
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe structural basis for Ras activation of PI3Kα lipid kinase†
Mingzhen
Zhang
 a,
Hyunbum
Jang
a and
Ruth
Nussinov
*ab
a,
Hyunbum
Jang
a and
Ruth
Nussinov
*ab
aComputational Structural Biology Section, Basic Science Program, Frederick National Laboratory for Cancer Research, Frederick, MD 21702, USA. E-mail: nussinor@mail.nih.gov
bDepartment of Human Molecular Genetics and Biochemistry, Sackler School of Medicine, Tel Aviv University, Tel Aviv 69978, Israel
First published on 28th May 2019
Abstract
PI3Kα is a principal Ras effector that phosphorylates PIP2 to PIP3 in the PI3K/Akt/mTOR pathway. How Ras activates PI3K has been unclear: is Ras’ role confined to PI3K recruitment to the membrane or does Ras activation also involve allostery? Recently, we determined the mechanism of PI3Kα activation at the atomic level. We showed the vital role and significance of conformational change in PI3Kα activation. Here, by a ‘best-match for hydrogen-bonding pair’ (BMHP) computational protocol and molecular dynamics (MD) simulations, we model the atomic structure of KRas4B in complex with the Ras binding domain (RBD) of PI3Kα, striving to understand the mechanism of PI3Kα activation by Ras. Point mutations T208D, K210E, and K227E disrupt the KRas4B–RBD interface in the models, in line with the experiments. We identify allosteric signaling pathways connecting Ras to RBD in the p110α subunit. However, the observed weak allosteric signals coupled with the detailed mechanism of PI3Kα activation make us conclude that the dominant mechanistic role of Ras is likely to be recruitment and restriction of the PI3Kα population at the membrane. Thus, RTK recruits the PI3Kα to the membrane and activates it by relieving its autoinhibition exerted by the nSH2 domain, leading to exposure of the kinase domain, which permits PIP2 binding. Ras recruitment can shift the PI3Kα ensemble toward a population where the kinase domain surface and the active site position and orientation favor PIP2 insertion. This work helps elucidate Ras-mediated PI3K activation and explores the structural basis for Ras–PI3Kα drug discovery.
Introduction
Phosphatidylinositol-4,5-bisphosphate 3-kinases (PI3Ks) are a family of important lipid kinases that control and deliver cellular signals in the PI3K/Akt/mTOR pathway by phosphorylating phosphatidylinositol 4,5-bisphosphate (PIP2) to phosphatidylinositol 3,4,5-trisphosphate (PIP3) on the membrane.1 They mediate a wide array of cellular activities, including cell growth, proliferation, differentiation, migration, mobility, and apoptosis. Dysfunction of PI3K is a hallmark of human cancer.2,3 PI3Ks consist of three classes, with different sequences, expressing tissues, substrate preferences, and functions.4,5 PI3Kα is the only Class I PI3K that has frequent oncogenic mutations in cancer. PI3Kα (PI3KCA) and its antagonist phosphatase and tensin homolog (PTEN) are the second and third most highly mutated oncogenes.6PI3Kα is an obligate heterodimer, which includes the p85α regulatory subunit and p110α catalytic subunit. Their interactions stabilize the overall structure and keep PI3Kα in the inactive state.7 The N-terminal SH2 domain (nSH2 domain) in the p85α regulatory subunit plays the dominant role in PI3Kα activation.8,9 The phosphorylated tyrosine (pY) motifs in the receptor tyrosine kinases (RTKs) possess high affinity to the nSH2 domain. It recruits PI3Kα to the membrane and activates it by releasing the nSH2 domain from the p110α catalytic subunit.10,11 Calmodulin (CaM) also targets SH2 domains, recruits and activates PI3Kα by a similar mechanism.12–14 nSH2 release promotes PI3Kα membrane interaction and increases its activity.15 We recently determined the atomic-level mechanism of PI3Kα activation, which emphasizes the critical role of the conformational change in the activation. Upon nSH2 release, the C-lobe of the kinase domain moves away from the C2 domain, making the PI3Kα membrane binding surface more exposed for the membrane interactions. The activation loop in the kinase domain becomes more flexible and approaches ATP, generating an active PI3Kα conformation for substrate catalysis.16
Ras is a key PI3Kα activator.17–20 Its catalytic domain binds effectors and the disordered hypervariable region (HVR) anchors, and can regulate, Ras attachment to the membrane.21–24 The HVR contains the hydrophobic palmitoyl motif, farnesyl motif or both, depending on the type of Ras isoform.25–28 Ras activates PI3Kα by promoting its membrane recruitment and gathering the PIP2 substrate.15,29 A single-molecule study showed that HRas interactions inhibit the activity of the membrane-bound, pY-activated PI3Kα, indicating that the role of Ras in PI3Kα activation is not confined to promoting membrane interactions.30 Ras recognizes the Ras binding domain (RBD) in PI3K. The crystal structure of Ras–PI3Kγ shows that HRas recognizes the PI3Kγ's RBD through the antiparallel β-sheet interactions and the surrounding residue contacts.31
Despite its significance, the structural basis for PI3Kα activation by Ras has been elusive. Initial attempts to characterize the Ras–PI3Kα complex by crystallography have been unsuccessful, probably due to the high structural complexity of PI3Kα and the low Ras–PI3Kα binding affinity. Biochemical and in vivo data have helped uncover key residues at the Ras–PI3Kα interface, including K227 and T208 in the RBD of PI3Kα. Their mutations reduce the Ras–PI3Kα interactions in vitro and in vivo;32,33 but in the absence of structure, exactly how has been unclear. Further, the absence of a structure left the question of how Ras activates PI3K unresolved.
Here, we modeled the atomic structure of KRas4B binding to the RBD of PI3Kα by a best-match for hydrogen-bonding pair (BMHP) computational protocol and molecular dynamics (MD) simulations, striving to understand PI3Kα activation by Ras. Different from Raf's activation which requires the kinase domain dimerization via a Ras dimer or nanocluster, PI3K activation is through single Ras interaction with single RBD.31,34 KRas4B shares a general scenario in recognizing PI3K via antiparallel β-sheet interactions but differs in the interfacial residue contacts. The modeled KRas4B–RBD structure exhibited a high structural stability in the simulation and was disrupted by the experimentally-verified mutations. We observed allosteric pathways from KRas4B to PI3Kα–RBD, albeit presenting weak, likely biologically irrelevant signals.
This suggests a scenario where membrane-anchored Ras recruits PI3Kα to the membrane. RTK also recruits PI3Kα to the membrane. RTK's phosphorylated motifs at the C-terminal relieve the autoinhibition exerted by the nSH2 domain of the p85α, exposing the catalytic site for the PIP2 substrate to bind. However, the population with the active site favorably positioned and oriented for substrate binding may fluctuate and be limited. Ras binding to the RBD may serve to recruit, restrict and shift the PI3Kα ensemble, promoting a preorganized PIP2-binding-favored state. Because the differences in stabilities between autoinhibited and active states are often relatively small, drugging these states can be challenging.35 This work provides the atomic-level structural and dynamic basis for physiological and mutant PI3Kα activation, and thereby deeper understanding of Ras-mediated PI3K/Akt/mTOR signaling and drug discovery targeting the Ras–PI3Kα interactions.
Results
Modeling of the KRas4B–PI3Kα–RBD complex
Ras is a small GTPase, regulating multiple cellular signaling pathways via downstream effectors including Raf in the Raf/MEK/ERK pathway, RalGDS in the RalGDS/Ral pathway, and PI3K in the PI3K/Akt pathway.36 Ras recognizes its effectors via the highly homologous RBDs.37 The antiparallel β-sheet interaction is the signature structural feature at the Ras–effector interface. The β2 region of Ras interacts with the exposed β-strand of RBD in effectors, forming intermolecular backbone hydrogen bonds (H-bonds). Like other Ras effectors, PI3Kα contains the typical RBD for Ras recognition, with the structure resembling other Ras effectors (Raf, RalGDS, PI3Kγ, etc.). Thus, antiparallel β-sheet interactions are expected at the interface between Ras and RBD of PI3Kα. Ras uses E37 and S39 for Raf recognition,37 E37, S39 and R41 for RalGDS recognition,38 and E37, S39, and R41 for PI3Kγ recognition at the antiparallel β-sheet interface.31 Here, we developed the BMHP protocol to provide possible modes of H-bond formations at the interface of KRas4B with PI3Kα–RBD. This protocol systematically screened the antiparallel β-sheet interface by sliding the residue pairs and performed a series of optimization for the H-bond pairs. Both KRas4B and PI3Kα–RBDPI3Kα–RBD β2 strands exhibit a bent conformation, allowing at most three residue pairs at the interface (Fig. 1A). Thus, five conformers were generated, covering the most likely interfacial residue pair matches (Fig. 1B). While the antiparallel β-sheet interface was established between KRas4B and PI3Kα–RBD, structural collapse existed at the other part. This indicates that Ras–PI3Kα recognition is not a rigid docking process solely mediated by the molecular interface; instead, structural ensembles and monomer dynamics may be at play, which, in part, explain the low KRas4B–RBD binding affinity. To eliminate the collapse, we conducted short molecular dynamics (MD) simulations with external constraints on the backbone H-bonds at the antiparallel β-sheet interface. Explicit-solvent all-atom MD simulations were then performed to evaluate their structural and energetic validity. In the simulations, four of the five KRas4B–RBD models (models 1, 2, 4, and 5) lost their interfaces, while model 3 showed excellent structural stability (Fig. 1C). We further enhanced the sampling by generating more systems with different initial atom velocities and ran additional 8 μs simulations (details can be found in Method and materials section). The overall structure and the antiparallel β-sheet interface of model 3 were maintained in a total of ∼10.4 μs simulations.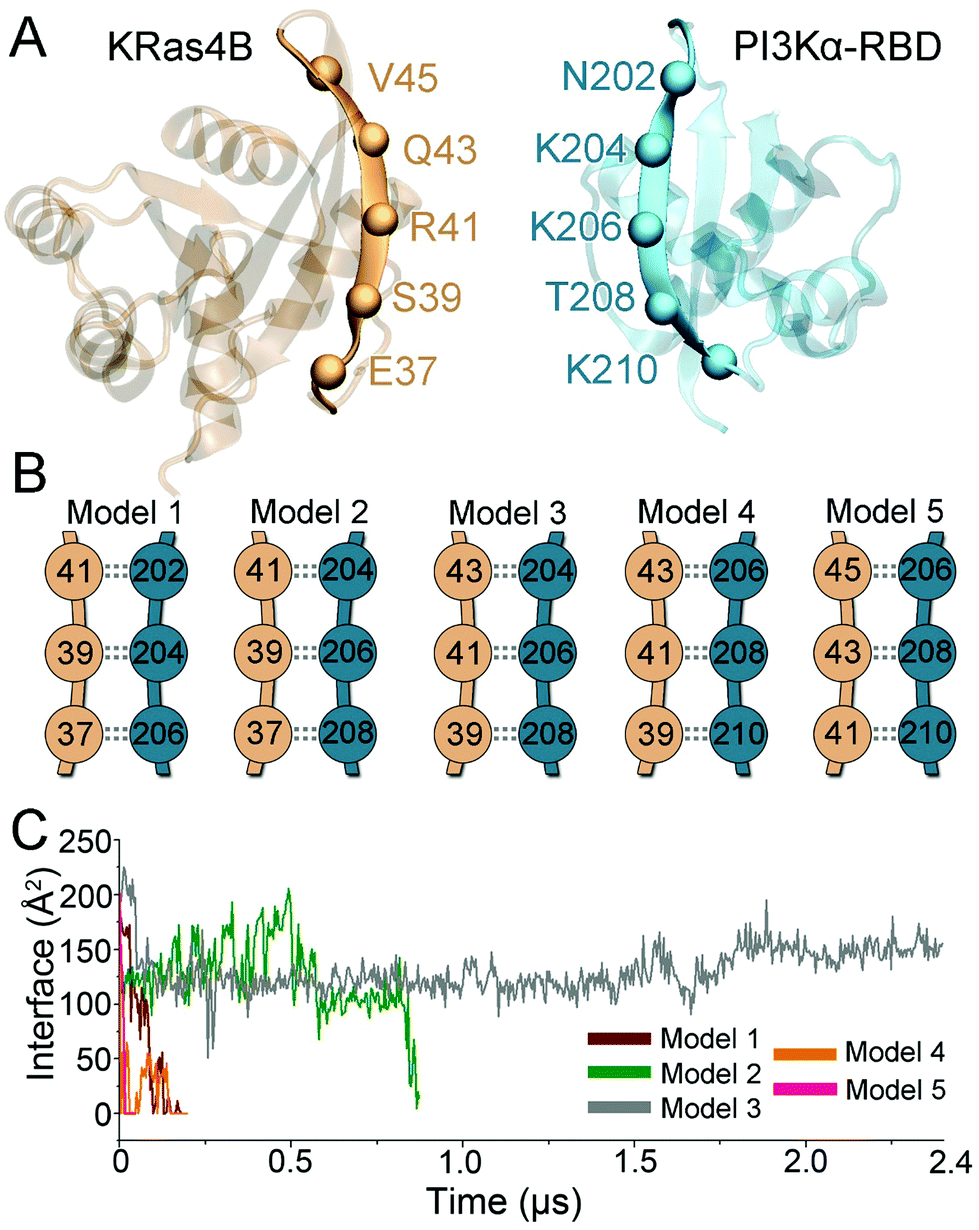 | ||
| Fig. 1 Modeling of the KRas4B–PI3Kα–RBD complex by best-match for hydrogen-bonding pair (BMHP) protocol. (A) The monomer structures for KRas4B and PI3Kα–RBD, (B) the generated models with different interfacial residue pairs and (C) the H-bond interface areas for the KRas4B–RBD conformers in the trajectories. | ||
KRas4B–PI3Kα–RBD interface
Model 3 initially had six backbone H-bonds at S39KRas4B–T208PI3Kα, R41KRas4B–K206PI3Kα, and Q43KRas4B–K204PI3Kα at the antiparallel β-sheet interface. Three of them at S39KRas4B–T208PI3Kα and R41KRas4B–K206PI3Kα, remained stable in the simulations, while the other three disappeared (Fig. 2A). A backbone H-bond also formed between E37KRas4B and K210PI3Kα due to the structural adjustment. The Switch I region of Ras shows a significant contribution to the KRas4B–RBD interface (Fig. 2B). The exposed acidic residues (D30, E31, D33, E37, and D38) in the Switch I region of KRas4B form a long negatively-charged surface, to which the basic residues in PI3Kα–RBD (K210, K227, and R230) fit. They establish strong electrostatic interactions. | ||
| Fig. 2 The structure of the stable KRas4B–PI3Kα–RBD complex is featured by (A) antiparallel β-sheets and (B) PI3Kα–RBD interaction with the Switch I region of KRas4B. | ||
We calculated the time-evolved residue distance profiles to evaluate the H-bonds and salt bridges at the KRas4B–RBD interface. Two H-bonds in the S39KRas4B–T208PI3Kα residue pair show a higher stability than others in E37KRas4B–K210PI3Kα and R41KRas4B–K206PI3Kα (Fig. 3A–D). The salt bridge between E37KRas4B and K210PI3Kα in the KRas4B–PI3Kα–RBD complex showed high stability, with very minor fluctuations (Fig. 3E). The dual salt bridge formed by D30KRas4B and E31KRas4B with R230PI3Kα was largely exposed to the solvent, resulting in a high fluctuation (Fig. 3F). K227PI3Kα interacted with D33KRas4B and D38KRas4B, forming dual salt bridges at the interface. They are more buried and exhibited higher stability (Fig. 3G). In the simulation, K227PI3Kα interacted with at least one of the two acidic residues (D33 and D38) in KRas4B.
 | ||
| Fig. 3 The distance profiles for residue pairs at the interface of KRas4B in complex with PI3Kα–RBD. | ||
Key point mutations at the KRas4B–PI3Kα–RBD interface
To verify the model, we tested mutations in the PI3Kα–RBD and explored their roles in KRas4B–RBD recognition. The first mutation was at K227PI3Kα. This mutation in PI3Kα–RBD, with charge reversal, has been shown to eliminate the Ras–PI3Kα interactions in vitro.33 The mutation also resulted in perinatal lethality in mouse with reduced PI3K/Akt/mTOR signals.32 We introduced the K227E mutation into KRas4B–PI3Kα–RBD complex and performed the simulations. In the wild-type KRas4B–RBD structure, K227PI3Kα formed a dual salt bridge with D33KRas4B and D38 KRas4B at the interface. The K227E mutation in PI3Kα–RBD resulted in repulsive forces to KRas4B Switch I region and disrupted the KRas4B–RBD interface in the simulation (Fig. 4A).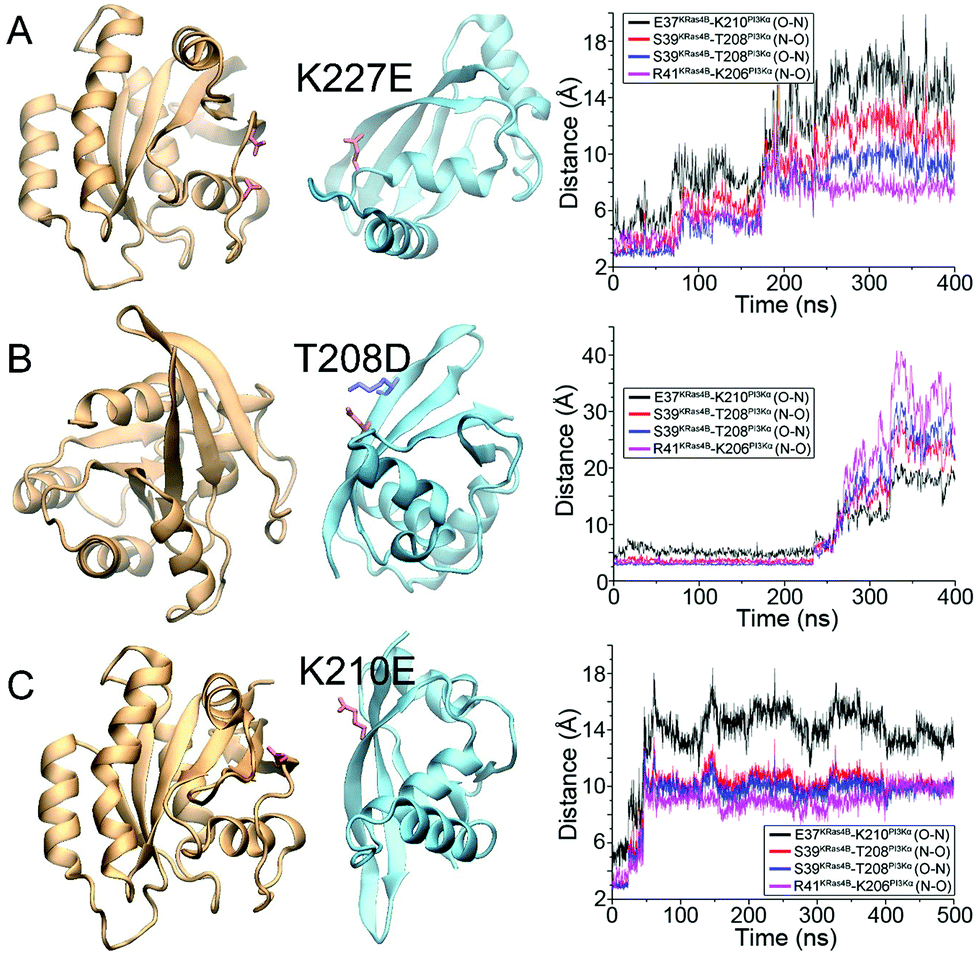 | ||
| Fig. 4 Point mutations of (A) K227E, (B) T208D, and (C) K210E in PI3Kα–RBD disrupt the KRas4B–PI3Kα–RBD complexes. The final snapshots for the mutated complexes are shown in the left panel, and the distance profiles for interfacial hydrogen bonds are shown in the right panel. | ||
The mutation T208D in PI3Kα–RBD showed a similar in vivo effect as K227E. It caused significant disorder in vivo, disrupting the PI3K/Akt/mTOR signaling in mouse.32 T208PI3Kα is a hydrophilic residue located at the β2 strand of PI3Kα–RBD. Its backbone formed key H-bonds with KRas4B at the antiparallel β-sheet interface (S39KRas4B–T208PI3Kα). When T208 was mutated to negatively charged D208PI3Kα, its side-chain formed electrostatic interactions with the adjacent residue K206PI3Kα. This led to a pronounced structural change at the β2 region of PI3Kα–RBD (Fig. S1, ESI†). The bent conformation became flat. This disrupted the antiparallel β-sheet interface and eventually destroyed the KRas4B–PI3Kα–RBD complex in the simulation (Fig. 4B).
The mutations at K227E and T208D in PI3Kα–RBD individually emphasized the importance of the interfacial salt bridges and β-sheet interactions in KRas4B–RBD recognition. We further eliminated the other three salt bridges (D30KRas4B–R230PI3Kα, E31KRas4B–R230PI3Kα, and E37KRas4B–K210PI3Kα) by mutating residues K210 and R230 in PI3Kα–RBD, exploring their roles in stabilizing the KRas4B–RBD interface. As expected, the mutation of PI3Kα at K210E disrupted the KRas4B–RBD interface in the simulation (Fig. 4C). However, the KRas4B–PI3Kα–RBD complex with the R230E mutation maintained its overall structural integrity through the 1 μs simulation, indicating that it is not as important for the KRas4B–RBD interface as other interfacial residues (Fig. S2, ESI†). This is in line with its low stability in the KRas4B–PI3Kα–RBD complex (Fig. 3F).
Allosteric signaling may propagate from KRas4B to PI3Kα
Ras activates PI3Kα and promotes PI3K/Akt signaling. Full activation of PI3Kα requires the release of autoinhibition (release of nSH2) and enhanced membrane localization.35 In PI3Kα, RBD connects to the helical domain (the domain mediating nSH2 autoinhibition), and the kinase domain (the domain that executes substrate catalysis). We performed weighted implementation of suboptimal path (WISP) analysis39 and identified multiple allosteric signaling pathways from KRas4B to PI3Kα–RBD (Fig. 5). The allosteric signaling pathways go through the antiparallel β-sheet interface, ending at the α-helix region (residue 265–280) in RBD. KRas4B residues S39, Y40, R41 and PI3Kα residues V192, V193, I194, Y207, T208, M278, L279, G280, R281, M282, P283 are in the allosteric pathways. The α-helix region in RBD connects to the C-lobe of the kinase domain in PI3Kα, and links to the helical domain via a random coil. This indicates that Ras may deliver allosteric signals to these domains via RBD. The helical domain is the main domain that accommodates the nSH2 domain of the p85α regulatory subunit. Its dynamics, which may be affected by the KRas4B, is crucial for the release of nSH2 in PI3Kα autoinhibition. The kinase domain in the p110 subunit catalyzes the reaction. We have shown that upon nSH2 release, the C-lobe of the kinase domain moved away from the C2 domain and became more accessible for membrane interactions.16 However the absence of observable RBD conformational change, coupled with the body of the experimental30 and computational results16 lead us to conclude that even though it may assist in PI3K activation, that contribution is likely to be minor.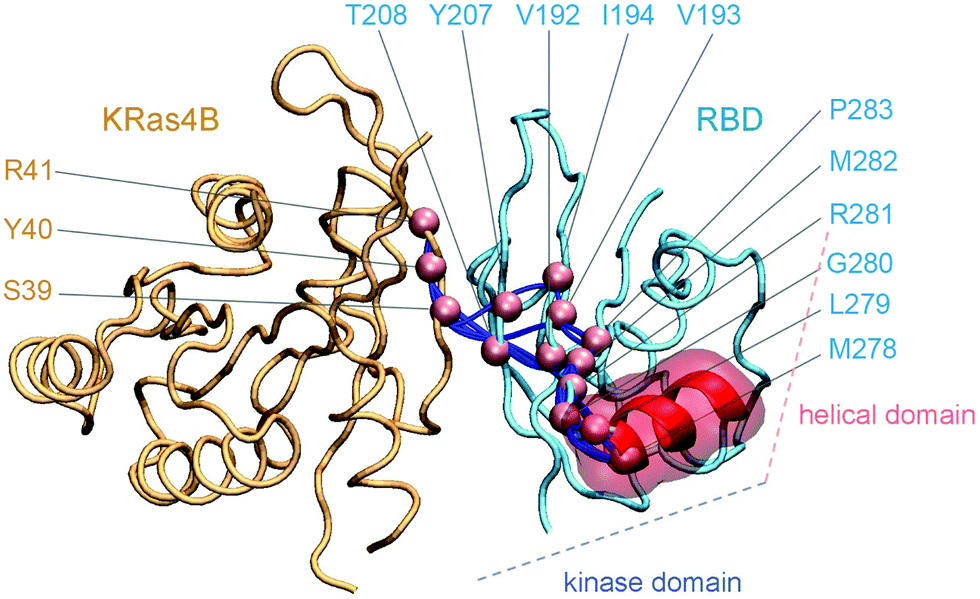 | ||
| Fig. 5 Allosteric signaling pathways through KRas4B to the RBD of PI3Kα. | ||
Discussion
PI3Kα is one of the primary Ras effectors.40 Its frequent mutations drive cancer.41–43 Despite its paramount significance in signaling and cancer drug discovery, the structural basis for PI3Kα activation by Ras has been elusive.44,45 Exploiting the BMHP protocol and MD simulations, we modeled the atomic structure of the KRas4B–PI3Kα–RBD complex, with properties in line with experiments. The antiparallel β-sheet interactions mediate the overall structural stability of the Ras–PI3Kα complex, and the salt bridges formed by the Switch I region in KRas4B with the RBD of PI3Kα contribute significantly to the Ras–PI3Kα interface. Ras follows a similar PI3Kγ recognition scenario but differs in the interfacial residue contacts. Ras interacts with both PI3Kα and PI3Kγ via the interfacial antiparallel β-sheet interactions and the extensive surrounding residue contacts.31 The structural alignment indicates that Ras employs identical residues (E37, S39, and R41) for the antiparallel β-sheet interactions with PI3Kα and PI3Kγ. Although the β2 region in PI3Kγ's RBD is shorter than PI3Kα, the antiparallel β-sheet interactions occur at the similar area (Fig. 6A). While the antiparallel β-sheet interactions show remarkable similarity, the surrounding residue contacts are different. This is largely due to sequence differences in RBD's β-sheet region and structural differences in α2 region (Fig. 6B). Notably, the interactions between Switch II region of HRas with PI3Kγ's RBD (E63HRas–K234PI3Kα) were not observed in the KRas4B–PI3Kα–RBD structure. However, when we modeled KRas4B into the full PI3Kα, the salt bridge (R73KRas4B–E888PI3Kα) was formed between Switch II region of KRas4B and the C-lobe of kinase domain in PI3Kα (Fig. 6C and D). | ||
| Fig. 6 Structural comparison of Ras interaction with PI3Kα and PI3Kγ. (A) The antiparallel β-sheet interaction and (B) the RBD α2 region in PI3Kα and PI3Kγ result in the different interfacial contacts for Ras interaction with (C) PI3Kγ and (D) PI3Kα. | ||
The Ras family of proteins includes the HRas, NRas and KRas (KRas4A and KRas4B) isoforms.28,46 All bind to PI3K.47,48 Ras isoforms share high sequence identity (∼80%). In this work, we selected KRas4B to explore the structural basis for PI3Kα activation by Ras, because of its frequent expression and thus significance in over-activating PI3Ks in cancer.49–53 The isoform-specific residues in Ras are all far away from the KRas4B–RBD interface, raising the possibility that these residues may have allosteric effects, albeit likely insignificant, since most of these isoform-specific residues are exposed on the Ras surface.54 Thus, the structure solved in this work is expected to represent a general Ras–PI3Kα interaction scenario.
Ras has frequent oncogenic mutations at G12.55 The cancer-driven mutations at G12 promote PI3K signaling but did not appear at the Ras–PI3Kα interface.56,57 We ran simulations of the KRas4B–PI3Kα–RBD complex with the G12D mutation and did not observe significant change at the KRas4B–RBD interface. Since the G12 mutation stabilizes the Switch I region of Ras and tends to allosterically release the HVR from the catalytic domain,48,58,59 it can promote Ras–PI3Kα recognition.
Ras is a membrane-attached protein with a hydrophobic HVR. The interaction of PI3Kα with Ras promotes its membrane localization, making PI3Kα more accessible to its lipid substrate.6 The RBD, to which Ras binds, connects to the helical and kinase domains in PI3Kα. Two domains are individually crucial for PI3Kα's autoinhibition and catalysis. The helical domain is the main domain in the p110α catalytic subunit that accommodates the nSH2 domain in the p85α regulatory subunit, whose release from p110α is the key event initiating PI3Kα activation. The kinase domain in the p110α subunit executes substrate catalysis. In PI3Kα activation, it moves away from the C2 domain and becomes more accessible for membrane interactions.16 The transition from an inactive to an active state executes signal transduction.22
A recent single molecule imaging study of PI3Kα activation by a receptor and HRas on a supported lipid bilayer observed synergistic activation,24 with a mechanism resembling our mechanism with phosphorylated CaM substituting for the receptor.12,13 However, contrary to the prevailing notion that Ras activates PI3K, as it does its other effectors (Raf, RalGDS, RASSF, and more), the authors observe that HRas binding inhibits PI3Kα, inhibition which the authors suggest is overcome by the membrane recruitment. HRas promotes membrane density of pY-activated PI3Kα by ∼20 fold but inhibit their activities by ∼2 fold, which, in total, lead to a ∼10-fold synergistic PI3Kα activation.30
Earlier we observed potential allosteric communication from HRas to Raf's RBD;37 however, it is questionable whether Ras is allosterically involved in Raf's activation given the long linker extending from the RBD/CRD (cysteine-rich domain) to the kinase domain. Thus, as in PI3K, Ras activates Raf by recruiting it to membrane; however, Ras assists in relieving Raf's autoinhibition not by allostery, but through high (nanomolar) binding affinity to Raf's RBD. This binding shifts the equilibrium toward Raf's open state, permitting dimerization and activation of the kinase domain.35,60–62 A similar membrane recruitment and equilibrium shift scenario was also suggested for tumor suppressor RASSF5, consistent with experimental observations.63,64 In PI3K the autoinhibition is relieved by the RTK's phosphorylated motif. This difference between Raf and PI3Kα reflects their functions: PI3K is a lipid kinase. The kinase domain has to be at the membrane. In contrast, Raf is a protein kinase. The long linker permits binding the MEK/ERK dimer complexes away from the membrane.65 At the same time, Ras assists in Raf's activation not only by recruiting the RBD to the membrane; but also, by restricting Raf's CRD fluctuations and stabilizing productive RBD–CRD orientations, a role resembling its recruitment and restriction of the PI3Kα active state at the membrane.66
Conclusions
Herein, we modeled the atomic structure of Ras binding to the PI3Kα, understanding the structural basis for PI3Kα activation by Ras. The structure presented excellent stability. The Ras–PI3Kα interface features antiparallel β-sheet interactions and residue contacts between the Switch I and II regions in Ras and PI3Kα. Mutations of key residues disrupted the Ras–PI3Kα interface, reproducing the experimental behavior. Ras isoforms have practically identical structures but with some distinctive sequences and conformational tendencies.67 Isoform-specific residues are away from the KRas4B–RBD interface, suggesting that the KRas4B–RBD complex may represent a general Ras–PI3Kα recognition scenario. The allosteric signaling pathways from KRas4B propagating to the RBD of PI3Kα appear an insignificant factor in PI3Kα activation. We favor a Ras role that involves PI3Kα recruitment to the membrane and assisting RTK in achieving full activation. RTKs act in relieving the PI3Kα autoinhibition via a conformational change that leads to exposure of the PI3K active site to the membrane, permitting the PIP2 substrate to bind there; however, it may not position and orient the kinase domain surface at a catalytically-favored state with respect to the membrane and the PIP2 substrate. Active Ras is at the membrane. Through binding to the PI3Kα RDB, it restricts the ensemble, thrusting it against the membrane in a PIP2-binding-favored state. Thus, even though RTKs can activate PI3K, coupling with Ras accomplishes full activation. The KRas4B–PI3Kα structure in this work sheds an atomistic-level light on how Ras activates PI3Kα and as such can offer the structural basis for the cancer drug discovery targeting the Ras–PI3Kα interactions.Materials and methods
KRas4B–PI3Kα–RBD complex
The initial coordinates of PI3Kα (PDB code: 4OVV) and GTP-bound KRas4B (PDB code: 3GFT) were obtained from the protein data bank. We extracted the RBD from the full PI3Kα structure and modeled it with KRas4B-GTP. The KRas4B–PI3Kα–RBD complex was modeled with antiparallel β-sheet interactions. The residues in β2Ras and β2RBD were aligned with different residue matches to generate five models. Each generated KRas4B–RBD model contained three residue pairs with six backbone hydrogen bonds. To test the structural validity of our model, we performed explicit-solvent all-atom MD simulations for five model complexes and confirmed one (M3 model) with excellent stability. To enhance the sampling, eight additional simulations were performed for the confirmed M3 model of KRas4B–RBD complex with different initial atom velocities. For each system, 1 μs simulation was performed. Meanwhile, the simulations of the KRas4B–RBD complex (M3 model) with five mutations were individually performed to explore their roles in KRas4B–PI3Kα recognition, i.e., T208D, K210E, K227E, and R230E mutations in PI3Kα, and a G12D mutation in KRas4B. The modeled KRas4B–RBD interface was then used to model KRas4B into the full PI3Kα.Simulation protocols
Production MD simulation runs were conducted by the NAMD package68 with the CHARMM all-atom additive force field (version C36).69 The NPT ensemble was controlled with the temperature of 310 K and pressure of 1 atm. The explicit TIP3 water model was used to solvate the systems in the isometric unit cell box. The system was neutralized by Na+ and Cl− ions, achieving a 0.15 mol L−1 ion concentration. Short-range van der Waals (vdW) and the long-rang electrostatic interactions were described by the switch function and the particle mesh Ewald (PME) algorithm. A time step of 2 fs was employed in the MD simulations. The allosteric pathways from KRas4B to PI3K's RBD were calculated by the weighted implementation of suboptimal paths (WISP) algorithm.39 WISP algorithm identified the primary residue communication path. The allosteric pathways are between R41KRas4B to M278PI3Kα for the aligned KRas4B–RBD complexes in the trajectories. The visualization of allosteric pathway was performed by VMD software.Author contributions
MZ, HJ, and RN conceived and designed the study. MZ conducted most of the simulations and analyzed the results. MZ, HJ, and RN wrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has been funded in whole or in part with Federal funds from the Frederick National Laboratory for Cancer Research, National Institutes of Health, under contract HHSN261200800001E. This research was supported (in part) by the Intramural Research Program of NIH, Frederick National Lab, Center for Cancer Research. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products or organizations imply endorsement by the high-performance computational facilities of the Biowuilf PC/Linux cluster at the National Institutes of Health, Bethesda, MD, USA (https://hpc.nih.gov/).References
- O. Vadas, J. E. Burke, X. Zhang, A. Berndt and R. L. Williams, Sci. Signaling, 2011, 4, re2 CrossRef PubMed.
- L. Stephens, R. Williams and P. Hawkins, Curr. Opin. Pharmacol., 2005, 5, 357–365 CrossRef CAS PubMed.
- L. M. Thorpe, H. Yuzugullu and J. J. Zhao, Nat. Rev. Cancer, 2015, 15, 7–24 CrossRef CAS PubMed.
- E. H. Walker, O. Perisic, C. Ried, L. Stephens and R. L. Williams, Nature, 1999, 402, 313–320 CrossRef CAS PubMed.
- Y. Ito, J. R. Hart, L. Ueno and P. K. Vogt, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 16826–16829 CrossRef CAS PubMed.
- M. S. Lawrence, P. Stojanov, C. H. Mermel, J. T. Robinson, L. A. Garraway, T. R. Golub, M. Meyerson, S. B. Gabriel, E. S. Lander and G. Getz, Nature, 2014, 505, 495–501 CrossRef CAS PubMed.
- L. M. Thorpe, J. M. Spangle, C. E. Ohlson, H. Cheng, T. M. Roberts, L. C. Cantley and J. J. Zhao, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 7095–7100 CrossRef CAS PubMed.
- J. Yu, C. Wjasow and J. M. Backer, J. Biol. Chem., 1998, 273, 30199–30203 CrossRef CAS PubMed.
- J. E. Burke, O. Perisic, G. R. Masson, O. Vadas and R. L. Williams, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15259–15264 CrossRef CAS PubMed.
- R. T. Nolte, M. J. Eck, J. Schlessinger, S. E. Shoelson and S. C. Harrison, Nat. Struct. Biol., 1996, 3, 364–374 CrossRef CAS PubMed.
- S. B. Gabelli, I. Echeverria, M. Alexander, K. C. Duong-Ly, D. Chaves-Moreira, E. T. Brower, B. Vogelstein and L. M. Amzel, Biophys. Rev., 2014, 6, 89–95 CrossRef CAS PubMed.
- M. Zhang, Z. Li, G. Wang, H. Jang, D. B. Sacks, J. Zhang, V. Gaponenko and R. Nussinov, J. Phys. Chem. B, 2018, 122(49), 11137–11146 CrossRef CAS PubMed.
- M. Zhang, H. Jang, V. Gaponenko and R. Nussinov, Biophys. J., 2017, 113, 1956–1967 CrossRef CAS PubMed.
- J. L. Joyal, D. J. Burks, S. Pons, W. F. Matter, C. J. Vlahos, M. F. White and D. B. Sacks, J. Biol. Chem., 1997, 272, 28183–28186 CrossRef CAS PubMed.
- W. C. Hon, A. Berndt and R. L. Williams, Oncogene, 2012, 31, 3655–3666 CrossRef CAS PubMed.
- M. Zhang, H. Jang and R. Nussinov, Chem. Sci., 2019, 10, 3671–3680 RSC.
- E. Castellano and J. Downward, Genes Cancer, 2011, 2, 261–274 CrossRef CAS PubMed.
- M. M. Murillo, S. Zelenay, E. Nye, E. Castellano, F. Lassailly, G. Stamp and J. Downward, J. Clin. Invest., 2014, 124, 3601–3611 CrossRef CAS PubMed.
- K. Tsutsumi, Y. Fujioka, M. Tsuda, H. Kawaguchi and Y. Ohba, Cell. Signalling, 2009, 21, 1672–1679 CrossRef CAS PubMed.
- J. Wang, Y. Yuan, Y. Zhou, L. Guo, L. Zhang, X. Kuai, B. Deng, Z. Pan, D. Li and F. He, J. Proteome Res., 2008, 7, 3879–3889 CrossRef CAS PubMed.
- R. Nussinov, C. J. Tsai and H. Jang, Semin. Cancer Biol., 2018, 54, 109–113 CrossRef PubMed.
- T. S. Chavan, H. Jang, L. Khavrutskii, S. J. Abraham, A. Banerjee, B. C. Freed, L. Johannessen, S. G. Tarasov, V. Gaponenko, R. Nussinov and N. I. Tarasova, Biophys. J., 2015, 109, 2602–2613 CrossRef CAS PubMed.
- H. Jang, A. Banerjee, T. S. Chavan, S. Lu, J. Zhang, V. Gaponenko and R. Nussinov, FASEB J., 2016, 30, 1643–1655 CrossRef CAS PubMed.
- T. C. Buckles, B. P. Ziemba, G. R. Masson, R. L. Williams and J. J. Falke, Biophys. J., 2017, 113, 2396–2405 CrossRef CAS PubMed.
- H. Jang, A. Banerjee, T. Chavan, V. Gaponenko and R. Nussinov, J. Biol. Chem., 2017, 292, 12544–12559 CrossRef CAS PubMed.
- H. Jang, S. J. Abraham, T. S. Chavan, B. Hitchinson, L. Khavrutskii, N. I. Tarasova, R. Nussinov and V. Gaponenko, J. Biol. Chem., 2015, 290, 9465–9477 CrossRef CAS PubMed.
- D. Abankwa, A. A. Gorfe, K. Inder and J. F. Hancock, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 1130–1135 CrossRef CAS PubMed.
- R. Nussinov, C. J. Tsai and H. Jang, Cancer Res., 2018, 78, 593–602 CrossRef CAS PubMed.
- A. Banerjee, H. Jang, R. Nussinov and V. Gaponenko, Curr. Opin. Struct. Biol., 2016, 36, 10–17 CrossRef CAS PubMed.
- T. C. Buckles, B. P. Ziemba, G. R. Masson, R. L. Williams and J. J. Falke, Biophys. J., 2017, 113, 2396–2405 CrossRef CAS PubMed.
- M. E. Pacold, S. Suire, O. Perisic, S. Lara-Gonzalez, C. T. Davis, E. H. Walker, P. T. Hawkins, L. Stephens, J. F. Eccleston and R. L. Williams, Cell, 2000, 103, 931–943 CrossRef CAS PubMed.
- S. Gupta, A. R. Ramjaun, P. Haiko, Y. Wang, P. H. Warne, B. Nicke, E. Nye, G. Stamp, K. Alitalo and J. Downward, Cell, 2007, 129, 957–968 CrossRef CAS PubMed.
- P. Rodriguez-Viciana, P. H. Warne, B. Vanhaesebroeck, M. D. Waterfield and J. Downward, EMBO J., 1996, 15, 2442–2451 CrossRef CAS PubMed.
- R. Nussinov, C. J. Tsai and H. Jang, Semin. Cancer Biol., 2018, 54, 114–120 CrossRef PubMed.
- R. Nussinov, M. Zhang, C. J. Tsai, T. J. Liao, D. Fushman and H. Jang, Biophys. Rev., 2018, 10, 1263–1282 CrossRef CAS PubMed.
- I. M. Ahearn, K. Haigis, D. Bar-Sagi and M. R. Philips, Nat. Rev. Mol. Cell Biol., 2011, 13, 39–51 CrossRef PubMed.
- S. K. Fetics, H. Guterres, B. M. Kearney, G. Buhrman, B. Ma, R. Nussinov and C. Mattos, Structure, 2015, 23, 505–516 CrossRef CAS PubMed.
- L. Huang, F. Hofer, G. S. Martin and S. H. Kim, Nat. Struct. Biol., 1998, 5, 422–426 CrossRef CAS PubMed.
- A. T. Van Wart, J. Durrant, L. Votapka and R. E. Amaro, J. Chem. Theory Comput., 2014, 10, 511–517 CrossRef CAS PubMed.
- L. C. Chang, H. M. Chiu, C. T. Shun, J. T. Liang, J. T. Lin, C. C. Chen, Y. C. Lee and M. S. Wu, BMC Gastroenterol., 2014, 14, 221 CrossRef PubMed.
- N. Miled, Y. Yan, W. C. Hon, O. Perisic, M. Zvelebil, Y. Inbar, D. Schneidman-Duhovny, H. J. Wolfson, J. M. Backer and R. L. Williams, Science, 2007, 317, 239–242 CrossRef CAS PubMed.
- L. Zhao and P. K. Vogt, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 2652–2657 CrossRef CAS PubMed.
- T. Pantsar, S. Rissanen, D. Dauch, T. Laitinen, I. Vattulainen and A. Poso, PLoS Comput. Biol., 2018, 14(9), e1006458 CrossRef PubMed.
- T. K. Owonikoko and F. R. Khuri, American Society of Clinical Oncology Educational Book, 2013 Search PubMed.
- E. Pons-Tostivint, B. Thibault and J. Guillermet-Guibert, Trends Cancer, 2017, 3, 454–469 CrossRef CAS PubMed.
- R. Nussinov, C. J. Tsai, M. Chakrabarti and H. Jang, Cancer Res., 2016, 76, 18–23 CrossRef CAS PubMed.
- P. Rodriguez-Viciana, P. H. Warne, R. Dhand, B. Vanhaesebroeck, I. Gout, M. J. Fry, M. D. Waterfield and J. Downward, Nature, 1994, 370, 527–532 CrossRef CAS PubMed.
- S. Lu, A. Banerjee, H. Jang, J. Zhang, V. Gaponenko and R. Nussinov, J. Biol. Chem., 2015, 290, 28887–28900 CrossRef CAS PubMed.
- R. Nussinov, G. Wang, C. J. Tsai, H. Jang, S. Lu, A. Banerjee, J. Zhang and V. Gaponenko, Trends Cancer, 2017, 3, 214–224 CrossRef CAS PubMed.
- R. Nussinov, S. Muratcioglu, C. J. Tsai, H. Jang, A. Gursoy and O. Keskin, Expert Opin. Ther. Targets, 2016, 20, 831–842 CrossRef CAS PubMed.
- R. Nussinov, C. J. Tsai, S. Muratcioglu, H. Jang, A. Gursoy and O. Keskin, Expert Rev. Proteomics, 2015, 12, 669–682 CrossRef CAS PubMed.
- R. Nussinov, H. Jang, C. J. Tsai, T. J. Liao, S. Li, D. Fushman and J. Zhang, Cell. Mol. Life Sci., 2017, 74, 3245–3261 CrossRef CAS PubMed.
- R. Nussinov, C. J. Tsai and H. Jang, Expert Rev. Proteomics, 2016, 13, 711–716 CrossRef CAS PubMed.
- R. Nussinov and C. J. Tsai, Cell, 2013, 153, 293–305 CrossRef CAS PubMed.
- G. A. Hobbs, C. J. Der and K. L. Rossman, J. Cell Sci., 2016, 129, 1287–1292 CrossRef CAS PubMed.
- J. A. Engelman, L. Chen, X. Tan, K. Crosby, A. R. Guimaraes, R. Upadhyay, M. Maira, K. McNamara, S. A. Perera, Y. Song, L. R. Chirieac, R. Kaur, A. Lightbown, J. Simendinger, T. Li, R. F. Padera, C. Garcia-Echeverria, R. Weissleder, U. Mahmood, L. C. Cantley and K. K. Wong, Nat. Med., 2008, 14, 1351–1356 CrossRef CAS PubMed.
- H. Y. Fan, M. Shimada, Z. Liu, N. Cahill, N. Noma, Y. Wu, J. Gossen and J. S. Richards, Development, 2008, 135, 2127–2137 CrossRef CAS PubMed.
- S. Lu, H. Jang, S. Muratcioglu, A. Gursoy, O. Keskin, R. Nussinov and J. Zhang, Chem. Rev., 2016, 116, 6607–6665 CrossRef CAS PubMed.
- S. Lu, H. Jang, S. Gu, J. Zhang and R. Nussinov, Chem. Soc. Rev., 2016, 45, 4929–4952 RSC.
- A. Tse and G. M. Verkhivker, PLoS One, 2016, 11(11), e0166583 CrossRef PubMed.
- G. M. Verkhivker, Mol. BioSyst., 2016, 12, 3146–3165 RSC.
- K. A. Marino, L. Sutto and F. L. Gervasio, J. Am. Chem. Soc., 2015, 137, 5280–5283 CrossRef CAS PubMed.
- T. J. Liao, H. Jang, C. J. Tsai, D. Fushman and R. Nussinov, Phys. Chem. Chem. Phys., 2017, 19, 6470–6480 RSC.
- T. J. Liao, C. J. Tsai, H. Jang, D. Fushman and R. Nussinov, Curr. Opin. Struct. Biol., 2016, 41, 217–224 CrossRef CAS PubMed.
- E. Santos and P. Crespo, Sci. Signaling, 2018, 11(11), eaav0917 CrossRef PubMed.
- S. Li, H. Jang, J. Zhang and R. Nussinov, Structure, 2018, 26(513-525), e512 Search PubMed.
- C. W. Johnson, D. Reid, J. A. Parker, S. Salter, R. Knihtila, P. Kuzmic and C. Mattos, J. Biol. Chem., 2017, 292, 12981–12993 CrossRef CAS PubMed.
- J. C. Phillips, R. Braun, W. Wang, J. Gumbart, E. Tajkhorshid, E. Villa, C. Chipot, R. D. Skeel, L. Kale and K. Schulten, J. Comput. Chem., 2005, 26, 1781–1802 CrossRef CAS PubMed.
- B. R. Brooks, C. L. Brooks, 3rd, A. D. Mackerell, Jr., L. Nilsson, R. J. Petrella, B. Roux, Y. Won, G. Archontis, C. Bartels, S. Boresch, A. Caflisch, L. Caves, Q. Cui, A. R. Dinner, M. Feig, S. Fischer, J. Gao, M. Hodoscek, W. Im, K. Kuczera, T. Lazaridis, J. Ma, V. Ovchinnikov, E. Paci, R. W. Pastor, C. B. Post, J. Z. Pu, M. Schaefer, B. Tidor, R. M. Venable, H. L. Woodcock, X. Wu, W. Yang, D. M. York and M. Karplus, J. Comput. Chem., 2009, 30, 1545–1614 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cp00101h |
| This journal is © the Owner Societies 2019 |