
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Detailed kinetic model for hexyl sulfide pyrolysis and its desulfurization by supercritical water†
Caleb A.
Class‡
 ,
AnGayle K.
Vasiliou§
,
Yuko
Kida¶
,
Michael T.
Timko||
and
William H.
Green
*
,
AnGayle K.
Vasiliou§
,
Yuko
Kida¶
,
Michael T.
Timko||
and
William H.
Green
*
Department of Chemical Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139, USA. E-mail: whgreen@mit.edu
First published on 26th April 2019
Abstract
A detailed reaction network is proposed for the pyrolysis and desulfurization of hexyl sulfide in the presence or absence of both supercritical water (SCW) and hexadecane, but without any added H2 or catalyst, for T = 400–450 °C. The new kinetic model is developed using the Reaction Mechanism Generator (RMG) software where most of the rate coefficients are derived from quantum chemical calculations. We previously reported that pentane, carbon monoxide and carbon dioxide are major products of hexyl sulfide desulfurization in SCW, but not in the anhydrous pyrolysis of hexyl sulfide. The observation of CO and CO2 in the reaction products indicates that water effectively acts as a hydrogen source; presumably this assists in sulfur reduction to H2S. Kinetic parameters for several of the important reactions are calculated using transition state theory and quantum chemical calculations at the CBS-QB3 level of theory and then further refined using CCSD(T)-F12//cc-pVTZ-F12 single point energies. Predictions from the new kinetic model agree with factor-of-2 accuracy with new and previously published experimental data for hexyl sulfide conversion and for yields of most major products, either neat or in a hexadecane solvent, both in the presence and absence of SCW. Flux analysis was then used to identify the most important reaction steps, and sensitivity analysis was used to propose reactions that should be studied further in the future to decrease the model's uncertainty. This study establishes the molecular role of water as diluent, hydrogen bond donor, and reductant in the decomposition of hexyl sulfide. Future work to add molecular weight growth pathways to the model would lead to a more complete mechanism, resulting in improved predictions of product yields.
1 Introduction
The ability to generate predictive kinetic models is valuable for many systems, ranging from process and product design1 to engines2,3 and large-scale atmospheric models.4 The primary goal of chemical kinetic models is to make predictions of the dynamic composition of a particular mixture held under specified reaction conditions. The model prediction can then be compared with experimental data for validation. In many systems of technological importance, the chemistry is complex, involving hundreds of reactive intermediates. For this reason, systematic construction of chemical kinetic models by computer algorithms and databases is becoming increasingly attractive. Several research groups have developed software that automates model generation.2,5–12 Our own efforts in this direction have resulted in the development of the Reaction Mechanism Generator (RMG), an open source software package for automatic mechanism generation.13 RMG offers several potentially useful features, including the ability to predict sulfur chemistry and reaction rates.14Automated mechanism generation might be particularly useful for modeling petroleum chemistry. Petroleum consists of many thousands of structures, meaning that manual generation of reaction mechanisms is prohibitively time consuming. Sulfur removal is an especially good test case, as sulfur is present as over 1000 different molecular structures, ranging from aliphatic sulfides and disulfides to aromatic thiophenes and larger polycyclic benzothiophenes15–18 and at concentrations from 0.1 wt% to 10 wt%.19 Sulfur content in crude oil negatively impacts the efficiency and cost of refinery processes, reducing the value of sour crude oils; moreover, very low sulfur levels are required in most fuels to reduce engine emissions and prevent poisoning of catalytic converters. However, the sulfur content of remaining crude oil reserves is trending upward, pushing oil companies to seek alternatives to standard hydrodesulfurization (HDS), which requires a heterogeneous catalyst and high pressure H2.20–23 Moreover, the heavy oils that are increasingly being used as petroleum feeds can foul the HDS catalyst, making the conventional approach even more challenging.
Because of these problems, pyrolysis (also called “thermal coking”) is commercially used to crack and partially desulfurize some heavy, sulfur-rich streams. The drawback of thermal coking is that much of the carbon is converted into a low-value solid (“coke”). Supercritical water (SCW) treatment of heavy sulfur-rich crude has the potential to reduce formation of low-value solid coke, improving carbon efficiency, and does not require catalyst or external hydrogen source. Previous research suggests that the desulfurization and cracking of heavy oils in SCW has the potential to be a sustainable, commercial-scale process.24–30 However, large-scale commercialization of processes using SCW has been impeded in part by a lack of mechanistic details and kinetic information of the upgrading and sulfur removal chemistry. As a step towards filling that gap, we undertook a study to elucidate the mechanisms and rates of hexyl sulfide decomposition with or without water present.
Patwardhan et al. measured the decomposition of hexyl sulfide diluted in hexadecane in supercritical water from 400 to 450 °C in a continuously stirred tank reactor (CSTR), and based on their measurements inferred that free radical chemistry must be important in this system.31 However, that paper did not present a detailed kinetic model or any quantum chemical calculations to support their conclusions. Moreover, although the presence of water can conceivably alter reaction rates due to solvation effects, understanding all of the observed effects of water are difficult if radical reactions are the only important pathways. More recently, batch experiments and quantum chemical calculations by Kida et al. demonstrated that beside being a solvent, supercritical water may directly participate in reacting with intermediates in sulfide decomposition.32 Kida et al. proposed a chain reaction for hexyl sulfide decomposition in the presence of water that included both radical and non-radical reactions. While the evidence for this chain reaction sequence is compelling, Kida et al. considered only a small fraction of the many reactions that must occur during hexyl sulfide decomposition. To date no comprehensive reaction mechanism describes hexyl sulfide decomposition, either in the presence or absence of supercritical water. Given the availability of high quality kinetic and product yield data, we choose hexyl sulfide decomposition as our model system to demonstrate the use of RMG to describe sulfur chemistry in supercritical water.
The accuracy of a desulfurization model requires accurate thermo-chemical and detailed rate information for sulfur compounds. Vandeputte et al.33–37 developed a detailed database for the thermochemistry and kinetics of sulfur compounds. Class et al. later extended the database to include reactions and thermochemistry estimations for compounds including both sulfur and oxygen, particularly for use in high-temperature models involving sulfur compounds and water.38 The sulfur database was validated in the Genesys kinetic model builder by modeling the high temperature thermal decomposition of diethyl sulfide and ethyl methyl sulfide, and it succeeded to predict all of the major compounds of these experiments with quantitative accuracy.39 This database has since been used successfully with RMG to model the decomposition of di-tert-butyl disulfide at 380 °C.14 These previous works establish that RMG should be capable of handling sulfur chemistry, and here we sought to apply RMG to hexyl sulfide decomposition as a logical extension of previous work.
The purpose of this paper is first to present the development and implementation of new sulfur thermochemistry and kinetic calculations into the RMG software and databases. Secondly, mechanisms are generated for the SCW desulfurization of neat hexyl sulfide or hexyl sulfide dissolved in hexadecane, as well as the pyrolysis of neat hexyl sulfide without water. Predictions from these models are compared and validated by comparison with recently published experimental data,31,32 as well as new data measured at different water loadings varying from 0 to 60 wt%. Sensitivity analysis is conducted to identify important reactions, and potential strategies for further model improvement are proposed. The results presented here extend the use of RMG to reaction mixtures containing a supercritical solvent and for larger molecules than have previously been considered for similar chemistries. Accordingly, we anticipate that this work will benefit future work to design new petrochemical processes involving supercritical water desulfurization.
2 Methods
2.1 Batch reactor experiment
Kinetic data and yields were obtained in a batch reactor, which has been described in detail elsewhere.32 In brief, a 316-stainless steel batch reactor constructed of SITEC components with an internal volume of 24 mL was used for all experiments. Heating was provided to the reactor by a fluidized sand bath. For the base experiment, hexyl sulfide and water were added to the reactor in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 ratio (wt/wt). To investigate the concentration effect of water on the production of pentane, the amount of hexyl sulfide loaded into the reactor was fixed, and the amount of water was adjusted from 0 to 60 wt%. A total of 8 water/sulfide ratios were investigated experimentally. The SCW experiments were conducted at 400 °C. The pressure in the reactor depends on the amount of water added; for the base case it was 28 ± 5 MPa, sufficient that the system is expected to be a homogeneous supercritical mixture.40,41 Air was flushed from the reactor by pressurization/depressurization by helium, and 20 bar of He was left in the headspace upon sealing. The same temperature was used for all experiments with different initial water loadings, and as expected, the pressure was observed to increase with increasing water load. Once the reaction was quenched, the gas-phase product and liquid phase products were collected and analyzed.
:3 ratio (wt/wt). To investigate the concentration effect of water on the production of pentane, the amount of hexyl sulfide loaded into the reactor was fixed, and the amount of water was adjusted from 0 to 60 wt%. A total of 8 water/sulfide ratios were investigated experimentally. The SCW experiments were conducted at 400 °C. The pressure in the reactor depends on the amount of water added; for the base case it was 28 ± 5 MPa, sufficient that the system is expected to be a homogeneous supercritical mixture.40,41 Air was flushed from the reactor by pressurization/depressurization by helium, and 20 bar of He was left in the headspace upon sealing. The same temperature was used for all experiments with different initial water loadings, and as expected, the pressure was observed to increase with increasing water load. Once the reaction was quenched, the gas-phase product and liquid phase products were collected and analyzed.
The gas-phase product was analyzed using a gas chromatograph (GC) equipped with a Shimadzu GC-FID for light hydrocarbon products (GC-2014 with RT-Q bond column), a 6890N Agilent GC-TCD for CO2 and H2S quantification (HP-1 column, 30 m × 250 μm × 0.25 μm). A De Jaye 5 Gas analyzer (NDIR) was used to measure the CO/CO2 ratio. After removing the gas phase, the water and oil phase were separated using a centrifuge. The oil phase was analyzed using a GC (7890A Agilent) with a flame ionization detector (FID) and HP-5MS column (30 m × 250 μm × 0.25 μm). Representative water samples were analyzed for sulfur and trace metals content.
Considerable precautions were necessary for these experiments, and researchers replicating them should use caution. In addition to usual hazards of high pressure reactors and temperature, if the reactions are run to high conversion using a high concentration of hexyl sulfide, a significant amount of toxic H2S with an unpleasant odor can be formed. Most of the H2S product will be present in the gas phase but some will be dissolved in the product liquids and might later evolve e.g., during centrifugation. Even small amounts of H2S that exit the GC exhaust may be irritating. Adequate ventilation is essential. For more details on the analytical instruments, reactor, and experimental techniques, see Kida et al.32
2.2 Principles of automated mechanism generation
Reaction Mechanism Generator (RMG-Java, version 4.0.1) employs advanced methods in thermochemistry and kinetic parameter estimation that allow the construction of complex reaction networks.13,42,43 RMG has been used to generate accurate kinetic models, including the pyrolysis of hydrocarbons and oxidation of radicals during autoignition.14,44,45More in-depth descriptions of the RMG software have been provided elsewhere,46 so only a brief summary is presented here. The heart of RMG is a rate-based algorithm that builds chemical kinetic models from an initial set of reactants and initial conditions; temperature, pressure, and species concentration.45 RMG systematically constructs reaction pathways between species present in the mixture, following a set of reaction templates and referencing kinetic and thermodynamic information stored in RMG's database. Likely products from the reaction mixture are added iteratively, and the resulting differential equations are solved for the updated reaction mechanism. In this sequential way, a reaction network for the given initial conditions is formed until some termination criterion (such as reactant conversion) is reached.
The accuracy of the model is determined by the accuracy of the kinetic and thermochemical parameters used in the model; RMG also uses these parameters to decide which chemical reaction pathways should be included in the model at all. High accuracy calculations are already available for many hydrocarbons and small molecules.47 More recently, a detailed database for the thermochemistry and kinetics of sulfur compounds was developed by Vandeputte et al.33–37 The sulfur database was then extended to include reactions and thermochemistry estimations for compounds containing both sulfur and oxygen, particularly for use in high-temperature models involving chemical reactions of sulfur compounds and water.38 Collectively, these data provide reasonably accurate rate estimations (within an order of magnitude) for a variety of reactions that are potentially relevant in our system, including the usual free radical reactions as well as more specific reaction types, such as the addition of water or hydrogen sulfide to a double-bond, ipso-additions to sulfides, and intra-hydrogen abstractions in organosulfur compounds (for a full list of reaction types in the RMG database, see the work by Gao et al.).46 However, the database only covers a very small percentage of all possible reactions that can occur, and many other reactions predicted by RMG will not have accurate rate parameters available. These other parameters are estimated by analogy, similar to Benson's method,48 but more typically using quantum chemical calculations rather than experimental data as the basis of the analogy, since there are relatively few experimental data on relevant reactions. Additional quantum chemical calculations are then conducted to improve the accuracy of rate or thermochemical parameters for reactions that are found to be important via sensitivity analysis, or for new reaction types that may be relevant but are not yet included in the RMG database.
2.3 Quantum chemical calculations
Thermochemical and kinetic data were calculated at the CBS-QB3 level of theory using the Gaussian 03 quantum chemistry package.49 Particularly important parameters were further refined using CCSD(T)-F12b/cc-pVTZ-F12 energies, which have been shown to be accurate to within about 2 kJ mol−1 of the basis set limit.38,50–52 All stable compounds were calculated in their singlet state, and radical compounds were calculated in their doublet states. Partition functions were calculated using the CanTherm software package,53 using the recommended scaling factor of 0.99 for the frequency analysis.54 One-dimensional hindered rotations were also included in the analysis, using scans at the B3LYP/6-311G(2d,p) level for each rotatable bond. Hindered rotor scans were stepped in 10 degree increments, and all other coordinates were allowed to re-optimize at each step. The effective moment of inertia I(2,3) for each hindered rotor was calculated at the equilibrated geometry.55 The rotor coordinates were projected out, and the remaining vibrational modes were approximated as harmonic oscillators.Thermochemistry data for most species were estimated using Benson group additivity and previously calculated parameters.35,38,56 Additional thermochemical parameters were calculated from vibrational frequencies using CanTherm. Enthalpy and entropy of formation were calculated at 298 K, and heat capacities were calculated at 300, 400, 500, 600, 800, 1000, 1500, 2000, and 2400 K. Bond additivity corrections were applied to obtain the enthalpy values. At the present time, no reliable correction is available for the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S double-bond, due to lack of experimental data.57 This introduces additional uncertainty of up to 4 kJ mol−1 in the relevant thermochemical estimations, and it suggests an avenue for future work in this area.
S double-bond, due to lack of experimental data.57 This introduces additional uncertainty of up to 4 kJ mol−1 in the relevant thermochemical estimations, and it suggests an avenue for future work in this area.
Tunneling corrections were applied to the rate constant calculations using the asymmetric Eckart method, which has been shown to provide accurate corrections for this type of chemistry.58,59 Rate constants were calculated in CanTherm using conventional transition state theory (TST) at 59 temperatures between 300 and 2000 K. These were fitted to the modified Arrhenius expression,
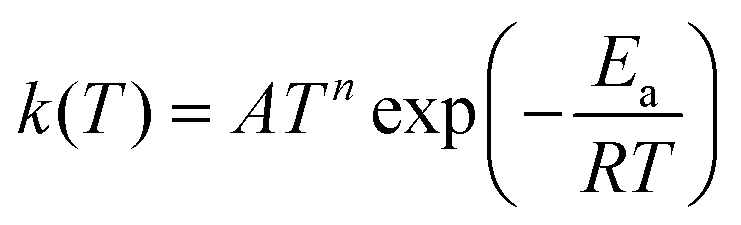
The modified Arrhenius expression generally fits the calculated rate constants with less than 20% error, although greater disagreement is sometimes observed at lower temperatures (300 to 400 K) when contributions from tunneling are significant. Additional caution should be taken if using these kinetics data at lower temperatures.
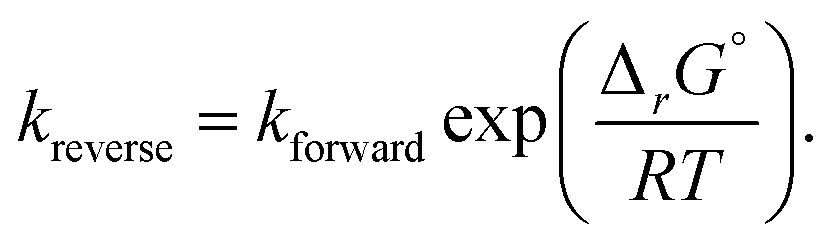
For reactions involving water as a reactant, we assumed its fugacity coefficient was φ = 0.5, based on a compressibility factor of 2 calculated at the supercritical conditions of Kida et al.32 This should be very close (within 10%) to the actual value in the main batch reactor simulation involving supercritical water, but it introduces additional uncertainty (potentially up to 50%) at some of the lower water concentrations used in the variable water experiment, where the mixture did not reach the critical point. However, the exact value of this coefficient had little impact on our product predictions for the low-water concentration experiments.
All other species were assumed to be ideal, i.e. to have a fugacity coefficient φ = 1. Thus, the reaction rate estimated by RMG for a bimolecular reaction between H2O and another species, R, will be
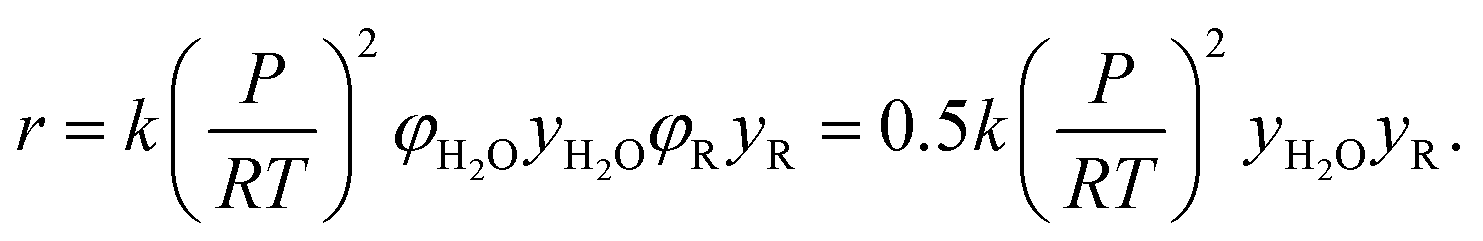
This ideal gas assumption will be accurate for very dilute species, which make up the vast majority of components in our predicted reaction mechanisms. However, more concentrated species (such as hexyl sulfide) may have a fugacity coefficient around 0.5 at these conditions, based on calculations conducted for a dodecane–water mixture. This introduces factor of 2 uncertainty in the relevant reaction rate estimates, although this nonideal effect will cancel out for most reactions of interest. In addition, we see that the fugacity coefficient of water shows slight temperature and pressure dependence at our conditions: the fugacity coefficient will increase with increasing temperature and decrease with increasing partial pressure. As these errors fall within our overall rate calculation uncertainty, we have not decided to apply detailed fugacity corrections in this work, but future modeling with accurate fugacity data would likely improve the overall model accuracy.
Additional rate constants were calculated using the same TST method based on the quantum chemical calculations by Deng et al. for the hydrolysis of carbonyl sulfide.60 These were then added to the RMG database.
2.4 Mechanism generation & reactor models
RMG was used to build a kinetic model valid at the reaction conditions of Patwardhan et al., Kida et al., and the new experiments reported here using varying amounts of water. The temperature, pressure, and reactant concentrations for each experiment were the main inputs for each simulation. Due to limited computational resources, the need to model reactants with greater than 13 heavy atoms (hexadecane and hexyl sulfide), and the fact that the number of minor reactions predicted by RMG increases dramatically with increasing conversion, the goal conversion of hexyl sulfide in the RMG model was set at 25% (likely leading to the omission of some products only formed at high conversion). In addition, the maximum number of carbon atoms allowed per species was set at 24, causing the omission of very large products that would overly tax the available computational resources. An edge tolerance of 0.01 was used.In all cases, the reaction network was implemented in CHEMKIN-PRO.54 Batch reactor experiments were simulated using the closed homogenous reactor model; this assumes that the reaction takes place in a single phase with perfect mixing, and that the reactor volume remains constant and equally occupied by the reaction mixture (i.e., there are no dead zones).61
The reactor was simulated as closely as possible to the experimental conditions, with a five-minute linear heat-up from ambient conditions to the experimental temperature of 400 °C. Initial reactant concentrations were calculated assuming a homogeneous mixture of hexyl sulfide, water, and inert helium. The CSTR experiments of Patwardhan et al. were modeled using the perfectly stirred reactor (PSR) model and taking as initial conditions the experimental feed compositions and reaction conditions. This model assumes a homogenous mixture throughout the reactor (including the outlet), as well as the other general assumptions described previously for the closed homogenous reactor model.
3 Results
The study by Kida et al. identified a possible mechanism for alkyl sulfide decomposition in the presence of SCW32 to explain the observation of carbon monoxide production. Since water was the only source of O atoms in the reaction mixture, the observation of a carbon monoxide product suggested that water had reacted with the sulfide or an intermediate in its decomposition. Quantum chemical calculations (presented here and in the ESI,† Section S1) identified a plausible mechanism to explain the observed products of hexane and hydrogen sulfide (which were observed in both the presence and absence of SCW) and pentane and carbon monoxide (which were only observed in the presence of SCW). More extensive modeling using RMG is useful in further elucidating the overall reaction mechanism and the specific role of water, by considering additional reactions involving minor products and reactive intermediates. Some additional quantum chemical calculations were conducted to expand the RMG database and explore hypothesized pathways. These are discussed in the next two sections, and additional reaction rate calculations are provided in the ESI.† RMG and CHEMKIN were then used to model the published CSTR and batch reactor data for hexyl sulfide decomposition with and without SCW and explore the reaction pathways involved in the different processes. Finally, we model new batch reactor data to investigate the dependence on water concentration.3.1 Quantum chemical calculations for the water-catalyzed elimination of H2S
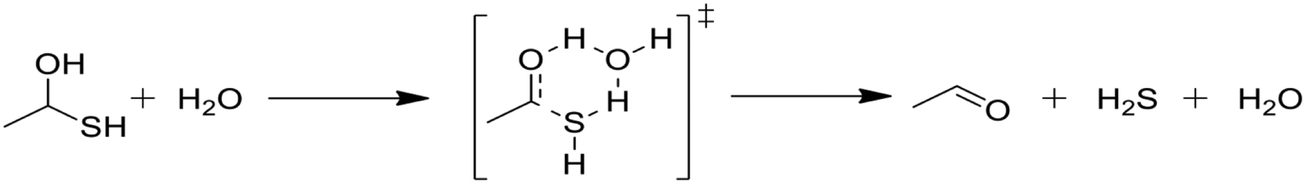
Kida et al. reported rate parameters for the water-catalyzed dehydration of geminal mercaptoalcohols, and these are shown in Table 1.32 Hydrogen bonding of the transition state to an additional water molecule increases the calculated first-order decomposition rate of the mercaptoalcohol by an order of magnitude. Water solvation effectively acts to catalyze this decomposition and subsequently affects product branching.| Rate parameters | ||||
|---|---|---|---|---|
| logA |
n | E a | ||
| (1) |
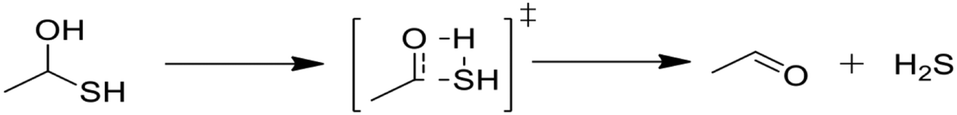
|
12.62 | 0.13 | 185.09 |
| k (673 K) = 0.041 1 s−1 | ||||
| (2) |
|
0.54 | 3.05 | 93.20 |
| k (673 K) × aH2O = 0.74 1 s−1 | ||||
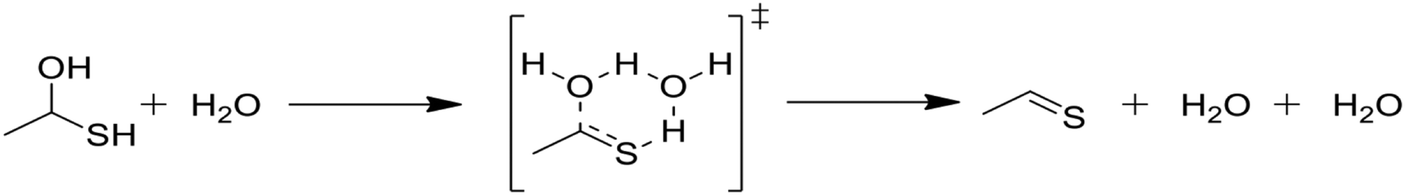
Due to the comparative speed of the water-catalyzed reaction in Table 1, a brief study was conducted to determine kinetic parameters for three additional reactions, which are presented in Table 2. The first reaction in Table 2, reaction (3), is the elimination of water from the mercaptoalcohol compound to form a thioaldehyde. A slightly lower activation energy was calculated for this reaction as compared with the H2S elimination reaction in Table 1, but under the conditions of this study reactions (2) and (3) will occur at similar rates. Thus, we expect a significant amount of pentane and CO production from the water-catalyzed elimination of H2S from the mercaptohexanol. This is a key contribution of water, as the lighter products formed in the presence of water decrease formation of heavy products and aromatic rings, as will be discussed in the next section.
| Rate parameters | ||||
|---|---|---|---|---|
| logA |
n | E a | ||
| (3) |
|
1.08 | 2.59 | 86.02 |
| (4) |
|
−0.79 | 3.44 | 115.06 |
| (5) |
|
0.32 | 2.94 | 119.75 |
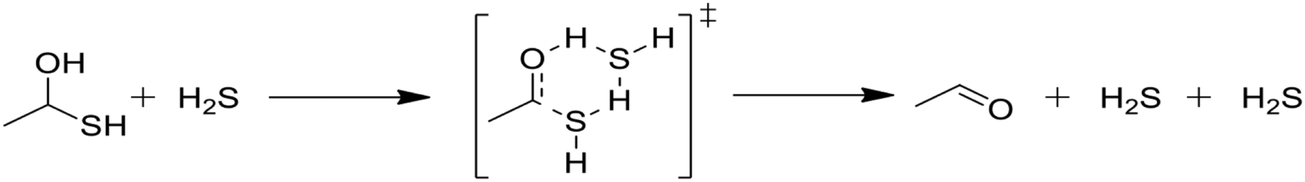
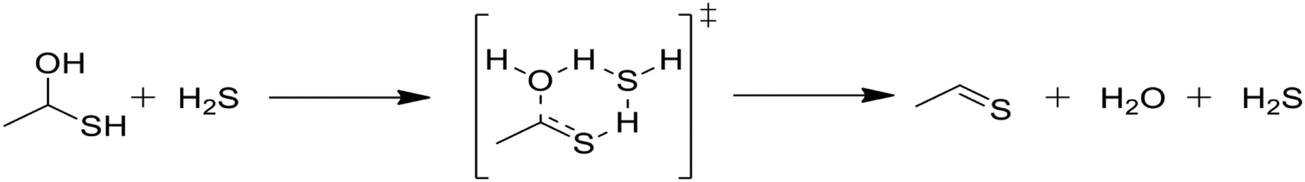
In Table 2, reactions (4) and (5) are analogous to reactions (2) and (3), respectively, but catalyzed by hydrogen sulfide instead of water. In both cases, the transition state is stabilized by the presence of the additional H2S molecule to form a six-member ring. This stabilization effect is about 30 kJ mol−1 less than in the water-catalyzed version of both cases. Due to the higher barrier and since H2S concentrations are less than water concentrations, the H2S-catalyzed reactions should be negligible compared with the water-catalyzed reactions. However, the H2S-catalyzed reaction may be important in situations where large amounts of H2S are present, including especially pyrolysis conditions in the absence of water. The analogous hexyl sulfide reactions for those presented in the two tables have been added to the RMG database in the direction presented, and the termolecular reverse reactions are also calculated by RMG using thermodynamic consistency.
3.2 Quantum chemical calculations for cyclic sulfide formation
Next, we considered formation of cyclic sulfides. A cyclic sulfide formation reaction is a unimolecular radical addition, where a radical attacks the sulfur atom of a thiocarbonyl group to form a cyclic sulfur compound, as shown in Fig. 1. Formation of cyclic sulfides is especially important to understand as it is a likely step in the formation of aromatic sulfides, an important class of highly stable sulfur-bearing compounds. Three cyclization reactions, listed in Table 3, were selected for this work as possible intermediate steps as they led to the generation of experimentally observed products ethyl-tetrahydrothiophene and ethyl-thiophene. We hypothesize that these reactions compete with the SCW addition reaction in the consumption of the aforementioned thioaldehyde intermediate. Accordingly, cyclization is expected to be a dominant pathway in the absence of water.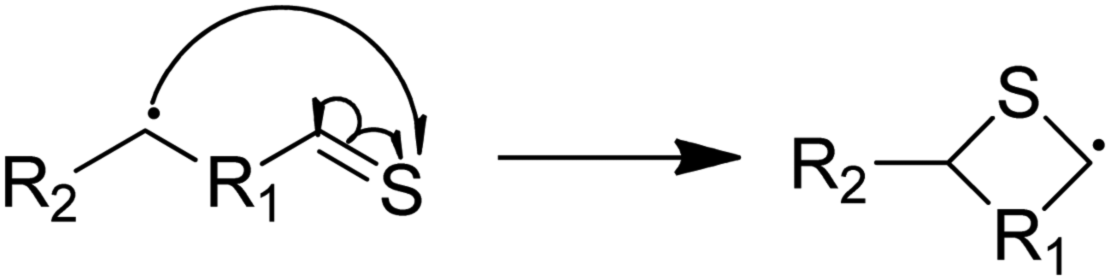 | ||
| Fig. 1 A cyclic sulfide formation reaction. | ||
| Rate parameters | Reverse | ||||||
|---|---|---|---|---|---|---|---|
| logA |
n | E a | logA |
n | E a | ||
| (6) |
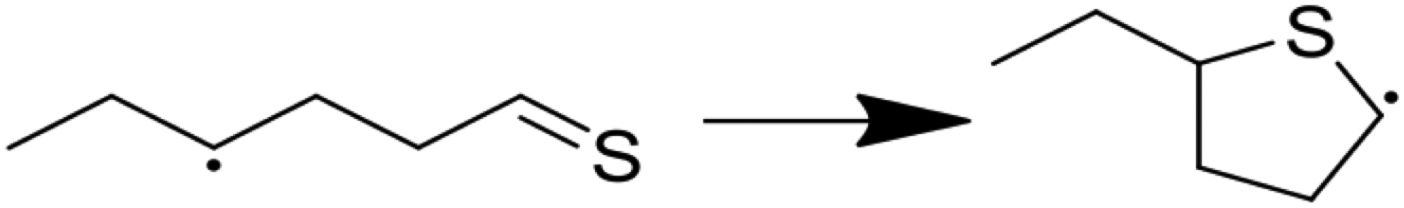
|
6.65 | 1.17 | 1.15 | 13.50 | 0.02 | 107.01 |
| (7) |
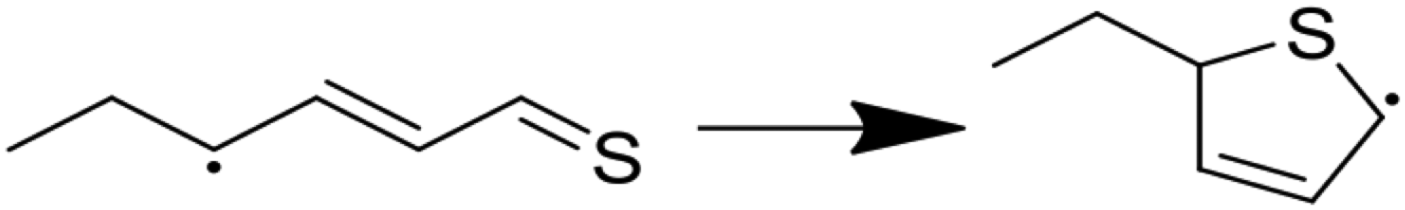
|
11.90 | 0.10 | 44.78 | 14.37 | −0.36 | 81.78 |
| (8) |
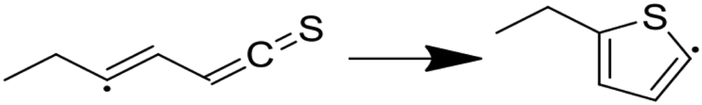
|
11.85 | −0.14 | 45.71 | 11.77 | 0.43 | 162.88 |
Modified Arrhenius parameters of the three proposed cyclization reactions are presented in Table 3, and the transition state geometries are presented in Fig. 2. Reaction (6) has a negligible activation energy in the forward direction, while the activation energies of reactions (7) and (8) agree with one another within the limits of computational precision. All three of these reactions are exothermic in the cyclization direction. Reaction (8) has the greatest barrier in the reverse direction, as it requires the breaking of a stable thiophenic ring to form an unstable thioketene. Reaction (6) in particular is very fast, with a nanosecond time constant at the reaction temperatures of interest.
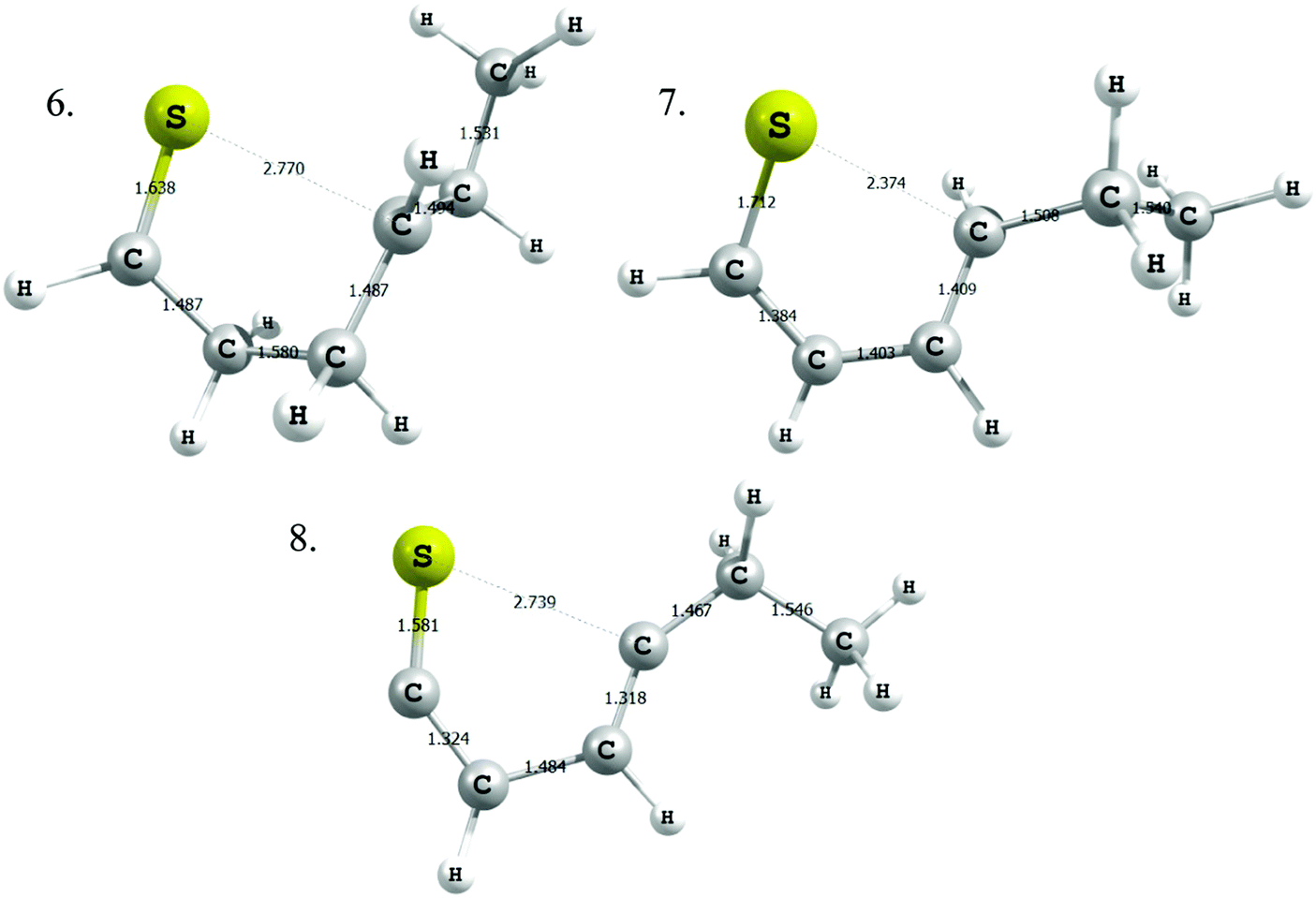 | ||
| Fig. 2 Transition state geometries for the three cyclic sulfide formation reactions. Distances (Ångstroms). | ||
3.3 Reaction path analysis using RMG
Having performed thermodynamic calculations on putative intermediates and kinetic calculations on plausible side reactions, the next step was to use RMG to develop a predictive kinetic model for hexyl sulfide decomposition. Mechanisms were generated by RMG in the presence and absence of water to simulate batch reactor experiments that were previously conducted at 400 °C.32 The SCW mechanism includes 273 species and 5971 reactions. Reaction path analysis of the RMG mechanisms, Fig. 3, confirms that the formation of the major products in the SCW treatment of hexyl sulfide occurs primarily via the reaction pathways proposed by Kida et al.32 The RMG model provides additional insight that the skeletal model proposed by Kida et al. does not,32 and we detail these new insights here.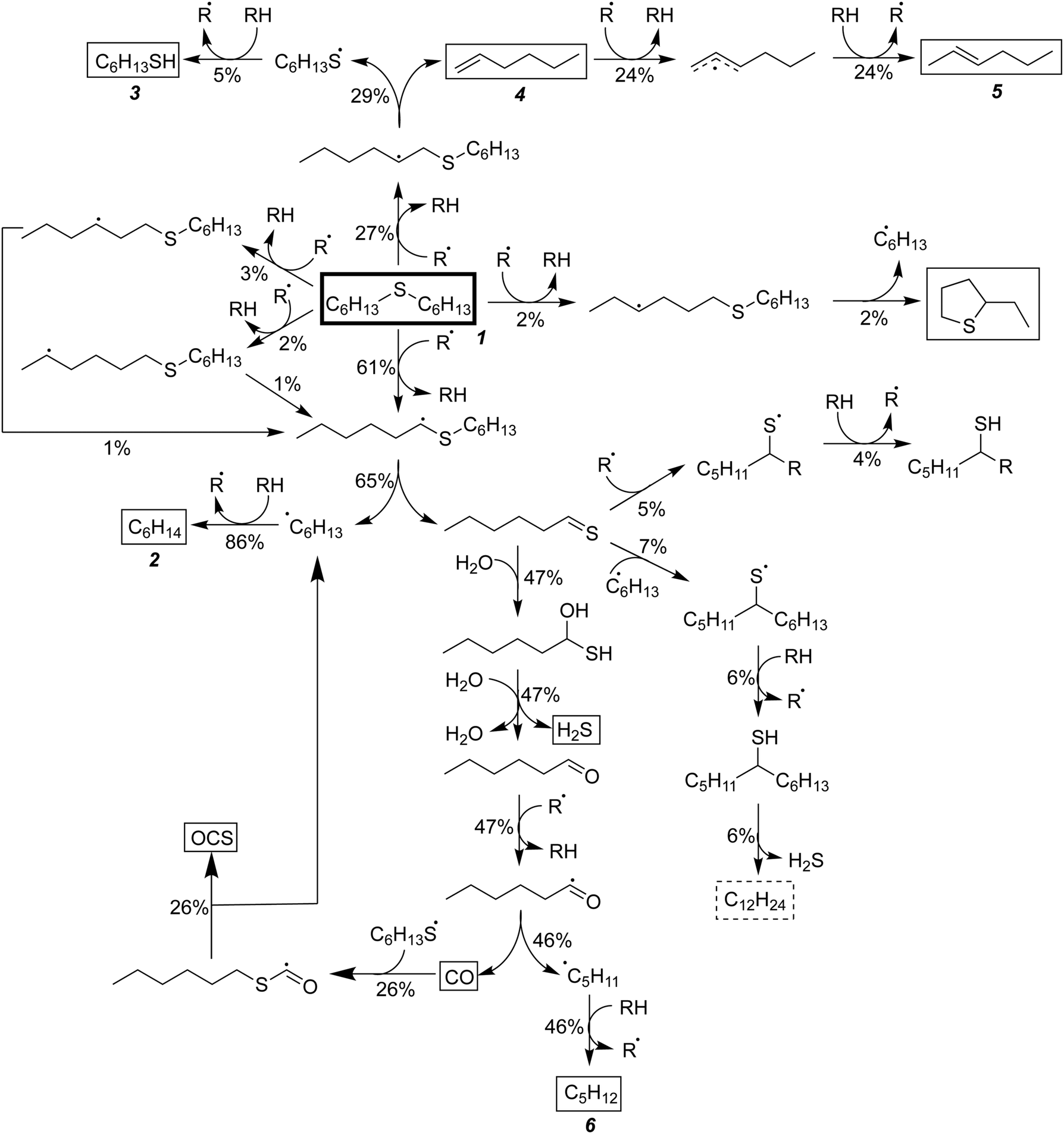 | ||
| Fig. 3 Predicted reaction fluxes for the SCW desulfurization of hexyl sulfide at 400 °C. The reactant and experimentally observed products are encased in boxes, while the unobserved product is encased in a dashed box. Percentages represent total proportion of reacted hexyl sulfide proceeding through a given pathway over 30 minutes. For clarity, lower flux pathways are not shown. | ||
As expected, the main initiation step is predicted to be the breaking of the relatively weak C–S bond (species 1 in Fig. 3). The full RMG model predicts that the highest flux channel for the cracking of hexyl sulfide starts with hydrogen abstract, which accounts for 61% of the total decomposition. H-abstraction occurs at the α-carbon (carbon adjacent to the sulfur atom), as the resulting α-radical is stabilized by its proximity to the sulfur atom. Production of small amounts of γ-, δ-, and ε-radicals are predicted, and these all are expected either to convert to the thermodynamically preferred α-radical via intramolecular hydrogen migration reactions or (mainly) react back to form the initial reactant. The alpha-radical then undergoes beta scission reaction (65% flux, relative to total hexyl sulfide decomposition) to form hexyl radical—which abstracts a hydrogen atom to become hexane (2)—and hexanethial, the latter of which will be discussed in more detail.
The second major reaction pathway (27% flux) involves a hydrogen abstraction from hexyl sulfide to form a β-radical, which quickly undergoes beta scission to form 1-hexene (4) and hexanethiyl radical (29% flux, as a small amount of beta radical is produced through other pathways, such as intramolecular hydrogen migration). Hexanethiyl radical further reacts to form hexanethiol (3). The 1-hexene intermediate is predicted to convert to 2-hexene (5) via hydrogen abstraction reactions.
Hexanethial produced by the beta scission of the alpha radical is predicted to be consumed by two pathways. The main one is the pentane (6) and CO production pathway proposed by Kida et al. (Path 1 in Fig. 3). Most of the CO is predicted to react with hexanethiyl radicals to form carbonyl sulfide (OCS), which would likely react further to form the experimentally detected CO2.60 RMG also predicts that hexanethial undergoes a variety of radical addition reactions, mainly attacking the carbon side of the CS double-bond and leading to the production of heavier byproducts, some of which continue to react. Additional hexanethial consumption pathways are also predicted, but with much smaller fluxes. While none of these byproducts are observed experimentally, these pathways could be early steps in the formation of large aromatic compounds, which may explain the dark color of the product solution observed in some of the experiments.32 These condensation pathways are discussed further in the analysis of the anhydrous decomposition mechanism.
This analysis has demonstrated that the two reactions (H2O addition and H2O-assisted H2S elimination) proposed by Kida et al. are likely steps in the mechanism forming carbon dioxide and pentane from hexyl sulfide, but we have only explored a few of the possible ways for SCW to participate in this reaction system. H2O has been demonstrated through quantum chemical calculations to decrease the activation barrier of a variety of intramolecular hydrogen-transfer reactions by about half (Section S2, ESI†).60,62–67 These barrier decreases could increase their respective reaction rates by multiple orders of magnitude at supercritical conditions, significantly changing the final product distributions. Thus, a thorough study of other H2O-catalyzed reactions involving hexyl sulfide decomposition intermediates may substantially improve our understanding of this chemistry.
In the absence of water, RMG constructed a mechanism for hexyl sulfide pyrolysis consisting of 277 species and 6155 reactions. For the most part, the major reaction pathways for pyrolysis in the absence of water are the same as in the SCW case, and the total flux analysis for this mechanism is presented in Fig. 4. The hydrolysis of the thioaldehyde shown in Fig. 3 is not possible in the absence of water, which necessarily increases the flux through the condensation pathways relative to those observed in the SCW mechanism. In addition, a significant amount of the thioaldehyde is predicted to form thiophene after a hydrogen abstraction and cyclization pathway; this is in contrast to the ethyl-thiophene that was observed experimentally but only predicted in trace amounts computationally.
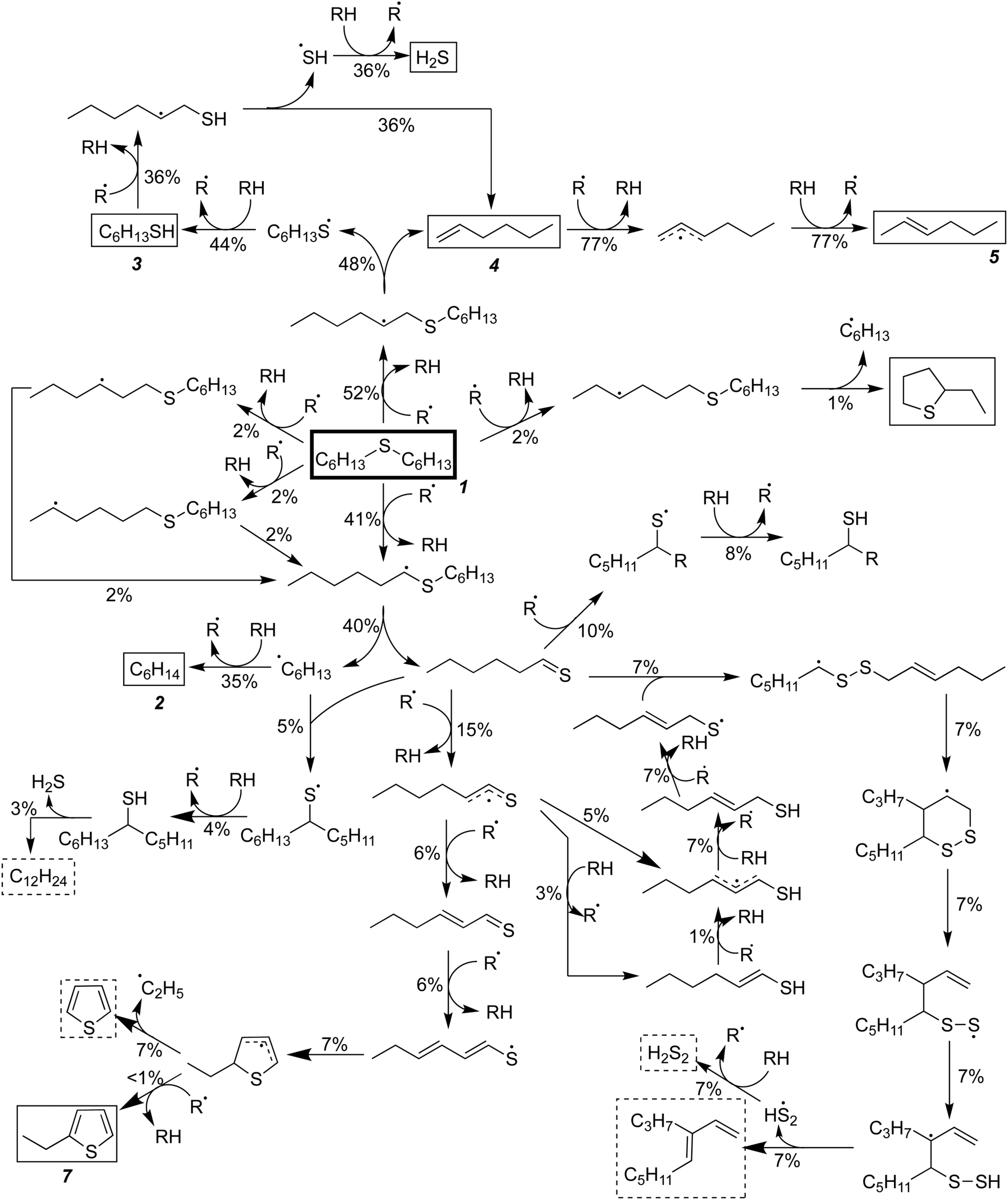 | ||
| Fig. 4 Predicted reaction fluxes for the pyrolysis of hexyl sulfide at 400 °C in the absence of water. The reactant and experimentally observed products are encased in boxes, while unobserved products are encased in dashed boxes. Percentages represent total proportion of reacted hexyl sulfide proceeding through a given pathway over 30 minutes. For clarity, lower flux pathways are not shown. | ||
In both models, the weak S–H single-bond provides an important source of hydrogen in an auto-catalytic manner, as described by Shum and Benson for the pyrolysis of dimethyl sulfide.68 In the presence of SCW, 65% of the predicted hexane is formed through hydrogen abstraction from H2S, and the resulting mercapto radical then regains its hydrogen from hexyl sulfide:
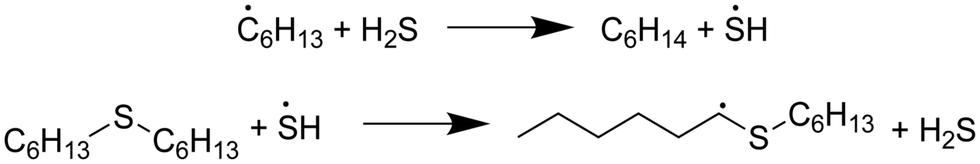
In the absence of SCW, a substantially higher concentration of hexanethiol and larger thiols are predicted—in addition to hydrogen disulfide—and these molecules provide hydrogen for 70% of the hexane production predicted by our model. The thioaldehyde also serves as a hydrogen donor, leading to slight production of 1-hexenethiol. A small amount of this thiol remains as a product, but some of it reacts further to become a thiyl radical, which adds back on to the thioaldehyde in the most prevalent (7% flux) condensation reaction predicted by RMG. This pathway leads to formation of a 12-carbon diene and hydrogen disulfide.
Dienes have been predicted as products in both models although they are not observed experimentally. This may correspond to the amount of coke—non-volatile products with a high aromatic content—that was observed in both experiments. Although coke yield was not quantified, it was clear that more coke was produced in the absence of water than in the presence, due to the darker color of the pyrolysis product and greater formation of carbon (about 30%) that could be not quantified by GC analysis of gas- and liquid-phase products.32 This 30% of unquantifiable carbon agrees reasonably well with the nearly 20% of dienes (27% when thiophene is included) predicted in the RMG mechanism in the absence of SCW. Thus, we propose that the production of dienes and cyclic species from the thioaldehyde is an early step in the production of larger compounds that are insufficiently volatile for detection using GC. Unfortunately, using RMG to track accumulation of heavy species in detailed mechanisms is extremely difficult, as there is no single “coke molecule,” but many different large species that will individually have very small concentrations. Using the current version of RMG with available computational resources, building a comprehensive mechanism including species with more than 30 heavy atoms is not practical. Improving the ability of mechanism generation software to capture large-molecule chemistry is a topic of on-going research.69
3.4 RMG model validation for hexyl sulfide conversion in a CSTR
The sulfur content of the batch experiments (effectively 15 wt%) is much greater than typical crude oils. Experiments with more realistic sulfur concentrations were required for more direct comparison with crude oils. Accordingly, an additional mechanism was generated using RMG to model a reaction mixture consisting of hexyl sulfide and hexadecane, simulating the SCW reaction conditions used by Patwardhan et al.31 The CSTR mechanism was generated for reaction temperatures between 400 and 450 °C, and it contains 140 species and 1957 reactions. The predicted reaction pathways for hexyl sulfide decomposition are largely similar to the hexyl sulfide + SCW case, although the radical pool is different in the hexadecane case. C–S bond scission remains the dominant initiation reaction, but as hexadecane was present in a much greater concentration than hexyl sulfide in the CSTR experiments, radicals produced by the hexadecane solvent exist in a higher concentration than do radicals produced directly from reactions of hexyl sulfide.The model predictions are compared with experimental data for hexyl sulfide conversion in Fig. 5. Overall, RMG predicts sulfide conversion within about 20%, with underprediction at the lowest temperature and a more significant overprediction at the highest. Due to constraints imposed by limited computational resources, the model does not include species only formed at high conversions, and it is likely to be inaccurate at the higher conversions observed in the 450 °C or long residence time CSTR experiments. Fig. 5, therefore, shows that even an incomplete RMG model can predict conversions to within about 20%, which suggests that the model includes all of the most important pathways and that future refinement can realistically bring the model into better quantitative agreement.
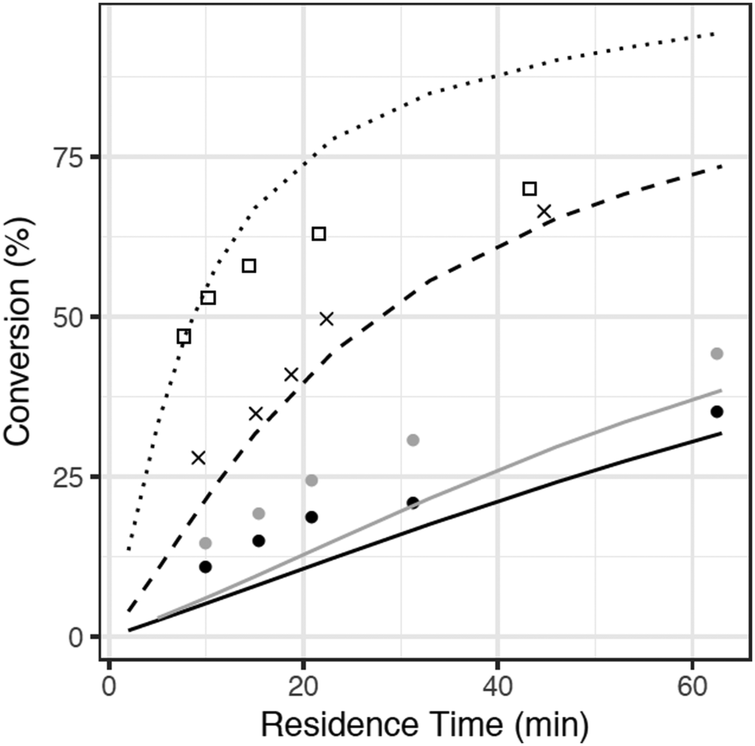 | ||
| Fig. 5 Comparison of RMG model predictions (curves) with data (points) of Patwardhan et al. for hexyl sulfide conversion in hexadecane/SCW mixtures in a CSTR at 235 bar, T = 400–450 °C. | ||
3.5 RMG model validation for product distributions in a batch reactor
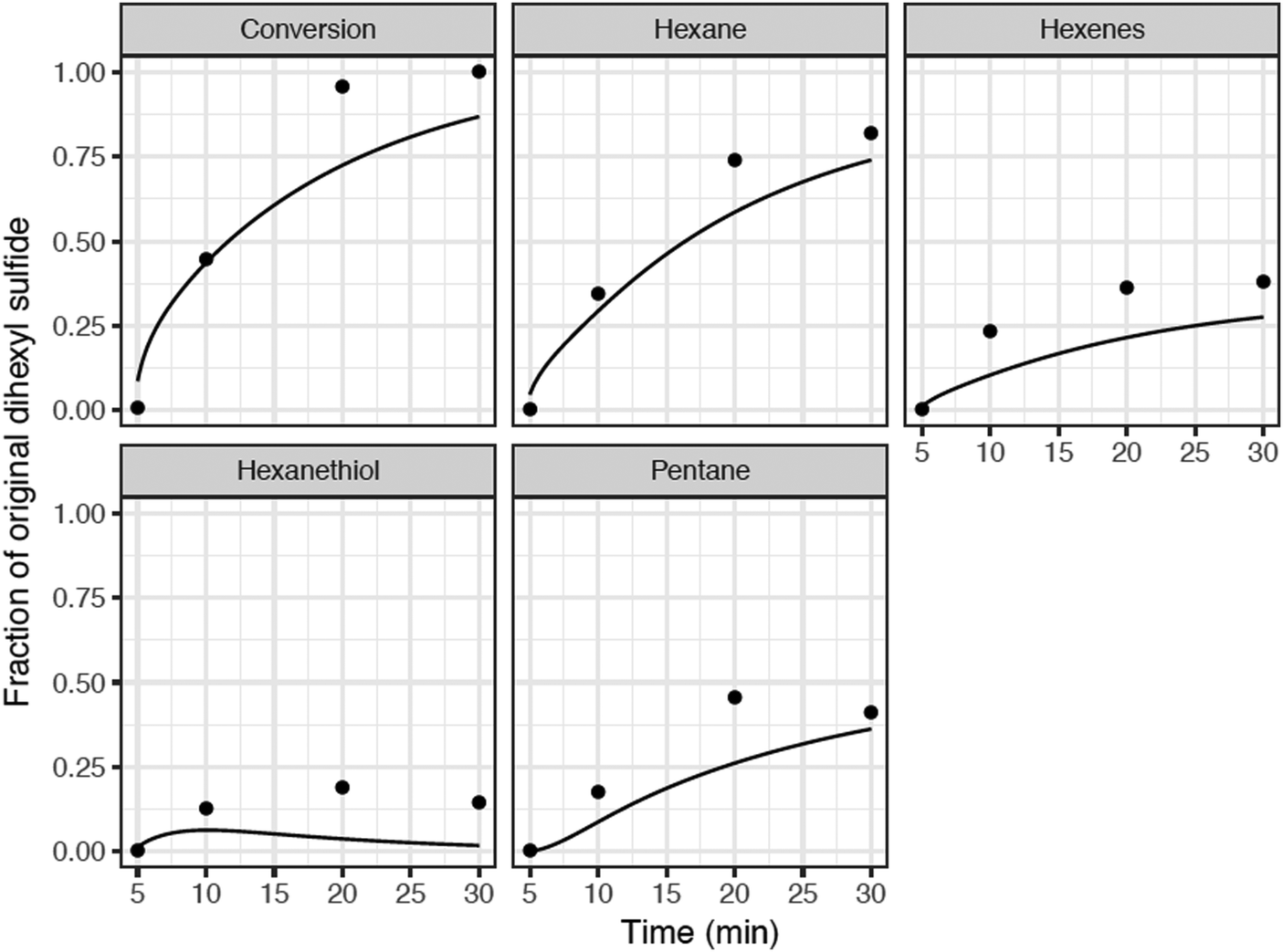 | ||
| Fig. 6 Comparison of detailed kinetic model predictions with batch reactor data for hexyl sulfide in SCW at 400 °C at the base-case composition. Model (curves), experimental data (•) from Kida et al.32 During the 5 minutes heat-up, there is negligible conversion of hexyl sulfide (both observed and predicted), so we start the time-axis at t = 5 minutes. | ||
Normalized sensitivity coefficients for hexyl sulfide concentration were calculated at a reaction time of 6 minutes, which was soon after thermal equilibration at the reaction temperature of 400 °C. The ten reactions with the highest sensitivity coefficients are presented in Fig. 7. At this reaction time, the most sensitive reaction for hexyl sulfide decomposition is the beta-scission of the alpha-radical to form hexanethial and hexyl radical, which are intermediates in the main predicted product pathway to form pentane and hexane, respectively. Hydrogen transfer reactions involving H2S and hexanethial are also important to the overall prediction of the sulfide decomposition rate. Although they are involved in minor pathways, the prediction is also sensitive to disproportionation reactions (and reverse disproportionations) involving hexanethial and subsequent intermediates, as these can have an important impact on the number of radical species available for hydrogen abstraction reactions. The rate estimates for these reactions have greater uncertainty than the other reactions in Fig. 7. In particular, two of these disproportionation reactions (the third and ninth most sensitive reactions) are only roughly estimated by the RMG database based on quantum chemical calculations for similar (but not identical) radical centers. We expect at least order-of-magnitude uncertainty in these reactions, and we recommend for them to be studied further to potentially improve the prediction accuracy of the model.
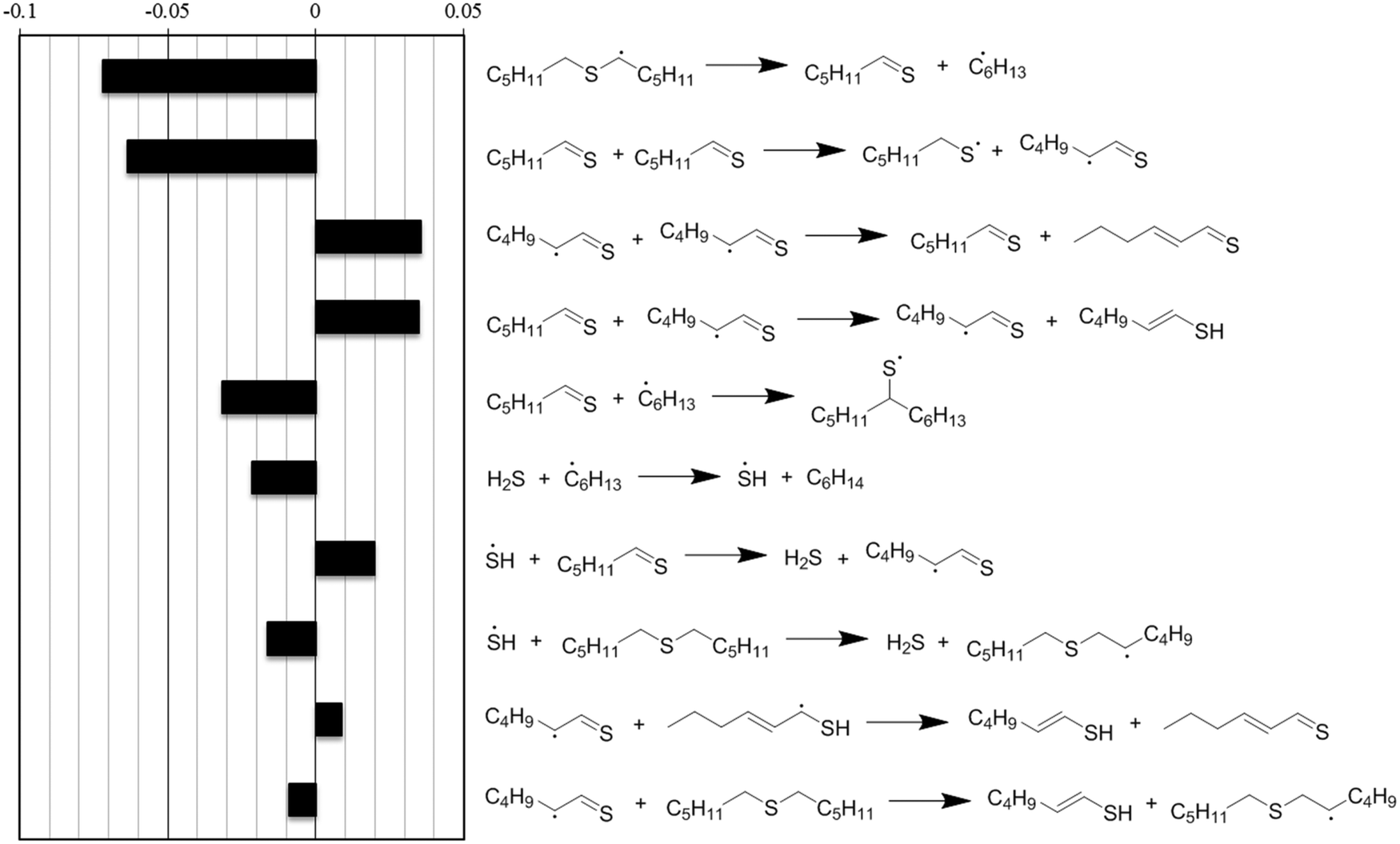 | ||
| Fig. 7 Normalized sensitivity coefficients for hexyl sulfide concentration, calculated at t = 360 s for the reaction conditions of Fig. 6. Negative sensitivity coefficients indicate that an increase in a given reaction's forward and reverse rate coefficient would increase the rate of hexyl sulfide consumption. All are reversible reactions, and the directions drawn indicate the net flux of the reactions at time t. | ||
Despite our best efforts, even the more precisely known rate constants also have uncertainties of a factor of 2 or more. Uncertainty in thermodynamic parameters is also a factor, particularly in the estimation of beta-scission reaction rates. Thus, order-of-magnitude uncertainty remains the norm for this type of complicated model, although cancellation of errors will sometimes result in more accurate predictions, like those observed for most of the products in this case.
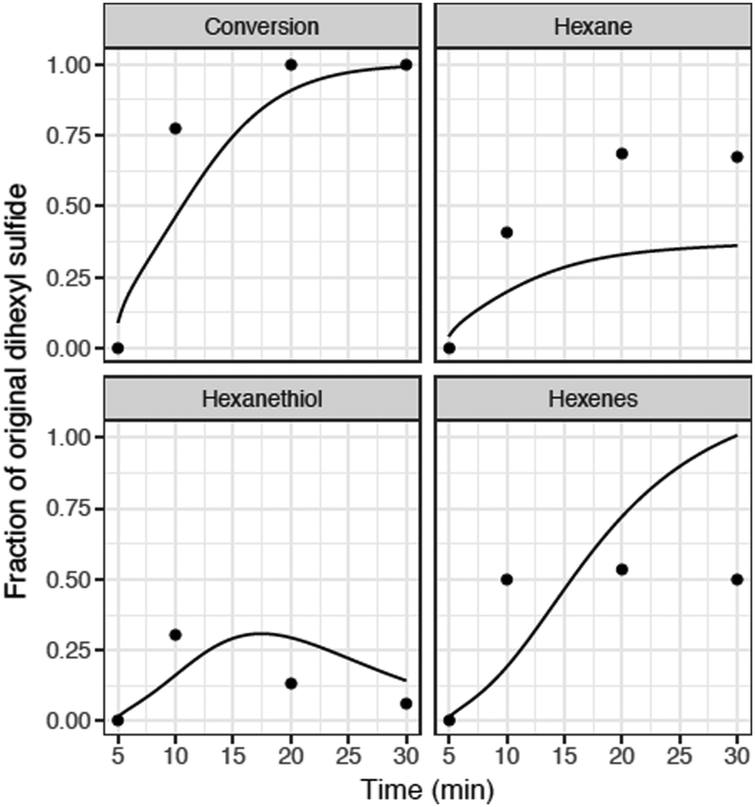 | ||
| Fig. 8 Comparison of RMG model with batch reactor data for hexyl sulfide pyrolysis at 400 °C. Full mechanism (solid), experimental data (•) from Kida et al.32 During the 5 minutes heat-up, there is negligible conversion of hexyl sulfide (both observed and predicted), so we start the time-axis at t = 5 minutes. | ||
3.6 Effect of water concentration
As a final test of the model, additional batch reactor experiments were conducted to investigate the dependence of pentane production on the initial concentration of water, and mechanisms were generated using RMG to model these experiments. The comparison of the model predictions and experimental data is presented in Fig. 9. The RMG model significantly underpredicts the production of pentane at low concentrations of water, and slightly overpredicts it at high concentrations. This disagreement underscores the complexity of the chemistry occurring in this system. Experiments below a water/sulfide ratio of 0.5 (the first 5 experimental points in Fig. 9) did not reach the critical pressure. These experiments were therefore conducted in superheated steam. At these low-water conditions, reaction (1) is more important than reaction (2). As the water loading increases so that the pressure exceeds water's critical point, the production of pentane is predicted to increase but then level off at a pentane yield greater than what is observed experimentally. The pentane overprediction could be due to the absence of important competing pathways for thioaldehyde consumption. Thioaldehyde is expected to be a major source of the experimentally observed “coke” (colored, nonvolatile material), which was produced in both the water and no-water cases (but decreased when supercritical water was present). This molecular-weight growth chemistry is extremely complex and currently very difficult to predict, but continued improvements in computational methods will make possible the examination of these important pathways.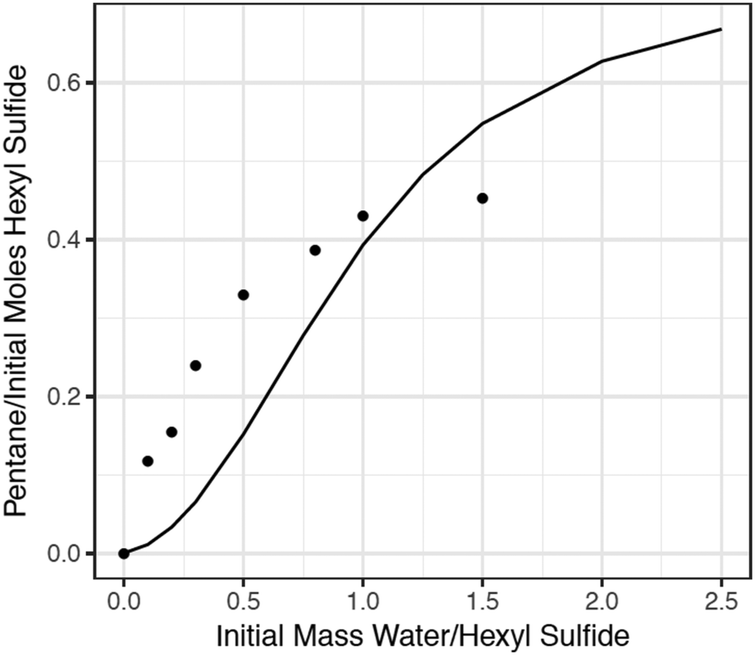 | ||
| Fig. 9 Comparison of experimental data and RMG simulations for production of pentane in 30 minutes in a 400 °C batch reactor, from different initial water/sulfide ratios. Model (curve), experimental data (•). | ||
4 Conclusions
Reaction mechanisms have been generated using the automated reaction mechanism generator to model the decomposition of hexyl sulfide, with and without the presence of hexadecane and water from 400 to 450 °C. Quantum chemical calculations have been completed to improve the rate constant and thermochemistry estimations used in mechanism generation. Calculations on the formation of carbon monoxide and pentane from thioaldehyde confirm the previous hypothesis that a water-catalyzed pathway accelerates CO formation at high water concentrations. Rate constants and thermochemistry parameters have also been calculated for intramolecular hydrogen migration reactions, as well as other reactions potentially relevant to the formation of thiophenic compounds from sulfide pyrolysis.CSTR models using the RMG mechanism provide excellent agreement with experimental data for the decomposition of hexyl sulfide in the presence of hexadecane and SCW. Good agreement is also observed between experimental measurements and model predictions for almost all of the products generated in the experiments performed in batch reactors. Sensitivity analysis for hexyl sulfide conversion shows that reasonable rate constant estimations are employed for the most important reactions in the simulation, but as always, more accurate and computationally expensive calculations could possibly improve agreement with the data. This work can help guide future calculations and experiments to focus on the thermochemical and kinetic parameters that are most important in controlling this chemistry. Overall, the work shows that we now have a semi-quantitative understanding of the details of alkylsulfide decomposition chemistry, and of the key reactions between sulfur-containing intermediates and water that lead to desulfurization.
While the detailed kinetic models presented here are generally in accord with experiment for the major reaction channels, some clear discrepancies persist; e.g., the model predicts significant thiophene yield under pyrolytic conditions, but this was not observed. Also, the models are not comprehensive, missing some important molecular weight growth reactions and other secondary reactions that become important at high conversions. Additionally, we have only explored a few of the many possible avenues for SCW to participate as a reactant in this system. Technical improvements in the modeling methodology over the next few years could overcome some of these limitations. It is also possible that our understanding of organosulfur chemistry is incomplete, and there is new chemistry to discover. Improved models for the known chemistry may make it easier to identify if additional types of reactions are important at these technologically important conditions.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank Azadeh Zaker (Worcester Polytechnic Institute) and Ping He (Lamar University) for assistance with fugacity coefficient calculations on the dodecane–water mixture. We gratefully acknowledge financial support from Saudi Aramco, a Founding Member of the MIT Energy Initiative.Notes and references
- S. Brarendregt, P. J. M. Valkenburg, E. S. Wagner, M. Dente and E. Ranzi, 14th Ethylene Producers' Conference, AIChE, New Orleans, LA, 2002, pp. 2497–2537 Search PubMed.
- Cleaner Combustion: Developing Detailed Chemical Kinetic Models, ed. F. Battin-Leclerc, J. M. Simmie and E. Blurock, Springer, London, 2013 Search PubMed.
- Y. Shi, H.-W. Ge and R. D. Reitz, Computational Optimization of Internal Combustion Engines, Springer, London, 2011 Search PubMed.
- Rethinking the Ozone Problem in Urban and Regional Air Pollution, ed. J. H. Seinfeld, P. G. Risser, J. A. Dutton and M. G. Wolman, National Research Council, National Academy of Press, Washington, DC, 1991 Search PubMed.
- W. H. Green, in Advances in Chemical Engineering, ed. G. B. Marin, Academic Press, 2007, vol. 32, pp. 1–50 Search PubMed.
- A. S. Tomlin, T. Turányi and M. J. Pilling, in Comprehensive Chemical Kinetics, ed. M. J. Pilling, Elsevier, 1997, vol. 35, pp. 293–437 Search PubMed.
- L. P. Wang, A. Titov, R. McGibbon, F. Liu, V. S. Pande and T. J. Martínez, Nat. Chem., 2014, 6, 1044–1048 CrossRef CAS PubMed.
- N. M. Vandewiele, K. M. Van Geem, M.-F. Reyniers and G. B. Marin, Chem. Eng. J., 2012, 207–208, 526–538 CrossRef CAS.
- R. G. Susnow, A. M. Dean, W. H. Green, P. Peczak and L. J. Broadbelt, J. Phys. Chem. A, 1997, 101, 3731–3740 CrossRef CAS.
- V. Warth, F. Battin-Leclerc, R. Fournet, P. A. Glaude, G. M. Côme and G. Scacchi, Comput. Chem., 2000, 24, 541–560 CrossRef CAS PubMed.
- M. Dente, E. Ranzi and A. G. Goossens, Comput. Chem. Eng., 1979, 3, 61–75 CrossRef CAS.
- P. J. Clymans and G. F. Froment, Comput. Chem. Eng., 1984, 8, 137–142 CrossRef CAS.
- W. H. Green, J. W. Allen, R. W. Ashcraft, G. J. Beran, C. A. Class, C. Gao, C. F. Goldsmith, M. R. Harper, A. Jalan, G. R. Magoon, D. M. Matheu, S. S. Merchant, J. D. Mo, S. Petway, S. Raman, S. Sharma, K. M. Van Geem, J. Song, J. Wen, R. H. West, A. Wong, H.-W. Wong, P. E. Yelvington and J. Yu, Reaction Mechanism Generator (RMG), 2013 Search PubMed.
- C. A. Class, M. Liu, A. G. Vandeputte and W. H. Green, Phys. Chem. Chem. Phys., 2016, 18, 21651–21658 RSC.
- J. Beens and R. Thijssen, J. High Resolut. Chromatogr., 1997, 20, 131–137 CrossRef CAS.
- A. G. Marshall and R. P. Rodgers, Acc. Chem. Res., 2004, 37, 53–59 CrossRef CAS PubMed.
- A. G. Marshall and R. P. Rodgers, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 18090–18095 CrossRef CAS PubMed.
- C. S. Hsu, C. L. Hendrickson, R. P. Rodgers, A. M. McKenna and A. G. Marshall, J. Mass Spectrom., 2011, 46, 337–343 CrossRef CAS PubMed.
- R. Hua, Y. Li, W. Liu, J. Zheng, H. Wei, J. Wang, X. Lu, H. Kong and G. Xu, J. Chromatogr. A, 2003, 1019, 101–109 CrossRef CAS.
- L.-Q. Zhao, Z.-M. Cheng, Y. Ding, P.-Q. Yuan, S.-X. Lu and W.-K. Yuan, Energy Fuels, 2006, 20, 2067 CrossRef CAS.
- K.-H. Choi, US Pat., US20130140214, 2013 Search PubMed.
- T. Kayukawa, T. Fujimoto, N. Inoue, S. Teratani and S. Nagamatsu, Canada Pat., CA2772095A1, 2012 Search PubMed.
- M. Morimoto, Y. Sugimoto, Y. Saotome, S. Sato and T. Takanohashi, J. Supercrit. Fluids, 2010, 55, 223 CrossRef CAS.
- A. R. Katritzky, R. A. Barcock, M. Balasubramanian, J. V. Greenhill, M. Siskin and W. N. Olmstead, Energy Fuels, 1994, 8, 498–506 CrossRef CAS.
- O. M. Ogunsola and N. Berkowitz, Fuel, 1995, 74, 1485 CrossRef CAS.
- T. Adschiri, R. Shibata, T. Sato, M. Watanabe and K. Arai, Ind. Eng. Chem. Res., 1998, 37, 2634–2638 CrossRef CAS.
- B. M. Vogelaar, M. Makkee and J. A. Moulijn, Fuel Process. Technol., 1999, 61, 265–277 CrossRef CAS.
- A. R. Katritzky, M. Balasubramanian and M. Siskin, Energy Fuels, 1992, 6, 431–438 CrossRef CAS.
- T. Sato, T. Adschiri, K. Arai, G. L. Rempel and F. T. T. Ng, Fuel, 2003, 82, 1231–1239 CrossRef CAS.
- M. Watanabe, S. Kato, S. Ishizeki, H. Inomata and R. L. Smith, Jr., J. Supercrit. Fluids, 2010, 52, 48–52 CrossRef.
- P. R. Patwardhan, M. T. Timko, C. A. Class, R. E. Bonomi, Y. Kida, H. H. Hernandez, J. W. Tester and W. H. Green, Energy Fuels, 2013, 27, 6108–6117 CrossRef CAS.
- Y. Kida, C. A. Class, A. J. Concepcion, M. T. Timko and W. H. Green, Phys. Chem. Chem. Phys., 2014, 16, 9220–9228 RSC.
- A. G. Vandeputte, PhD thesis, University of Ghent, 2012.
- A. G. Vandeputte, M. K. Sabbe, M.-F. Reyniers and G. B. Marin, Phys. Chem. Chem. Phys., 2012, 14, 12773–12793 RSC.
- A. G. Vandeputte, M. K. Sabbe, M.-F. Reyniers and G. B. Marin, Chem. – Eur. J., 2011, 17, 7656–7673 CrossRef CAS PubMed.
- A. G. Vandeputte, M.-F. Reyniers and G. B. Marin, ChemPhysChem, 2013, 14, 1703–1722 CrossRef CAS PubMed.
- A. G. Vandeputte, M.-F. Reyniers and G. B. Marin, ChemPhysChem, 2013, 14, 3751–3771 CrossRef CAS PubMed.
- C. A. Class, J. Aguilera-Iparraguirre and W. H. Green, Phys. Chem. Chem. Phys., 2015, 17, 13625–13639 RSC.
- R. Van de Vijver, N. M. Vandewiele, A. G. Vandeputte, K. M. Van Geem, M.-F. Reyniers, W. H. Green and G. B. Marin, Chem. Eng. J., 2015, 278, 385–393 CrossRef CAS.
- S. Dabiri, G. Wu, M. T. Timko and A. F. Ghoniem, J. Supercrit. Fluids, 2012, 67, 29–40 CrossRef CAS.
- G. Wu, S. Dabiri, M. T. Timko and A. F. Ghoniem, J. Supercrit. Fluids, 2012, 72, 150–160 CrossRef CAS.
- J. Song, S. Raman, J. Yu, C. D. Wijaya, G. Stephanopoulos and W. H. Green, Abstr. Pap. Am. Chem. Soc., 2003, 226, U530–U531 Search PubMed.
- J. Song, R. Sumathi, J. Yu and W. H. Green, Abstr. Pap. Am. Chem. Soc., 2004, 228, U233 Search PubMed.
- K. M. Van Geem, M.-F. Reyniers, G. B. Marin, J. Song, W. H. Green and D. M. Matheu, AIChE J., 2006, 52, 718–730 CrossRef CAS.
- M. R. Harper, K. M. Van Geem, S. P. Pyl, G. B. Marin and W. H. Green, Combust. Flame, 2011, 158, 16–41 CrossRef CAS.
- C. Gao, J. W. Allen, W. H. Green and R. H. West, Comput. Phys. Commun., 2016, 203, 212–225 CrossRef CAS.
- C. F. Goldsmith, G. R. Magoon and W. H. Green, J. Phys. Chem. A, 2012, 116, 9033–9057 CrossRef CAS PubMed.
- S. W. Benson, Chem. Rev., 1978, 78, 23–35 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. Montgomery, T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 03, Gaussian, Inc., Wallingford CT, 2004 Search PubMed.
- W. Klopper, R. A. Bachorz, D. P. Tew, J. Aguilera-Iparraguirre, Y. Carissan and C. Hättig, J. Phys. Chem. A, 2009, 113, 11679–11684 CrossRef CAS PubMed.
- J. Aguilera-Iparraguirre, H. J. Curran, W. Klopper and J. M. Simmie, J. Phys. Chem. A, 2008, 112, 7047–7054 CrossRef CAS.
- J. Aguilera-Iparraguirre, A. D. Boese, W. Klopper and B. Ruscic, Chem. Phys., 2008, 346, 56–68 CrossRef CAS.
- S. Sharma, M. R. Harper and W. H. Green, CanTherm open-source software package, 2010 Search PubMed.
- A. P. Scott and L. Radom, J. Phys. Chem., 1996, 100, 16502–16513 CrossRef CAS.
- A. L. L. East and L. Radom, J. Chem. Phys., 1997, 106, 6655–6674 CrossRef CAS.
- S. W. Benson, F. R. Cruickshank, D. M. Golden, G. R. Haugen, H. E. O'Neal, A. S. Rodgers, R. Shaw and R. Walsh, Chem. Rev., 1969, 69, 279–324 CrossRef CAS.
- G. A. Petersson, D. K. Malick, W. G. Wilson, J. W. Ochterski, J. A. Montgomery and M. J. Frisch, J. Chem. Phys., 1998, 109, 10570–10579 CrossRef CAS.
- A. G. Vandeputte, M. K. Sabbe, M.-F. Reyniers, V. Van Speybroeck, M. Waroquier and G. B. Marin, J. Phys. Chem. A, 2007, 111, 11771–11786 CrossRef CAS PubMed.
- C. Eckart, Phys. Rev., 1930, 35, 1303–1309 CrossRef CAS.
- C. Deng, Q.-G. Li, Y. Ren, N.-B. Wong, S.-Y. Chu and H.-J. Zhu, J. Comput. Chem., 2007, 29, 466–480 CrossRef.
- CHEMKIN-PRO 10131, ANSYS, San Diego, 2013 Search PubMed.
- C. Deng, X.-P. Wu, X.-M. Sun, Y. Ren and Y.-H. Sheng, J. Comput. Chem., 2008, 30, 285–294 CrossRef.
- A.-P. Fu, H.-L. Li, D.-M. Du and Z.-Y. Zhou, Chem. Phys. Lett., 2003, 382, 332–337 CrossRef CAS.
- X.-C. Wang, J. Nichols, M. Feyereisen, M. Gutowski, J. Boatz, A. D. J. Haymet and J. Simons, J. Phys. Chem., 1991, 95, 10419–10424 CrossRef CAS.
- Q.-G. Li, Y. Xue and G.-S. Yan, Theochem, 2008, 868, 55–64 CrossRef CAS.
- A. R. Hajipour, M. Karimzadeh, S. Ghorbani, H. Farrokhpour and A. N. Chermahini, Struct. Chem., 2016, 27, 1345–1362 CrossRef CAS.
- C. Trujillo, O. Mó and M. Yáñez, Org. Biomol. Chem., 2007, 5, 3092–3099 RSC.
- L. G. S. Shum and S. W. Benson, Int. J. Chem. Kinet., 1985, 17, 749–761 CrossRef CAS.
- K. Han and W. H. Green, Ind. Eng. Chem. Res., 2018, 57, 14022–14030 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cp00234k |
| ‡ Present address: MD Anderson Cancer Center, Houston TX, USA. |
| § Present address: Middlebury College, Middlebury, VT, USA. |
| ¶ Present address: Dow Chemical Company, Freeport, TX, USA. |
| || Present address: Worcester Polytechnic Institute, Worcester, MA, USA. |
| This journal is © the Owner Societies 2019 |