
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDetermination of element–deuterium bond lengths in Zintl phase deuterides by 2H-NMR†
Robin
Guehne
ab,
Henry
Auer
 *c,
Holger
Kohlmann
c,
Jürgen
Haase
a and
Marko
Bertmer
*a
*c,
Holger
Kohlmann
c,
Jürgen
Haase
a and
Marko
Bertmer
*a
aFelix Bloch Institute, Leipzig University, Linnéstrasse 5, 04103 Leipzig, Germany. E-mail: bertmer@physik.uni-leipzig.de
bMacDiarmid Institute, School of Chemical and Physical Sciences, Victoria University of Wellington, P.O. Box 600, Wellington 6140, New Zealand
cDepartment of Inorganic Chemistry, Leipzig University, 04103 Leipzig, Germany. E-mail: henry.auer@uni-leipzig.de
First published on 3rd May 2019
Abstract
The Zintl phase deuterides CaSiD4/3, SrSiD5/3, BaSiD2, SrGeD4/3, BaGeD5/3 and BaSnD4/3 were investigated by nuclear magnetic resonance (NMR) spectroscopy and density functional theory (DFT) calculations to reliably determine element–deuterium bond lengths. These compounds show deuterium bound to the polyanion and deuteride ions in tetrahedral cationic voids. With 2H-NMR experiments we characterised the individual signals of the two distinct crystal sites. Quadrupolar coupling constants (CQ) of the anion-binding site were determined as 58 to 78 kHz (Si compounds), 51 to 61 kHz (Ge compounds) and 38 kHz (Sn compound). These values agree well with the quadrupole couplings derived from DFT using optimized structural models. We further calculated the general element–deuterium distance dependency of CQ using DFT methods that allow an accurate determination of bond lengths via the 2H quadrupole interaction. The thus determined bond lengths are evaluated as d(Si–D) = 1.53–1.59 Å, d(Ge–D) = 1.61–1.65 Å and d(Sn–D) = 1.86 Å. Chemical shifts of the anion-binding site range from 0.3 to 1.3 ppm. The isotropic chemical shifts of the tetrahedral sites are 5.1 ppm (CaSiD4/3), 7.0 to 10.0 ppm (Sr compounds) and 10.7 to 11.6 ppm (Ba compounds).
1 Introduction
Zintl phases are polyanionic compounds. They consist of an electropositive metal from the s-block (or a rare earth metal) and a more electronegative element from group 13 to 16. The compounds can be formally described by an ionic picture. The anions do not reach an electron octet thus forming covalent element–element bonds according to the general 8-N rule.1–4Zintl phase hydrides can be described by the same formalism. Hydrogen shows a similar or even higher electronegativity than the p-block element. It can be incorporated in two ways: (i) hydrogen can form an anion (H−) that is exclusively coordinated by cations (interstitial hydride), or (ii) it can form a heteropolar bond towards the polyanion (Hδ−, formally H0, polyanionic hydride).5
There are several examples of group 13 polyanionic hydrides. For example, SrAlH2,6AeGaH2,7 and AeTrTtH8–11 (Ae = Ca, Sr, Ba, Tr = Al, Ga, In, Tt = Si, Ge, Sn) show puckered, honey-comb like six-rings of Tt− and (TrH)− anions. According to inelastic neutron scattering and quantum chemical calculations these compounds show a covalent Tr–hydrogen interaction.10–13 Further examples are polyethylene-like (GaH2)−,14,15 propane-like (Ga3H8)3−,16 or neo-pentane-like Ga(Ga4H12)5−![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 14 polyanions. In contrast the polyanions in NdGaH1.66 and GdGaH1.66 show only a weak element–hydrogen interaction according to diffraction-derived bond lengths and calculations.17–19
14 polyanions. In contrast the polyanions in NdGaH1.66 and GdGaH1.66 show only a weak element–hydrogen interaction according to diffraction-derived bond lengths and calculations.17–19
The situation for group 14 polyanionic hydrides is still ambiguous. There are contradictory works on CaSiHy (1.0 < y < 1.33) claiming a covalent bond (d(Si–H) = 1.58 Å)20 or only a weak interaction (d(Si–H) = 1.84 Å).21 Recently, some of us determined the crystal structures of the homologue systems BaSiH1.9, SrGeH1.2 and BaSnH1.322 as well as SrSiH1.5 and BaGeH1.6.23,24 The compounds exhibit hydride anions in tetrahedral cationic voids and additional hydrogen binding to the polyanion. Due to short bond lengths, there is strong indication for a covalent interaction, but the estimated uncertainties of a Rietveld based analysis are rather high. This work investigates deuterated samples of Zintl phase hydrides. We apply 2H nuclear magnetic resonance (NMR) and interpret the results based also on density functional theory (DFT) calculations, to improve the bonding picture.
In 2H NMR, the electric quadrupole interaction is a very sensitive probe for the local distribution of charges surrounding the nucleus.25 The quadrupole coupling constant, CQ = e2qQ/h sets the peak distance, Δ (in frequency units), observed in the powder spectra for this I = 1 nucleus for uniaxial symmetry (η = 0) of the quadrupole tensor, as expected from a covalent bond, i.e.,
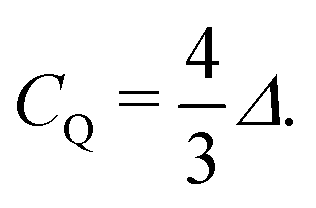 | (1) |
As usual, eQ is the known electric quadrupole moment of 2H and h denotes the reduced Planck's constant, while eq = VZZ (and η = 0) defines the symmetric electric field gradient (EFG) at the nucleus' site that was calculated with DFT.
Based on our measurements we determine group 14 element–deuterium bond lengths with substantially improved accuracy through a combination of experimentally determined quadrupole interactions and density functional theory (DFT) calculations. We further develop a more comprehensive picture of the NMR of Zintl phase deuterides and its relation to their general chemistry.
2 Experimental
2.1 Synthesis
All manipulations were done in an argon filled glove box. Moisture and oxygen content were kept below 1 ppm each. The Zintl phases were prepared by stoichiometric mixtures of the respective elements using tantalum metal jackets which were subsequently sealed and placed in fused silica ampoules at reduced pressure. The deuterides were prepared in a Nicrofer 5219Nb-alloy 718 autoclave at typical D2 gas pressures of 5 to 9 MPa and 473 K. Details are given elsewhere (SrSiD5/3 and BaGeD5/3;23,24 BaSiD2, SrGeD4/3 and BaSnD4/322).The Zintl phase CaSi was prepared from the elements as well (Si: abcr, 99.9999%; Ca: Alfa Aesar, 99.5%). The reactants were placed in tantalum jackets which were sealed and put into fused silica ampoules under reduced argon pressure. The samples were heated with 2.2 K min−1 to 1573 K and kept there for one hour. CaSi was annealed at 1473 K for 12 h. In the literature CaSi typically is fast quenched in cold water.26,27 We want to emphasize that this step is crucial to obtain samples reactive towards hydrogen. CaSiD4/3 was prepared in an autoclave reaction of CaSi under 15 MPa D2 pressure at 523 K for 72 h. The sample was annealed for 72 h.
2.2 Nuclear magnetic resonance
NMR measurements have been carried out on powders sealed in evacuated quartz tubes. Magic angle spinning (MAS) experiments were carried out using MAS zirconia rotors equipped with airtight caps. External static magnetic fields of 9.4 T, 11.7 T, and 17.6 T in combination with Bruker (Bruker Corporation, Billerica, MA, USA) spectrometers (AVANCE 400/750 and AVANCE III HD) were used. Homebuilt static and 4 mm Bruker MAS probes were employed. Spectra were recorded with free induction decay (FID – static and MAS) and solid-echo (π/2–τ–π/2) pulse sequences28,29 (only static) with typical π/2-pulse lengths of 3 to 5 μs and pulse separation times τ = 30 μs. Recycle delays were set to 500 s due to spin lattice relaxation times (T1) of 50 to 100 s depending on sample, crystal site and external magnetic field.Low MAS spinning frequencies were used to generate sufficiently detailed spectra that allow accurate fitting of the quadrupolar lineshapes. For isolating isotropic signals, higher spinning frequencies up to 8 kHz were used. Data analysis was done with Bruker Topspin, dmfit,30 and OriginPro 7.5G software package (OriginLab Corporation, Northampton, MA, USA). Deuterated Water (D2O) was used for optimizing measurement conditions and referencing.
2.3 Density functional theory (DFT) calculations
The Abinit package31–35 was used to calculate quadrupole splittings. Calculations were performed using the generalized gradient approximation (GGA) as implemented in the Perdew–Burke–Ernzerhof (PBE) functional.36,37 Projector-augmented wave (PAW)38,39 atomic data were taken from the JTH PAW atomic dataset table.40,41 Electron densities and, therefore, the EFGs at the corresponding deuterium sites were calculated with a SCF threshold of 10−9 Hartree regarding total energy (toldfe) on a 4 × 12 × 12 (Pnma-BaSiH2), 4 × 12 × 4 (Pnma-CaSiH4/3, -SrGeH4/3, -BaSnH4/3) or 4 × 12 × 2 (Pnma-SrSiH5/3, -BaGeH5/3) Monkhorst–Pack42k-point grid which corresponds to about 0.01 Bohr−1k-point accuracy. For molecular calculations SiH4, GeH4 and SnH4 were placed in a large box (a = 20 Å) with Γ as a single k-point. Since previous density of states (DOS) calculations showed no or just pseudo band gaps, the systems were treated metallic with 0.001 Hartree Gaussian smearing. This value was tested for chemically similar systems and seemed appropriate. The kinetic energy cut-off (ecut) was set to 25 Hartree which is the suggested high value for hydrogen. The corresponding pawecutdg was set to 2·ecut. The convergence of CQ was tested with regard to ecut in detail for the systems SiH4, GeH4 and BaSiH2. Convergence regarding the k-grid was tested in detail for BaSiH2. The used values show an agreement better than 0.25 kHz in CQ. For deuterium an electric quadrupole moment of 2.86 × 10−3 barn43,44 was used.3 Results
As the structural properties of the materials under consideration are crucial for the interpretation of their NMR results, we will first review the crystallographic details that are published already elsewhere.20–24Within the homologue series of the Zintl phase deuterides AeTtD(1+y), Ae = Ca–Ba, Tt = Si–Sn, three distinct structure types occur (Fig. 1). All structure types show one deuterium atom per formula unit in pseudo-tetrahedral Ae4 voids (interstitial hydride). The remaining deuterium atoms bind to Tt atoms (polyanionic hydride). According to the Zintl concept, the compounds can be formulated as (Ae2+ D−)(Tt−)1−y(TtD−)y. Therefore, the Tt atoms can be expected to be three-binding. This is realized by the formation of Tt zigzag chains in crystallographic b direction (2 bonds). The remaining bond is saturated by deuterium (BaSiD2 structure type) or by an additional connection of the chains in crystallographic c direction. The CaSiD4/3 structure type is formed by a deuterium termination after three of these chains. The SrSiD5/3 structure type shows two different polyanions. One is a Tt chain where each atom is saturated by deuterium as found for BaSiD2. The other one exhibits an additional Tt–Tt bond connecting two chains. The remaining Tt atoms are bound to deuterium. Furthermore, diffraction showed that the chain-coordinating deuterium site is always under-occupied (details were given elsewhere22–24) which is indicated by a correction term x in the chemical formula. The compounds CaSiD4/3−x,20,21 SrGeD4/3−x22 and BaSnD4/3−x22 crystallize in the CaSiD4/3 type. SrSiD5/3−x23,24 and BaGeD5/3−x23,24 adopt the SrSiD5/3 structure type. The compound BaSiD2−x22 shows its own structure type (BaSiD2 type).
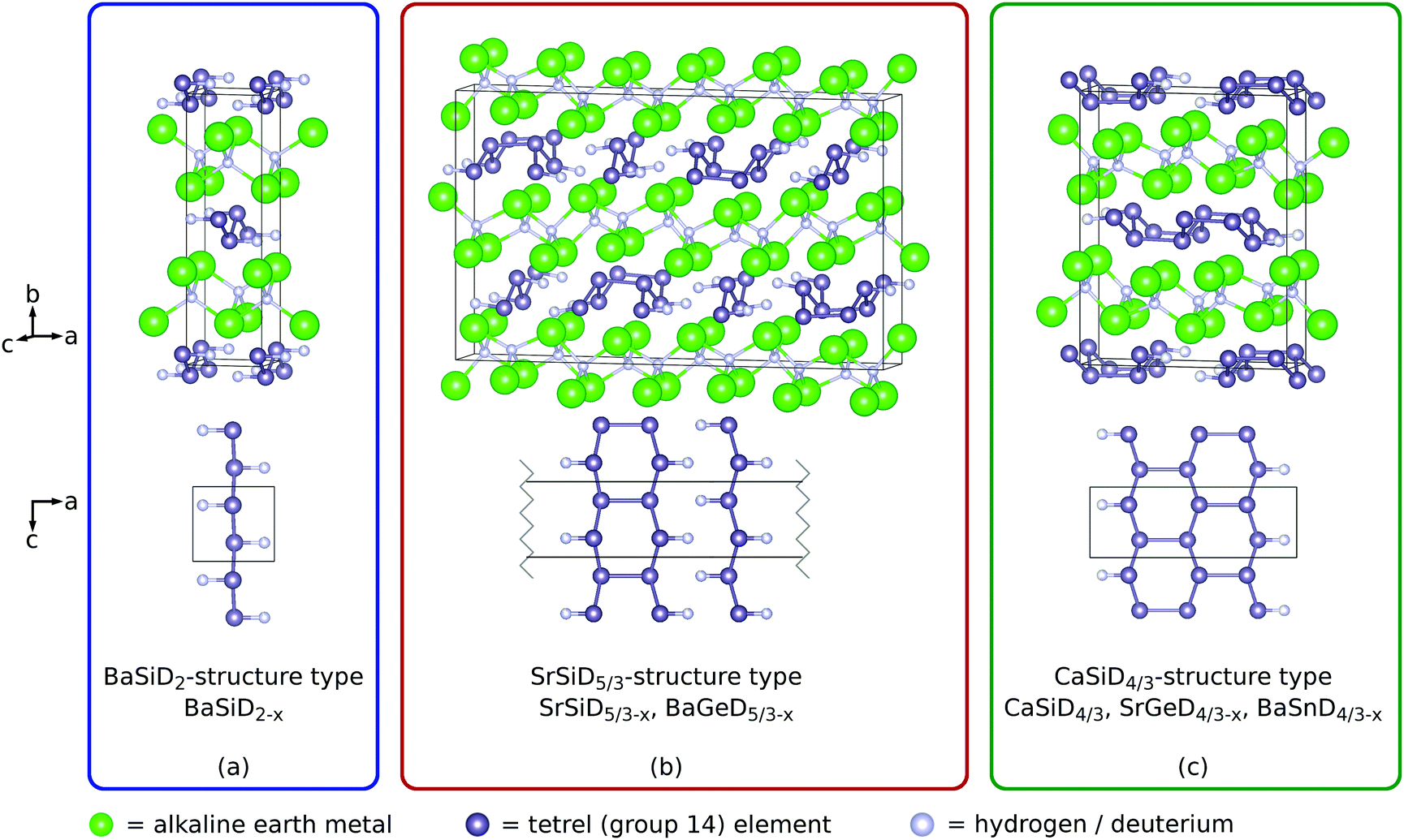 | ||
| Fig. 1 The three structure types (space-group type Pnma for all of them) adopted by the Zintl phase deuterides under investigation and a projection of the polyanions. | ||
3.1 NMR
From the 2H NMR spectra, we determined quadrupole coupling constants (CQs), isotropic shifts, linewidths and estimated the relative intensity of resonances in the present samples. The corresponding static spectra are given in Fig. 2, with more NMR parameters listed in Table 1.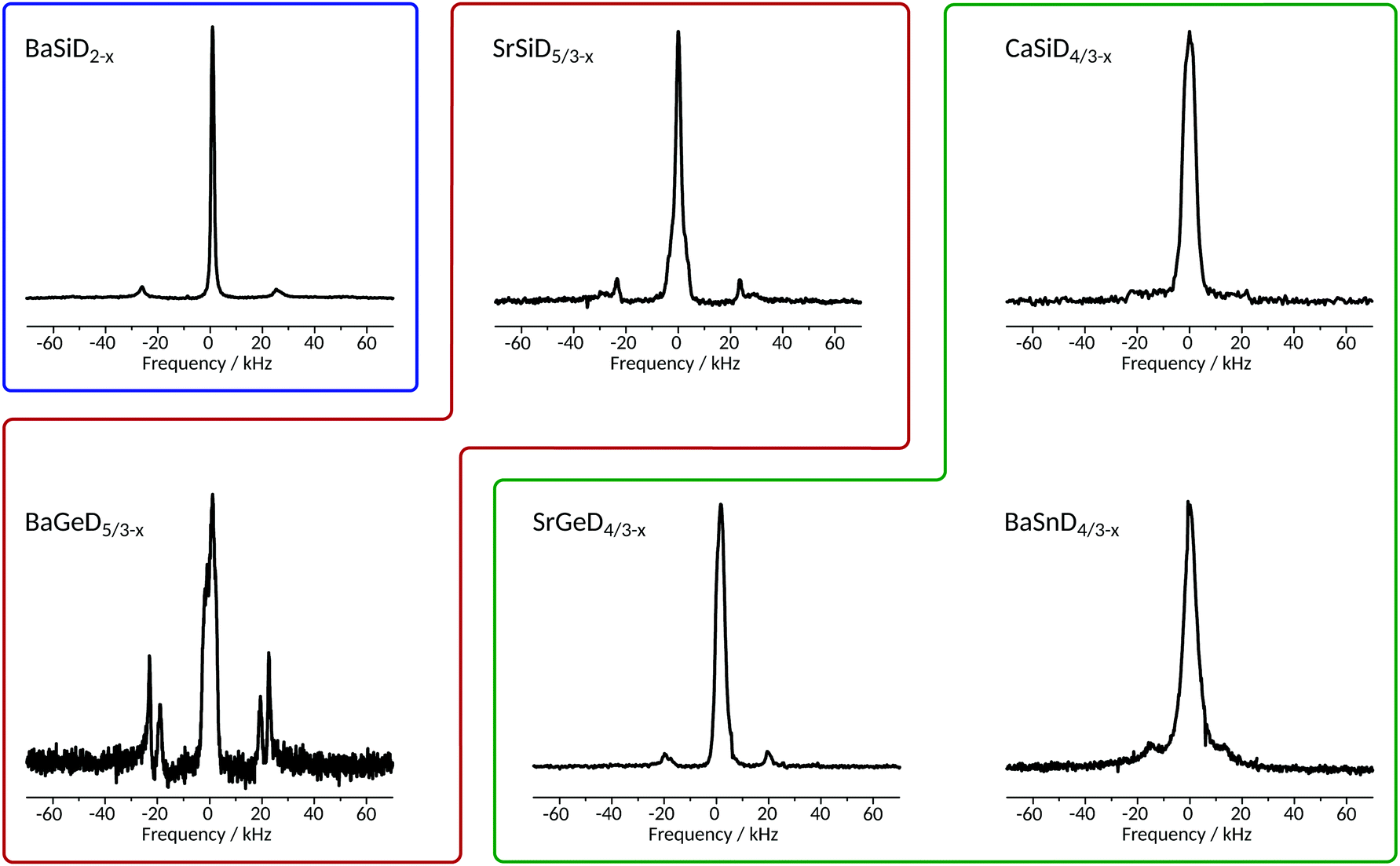 | ||
| Fig. 2 Static 2H spectra of Zintl family deuterides obtained at a magnetic field of 17.6 T (except for CaSiD4/3−x obtained at 11.7 T) with a π/2-pulse length of 5.7 μs (3 μs for CaSiD4/3−x) averaging between 144 and 304 scans (depending on sample). Each spectrum consists of a narrow central line and a broad, quadrupolar split one. The quadrupole interaction can be evaluated by the distance in frequency units of the two horns which is directly related to CQ as defined in the introduction. Coloured frames correspond to isotypic structures as indicated in Fig. 1. | ||
| Compound | x neutron | C chainQ,exp | C chainQ,DFT | η chainDFT | Shiftchain | Λ void | C voidQ,DFT | η voidDFT | Shiftvoid |
|---|---|---|---|---|---|---|---|---|---|
| BaSiD2−x | 0.13(2) | 69(1) | 65.3 | 0.06 | 1.0 | 1.6(1) | 0.81 | 0.87 | 10.7 |
| SrSiD5/3−x | 0.17(2) | 63(1)/78(3) | 57.7/63.5 | 0.02/0.11 | 1.4 | 1.9(1)/7.2(2) | 0.95–(−7.14) | 0.14–0.64 | 7.0 |
| CaSiD4/3−x | 0.03–0.13 | 58(2) | 57.6 | 0.01 | 0.3 | 5.4(2) | 3.92–8.36 | 0.24–0.32 | 5.1 |
| SrGeD4/3−x | 0.139(3) | 52(2) | 47.7 | 0.02 | 0.5 | 3.9(3) | 2.3–(−5.9) | 0.03-0.64 | 10.0 |
| BaGeD5/3−x | 0.10(3) | 51(2)/61(2) | 50.1/55.5 | 0.02/0.04 | 1.3 | 5.7(5) | 0.31–(−4.31) | 0.4–0.86 | 11.6 |
| BaSnD4/3−x | 0.055(2) | 38(2) | 36.9 | 0.02 | 0.7 | 5.1(3) | −1.3–(−7.06) | 0.41–0.89 | 11.2 |
The quadrupole splitting constant CQ is directly accessible via the distance of the horns of the axial symmetric quadrupole powder patterns (or peaks in Fig. 2), see eqn (1). The results were confirmed by fitting slow MAS spectra. Field dependent measurements were used to separate contributions from field dependent (shift anisotropy) and field independent (quadrupole and dipole interactions) effects. It was found that both lines, the narrow as well as the quadrupole split one, are essentially magnetic field independent.
The NMR spectrum of each compound is composed of a narrow resonance line in the middle of a broad quadrupole powder pattern that becomes visible through the two horns of the quadrupole doublet. We carefully examined the spectra of static as well as MAS experiments and found no evidence for a deviation from axial symmetry, i.e. η = 0. Note that there is no central transition in quadrupolar split deuterium spectra (contrary to half-integer spin systems under quadrupole interaction). As expected, in MAS spectra, the broad quadrupole pattern is broken up into a manifold of spinning sidebands (data shown in ESI†). The reduced linewidths of the MAS spectra also reveal a shift difference between the narrow and the broad resonances (see below).
These two signals, different in shape and shift, represent very different local environments of the related nuclei, in agreement with the crystal structures as described above. On the basis of the general similarities of the crystal structures investigated within this work, i.e. a high symmetry environment (cubic pseudo-symmetry) and a low symmetry environment (no pseudo-symmetry) for D atoms,23,24 we find consistently with our report on BaSiD2−x and SrGeD4/3−x,22 that the narrow resonance lines in Fig. 2 can be assigned to 2H nuclei residing in the Ae4 voids (high symmetry), whereas the quadrupolar split ones belong to Tt chain sites (low symmetry). Measured shifts confirm this assignment which is further substantiated through the relative intensity of both lines reproducing in good approximation the relative occupation of both crystal sites. The quadrupole splitting being consistently axially symmetric for each sample reflects the cylinder symmetry of the chemical bond that gives rise to the EFG at the nucleus' site.
In BaSiD2−x with its separated, unconnected chains, there is one crystallographic site of deuterium binding to the Si-chain and another located within the Ba4 voids. This structure allows the maximum amount of hydrogen/deuterium to be brought into the system, i.e. AeTtD2. Consistently, we identify a well defined signal for each crystal site (cf.Table 1) of comparable intensity.
In the CaSiD4/3−x structure type three chains are interconnected. These systems also exhibit only one crystallographic deuterium site saturating the non-connected Tt atoms, however, there are three crystallographic deuterium sites within Ae4 voids which are no longer as regular as found for BaSiD2−x. The maximum number of H/D atoms is necessarily reduced, i.e. AeTtD4/3. NMR spectra consist of a quadrupole split resonance and a narrow line in the middle. The relative intensity of the former resonance is significantly reduced compared to the latter one, a result much different to what is found in the case of BaSiD2−x. This reflects the altered relative occupation due to larger polyanions.
The SrSiD5/3−x structure type, realized in SrSiD5/3−x and BaGeD5/3−x, is crystallographically the richest case in the present study (Fig. 1). Single chains as in BaSiD2−x alternate in c direction with a deuterium saturated double row (two connected chains). There are four crystallographically independent chain-binding deuterium sites present (D1 to D4; see Fig. 5), two for each polyanion. Due to molecular pseudo-symmetry there are only two quadrupole couplings that differ in CQ and η. For deuterium ions within Ae4 voids, six crystallographic sites can be identified. Hence, these voids are even more irregular than in the CaSiD4/3−x case. For the two samples we do in fact find two different quadrupole splittings of comparable intensity (Fig. 2) and a substantially broadened resonance in the center.
3.2 DFT calculations
As shown below, CQ values only depend on the Tt–D distances. Thus, we constrain our discussion to the DFT optimised models here.
DFT calculations of the optimized structural models clearly show that deuterium in Ae4 voids exhibit a very small EFG (CQ < 9 kHz), while for the Tt coordinating sites a considerable EFG (CQ > 35 kHz) is present. The tetrahedral voids are a pseudo-cubic environment for deuterium. Still, crystallographic symmetry (point symmetry m) is lower and the asymmetry parameter η deviates from zero considerably (Table 1). The deuterium atoms at the Tt chains are also located on a low symmetric site (point symmetry m). They show vanishing asymmetry parameters smaller than 0.1 (in most cases even η < 0.04; Table 1) indicating almost perfect axial symmetry. The direction of the main component of the EFG deviates less than 5° from the symmetry axis of the bond which is the connecting line of the atoms. The quadrupole splittings of the Tt-binding site can be compared to the experimental values. The CQ values agree very well (Table 1).
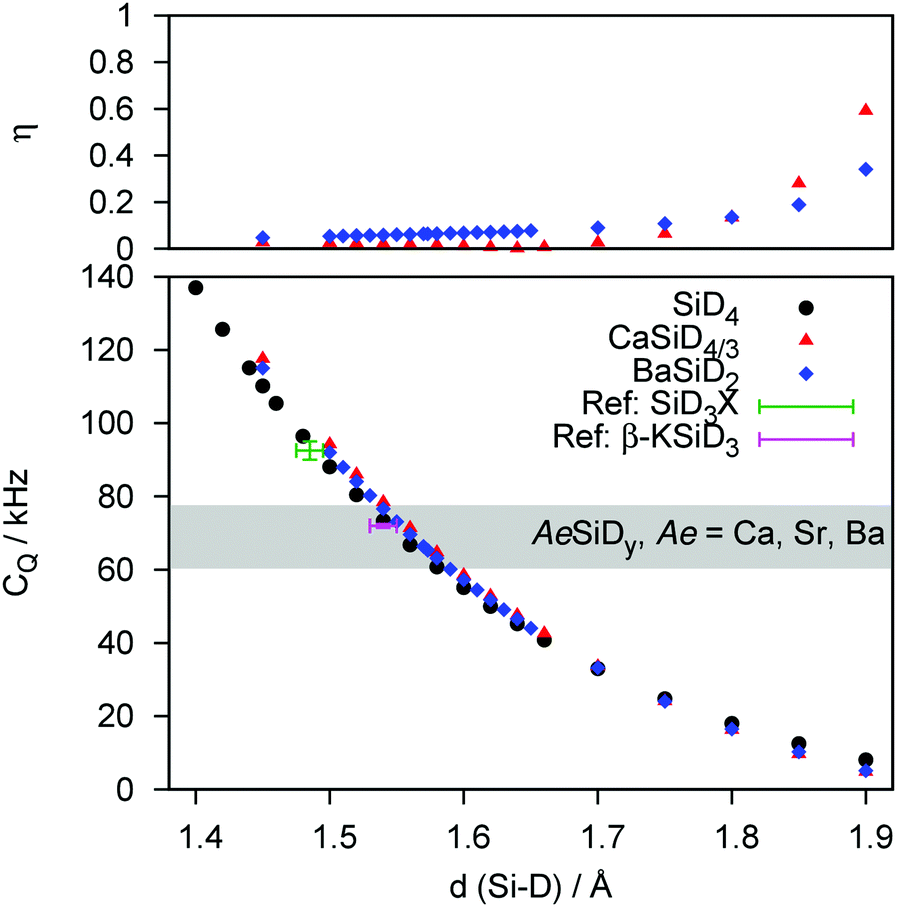 | ||
| Fig. 3 DFT derived distance dependence of CQ and the corresponding asymmetry parameter η of SiD4, CaSiD4/3, and BaSiD2. Experimental data of silane derivatives SiD3X (X = D, CH3, C5H6) and β-KSiD3 taken from the literature are added for reference (see text for details). The range of experimental CQ-values is shown as a grey bar. | ||
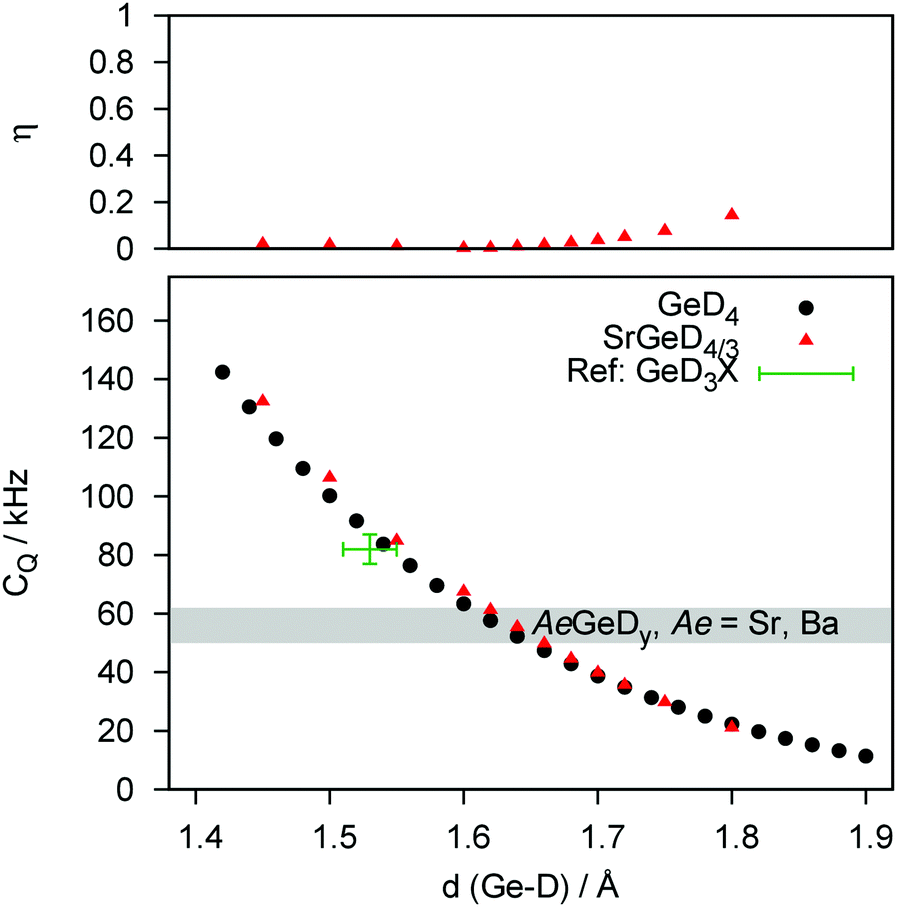 | ||
| Fig. 4 DFT derived distance dependence CQ and the corresponding asymmetry parameter η of GeD4, and SrGeD4/3. Experimental data of germane derivatives GeD3X (X = D, CH3) taken from the literature are added for reference (see text for details). The range of experimental CQ-values is shown as a grey bar. | ||
The distance dependency of CQ regarding the Tt–D bond length for covalent, molecular deuterides and the Zintl phase deuterides agree well, suggesting similar electronic structures, i.e., a covalent character of the Tt–D bond. Furthermore, while the surrounding of a given polyanionic deuteride seems not to affect the quadrupolar interaction considerably, effects on the asymmetry parameter η are small as well. It starts to increase significantly for Tt–D distances much larger than expected for the AeTtDy systems, i.e. larger than 1.80 Å (Si and Ge compounds). The dependency of CQ and η in regard to the dihedral angle of deuterium towards the plane of the zigzag chain was evaluated for BaSiD2 (see ESI,† Fig. S1). There is hardly an effect on the magnitude of the quadrupolar interaction (less than 5 kHz for ±10°). Decreasing the dihedral angle increases CQ, while increasing the angle strongly affects the asymmetry parameter. Bond angles smaller than 90° are unlikely due to chemical reasoning in agreement with experimental results where no evidence for an asymmetric quadrupole interaction could be found.
4 Discussion
This work takes advantage of NMR spectroscopy and DFT calculations to improve the precision of diffraction derived data. The local approach aims to overcome limitations of the structurally averaging character of powder diffraction and to create a comprehensive understanding of the materials under investigation. We mainly focus on the relation between quadrupolar coupling strengths and the corresponding bond lengths. We further discuss other NMR features such as linewidths and isotropic shifts.We use three independent approaches:
(i) an experimental determination of CQ values
(ii) a theoretical calculation of CQ values based on DFT optimised models. They are based on the crystallographic models without non-stoichiometry effects.
(iii) a DFT-modelling approach towards an element–deuterium distance dependency which is verified against an independent model system, i.e., DFT calculations of the molecular TtD4 compounds.
The synthesis of these approaches allows us to recalculate bond lengths from experimental CQ values.
4.1 Quadrupole interactions
Quadrupole interactions of those 2H nuclei that bind to Tt atoms were measured in terms of CQ in all samples. The results agree well with those values obtained from DFT optimized structures (Table 1). It is important to point out again, that diffraction based structural models suffer from large estimated uncertainties. Therefore, the precision of the actual bond lengths is not very high. This underlines the importance of combining different methods, experiment and modelling, to improve the structural and especially the bonding picture we have of these compounds.In a first step, we can consider some general trends comparing calculated NMR parameters from the DFT optimised structure and experimental ones. The only mismatch between calculated and measured CQ occurs for the second chain site in SrGeD4/3−x and BaGeD5/3−x. DFT clearly underestimates the quadrupolar coupling strength, which indicates that the bond length is even shorter than expected from DFT calculations. According to DFT, the uncertainty must be assigned to the single-chain polyanions as depicted in Fig. 5. From the sole experimental (NMR) point of view, it is not possible to differentiate between these two main chain sites.
 | ||
| Fig. 5 Single-chain (left) and double-chain polyanions (right) of AeTtD5/3−x. There are four crystallographically independent deuterium sites (D1 to D4). Due to molecular pseudo-symmetry NMR parameters only deviate for the different polyanions. Large grey spheres are Si or Ge; small white spheres are D. | ||
In a second step, the experimental quadrupole splittings will be assigned to discrete bond length values by DFT modelling. The quadrupole coupling of D is a most sensitive tool to probe electronic properties of a nucleus' closest environment. DFT confirms that for different Tt elements the interatomic distance to D changes systematically. And since these bond lengths or even the binding character are still a subject of controversy as mentioned in the beginning of this paper, we calculated a general distance dependence of CQ for Si (Fig. 3) and Ge (Fig. 4) as bonding partners (Sn as bonding partner is given in the ESI,† Fig. S4). We propose, that the observed trend is independent of the model system. This is underlined by the fact, that the curves determined for a molecular system (TtD4) and for the Zintl phase deuterides behave quantitatively equal.
The DFT-derived curves can be used to calculate values for Tt–D bond lengths from experimental quadrupole splittings (see ESI,† Fig. S2–S4; Tables S1 and S2). The distance dependency can be verified against literature values at first. For a couple of silane derivatives, SiD3X quadrupole coupling constants were determined.45–47 We can use them to calculate bond lengths of 1.49(1) Å (X = D), 1.50(1) Å (X = CH3) and 1.50(1) Å (X = C6H5). Typical Si–D bond lengths in molecular compounds are 1.47–1.49 Å as determined for disilane.48,49 For the Zintl-like deuteride β-KSiD3, which exhibits monomeric SiD3−-moieties, bond lengths (1.537(8) and 1.545(6) Å at 4 K50) and the quadrupole splitting (CQ = 72.0(5) kHz at 200 K51) are known. From our curves we can estimate the bond length as 1.55(1) Å which fits the literature value. For germane52 and methylgermane53 the bond lengths can be estimated as 1.55(2) Å which is a typical value, i.e. 1.517 Å (GeD4 at 4 K54) to 1.541 Å (Ge2H655). For stannane56 the bond length can be estimated as 1.72(2) Å which fit the neutron diffraction derived bond length of 1.706(3) Å.54 From our experimental CQ values we can evaluate the following bond lengths:
| CaSiD4/3−x | 1.56(1) Å |
| SrSiD5/3−x | 1.58(1) Å (single chain polyanion) |
| 1.53(1) Å (double chain polyanion) | |
| BaSiD2−x | 1.59(1) Å |
| SrGeD4/3−x | 1.65(1) Å |
| BaGeD5/3−x | 1.65(1) Å (single chain polyanion) |
| 1.61(1) Å (double chain polyanion) | |
| BaSnD4/3−x | 1.84(1) Å |
Single and double chain polyanions are assigned according to the trend derived from DFT structures (cf.Fig. 5). A comparison to diffraction- and DFT-derived bond lengths is given in Table 2.
| Compound | NPD d(Tt–D)/Å | DFT d(Tt–D)/Å | NMR d(Tt–D)/Å | |
|---|---|---|---|---|
| BaSiD2−x | 1.641(5)22 | 1.57322 | 1.59(1) | |
| SrSiD5/3−x | (D1) | 1.43(4)23,24 | 1.57623,24 | 1.53(1) |
| (D2) | 1.58(4)23,24 | 1.57623,24 | ||
| (D3) | 1.82(4)23,24 | 1.60123,24 | 1.58(1) | |
| (D4) | 1.50(4)23,24 | 1.59923,24 | ||
| CaSiD4/3−x | 1.8421 | 1.5820 | 1.56(1) | |
| SrGeD4/3−x | 1.521(9)22 | 1.66722 | 1.65(1) | |
| BaGeD5/3−x | (D1) | 1.61(5)23,24 | 1.63823,24 | 1.61(1) |
| (D2) | 1.75(5)23,24 | 1.63923,24 | ||
| (D3) | 1.57(5)23,24 | 1.66323,24 | 1.65(1) | |
| (D4) | 1.55(6)23,24 | 1.66223,24 | ||
| BaGeD4/3−x | 1.858(8)22 | 1.86722 | 1.84(1) |
4.2 Linewidths
DFT calculations and NMR offer further insight into the nature of the narrow lines shown in Fig. 2. The linewidth in particular tells us a lot about the void's geometry in which the nuclei are placed. First, even though we are investigating powdered samples, broadening effects due to shift anisotropy in 1H and 2H are known to be small because of the low number of electrons and can therefore be neglected.Second, the samples under investigation not only show comparatively large widths, we also notice rather significant differences when comparing different samples (Fig. 2). Especially under fast spinning MAS (Fig. 6), the remaining widths vary substantially, sometimes being apparently structured like several lines in superposition. This implies that the Ae environment is slightly variable. Being essentially independent of the external magnetic field (data not shown) further indicates the broadening must stem from dipolar or quadrupolar interactions.
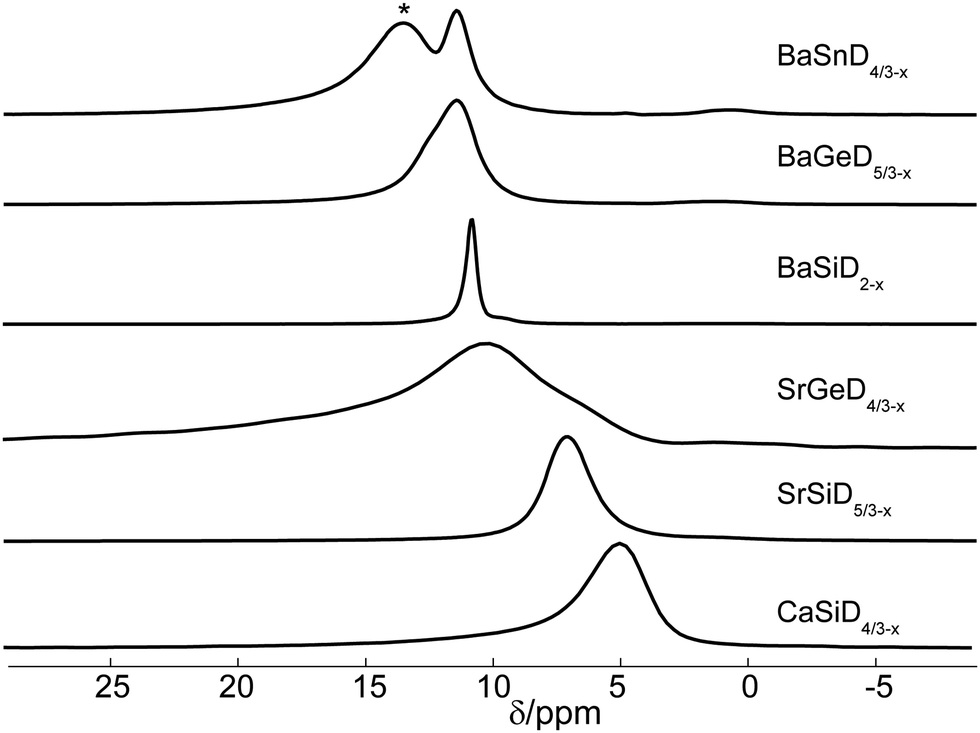 | ||
| Fig. 6 Isotropic region of the 2H MAS spectra. The asterisk indicates signal from a side product. | ||
Magnetic dipole interaction can easily be calculated using the method of moments.25,57 The coupling is chiefly homonuclear, i.e.2H will interact with other 2H in its vicinity, because other NMR active nuclei in our systems have a low abundance. We receive consistently a broadening from magnetic dipole interactions of about 1–1.5 kHz for each sample.
In BaSiD2−x, the linewidth is only 1.5(1) kHz, in agreement with the calculated dipole–dipole coupling. This is confirmed by diffraction and DFT calculations where only a single, highly symmetric crystallographic site is found for the Ae voids.
For the other samples, static NMR linewidths are much broader, up to 6–7 kHz. Dipolar coupling is not strong enough to account for such a broadening. However, the crystallographic models show three (CaSiD4/3 structure type) to six (SrSiD5/3 structure type) distinct crystallographic sites. The corresponding tetrahedra deviate from their equilateral shape leaving a slightly non-cubic environment to the 2H nuclei in their middle with non-vanishing EFGs, yielding small quadrupolar splittings.
These quadrupole couplings cannot be resolved experimentally, because the present broadening from dipole interactions and additional effects smooth out individual features. DFT calculates CQ values between 0.31 and 8.36 kHz. The interactions' symmetry (η) further varies between 0.03 and 0.86. Thus, in all samples except for BaSiD2−x, DFT reveals that the narrow lines are composed of three to six components differing in CQ and η. The combined effects, including magnetic dipole broadening and a small contribution from shift anisotropies, will then result in a NMR line broadening of several kHz. Possible differences in isotropic shift will also contribute to the linewidth. In Table 1 we give for each sample with multiple Ae sites the range of the calculated CQ values and compare them to the linewidths (Λ) obtained by static FID measurements.
4.3 Shifts
Though the total range of 2H shifts is rather small, significant differences in the sample series can be identified. The isotropic lines under MAS of the six samples are given in Fig. 6. For BaSnD4/3−x an additional signal is present around 13 ppm. It seems to be stemming from a side product as its shift together with the fact that intense spinning sidebands indicating a large quadrupolar splitting are present that don't go along with the identification of a deuteride in a tetrahedral void. Maybe the signal is due to decomposition within the MAS rotor.Because of the large frequency distribution for the spinning sidebands of the quadrupolar lineshape, their central line is barely visible in comparison to the narrow line. As can be seen in Table 1, there are some small variations for the isotropic shifts of chain sites across the samples which will not be discussed further. Bonding partner and bond length are expected to have an influence on the chemical shift.
More interestingly, the isotropic shifts of the narrow lines seem to be a function of the surrounding atoms. Going from calcium to strontium and barium, an increase in isotropic shift is observed. A similar observation was found for CaH2, SrH2 and BaH258 where the 1H shift was investigated by line narrowing techniques on static samples. Here, it was concluded that the shift correlates with the electronegativity of the alkaline-earth element. Our investigations of SrD2 and BaD2 yielded isotropic shifts of 5.6 and 10.5 ppm, respectively (data not shown). Though there is only a limited number of samples, there seems to be a systematic effect. The large linewidth of SrGeD4/3−x prevents a more accurate assignment and the shift given in Table 1 represents only that of the maximum intensity. Whereas calculations of quadrupolar interactions are typically quite reliable, calculation of chemical shifts is more demanding especially for small shift ranges. In the future, these calculations might be useful to give a more detailed picture on expected isotropic shifts.
5 Conclusions
We combine NMR and DFT techniques to further improve the understanding of structural and bonding properties of the homologue series of Zintl phase deuterides AeTtD(1+y), Ae = Ca–Ba, Tt = Si–Sn, y ≈ 1/3, 2/3 or 1. NMR is well suited to study Zintl phase deuterides since it allows to distinguish and characterize the individual features of the ionic (D−) and the covalent deuterium-bound polyanionic moieties.We further derived a generalized deuterium-element bond length dependency of the 2H quadrupole interaction. Typically, the determination of hydrogen/deuterium positions can only be done reliably from neutron diffraction data. Complex crystal structures or poorly crystalline samples can complicate the crystallographic approach as was the case for the AeTtD(1+y) family.
NMR measurements allow a comparatively fast and highly efficient lab-based approach to evaluate covalent element–deuterium bond length. Preparation, optimization and measurements can be done within hours. This approach can thus be used, e.g. to assign possible deuterium sites in a crystallographic model from X-ray data that are insensitive to hydrogen/deuterium. It might be even suitable to determine X–D bond lengths (X = Si, Ge, Sn) reliably without crystallographic model. The evaluation of quadrupolar interactions thus expands the toolbox of NMR crystallography. It allowed us to refine Tt–D covalent bond lengths of Zintl phase deuterides which were only described with low precision by diffraction models so far. To verify this general approach, further studies on similar systems would be beneficial.
Furthermore, DFT calculations confirm the presence of small and distributed quadrupole interactions for those 2H nuclei occupying the Ae void sites. Together with experimental evidence from static and MAS NMR, the findings provide a comprehensive local chemical picture of the present materials. The isotropic chemical shifts of the hydridic signals, even though rather broad due to the scattered crystal sites, indicate an increasing shift with larger surrounding atom.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
R. G. acknowledges the funding by the MacDiarmid Institute and Leipzig University. R. G. and M. B. acknowledge the help of M. Lindel. H. K. and H. A. thank the Deutsche Forschungsgemeinschaft (DFG, grant Ko1803/8-1) and the Fonds der Chemischen Industrie (Grant 194371) for financial support.Notes and references
- E. Zintl, Angew. Chem., 1939, 52, 1–6 CrossRef CAS.
- R. Nesper, Angew. Chem., 1991, 103, 805–834 CrossRef CAS.
- O. Janka and S. M. Kauzlarich, Zintl Compounds, John Wiley & Sons, Ltd, 2011 Search PubMed.
- R. Nesper, Z. Anorg. Allg. Chem., 2014, 640, 2639–2648 CrossRef CAS.
- U. Häussermann, V. F. Kranak and K. Puhakainen, Struct. Bonding, 2010, 139, 143–161 CrossRef.
- F. Gingl, T. Vogt and E. Akiba, J. Alloys Compd., 2000, 306, 127–132 CrossRef CAS.
- T. Björling, D. Noréus and U. Häussermann, J. Am. Chem. Soc., 2006, 128, 817–824 CrossRef.
- M. H. Lee, T. Björling, B. C. Hauback, T. Utsumi, D. Moser, D. Bull, D. Noréus, O. F. Sankey and U. Häussermann, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 195209 CrossRef.
- M. J. Evans, G. P. Holland, F. J. Garcia-Garcia and U. Häussermann, J. Am. Chem. Soc., 2008, 130, 12139–12147 CrossRef CAS.
- V. F. Kranak, M. J. Evans, L. L. Daemen, T. Proffen, M. H. Lee, O. F. Sankey and U. Häussermann, Solid State Sci., 2009, 11, 1847–1853 CrossRef CAS.
- M. J. Evans, V. F. Kranak, F. J. Garcia-Garcia, G. P. Holland, L. L. Daemen, T. Proffen, M. H. Lee, O. F. Sankey and U. Häussermann, Inorg. Chem., 2009, 48, 5602–5604 CrossRef CAS.
- M. H. Lee, M. J. Evans, L. L. Daemen, O. F. Sankey and U. Häussermann, Inorg. Chem., 2008, 47, 1496–1501 CrossRef CAS.
- M. J. Evans, M. H. Lee, G. P. Holland, L. L. Daemen, O. F. Sankey and U. Häussermann, J. Solid State Chem., 2009, 182, 2068–2073 CrossRef CAS.
- H. Fahlquist, D. Noréus, S. Callear, W. I. F. David and B. C. Hauback, J. Am. Chem. Soc., 2011, 133, 14574–14577 CrossRef CAS.
- H. Fahlquist, D. Noréus and M. H. Sørby, Inorg. Chem., 2013, 52, 4771–4773 CrossRef CAS PubMed.
- H. Fahlquist and D. Noréus, Inorg. Chem., 2013, 52, 7125–7129 CrossRef CAS.
- J. Ångström, R. Johansson, T. Sarkar, M. H. Sørby, C. Zlotea, M. S. Andersson, P. Nordblad, R. H. Scheicher, U. Häussermann and M. Sahlberg, Inorg. Chem., 2016, 55, 345–352 CrossRef.
- R. Nedumkandathil, V. F. Kranak, R. Johansson, J. Ångström, O. Balmes, M. S. Andersson, P. Nordblad, R. H. Scheicher, M. Sahlberg and U. Häussermann, J. Solid State Chem., 2016, 239, 184–191 CrossRef CAS.
- H. Auer, R. Nedumkandathil, U. Häussermann and H. Kohlmann, Z. Anorg. Allg. Chem., 2019, 645, 175–181 CrossRef CAS.
- N. Ohba, M. Aoki, T. Noritake, K. Miwa and S.-I. Towata, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 075104 CrossRef.
- H. Wu, W. Zhou, T. J. Udovic, J. J. Rush and T. Yildirim, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 224101 CrossRef.
- H. Auer, R. Guehne, M. Bertmer, S. Weber, P. Wenderoth, T. C. Hansen, J. Haase and H. Kohlmann, Inorg. Chem., 2017, 56, 1061–1071 CrossRef CAS.
- H. Auer, R. Schlegel, O. Oeckler and H. Kohlmann, Angew. Chem., Int. Ed., 2017, 56, 12344–12347 CrossRef CAS.
- H. Auer, R. Schlegel, O. Oeckler and H. Kohlmann, Angew. Chem., 2017, 129, 12515–12518 CrossRef.
- A. Abragam, The Principles of Nuclear Magnetism, Oxford University Press, 1983 Search PubMed.
- M. Aoki, N. Ohba, T. Noritake and S. Towata, Appl. Phys. Lett., 2004, 85, 387–388 CrossRef CAS.
- M. Armbruster, M. Wörle, F. Krumeich and R. Nesper, Z. Anorg. Allg. Chem., 2009, 635, 1758–1766 CrossRef CAS.
- E. L. Hahn, Phys. Rev., 1950, 80, 580 CrossRef.
- J. Powles and P. Mansfield, Phys. Lett., 1962, 2, 58–59 CrossRef CAS.
- D. Massiot, F. Fayon, M. Capron, I. King, S. L. Calvé, B. Alonso, J.-O. Durand, B. Bujoli, Z. Gan and G. Hoatson, Magn. Reson. Chem., 2002, 40, 70–76 CrossRef CAS.
- X. Gonze, J.-M. Beuken, R. Caracas, F. Detraux, M. Fuchs, G.-M. Rignanese, L. Sindic, M. Verstraete, G. Zerah, F. Jollet, M. Torrent, A. Roy, M. Mikami, P. Ghosez, J.-Y. Raty and D. Allan, Comput. Mater. Sci., 2002, 25, 478–492 CrossRef.
- X. Gonze, Z. Kristallogr., 2005, 220, 558–562 CAS.
- X. Gonze, B. Amadon, P.-M. Anglade, J.-M. Beuken, F. Bottin, P. Boulanger, F. Bruneval, D. Caliste, R. Caracas, M. Côté, T. Deutsch, L. Genovese, P. Ghosez, M. Giantomassi, S. Goedecker, D. Hamann, P. Hermet, F. Jollet, G. Jomard, S. Leroux, M. Mancini, S. Mazevet, M. Oliveira, G. Onida, Y. Pouillon, T. Rangel, G.-M. Rignanese, D. Sangalli, R. Shaltaf, M. Torrent, M. Verstraete, G. Zerah and J. Zwanziger, Comput. Phys. Commun., 2009, 180, 2582–2615 CrossRef CAS.
- X. Gonze, F. Jollet, F. A. Araujo, D. Adams, B. Amadon, T. Applencourt, C. Audouze, J.-M. Beuken, J. Bieder, A. Bokhanchuk, E. Bousquet, F. Bruneval, D. Caliste, M. Côté, F. Dahm, F. D. Pieve, M. Delaveau, M. D. Gennaro, B. Dorado, C. Espejo, G. Geneste, L. Genovese, A. Gerossier, M. Giantomassi, Y. Gillet, D. Hamann, L. He, G. Jomard, J. L. Janssen, S. L. Roux, A. Levitt, A. Lherbier, F. Liu, I. Lukačević, A. Martin, C. Martins, M. Oliveira, S. Poncé, Y. Pouillon, T. Rangel, G.-M. Rignanese, A. Romero, B. Rousseau, O. Rubel, A. Shukri, M. Stankovski, M. Torrent, M. V. Setten, B. V. Troeye, M. Verstraete, D. Waroquiers, J. Wiktor, B. Xu, A. Zhou and J. Zwanziger, Comput. Phys. Commun., 2016, 205, 106–131 CrossRef CAS.
- http://www.abinit.org, Abinit v. 8.2.2, GNU General Public License.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- M. A. Marques, M. J. Oliveira and T. Burnus, Comput. Phys. Commun., 2012, 183, 2272–2281 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- M. Torrent, F. Jollet, F. Bottin, G. Zérah and X. Gonze, Comput. Mater. Sci., 2008, 42, 337–351 CrossRef CAS.
- https://www.abinit.org/downloads/PAW2, JTH PAW atomic datasets, version 1.0.
- F. Jollet, M. Torrent and N. Holzwarth, Comput. Phys. Commun., 2014, 185, 1246–1254 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- D. M. Bishop and L. M. Cheung, Phys. Rev. A: At., Mol., Opt. Phys., 1979, 20, 381–384 CrossRef CAS.
- N. Stone, At. Data Nucl. Data Tables, 2005, 90, 75–176 CrossRef CAS.
- U. Lähteenmäki, L. Niemelä and P. Pyykkö, Phys. Lett. A, 1967, 25, 460–461 CrossRef.
- R. Ader and A. Loewenstein, Mol. Phys., 1974, 27, 1113–1116 CrossRef CAS.
- B.-M. Fung and I. Y. Wei, J. Am. Chem. Soc., 1970, 92, 1497–1501 CrossRef CAS.
- L. O. Brockway and J. Y. Beach, J. Am. Chem. Soc., 1938, 60, 1836–1846 CrossRef CAS.
- B. Beagley, A. Conrad, J. Freeman, J. Monaghan, B. Norton and G. Holywell, J. Mol. Struct., 1972, 11, 371–380 CrossRef CAS.
- W. S. Tang, J.-N. Chotard, P. Raybaud and R. Janot, J. Phys. Chem. C, 2014, 118, 3409–3419 CrossRef CAS.
- R. Nedumkandathil, A. Jaworski, A. Fischer, C. Österberg, Y.-C. Lin, M. Karlsson, J. Grins, A. J. Pell, M. Edén and U. Häussermann, J. Phys. Chem. C, 2017, 121, 5241–5252 CrossRef CAS.
- V. Hovi, U. Lähteenmäki and R. Tuulensuu, Phys. Lett. A, 1969, 29, 520–521 CrossRef CAS.
- R. Ader and A. Loewenstein, J. Am. Chem. Soc., 1974, 96, 5336–5340 CrossRef CAS.
- I. J. Maley, D. H. Brown, R. M. Ibberson and C. R. Pulham, Acta Crystallogr., Sect. B: Struct. Sci., 2008, 64, 312–317 CrossRef CAS.
- B. Beagley and J. J. Monaghan, Trans. Faraday Soc., 1970, 66, 2745 RSC.
- L. Niemela and J. Mäkelä, Phys. Lett. A, 1973, 43, 343–344 CrossRef CAS.
- J. V. Vleck, Phys. Rev., 1948, 116–183 Search PubMed.
- A. T. Nicol and R. W. Vaughan, J. Chem. Phys., 1978, 69, 5211–5213 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cp00292h |
| This journal is © the Owner Societies 2019 |