
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFirst principles calculations of surface dependent electronic structures: a study on β-FeOOH and γ-FeOOH†
Yuki
Sakamoto
ab,
Yusuke
Noda
ac,
Kaoru
Ohno
 ad,
Kayo
Koike
e,
Katsushi
Fujii
e,
Tomiko M.
Suzuki
f,
Takeshi
Morikawa
f and
Shinichiro
Nakamura
*a
ad,
Kayo
Koike
e,
Katsushi
Fujii
e,
Tomiko M.
Suzuki
f,
Takeshi
Morikawa
f and
Shinichiro
Nakamura
*a
aCluster for Science, Technology and Innovation, Nakamura Laboratory, RIKEN, 2-1, Hirosawa, Wako, Saitama 351-0198, Japan. E-mail: snakamura@riken.jp; Fax: +81-48-467-8503; Tel: +81-48-467-9477
bDepartment of Biological Information, Tokyo Institute of Technology, 4259 Nagatsuta, Midori-ku, Yokohama 226-8501, Japan
cDepartment of Materials Physics, Nagoya University, Furo, Chikusa, Nagoya, Aichi 464-8603, Japan
dDepartment of Physics, Graduate School of Engineering, Yokohama National University, 79-5, Tokiwadai, Hodogaya, Yokohama 240-8501, Japan
ePhotonics Control Technology Team, RIKEN Center for Advanced Photonics, 2-1 Hirosawa, Wako, Saitama 351-0198, Japan
fToyota Central R&D Labs., Inc., 41-1 Yokomichi, Nagakute 480-1192, Japan
First published on 15th May 2019
Abstract
We report a theoretical study on iron oxyhydroxide (FeOOH). The FeOOH surface is expected to act as an efficient electrochemical catalyst for the oxygen evolution reaction (OER), because it is based on iron, an element of the fourth highest Clarke number. Experimentally, the OER activity of β-FeOOH is known to be higher than that of γ-FeOOH. However, the details of the OER mechanism and the surface reactivities of the FeOOH polymorphs have not yet been fully understood. We performed first-principles calculations of bulk and surfaces of β-FeOOH and γ-FeOOH using density functional theory, to investigate their electronic structures and catalytic activities. The calculations suggest that depending on the surface indices, several surfaces may be favored for catalytic activities.
1. Introduction
To realize a sustainable energy society, it is crucially important to develop powerful catalysts aiming at artificial photosynthesis, which generates useful chemicals such as hydrogen and organic compounds from water and CO2 using sunlight. Among the relevant reactions, the electrochemical water oxidation reaction and oxygen evolution reaction (hereafter referred to as OER) are the most important reactions for energy conversion and storage to utilize water molecules as an electron source for producing useful chemical substances. So far, many research works on various OER catalysts have been reported.1–3 Among them, IrOx and RuOx show the highest catalytic activity.2,4 However, to realize the OER on an industrial scale, it is more beneficial to develop OER catalysts composed of earth-abundant metals such as 3d transition metals, instead of precious metals.Among these studies on 3d transition metal OER catalysts, Burke et al. reported remarkable results; iron doped nickel hydroxides showed the highest OER activity among 3d transition metal (oxy) hydroxides.5 To enhance the activity of nickel hydroxides, it is crucial to dope them with iron.6 Therefore, doping with a high concentration of iron is expected to provide a higher OER catalytic activity. However, subsequent studies have revealed that the reactivity increases only up to about 30% of iron content,7 which indicates that iron itself is the active site for the OER on these Ni-rich Ni–Fe–O systems. In contrast, when the iron content is greater than 30%, the catalytic activity decreases and accordingly iron oxyhydroxides themselves have been considered to possess a low OER activity.
Recently two of the authors, Suzuki and Morikawa, including others synthesized a highly crystalline β-FeOOH(Cl) nanorod, which is another polymorph of FeOOH8 with a much smaller size of 3 × 13 nanometers, and reported a remarkably high OER activity. According to the results of the OER activity in alkaline electrolytes, the β-FeOOH nanorod catalysts showed the highest OER activity among FeOOH polymorphs such as amorphous, α-, and γ-FeOOH.8,9 Furthermore, doping Ni into β-FeOOH highly enhanced its OER activity. Therefore, further understanding of electronic structures of bulk and surfaces of the FeOOH polymorphs will provide important clues for further improvement of the OER. The OER profiles of these catalysts must be compared in detail by various methods such as ECSA methods,10 in order to develop ideal OER electrocatalytic devices.
To the best of our knowledge, FeOOH polymorphs have been studied theoretically as well as experimentally by researchers mainly in the field of mineralogy.11–13 According to their reports, FeOOH consists of four polymorphs; α-FeOOH (goethite), β-FeOOH (akaganeite), γ-FeOOH (lepidocrocite), and ε-FeOOH. Among these polymorphs, ε-FeOOH is found under high pressure.12 These crystal structures are shown in Fig. 1. In these theoretical studies, several physical properties of FeOOH polymorphs such as magnetic configurations and electronic conduction mechanisms, have been revealed.12,14 On the other hand, theoretical investigations focusing on the catalyst, especially on their electrocatalytic activities and the origin of these differences, have been very limited.7,15
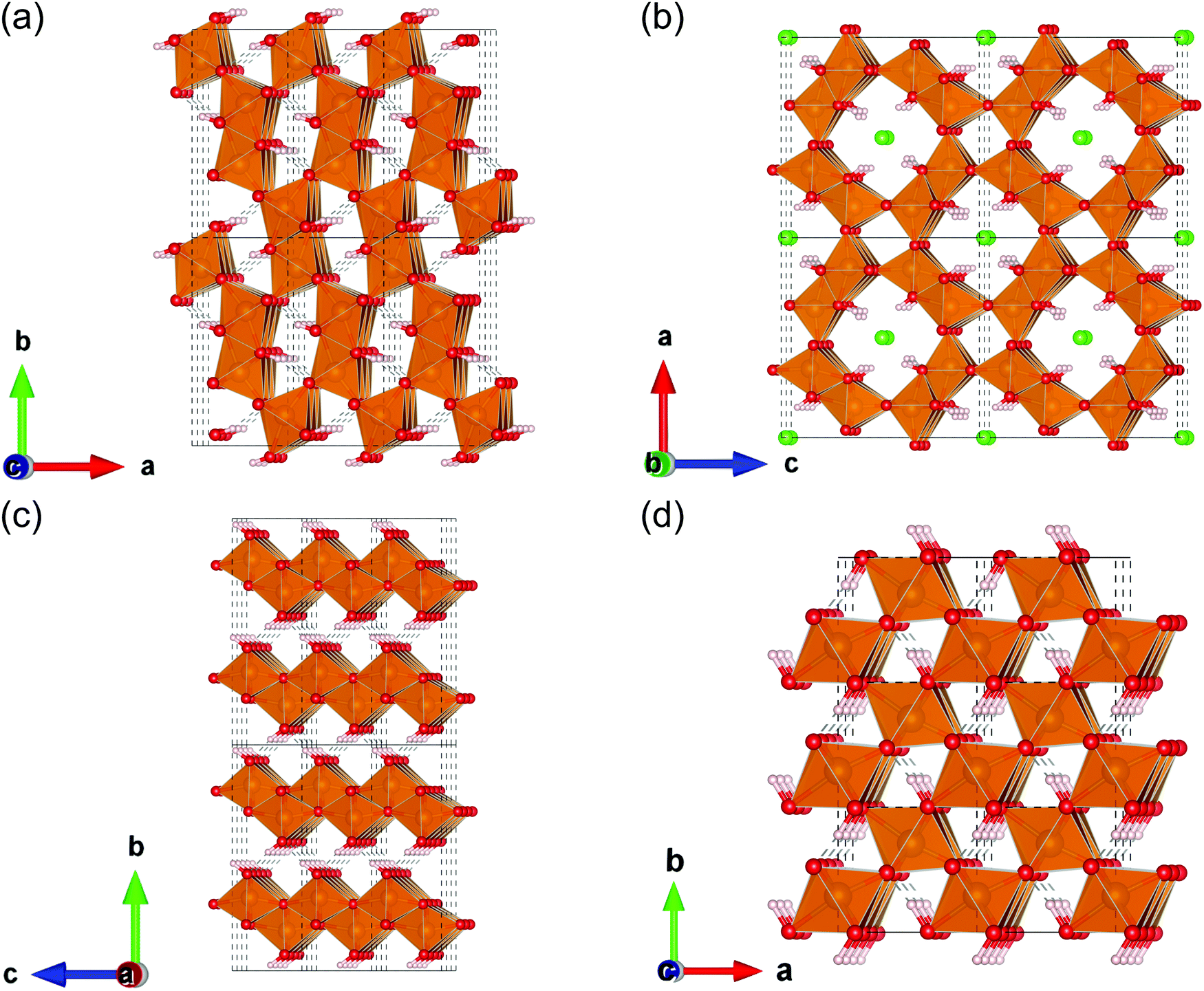 | ||
| Fig. 1 FeOOH polymorphs; (a) α-FeOOH (goethite), (b) β-FeOOH (akageneite), (c) γ-FeOOH (lepidocrocite), and (d) ε-FeOOH. The polyhedron colored in gold indicates that its centered atom is iron. The balls colored in red, green, and white denote oxygen, chloride, and hydrogen, respectively. | ||
Previously, we reported a study on the band structures of various polymorphs of manganese oxides (MnO2) by density functional theory,16 as an approach to mimic natural photosynthesis, which follows up on our previous studies on the MnCaO5 cluster.17,18 We also reported a theoretical study on the electronic structure of iron doped vs. un-doped nickel hydroxides.19 In a previous study,19 we investigated the solid and surface electronic properties (obtained by periodic system calculations) in terms of molecular orbital interactions.20
As a natural extension of this study, in the present report we focus on the electronic structures of bulk and surfaces of the FeOOH catalyst, using first principles calculations. Here it is important to note that the catalytic activities are determined by a number of factors such as bulk and exposed structures, and orientation and electronic features. Amongst all, for the electrocatalysts of FeOOH polymorphs, the key factor determining its catalytic activities has not yet been understood. Therefore, the present study is aimed to obtain useful chemical insights to investigate electrocatalytic reactions. As will be explained in detail below, a series of calculations performed revealed that there are qualitative differences in the surface electronic structures between β-FeOOH and γ-FeOOH, which possess excellent and moderate OER activity among FeOOH polymorphs as described later. The result provides useful information on the relation of the OER catalytic activities with the crystal polymorphs and strategies for further improvement in the activities.
2. Experimental and computational details
2.1 Experimental details
α-FeOOH and γ-FeOOH (powder) were purchased from Alfa Aesar. β-FeOOH nanorod colloidal solution was synthesized by a previously reported method.8 α-FeOOH and γ-FeOOH on carbon paper (CP) electrodes were prepared by depositing 1000 μL of the suspension in H2O and ethanol (α-FeOOH and γ-FeOOH) on CP (TORAY, TGP-H-060, 1.8 × 2.2 cm), followed by storage at room temperature for 6 h and further drying under vacuum at 313 K. The β-FeOOH on CP electrode was prepared by a previously reported method.8 In this case, ca. 1.0 mg of various FeOOHs was loaded per 1 cm2 of the CP.The crystal structures of the electrodes were assessed by X-ray diffraction (XRD: Rigaku, Ultima IV) using CuKα radiation at 40 kV and 40 mA. The electrochemical characteristics of the electrodes (1 × 1 cm) were investigated in 1 M KOH aqueous solution (pH 13.6) with a three-electrode configuration using an Ag/AgCl reference electrode and a Pt-wire counter electrode.
2.2 Computational details
We performed DFT calculations using an ultrasoft pseudopotential implemented on the PWscf package of the Quantum ESPRESSO program.21 The Perdew–Burke–Ernzerhof (PBE) generalized gradient approximation functional22 and Hubbard-U corrections23 (5.0 eV for Fe14) were used. The cutoff energies for the wave function and charge density were set at 40 Ry and 400 Ry, respectively. The magnetic moment of each iron atom was initially set at ±5.00 μB, corresponding to the high spin state of Fe(III).The geometries of the β and γ phases of FeOOH were prepared and optimized, owing to the references by Post et al.24 and Christensen et al.,25 respectively. For the sake of simplicity and to focus on the crystalline framework, we assumed that β-FeOOH contains neither chloride anions (Cl−) nor additional protons to neutralize the Cl− in the unit cell. As for the magnetic configuration, we assumed the antiferromagnetic configuration, following the previous studies by Alexandrov et al.14 and Guo et al.11 Notice that the 57Fe Mössbauer spectra revealed that pure β-FeOOH was paramagnetic at room temperature.8 To perform the band structure calculation, we prepared initial geometries with a magnetic primitive cell, which is the minimum cell to describe the specified magnetic configurations, based on these structures. For the k-point sampling in the geometrical optimization, we used a Monkhorst–Pack grid26 with 3 × 9 × 3 and 6 × 8 × 4 for β-FeOOH and γ-FeOOH, respectively.
To calculate the surface properties of β-FeOOH and γ-FeOOH, initial geometries were prepared using fully relaxed conventional unit cells. We take the unit cell so that the exposed surfaces were perpendicular to the z-axis (to be shown later). We used supercells including the vacuum region of at least 12 Å. The surface iron atoms were assumed to be six-coordinated and terminated by the OH− group, following previous studies.13 For structural relaxation, cell axes were fixed. Moreover, iron atoms located on the layers deeper than the first and second layers from the top surface were also fixed. For geometrical optimizations of these slab structures, we used 9 × 3 × 1 and 3 × 3 × 1 k-point grids for the (100) and the (010) surface exposed structure of β-FeOOH, whereas 3 × 4 × 1 and 8 × 2 × 1 k-point grids for the (010) and the (001) surface exposed structure of γ-FeOOH, respectively.
For the density of states (DOS) and partial DOS calculations, twice as many k-points along each axis were sampled. All symmetry k-points and symmetry k-lines in Brillouin zone were determined by the automatic flow program (AFLOW).27,28 The structures and surfaces were visualized with VESTA.29
3. Results and discussion
3.1 Electrocatalytic OER activities of various FeOOH polymorphs
In order to compare the OER activity among FeOOH polymorphs, the electrodes with α-, β-, and γ-FeOOH deposited on CP were prepared. α- and γ-FeOOH were used as received, while β-FeOOH was synthesized according to our previous literature,8 since it is not commercially available. We confirmed crystal structures of the electrodes by XRD (as shown in Fig. S1, ESI†).The current–potential characteristics of the FeOOH polymorphs on CP were evaluated in 1 M KOH aqueous solutions. As shown in Fig. 2(a), the overpotential during the OER was significantly different among the FeOOH polymorphs. The overpotential of OER was the lowest on β-FeOOH, followed by γ-FeOOH and α-FeOOH. The time course of the reaction current measured at +1.60 V (vs. RHE) in Fig. 2(b) show that the OER current density of β-FeOOH was 1.3 mA cm−2, which was 7.2 times and 130 times higher than that of the γ-phase and α-phase, respectively. These results indicate that β-FeOOH exhibits the most excellent OER activity depending on its lowest overpotential among typical FeOOH polymorphs as previously reported.8
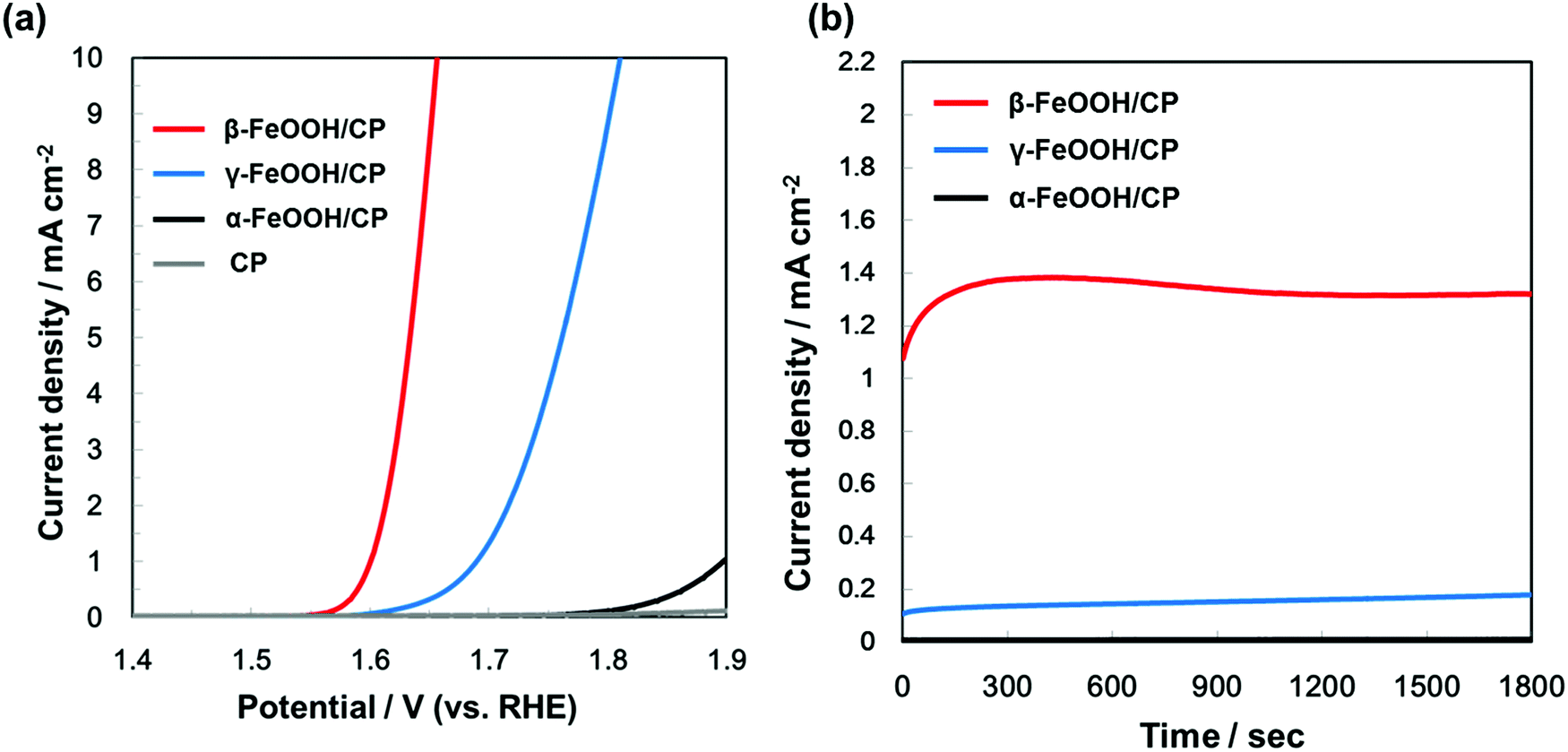 | ||
| Fig. 2 (a) Current–potential characteristics and (b) time courses of currents (at +1.60 V vs. RHE) of β-FeOOH, γ-FeOOH, and α-FeOOH/CP electrodes, acquired in 1 M KOH aqueous solution. | ||
3.2 The band structure of β and γ-FeOOH
We calculated the band structure of β- and γ-FeOOH, which showed the first and second highest OER activities as shown in Fig. 2, in order to compare their electronic structures, with those of their surfaces. The calculated unit cells with the spin magnetic configuration are shown in Fig. 3(a and b) for β-FeOOH and γ-FeOOH, respectively. The shapes of their first Brillouin zones are the same as shown in Fig. 3(c and d) for β-FeOOH and γ-FeOOH, respectively. For β-FeOOH, we prepared the initial coordinate based on the I2/m structure as reported by Post et al.24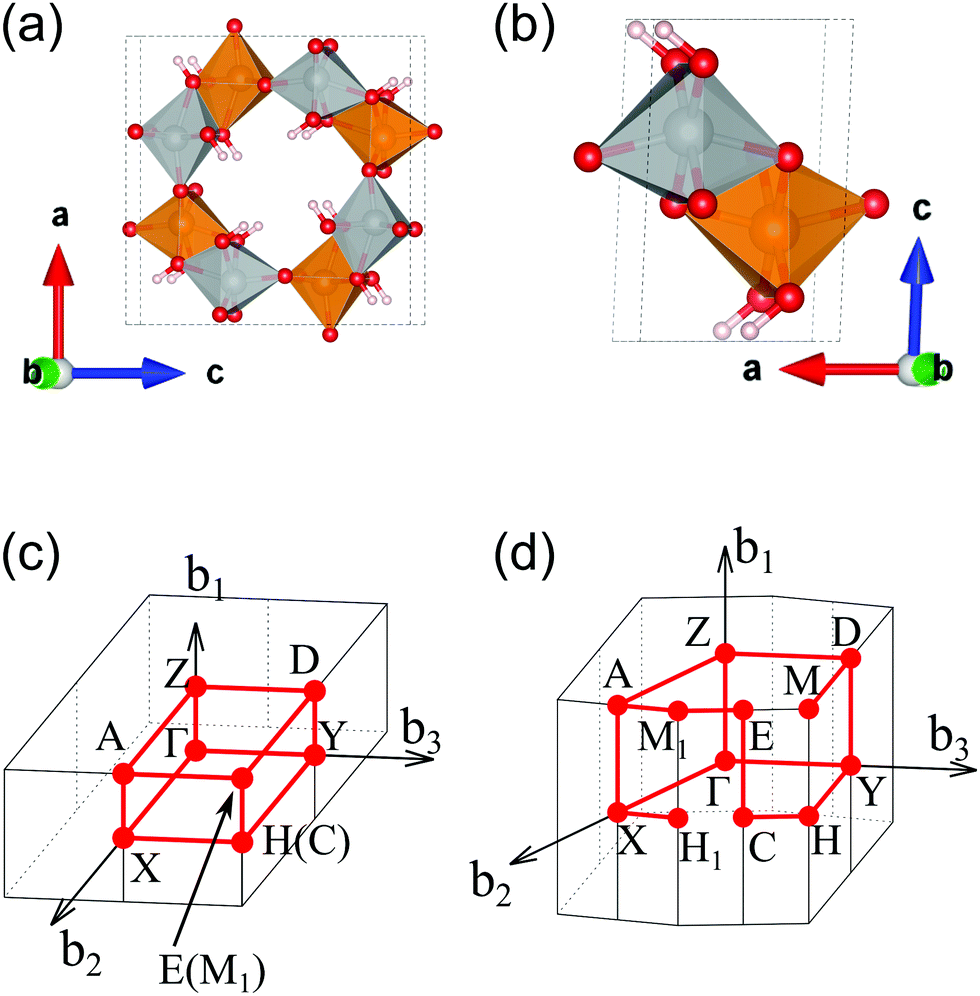 | ||
| Fig. 3 Calculated unit cells of (a) β-FeOOH and (b) γ-FeOOH. Their first Brillouin zones are shown in (c and d) for β-FeOOH and γ-FeOOH, respectively. In (a and b), the gold and silver colored polyhedrons indicate the up-spin and down-spin configurations of the central iron atom, respectively. Other atoms colored in red and white denote oxygen and hydrogen, respectively. | ||
The calculated total and partial DOS and the band structure of β-FeOOH are shown in Fig. 4(a and b), respectively. According to the calculated band structure, there is almost no electronic interaction around the tunnel structure (plane normal to the tunnel axis; see Fig. 3(a)), since the band shows almost no large dispersion along the Γ–Y and Γ–Z lines in the reciprocal space as shown in Fig. 4(b). On the other hand, valence bands have larger dispersion along the Γ–X line (white blurred regions). The X point is at a distance half of the reciprocal vector b2 (Fig. 3(c)). These bands indicate that electronic interactions mainly exist along the tunnel structure. The bulk β-FeOOH has a band gap of 1.74 eV.
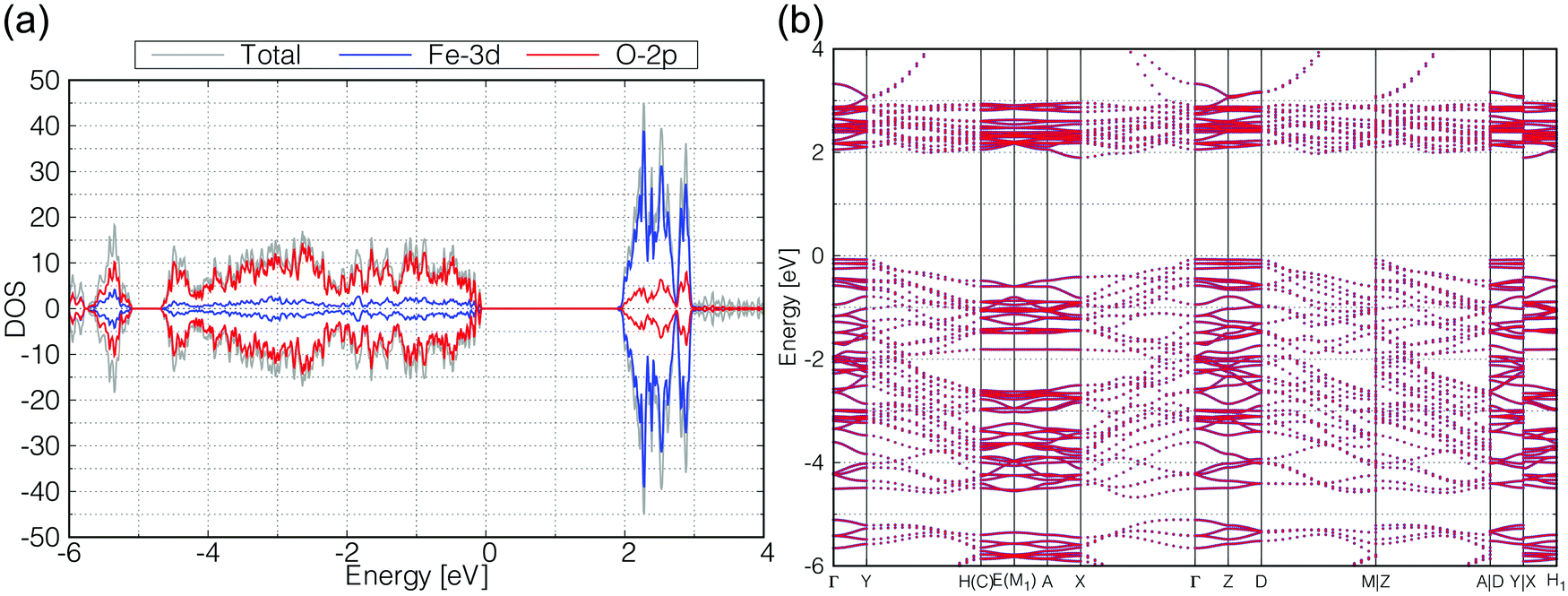 | ||
| Fig. 4 Calculated (a) total DOS (gray), Fe-3d (blue) and O-2p (red) partial DOS, and (b) the band structure of β-FeOOH. In (b), all up- and down-spin bands are degenerated on the calculated reciprocal points. | ||
As for γ-FeOOH, there are two possibilities for its crystal system; Cmcm and Cmc21. Previously, the magnetic configuration of γ-FeOOH was reported by Alexandrov et al. using DFT calculations.14 In their study, they adopted the Cmc21 space group, which is non-centrosymmetric and has a lower symmetry than the Cmcm space group. Furthermore, they concluded that γ-FeOOH prefers the antiferromagnetic configuration, as shown in Fig. 3(b). Following their preceding study, we also assumed that γ-FeOOH has the Cmc21 space group and the antiferromagnetic configuration. The calculated total and partial DOS and band structure of γ-FeOOH are shown in Fig. 5(a and b), respectively. The valence band maximum (VBM) is located on the symmetry line connecting the E and M1 points, but not at the Γ point. The conduction band minimum (CBM) is located at the H point with the 1.18 eV band gap.
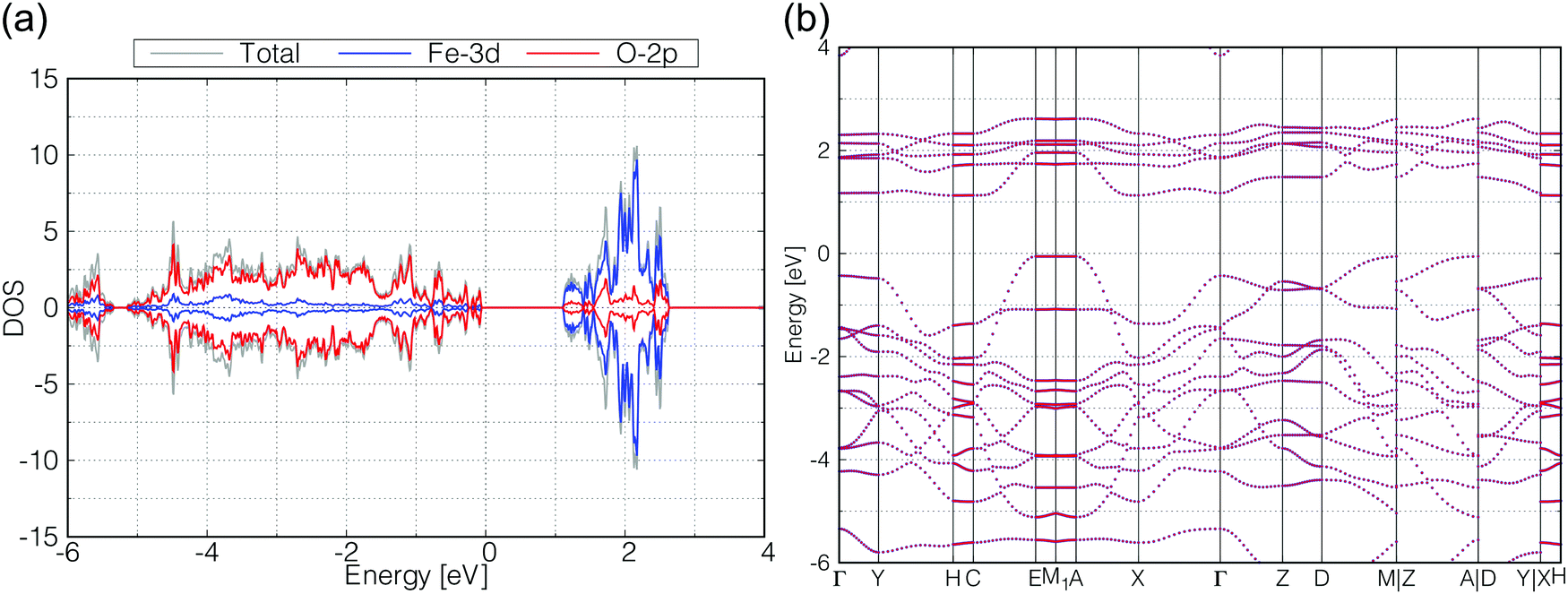 | ||
| Fig. 5 Calculated (a) total DOS (gray), Fe-3d (blue) and O-2p (red) partial DOS, (b) band structure of γ-FeOOH. In (b), all up- and down-spin bands are degenerated on the calculated reciprocal points. | ||
In order to check the influence of the Hubbard-U parameter for the results,30 we calculated the electronic structures of β-FeOOH and γ-FeOOH also with 3.0 eV of Hubbard-U parameter for iron. The results are shown in Fig. S2 and S3 (ESI†). The resulting electronic structures are almost the same as described above although their band gaps become slightly narrower. Therefore, we adopted the 5.0 eV for Hubbard-U value for iron in the later calculations.
3.3 Surface electronic structure of β-FeOOH
We then calculated the electronic structures of these crystal surfaces. In this section, we focus on β-FeOOH, especially on its (100) and (010) surfaces. They are representative in a sense that both are independent and simplest surfaces (Fig. 6) among numerous surface indices. The calculated structures are shown in Fig. 6(a and b), respectively. These geometries contain eight layers of iron atoms along the z axis. Additional H atoms and OH groups are connected to the surface layers to keep OH terminations, following the theoretical study reported by Otte et al.13 In the figures of these calculated surface structures, note that the hydrogen atoms of the surface OH− groups moved to the surface side to form hydrogen bonds with other oxygen atoms during the optimizations, due to the absence of the electrolytes in these models.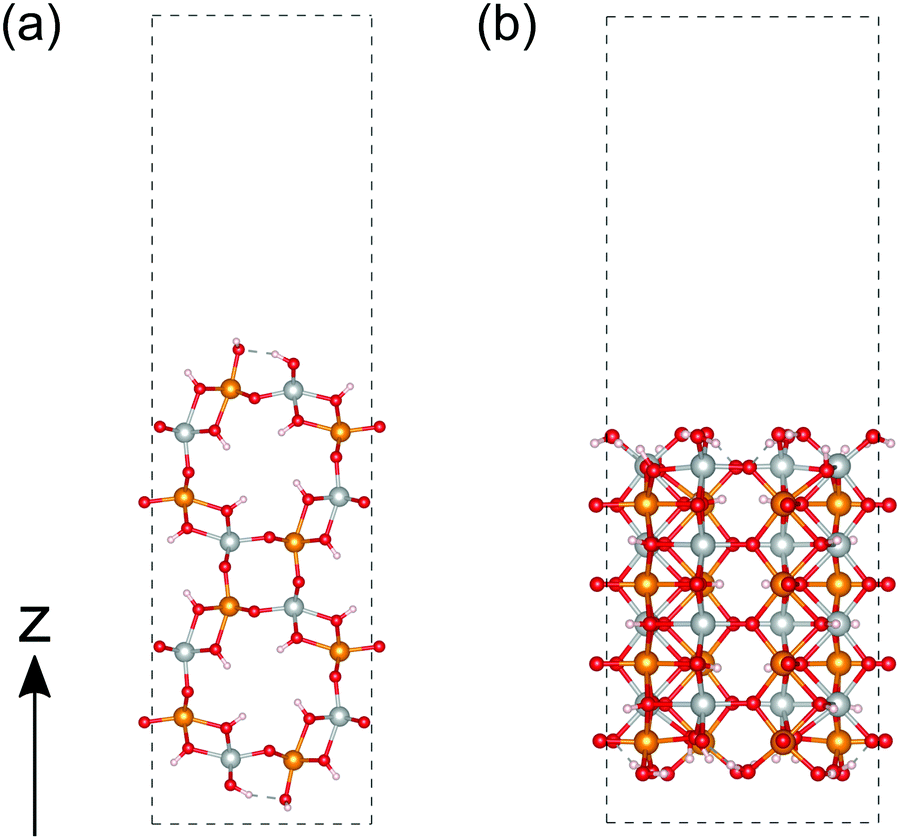 | ||
| Fig. 6 Calculated structure of β-FeOOH. (a and b) are the (100) and (010) surfaces, respectively. The atoms colored in gold, silver, red and white indicate iron with up-spin, iron with down-spin, oxygen, and hydrogen, respectively. | ||
The calculated total DOS and the Fe-3d and O-2p partial DOS of the (100) and (010) surface exposed structures are shown in Fig. 7(a and b), respectively. As seen from these calculated DOS, the (010) surface of β-FeOOH has a narrower band gap (0.6 eV) than the bulk structure, while the (100) surface exposed structure has almost the same band gap as that of the bulk. From these observations, the (010) surface may possibly have a higher reactivity than the (100) surface due to the narrower bandgap.
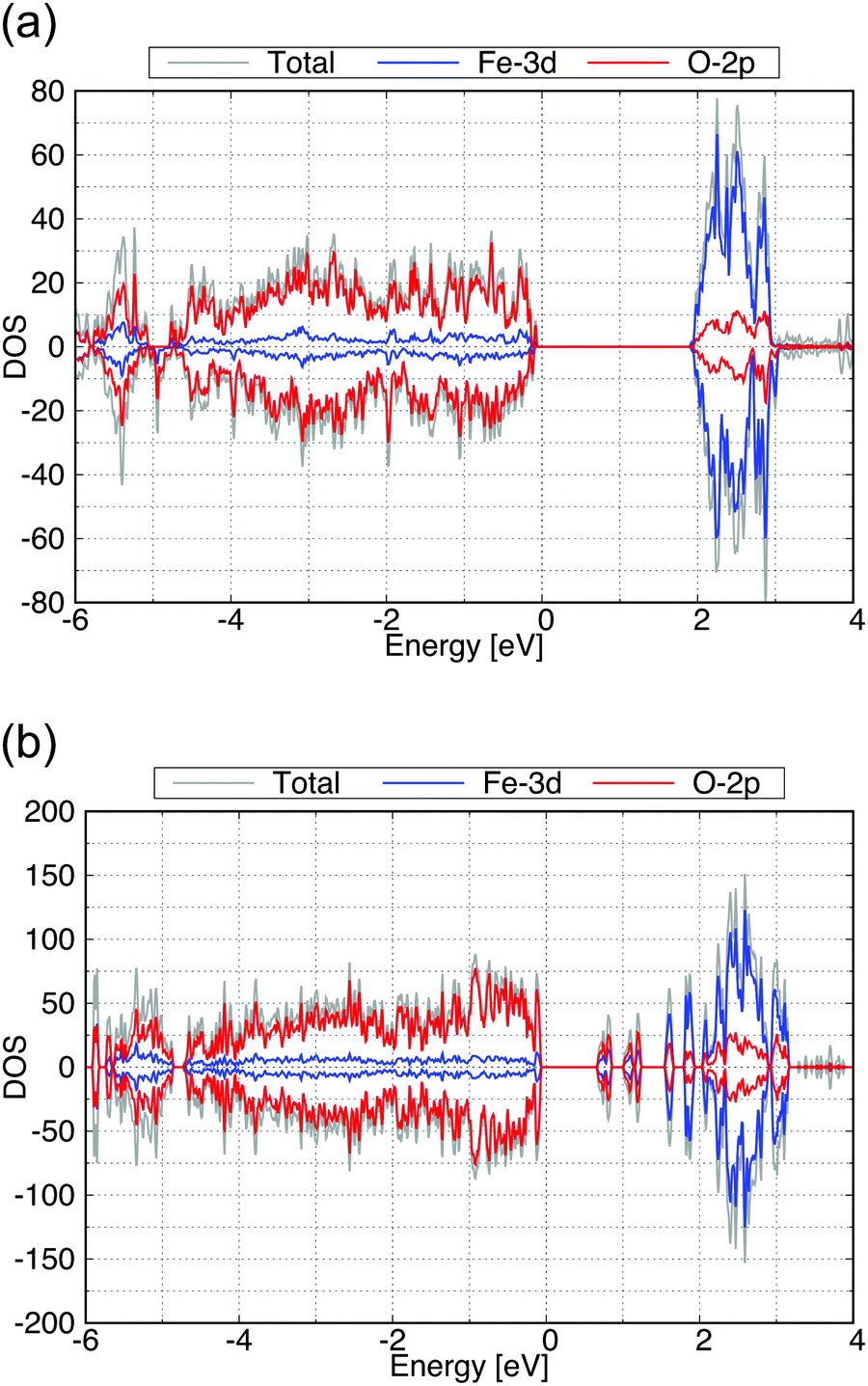 | ||
| Fig. 7 Calculated total DOS (gray), the Fe-3d (blue) and O-2p (red) partial DOS of (a) for the (100) and (b) for the (010) surface exposed structures of β-FeOOH. | ||
In addition, we compared their surface stabilities from our calculations. Since the chemical composition ratio of these surface structures is different from the stoichiometric composition (Fe8O16H8 for the bulk structure) due to the OH surface terminations, we used the following expression to evaluate their surface stabilities ε.
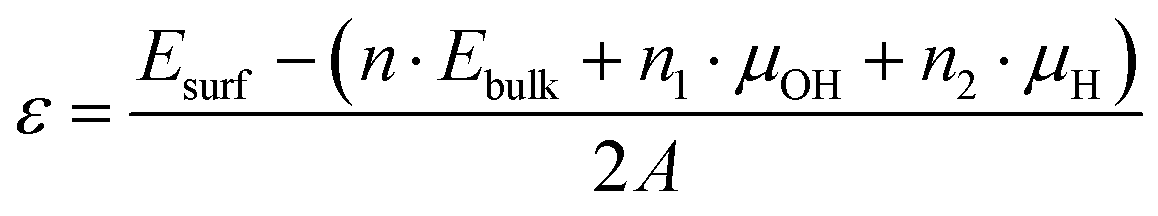
The calculated surface stabilities of (100) and (010) surfaces of β-FeOOH are +0.018 and +0.197 eV Å−2, respectively. These results show that the stable (100) surface possesses a similar electronic structure to that of the bulk structure, on the other hand, the less stable (010) surface shows the electronic structure different from that of the bulk.
3.4 Surface electronic structure of γ-FeOOH
We then performed a similar calculation for the (010) and (001) surfaces of γ-FeOOH. The calculated structures are shown in Fig. 8. The (010) surface of γ-FeOOH has an exfoliated structure of the bulk layered structure. Their calculated total DOS and the Fe-3d and O-2p partial DOS are shown in Fig. 9.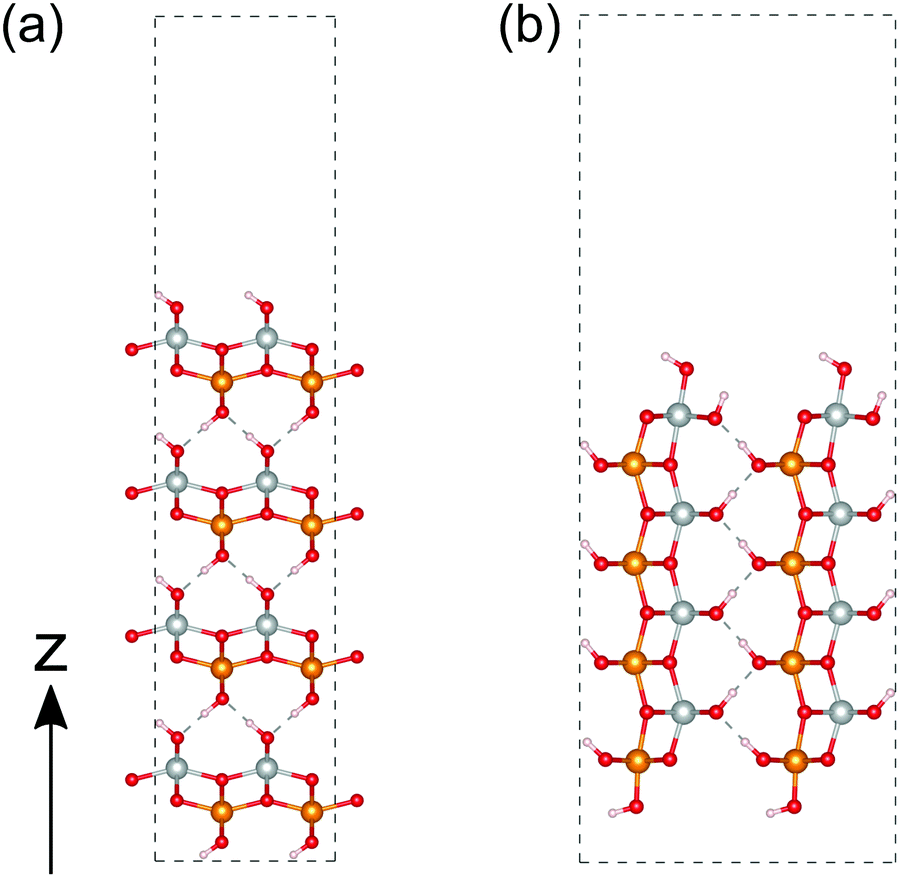 | ||
| Fig. 8 Calculated structures of γ-FeOOH. (a and b) correspond to the (010) and (001) surfaces, respectively. The atoms colored in gold, silver, red and white indicate iron with up-spin, iron with down-spin, oxygen, and hydrogen, respectively. | ||
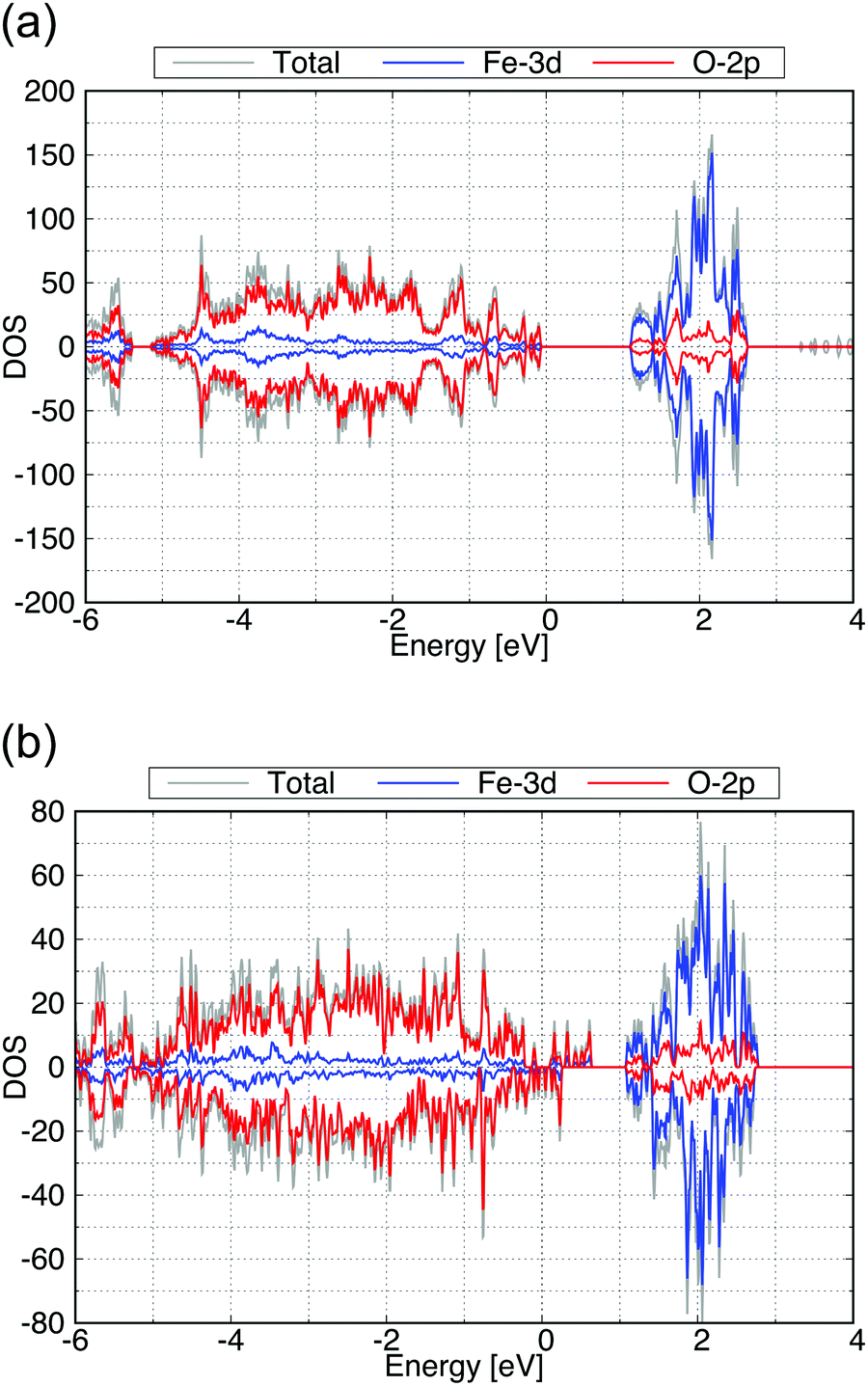 | ||
| Fig. 9 Calculated total DOS (gray), Fe-3d (blue) and O-2p (red) partial DOS of (a) for the (010) and (b) for the (001) surface exposed structure of γ-FeOOH. | ||
The band gap of the (010) surface is almost identical to that of the bulk. On the other hand, in the (001) surface, the valence band crosses the Fermi energy. From these observations, the (001) surface may show a higher reactivity than the (010) surface.
We evaluated the surface stabilities in the same way as β-FeOOH surfaces. The calculated surface stabilities of (010) and (001) surfaces are +0.021 and +0.150 eV Å−2, respectively. As in the case of β-FeOOH, the less stable (001) surface showed the electronic structure different from that of the bulk structure, while the stable (010) surface showed an electronic structure similar to it.
3.5 DOS decomposition of the surface exposed structure
As we discussed in the preceding sections, the DOS of the (010) surface exposed structure of β-FeOOH and the (001) surface exposed structure of γ-FeOOH are qualitatively different from those of the bulk structures. In general, it is widely known that the surface stabilities are important for crystal habits and crystal growths. However, the relation between surface stabilities and catalytic reactivities (such as OER) has not yet been established. The current results suggest that there is a possibility that surfaces that are energetically less stable may remain on the nanometer scale and may participate in oxygen evolution reactions. However, the details are a subject of future study. Therefore, the details of these two are worthwhile to be examined. Although their catalytic reaction mechanism from the dynamical point of view is an interesting subject, it is beyond the scope of the current study. In order to understand this feature in more detail from the surface physics point of view, we decomposed the DOS into the local DOS of Fe-3d orbitals at each layer of iron atoms, as shown in Fig. 10. The layers in real space are numbered 1, 2, 3, and 4 as shown in Fig. 10(a and c). Note that the top and the bottom are the surfaces.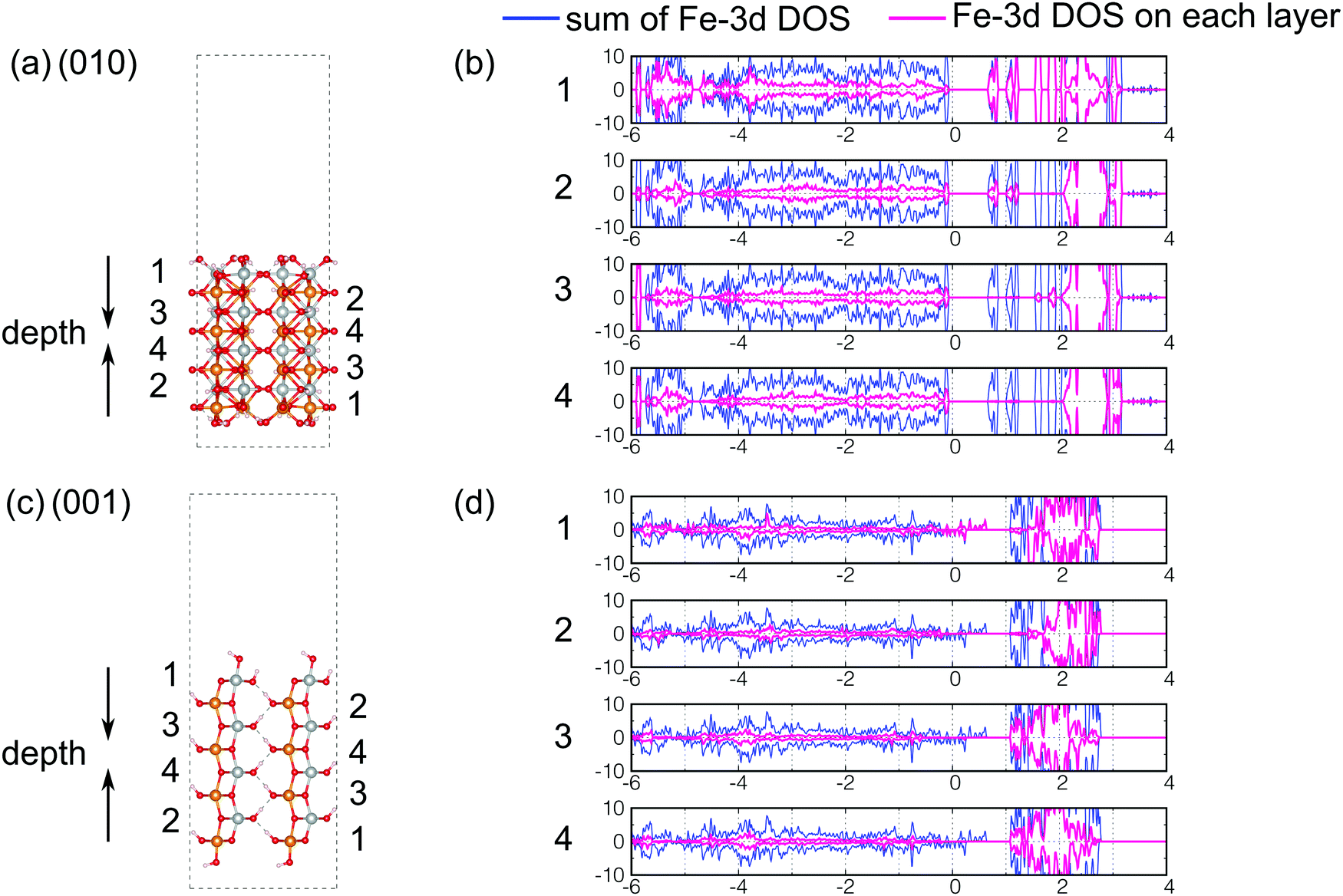 | ||
| Fig. 10 Index of iron atoms (a) for the (010) surface exposed structure of β-FeOOH and (c) for the (001) surface exposed structure of γ-FeOOH. The decomposed local DOS of Fe-3d orbitals are shown in pink for (b) the (010) surface exposed structure of β-FeOOH and (d) for the (001) surface exposed structure of γ-FeOOH. In (b and d), the numbers correspond to the layers in (a and c), respectively. In (b and d), the blue curves mean the sum of the partial DOS of Fe-3d orbitals. (They are the same as the Fe-3d DOS presented in Fig. 7(b) and 9(b).) | ||
According to Fig. 10(b), in the (010) surface exposed structure of β-FeOOH, the lowest conduction band is mainly assigned to the iron atoms located on layers 1 and 2, i.e., on the first and second layers from the top surface. This indicates that the (010) surface of β-FeOOH can accept electrons better than the other surfaces. On the other hand, in γ-FeOOH (Fig. 10(d)), the highest valence band, which crosses the Fermi energy, is mainly comprised of iron atoms located on the first layer, the exposed iron atoms. Thus, in both cases, the local DOS of iron atoms reveals a peculiar behavior of the surfaces, which is different from the bulk, even though all iron atoms are six-coordinated. According to the studies on the OER mechanisms under alkaline conditions, most of the proposed mechanisms contain the reactions to form intermediates, as shown below.2 On the other hand, as for the later steps, several O2 forming mechanisms are proposed.
| M + OH− → MOH |
| MOH + OH− → MO + H2O(l) |
Focusing on the (010) surface of β-FeOOH, we show the spatial distribution of the Kohn–Sham wave functions at the Γ point in real space. In Fig. 11, the up-spin pseudo wave functions of the highest occupied level and the lowest unoccupied level of the β-FeOOH(010) surface exposed structure are visualized analogous to the HOMO/LUMO visualization in molecular quantum chemistry. As shown in Fig. 11(a), the highest occupied orbital is widely distributed on the oxygen atoms. On the other hand, the lowest unoccupied orbital is localized on the up-spin polarized iron atoms located on the surface. The down-spin pseudo wave functions have a similar behavior due to the antiferromagnetic configuration. The distributions of these pseudo wave functions are consistent with the behavior described by the decomposed local DOS shown in Fig. 10(b), indicating again that the (010) of β-FeOOH is favorable for the OER activity. The DOS and pseudo wave function of the lowest conduction bands of the β-FeOOH(010) surface exposed structure suggest that the accepter site is localized on the atoms near the surface. On the other hand, the highest valence band distributes widely in the whole structure, not localized on specific location. In view of the fact that a chemical reaction is an event occurring at the localized sites, the (010) surface may prefer more electron accepting reactions than electron donating reactions.
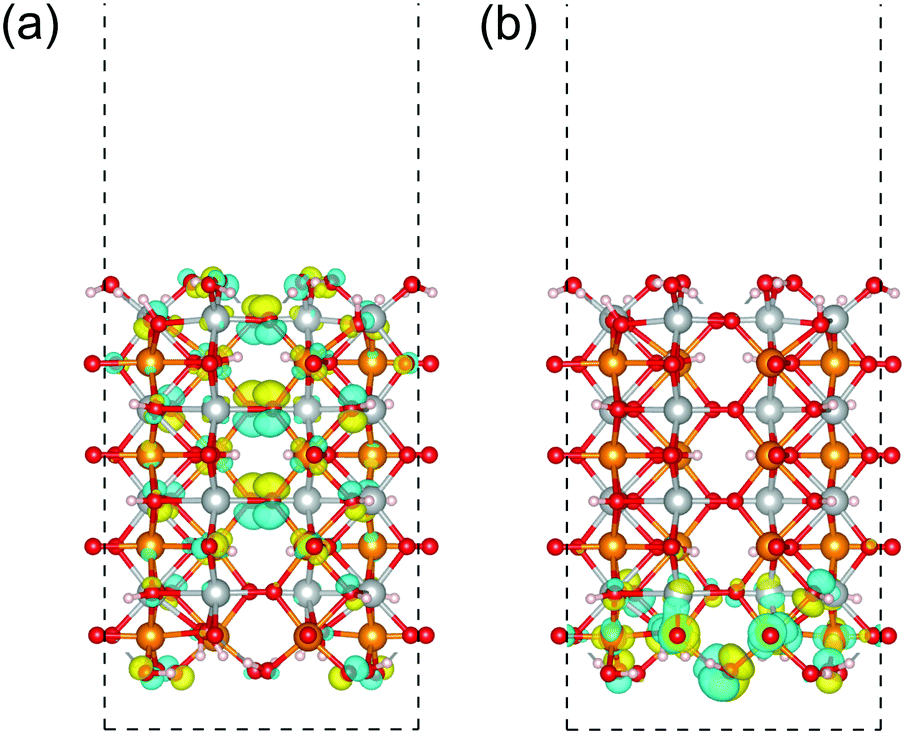 | ||
| Fig. 11 Up-spin pseudo wave functions of the (010) surface exposed structure of β-FeOOH at the Γ point. The highest occupied orbital and lowest unoccupied orbital are displayed in (a and b), respectively. The isosurfaces colored in yellow and sky blue indicate the positive and negative phase regions of the wave functions. The atoms colored in gold, silver, red and white indicate iron with up-spin, iron with down-spin, oxygen, and hydrogen, respectively. | ||
In the previous DFT study, Otte et al.13 compared the most common surface of α-FeOOH, β-FeOOH and γ-FeOOH with various termination structures. They concluded that the electronic structures of surface iron atoms mainly depend on their coordination numbers and termination structures, but not on the crystalline phase. In the present study also, the calculated electronic structures of the same surface: the (100) surface of β-FeOOH and the (010) surface of γ-FeOOH, are consistent in their results qualitatively. On the other hand, our calculations show that some surfaces have distinct properties depending on the surface indices even in the same crystal structures. It is natural to infer that the catalytic activity depends on the surface properties. Therefore, the stability of the active surface is a critical problem for the catalysts. In general, the most exposed surface of crystals is relatively more stable than the others. On the other hand, the catalytic activity may not always belong to the most stable surface.
As seen above, the (010) surface of β-FeOOH have lower conduction bands than the bulk, while the (001) surface of γ-FeOOH has higher valence bands than the bulk. These features are contrasting with each other. Since the highest occupied states are localized near the (001) surface in γ-FeOOH, it is expected that the electrolytes existing nearby can easily access the surface (of the electronic structure) and may cause interaction. On the other hand, since the lowest unoccupied states are localized on the (010) surface of β-FeOOH, the surface electronic structure is expected to be suitable for the initial OER steps. This observation can be one explanation of the difference in the catalytic activities and stabilities of the β-FeOOH and γ-FeOOH.
4. Conclusions
We studied the electronic states of bulk and low-indexed surfaces of β-FeOOH and γ-FeOOH, which showed excellent and moderate OER activities. These comparative results showed that the electronic structure at the surfaces has qualitatively distinct features. In particular, the energy level of the conduction band minimum is lower at the β-FeOOH(010) surface than in the bulk, while the valence band maximum is almost identical. On the other hand, the valence band maximum is higher on the γ-FeOOH(001) surface than in the bulk. Although there are many problems to be solved to understand these catalytic activities, especially quantum chemical reaction analysis that is left as a subject of future studies, the difference of these surface properties suggests a possible mechanism for the experimentally obtained remarkable catalytic stability of β-FeOOH.Conflicts of interest
There are no conflicts to declare.Acknowledgements
All calculations were performed on the HOKUSAI computing system of RIKEN. This work was supported by the RIKEN Junior Research Associate (JRA) Program for Y. S.References
- T. R. Cook, D. K. Dogutan, S. Y. Reece, Y. Surendranath, T. S. Teets and D. G. Nocera, Chem. Rev., 2010, 110, 6474–6502 CrossRef CAS PubMed.
- N.-T. Suen, S.-F. Hung, Q. Quan, N. Zhang, Y.-J. Xu and H. M. Chen, Chem. Soc. Rev., 2017, 46, 337–365 RSC.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS PubMed.
- Y. Lee, J. Suntivich, K. J. May, E. E. Perry and Y. Shao-Horn, J. Phys. Chem. Lett., 2012, 3, 399–404 CrossRef CAS PubMed.
- M. S. Burke, S. Zou, L. J. Enman, J. E. Kellon, C. A. Gabor, E. Pledger and S. W. Boettcher, J. Phys. Chem. Lett., 2015, 6, 3737–3742 CrossRef CAS PubMed.
- L. Trotochaud, S. L. Young, J. K. Ranney and S. W. Boettcher, J. Am. Chem. Soc., 2014, 136, 6744–6753 CrossRef CAS.
- D. Friebel, M. W. Louie, M. Bajdich, K. E. Sanwald, Y. Cai, A. M. Wise, M.-J. Cheng, D. Sokaras, T.-C. Weng and R. Alonso-Mori, J. Am. Chem. Soc., 2015, 137, 1305–1313 CrossRef CAS PubMed.
- T. M. Suzuki, T. Nonaka, A. Suda, N. Suzuki, Y. Matsuoka, T. Arai, S. Sato and T. Morikawa, Sustainable Energy Fuels, 2017, 1, 636–643 RSC.
- Y. Liang, Y. Yu, Y. Huang, Y. Shi and B. Zhang, J. Mater. Chem. A, 2017, 5, 13336–13340 RSC.
- C. C. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977–16987 CrossRef CAS PubMed.
- H. Guo and A. S. Barnard, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 094112 CrossRef.
- K. Otte, R. Pentcheva, W. W. Schmahl and J. R. Rustad, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 205116 CrossRef.
- K. Otte, W. W. Schmahl and R. Pentcheva, Surf. Sci., 2012, 606, 1623–1632 CrossRef CAS.
- V. Alexandrov and K. M. Rosso, J. Chem. Phys., 2014, 140, 234701 CrossRef PubMed.
- J. Yan, P. Li, Y. Ji, H. Bian, Y. Li and S. F. Liu, J. Mater. Chem. A, 2017, 5, 21478–21485 RSC.
- Y. Noda, K. Ohno and S. Nakamura, Phys. Chem. Chem. Phys., 2016, 18, 13294–13303 RSC.
- M. Hatakeyama, K. Ogata, K. Fujii, V. K. Yachandra, J. Yano and S. Nakamura, Chem. Phys. Lett., 2016, 651, 243–250 CrossRef CAS PubMed.
- K. Ogata, T. Yuki, M. Hatakeyama, W. Uchida and S. Nakamura, J. Am. Chem. Soc., 2013, 135, 15670–15673 CrossRef CAS PubMed.
- Y. Sakamoto, Y. Noda, K. Ohno and S. Nakamura, J. Phys. Chem. C, 2017, 121, 24603–24611 CrossRef CAS.
- R. Hoffmann, Angew. Chem., Int. Ed. Engl., 1987, 26, 846–878 CrossRef.
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni and I. Dabo, J. Phys.: Condens. Matter, 2009, 21, 395502 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- M. Cococcioni and S. De Gironcoli, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 035105 CrossRef.
- J. E. Post, P. J. Heaney, R. B. V. Dreele and J. C. Hanson, Am. Mineral., 2003, 88, 782–788 CrossRef CAS.
- H. Christensen and A. N. Christensen, Acta Chem. Scand., Ser. A, 1978, 32, 87–88 CrossRef.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188 CrossRef.
- S. Curtarolo, W. Setyawan, G. L. W. Hart, M. Jahnatek, R. V. Chepulskii, R. H. Taylor, S. Wang, J. Xue, K. Yang, O. Levy, M. J. Mehl, H. T. Stokes, D. O. Demchenko and D. Morgan, Comput. Mater. Sci., 2012, 58, 218–226 CrossRef CAS.
- W. Setyawan and S. Curtarolo, Comput. Mater. Sci., 2010, 49, 299–312 CrossRef.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- Y.-F. Li and A. Selloni, ACS Catal., 2014, 4, 1148–1153 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. Relaxed coordinates of all the calculated structures are presented. See DOI: 10.1039/c9cp00157c |
| This journal is © the Owner Societies 2019 |