T. Atkins, Nature, New Biol., 1973, 244, 252–253 CrossRef CAS.
- J. J. Cael, W. T. Winter and S. Arnott, J. Mol. Biol., 1978, 125, 21–42 CrossRef CAS PubMed.
- W. T. Winter, S. Arnott, D. H. Isaac and E. D. T. Atkins, J. Mol. Biol., 1978, 125, 1–19 CrossRef CAS PubMed.
- R. P. Millane, A. K. Mitra and S. Arnott, J. Mol. Biol., 1983, 169, 903–920 CrossRef CAS PubMed.
- E. D. T. Atkins, R. Gaussen, D. H. Isaac, V. Nandanwar and J. K. Sheehan, J. Polym. Sci., Polym. Lett. Ed., 1972, 10, 863–865 CrossRef.
- S. Arnott, J. M. Guss, D. W. L. Hukins and M. B. Mathews, Biochem. Biophys. Res. Commun., 1973, 54, 1377–1383 CrossRef CAS PubMed.
- S. Arnott, J. M. Guss, D. W. L. Hukins and M. B. Mathews, Science, 1973, 180, 743–745 CrossRef CAS PubMed.
- S. Yamada, K. Yoshida, M. Sugiura and K. Sugahara, J. Biochem., 1992, 112, 440–447 CrossRef CAS PubMed.
- K. Sugahara, Y. Tanaka and S. Yamada, Glycoconjugate J., 1996, 13, 609–619 CrossRef CAS PubMed.
- F. Yu, J. J. Wolff, I. J. Amster and J. H. Prestegard, J. Am. Chem. Soc., 2007, 129, 13288–13297 CrossRef CAS PubMed.
- B. M. Sattelle, J. Shakeri, I. S. Roberts and A. Almond, Carbohydr. Res., 2010, 345, 291–302 CrossRef CAS PubMed.
- V. Blanchard, F. Chevalier, A. Imberty, B. R. Leeflang, Basappa, K. Sugahara and J. P. Kamerling, Biochemistry, 2007, 46, 1167–1175 CrossRef CAS PubMed.
- A. Almond and J. K. Sheehan, Glycobiology, 2000, 10, 329–338 CrossRef CAS PubMed.
- G. Cilpa, M. T. Hyvönen, A. Koivuniemi and M. L. Riekkola, J. Comput. Chem., 2010, 31, 1670–1680 CAS.
- M. A. Rodríguez-Carvajal, A. Imberty and S. Pérez, Biopolymers, 2003, 69, 15–28 CrossRef PubMed.
- V. H. Pomin, Prog. Biophys. Mol. Biol., 2014, 114, 61–68 CrossRef CAS PubMed.
- A. Imberty and S. Pérez, Chem. Rev., 2000, 100, 4567–4588 CrossRef CAS PubMed.
- T. R. Rudd, M. A. Skidmore, S. E. Guimond, C. Cosentino, G. Torri, D. G. Fernig, R. M. Lauder, M. Guerrini and E. A. Yates, Glycobiology, 2009, 19, 52–67 CrossRef CAS PubMed.
- K. Matsuo, H. Namatame, M. Taniguchi and K. Gekko, Biosci., Biotechnol., Biochem., 2009, 73, 557–561 CrossRef CAS PubMed.
- W. Garnjanagoonchorn, L. Wongekalak and A. Engkagul, Chem. Eng. Process., 2007, 46, 465–471 CrossRef CAS.
- N. Mainreck, S. Brézillon, G. D. Sockalingum, F. X. Maquart, M. Manfait and Y. Wegrowski, J. Pharm. Sci., 2011, 100, 441–450 CrossRef CAS PubMed.
- R. Ellis, E. Green and C. P. Winlove, Connect. Tissue Res., 2009, 50, 29–36 CrossRef PubMed.
- R. Bansil, I. V. Yannas and H. E. Stanley, Biochim. Biophys. Acta, 1978, 541, 535–542 CrossRef CAS.
- G. J. Miller, S. U. Hansen, M. Baráth, C. Johannessen, E. W. Blanch, G. C. Jayson and J. M. Gardiner, Carbohydr. Res., 2014, 400, 44–53 CrossRef CAS PubMed.
- L. D. Barron and A. D. Buckingham, Mol. Phys., 1971, 20, 1111–1119 CrossRef CAS.
- L. D. Barron, M. P. Bogaard and A. D. Buckingham, Nature, 1973, 241, 113–114 CrossRef CAS.
- L. D. Barron, M. P. Bogaard and A. D. Buckingham, J. Am. Chem. Soc., 1973, 95, 603–605 CrossRef CAS.
- Z. Q. Wen, L. D. Barron and L. Hecht, J. Am. Chem. Soc., 1993, 20, 285–292 CrossRef.
- A. F. Bell, L. Hecht and L. D. Barron, J. Am. Chem. Soc., 1994, 116, 5155–5161 CrossRef CAS.
- A. F. Bell, L. Hecht and L. D. Barron, J. Raman Spectrosc., 1993, 24, 633–635 CrossRef CAS.
- A. F. Bell, L. Hecht and L. D. Barron, J. Raman Spectrosc., 1995, 26, 1071–1074 CrossRef CAS.
- F. Zhu, N. W. Isaacs, L. Hecht and L. D. Barron, J. Am. Chem. Soc., 2005, 127, 6142–6143 CrossRef CAS PubMed.
- C. Johannessen, R. Pendrill, L. Hecht, G. Widmalm and L. D. Barron, AIP Conf. Proc., 2010, 1267, 104–105 CrossRef.
- C. Johannessen, R. Pendrill, G. Widmalm, L. Hecht and L. D. Barron, Angew. Chem., Int. Ed., 2011, 50, 5349–5351 CrossRef CAS PubMed.
- N. R. Yaffe, A. Almond and E. W. Blanch, J. Am. Chem. Soc., 2010, 132, 10654–10655 CrossRef CAS PubMed.
- A. Rüther, A. Forget, A. Roy, C. Carballo, F. Mießmer, R. K. Dukor, L. A. Nafie, C. Johannessen, V. P. Shastri and S. Lüdeke, Angew. Chem., Int. Ed., 2017, 56, 4603–4607 CrossRef PubMed.
- T. R. Rudd, R. Hussain, G. Siligardi and E. A. Yates, Chem. Commun., 2010, 46, 4124–4126 RSC.
- W. Hug and G. Hangartner, J. Raman Spectrosc., 1999, 30, 841–852 CrossRef CAS.
- J. Palacký, P. Mojzeš and J. Bok, J. Raman Spectrosc., 2011, 42, 1528–1539 CrossRef.
- V. Profant, M. Pazderková, T. Pazderka, P. Maloň and V. Baumruk, J. Raman Spectrosc., 2014, 45, 603–609 CrossRef CAS.
- E. R. Malinovski, Factor analysis in Chemistry, Wiley, New York, 3rd edn, 2002 Search PubMed.
- V. Profant, V. Baumruk, X. Li, M. Šafařík and P. Bouř, J. Phys. Chem. B, 2011, 115, 15079–15089 CrossRef CAS PubMed.
- V. Profant, M. Šafařík, P. Bouř and V. Baumruk, Spectroscopy, 2010, 24, 213–217 CrossRef.
- D. Cremer and J. A. Pople, J. Am. Chem. Soc., 1975, 97, 1354–1358 CrossRef CAS.
- M. Zsiska and B. Meyer, Carbohydr. Res., 1993, 243, 225–258 CrossRef CAS.
- F. Zhu, N. W. Isaacs, L. Hecht, G. E. Tranter and L. D. Barron, Chirality, 2006, 18, 103–115 CrossRef CAS PubMed.
- B. M. Sattelle, S. U. Hansen, J. Gardiner and A. Almond, J. Am. Chem. Soc., 2010, 132, 13132–13134 CrossRef CAS PubMed.
- P. Bouř, J. Sopková, L. Bednárová, P. Maloň and T. A. Keiderling, J. Comput. Chem., 1997, 18, 646–659 CrossRef.
- N. S. Bieler, M. P. Haag, C. R. Jacob and M. Reiher, J. Chem. Theory Comput., 2011, 7, 1867–1881 CrossRef CAS PubMed.
- J. R. Cheeseman, M. S. Shaik, P. L. A. Popelier and E. W. Blanch, J. Am. Chem. Soc., 2011, 133, 4991–4997 CrossRef CAS PubMed.
- A. Melcrová, J. Kessler, P. Bouř and J. Kaminský, Phys. Chem. Chem. Phys., 2016, 18, 2130–2142 RSC.
- J. Kaminský, J. Kapitán, V. Baumruk, L. Bednárová and P. Bouř, J. Phys. Chem. A, 2009, 113, 3594–3601 CrossRef PubMed.
- H. M. Wang, D. Loganathan and R. J. Linhardt, Biochem. J., 1991, 278, 689–695 CrossRef CAS PubMed.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Further experimental data and details of spectral analysis. See DOI: 10.1039/c9cp00472f |
| This journal is © the Owner Societies 2019 |