Rethinking the X− + CH3Y [X = OH, SH, CN, NH2, PH2; Y = F, Cl, Br, I] SN2 reactions†
Received
27th December 2018
, Accepted 15th March 2019
First published on 18th March 2019
Abstract
Moving beyond the textbook mechanisms of bimolecular nucleophilic substitution (SN2) reactions, we characterize several novel stationary points and pathways for the reactions of X− [X = OH, SH, CN, NH2, PH2] nucleophiles with CH3Y [Y = F, Cl, Br, I] molecules using the high-level explicitly-correlated CCSD(T)-F12b method with the aug-cc-pVnZ(-PP) [n = D, T, Q] basis sets. Besides the not-always-existing traditional pre- and post-reaction ion-dipole complexes, X−⋯H3CY and XCH3⋯Y−, and the Walden-inversion transition state, [X–CH3–Y]−, we find hydrogen-bonded X−⋯HCH2Y (X = OH, CN, NH2; Y ≠ F) and front-side H3CY⋯X− (Y ≠ F) complexes in the entrance and hydrogen-bonded XH2CH⋯Y− (X = SH, CN, PH2) and H3CX⋯Y− (X = OH, SH, NH2) complexes in the exit channels depending on the nucleophile and leaving group as indicated in parentheses. Retention pathways via either a high-energy front-side attack barrier, XYCH3−, or a novel double-inversion transition state, XH⋯CH2Y−, having lower energy for X = OH, CN, and NH2 and becoming submerged (barrier-less) for X = OH and Y = I as well as X = NH2 and Y = Cl, Br, and I, are also investigated.
I. Introduction
Since the discovery of an optical inversion in a chemical reaction by Paul Walden in 1896,1 bimolecular nucleophilic substitution (SN2) has become a widely known reaction class in organic chemistry. The atomic-level mechanisms of SN2 reactions were already described in the 1930s by Ingold and co-workers.2 The traditional SN2 Walden-inversion pathway goes through a double-well potential featuring ion-dipole complexes in the entrance- and exit-channel wells separated by a central transition state (first-order saddle point). In a typical SN2 reaction X− + CH3Y, the nucleophile (X−) attacks the back side (methyl side) of the CH3Y molecule and while a new X–C bond forms and the C–Y bond breaks an umbrella motion of the H atoms inverts the configuration around the carbon center. The above-described Walden-inversion mechanism of SN2 reactions has become textbook material and is probably the best-known stereo-specific reaction pathway in chemistry. However, in 2016 Xie and Hase3 published a perspective article “rethinking the SN2 reaction”, because “the gas-phase dynamics of a paradigm organic reaction are more complex than expected”. This statement on the complexity of SN2 reactions was based on recent experimental and theoretical findings revealing several novel and unexpected reaction mechanisms for these paradigm systems.4–9 For example, SN2 reactions can proceed with direct rebound, stripping, and front-side attack mechanisms as well as via indirect ion-dipole, hydrogen-bond, and front-side complex forming, roundabout,4 and double-inversion7 pathways.3 Among the above mechanisms front-side attack is a retention pathway, which proceeds via a high-energy XYCH3−-like transition state with X–C–Y angle of around 80°.10 This front-side attack retention pathway has been known from the early 1930s;2 however, a quantitative description of the front-side attack dynamics was just recently achieved in our group utilizing newly developed analytical ab initio potential energy surfaces.7,11,12 Furthermore, our reaction dynamics simulations on these analytical potentials revealed a new retention mechanism called double inversion,7 where a proton-abstraction induced inversion (first inversion) is followed by a Walden inversion (second inversion) resulting in retention of the initial configuration. The non-traditional front-side complex (H3CY⋯X−) formation was also quantitatively characterized by our trajectory simulations13 motivated by the joint work with the experimental group of Wester and co-workers.8 Another non-traditional pre-reaction complex stabilized by a hydrogen-bond between X− and the methyl group of CH3Y was also found first by Hase and co-workers14 and later in our theoretical studies.7,11,12,15
In the present work we focus on “rethinking” the gas-phase X− + CH3Y [X = OH, SH, CN, NH2, PH2; Y = F, Cl, Br, I] SN2 reactions using high-level ab initio methods. Among the 20 possible fundamental SN2 reactions with the above-defined 5 different nucleophiles (X−) and 4 leaving groups (Y), there are only a few which were studied previously in the gas16–24 and/or condensed25–29 phases, and almost none of them in view of the recent non-traditional mechanisms. Focusing on the gas-phase studies, in the case of Y = F Gonzales et al.16,17 characterized 3 stationary points, i.e., pre- and post-reaction complexes and a Walden-inversion transition state, for each reaction; however, front-side complex formation, a front-side attack transition state, and double inversion were not investigated, partially due to the fact that some of these were not known at that time. For OH− + CH3F Hase and co-workers18,19 described the same 3 stationary points as Gonzales et al.16,17 and reported interesting direct dynamics results showing that trajectories usually avoid the deep post-reaction minimum.18 Considering ligands other than Y = F, Tachikawa and co-workers20 investigated again 3 stationary points for the OH− + CH3Cl reaction and recently Wester and co-workers21 reported crossed-beam experiments for the CN− + CH3I system and characterized 3 stationary points for each of the C–C and C–N bond forming SN2 reactions. In the case of X = OH and SH, the X− + CH3Y [Y = F, Cl, Br] reactions were investigated by Longo and co-workers22 reporting again 3 stationary points for each system. Perhaps the most thoroughly studied reaction is OH− + CH3I, for which H-bonded and front-side complex formation were studied in the Hase group24 and experiments were performed in the groups of Viggiano23 and Wester.24 In 2018 we reported a high-level ab initio study on the OH− + CH3Y [Y = F, Cl, Br, I] SN2 reactions applying core- and post-CCSD(T) correlation corrections, which are usually neglected in the literature, and revealing many stationary points involving H-bonded and front-side complexes as well as front-side attack and double-inversion transition states.30 In the present work we extend our previous studies9,30 considering SH−, CN−, NH2−, and PH2− nucleophiles besides OH−, which is described again for completeness and comparison. For the first time for X = SH, CN, NH2, and PH2 we use the high-level modern explicitly-correlated CCSD(T)-F12b method31 to obtain benchmark structures and relative energies for the stationary points. However, instead of presenting technical details of a high-level electronic structure study, the present work aims to provide new qualitative insights into the reaction pathways of 20 different SN2 reactions, showing that these systems “are more complex than expected”,3 supporting the views of the above-mentioned “rethinking” perspective article.3
II. Computational details
The energies, geometries, and harmonic vibrational frequencies of the stationary points are computed using second-order Møller–Plesset perturbation theory32 (MP2) with the correlation-consistent aug-cc-pVDZ basis set33 and the explicitly correlated coupled cluster singles, doubles, and perturbative triples CCSD(T)-F12b method31 with the aug-cc-pVDZ and the aug-cc-pVTZ basis sets.33 To obtain the benchmark classical relative energies at the CCSD(T)-F12b/aug-cc-pVTZ geometries single-point computations are performed using the CCSD(T)-F12b method with the quadruple-ζ aug-cc-pVQZ basis set.33 For bromine and iodine relativistic small-core effective core potentials34 and the corresponding pseudo-potential basis sets34 are employed.
The benchmark adiabatic relative energies are determined as:
| | | ΔE[CCSD(T)-F12b/aug-cc-pVQZ] + ΔZPE[CCSD(T)-F12b/aug-cc-pVTZ] | (1) |
where Δ
E is the benchmark classical relative energy and ΔZPE is the harmonic zero-point energy correction. Note that if the CCSD(T)-F12b/aug-cc-pVTZ or the CCSD(T)-F12b/aug-cc-pVDZ geometry optimizations do not converge the CCSD(T)-F12b/aug-cc-pVDZ or the MP2/aug-cc-pVDZ structures are utilized, respectively.
All the structural parameters, harmonic frequencies, absolute energies, and relative energies can be found in the ESI.† For the best stationary-point geometries Cartesian coordinates are also given in the ESI† in a software-friendly format. We note that in the case of some minima small artificial imaginary frequencies (<100i cm−1) are found most likely due to the uncertainty of the numerical gradient and/or Hessian computations. For the present ab initio study the Molpro program package35 is used.
III. Results and discussion
OH− + CH3Y [Y = F, Cl, Br, I]
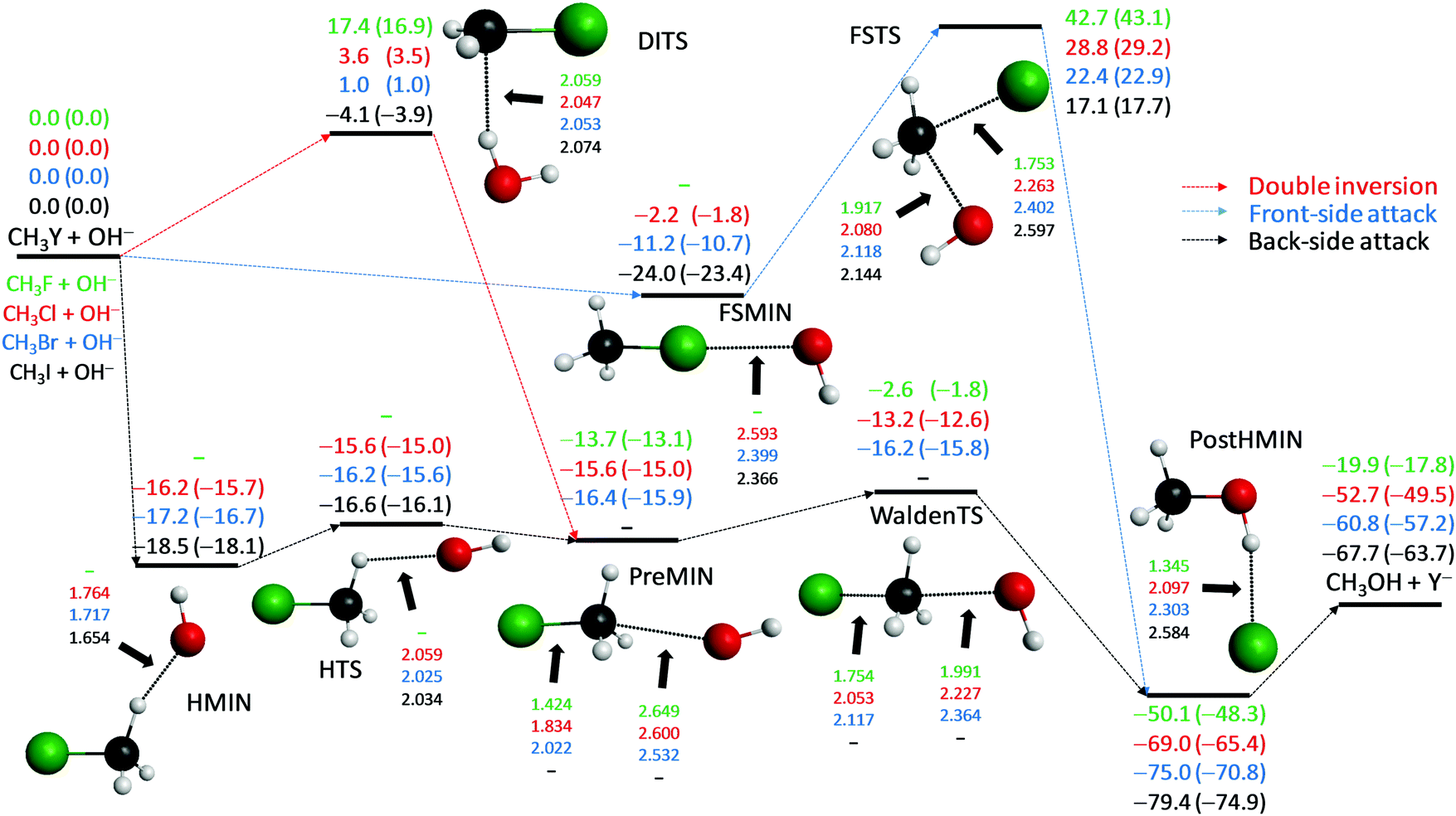
The OH− + CH3Y [Y = F, Cl, Br, I] SN2 reactions are exothermic and may proceed via back-side attack inversion, front-side attack retention, or double-inversion retention pathways as shown in Fig. 1. Back-side attack is a collective name for several direct and indirect inversion mechanisms such as rebound, stripping, ion-dipole complex formation, hydrogen-bond complex formation, front-side complex formation, and roundabout. The traditional picture says that back-side attack Walden inversion proceeds via a pre-reaction ion-dipole complex (PreMIN), a Walden-inversion transition state (WaldenTS), and a post-reaction ion-dipole complex HOCH3⋯Y−. However, instead of the traditional HOCH3⋯Y− complex, Y− connects to the OH group with a single hydrogen bond forming a PostHMIN complex for all the four leaving groups, as shown in Fig. 1. Thus, Walden inversion of OH− + CH3F proceeds as PreMIN → WaldenTS → PostHMIN. (Note that besides PostHMIN a hydrogen-bonded F−⋯HCH2OH complex also exists, which is not shown in Fig. 1, but presented in the ESI.†) This is not the end of the nontraditional mechanisms, because for Y = Cl, Br, and I a hydrogen-bonded pre-reaction complex (HMIN), where OH− connects to one of the H atoms of the methyl group, and a transition state (HTS) connecting HMIN and PREMIN come into play, where HMIN is below PREMIN by about 2 kcal mol−1. In the case of Y = Cl and Br, OH− + CH3Y Walden inversion occurs via HMIN → HTS → PreMIN → WaldenTS → PostHMIN. However, for Y = I the traditional PreMIN and WaldenTS do not exist and the OH− + CH3I inversion proceeds as HMIN → HTS → PostHMIN. Furthermore, for Y = Cl, Br, and I a front-side complex (FSMIN), where OH− connects to Y, can also be found, which in the case of Y = I is actually a significantly deeper minimum than HMIN, suggesting a steering effect into a nonreactive orientation, thereby making the OH− + CH3I reaction indirect as previous experiments5,8 and our dynamics simulations13 suggested for F− + CH3I. Unlike back-side attack inversion which has submerged stationary points, front-side attack retention proceeds via a high-energy transition state (FSTS) with adiabatic barrier heights of 43.1 (F), 29.2 (Cl), 22.9 (Br), and 17.7 (I) kcal mol−1, whereas the corresponding double-inversion transition state (DITS) barrier heights are only 16.9 (F), 3.5 (Cl), 1.0 (Br), and −3.9 (I) kcal mol−1. Thus, double inversion opens a barrier-less retention pathway for the OH− + CH3I SN2 reaction.
 |
| | Fig. 1 Schematic potential energy surfaces of the OH− + CH3Y [Y = F, Cl, Br, I] SN2 reactions showing the classical (adiabatic) CCSD(T)-F12b/aug-cc-pVQZ (+ΔZPE[CCSD(T)-F12b/aug-cc-pVTZ]) relative energies, in kcal mol−1, and the most important CCSD(T)-F12b/aug-cc-pVTZ structural parameters, in Å, of the stationary points along the different reaction pathways. For core and post-CCSD(T) correlation corrected energies and their comparison with experimental reaction enthalpies see ref. 30. | |
SH− + CH3Y [Y = F, Cl, Br, I]
If we replace the OH− nucleophile with SH−, the SN2 reactions become less exothermic and, moreover, endothermic for Y = F, as shown in Fig. 2. Unlike OH−, SH− does not form a hydrogen-bonded pre-reaction complex, except for Y = I as shown in the ESI,† and the back-side attack inversion pathway proceeds as PreMIN → WaldenTS → PostHMIN. The WaldenTSs are below the reactants by 1.4, 6.0, and 8.5 kcal mol−1, for Y = Cl, Br, and I, respectively, whereas the SH− + CH3F SN2 reaction has a positive adiabatic barrier height of 13.7 kcal mol−1. Similar to the reactions of OH−, the global minimum of the potential energy surfaces of the SH− + CH3Y systems is PostHMIN, but in the present case Y−⋯HC bonded (PostHMIN2) and traditional ion-dipole post-reaction (WaldenPostMIN) complexes are also found with similar energies within 1 kcal mol−1 and above POSTHMIN by a few kcal mol−1, except for Y = F, where PostHMIN2 is just slightly bonded and WaldenPostMIN does not exist, as shown in Fig. 2. The dissociation energies of the PostHMIN complexes are around 10 kcal mol−1 for Y = Cl, Br, and I, whereas much larger, about 38 kcal mol−1, for Y = F. In the case of X = OH, the Y = Cl, Br, and I PostHMIN complexes are more stable by a few kcal mol−1, however, the Y = F complex has a dissociation energy of only about 30 kcal mol−1. These energy trends are in accord with the Y−⋯HX distances, as these are slightly longer for X = SH than OH if Y = Cl, Br, and I, whereas the F−⋯HS distance (0.985 Å) is significantly shorter than the F−⋯HO distance (1.345 Å) as shown in Fig. 1 and 2. Similar to the OH− + CH3Y [Y = Cl, Br, I] reactions, FSMIN complexes are also found for X = SH systems, but with longer Y⋯X− distances and consequently with less stability. Here the H3CI⋯SH− complex is just similarly stable to PreMIN, whereas FSMIN is a significantly deeper minimum than PREMIN in the case of the OH− complex. We find qualitatively similar DITS and FSTS structures for the reactions of OH− and SH−, with significantly higher barrier heights for SH−. Furthermore, unlike in the case of the OH− systems, the DITSs of the SH− nucleophile are always above the corresponding FSTSs.
 |
| | Fig. 2 Schematic potential energy surfaces of the SH− + CH3Y [Y = F, Cl, Br, I] SN2 reactions showing the classical (adiabatic) CCSD(T)-F12b/aug-cc-pVQZ (+ΔZPE[CCSD(T)-F12b/aug-cc-pVTZ]) relative energies, in kcal mol−1, and the most important CCSD(T)-F12b/aug-cc-pVTZ structural parameters, in Å, of the stationary points along the different reaction pathways. Results indexed by * correspond to CCSD(T)-F12b/aug-cc-pVDZ structures. | |
CN− + CH3Y [Y = F, Cl, Br, I]
The CN− nucleophile can form either a C–C or a C–N bond in the SN2 reactions with the CH3Y molecules. In the present work we study the energetically favored, more exothermic C–C bond formation channel leading to Y− + CH3CN products. The CN− + CH3Y reactions are more and less exothermic than the corresponding SH− and OH− reactions, respectively. Since the CN ligand does not have a hydrogen atom, PostHMIN-type post-reaction complexes do not exist. Here the global minimum is either a traditional ion-dipole WaldenPostMIN (C3v symmetry) or a hydrogen-bonded PostHMIN2 (Cs symmetry) complex, whose energies agree within 1 kcal mol−1 as shown in Fig. 3. Note that for Y = F, only PostHMIN2 is found. Traditional PreMINs and WaldenTSs are found for all the four leaving groups and unlike in the case of OH− and SH−, these stationary points have C3v symmetry with collinear Y–C–C–N arrangements. WaldenTSs are clearly submerged for Y = Br and I with classical (adiabatic) energies of −4.2 (−3.7) and −6.1 (−5.6) kcal mol−1, respectively, relative to the reactants. For Y = Cl the barrier height is close to zero, i.e. −0.2 (0.2) kcal mol−1, whereas for Y = F Walden inversion has a positive barrier of 12.2 (12.3) kcal mol−1, similar to the SH− + CH3F reaction. Besides the ion-dipole PreMIN complexes, we find hydrogen-bonded complexes (HMIN1) for Y = Br and I and a transition state connecting HMIN1 and PreMIN for Y = I as seen in Fig. 3. Unlike in the OH− case, here HMIN1 is slightly above PreMIN. Furthermore, for all the four CN− + CH3Y systems we find pre-reaction complexes (HMIN2), where the C and N atoms of CN− are connected to the H and C atoms of CH3Y, respectively, as seen in Fig. 3. These HMIN2 complexes are slightly more stable than the PREMINs; thus, HMIN2 configurations are the deepest regions of the entrance channels. Similar to the OH− and SH− reactions, front-side FSMIN complexes are found for Y = Cl, Br, and I, but unlike in the former cases, the H3CY⋯CN− complexes have C3v symmetry. In the Y = I case the depth of FSMIN is similar, 10 ± 1 kcal mol−1, to the other minima in the entrance channel, whereas for Y = Br the classical (adiabatic) FSMIN depth is only 3.4 (3.1) kcal mol−1. For Y = Cl FSMIN is unbonded, with an energy slightly above the reactant asymptote. The retention pathways have large barriers between 30 and 60 kcal mol−1 depending on the leaving group. Similar to the OH− + CH3Y systems, DITSs are always below the corresponding FSTSs, but whereas the OH− + CH3I reaction has a negative double-inversion barrier, the classical (adiabatic) barrier height of the CN− + CH3I reaction is 31.9 (29.9) kcal mol−1.
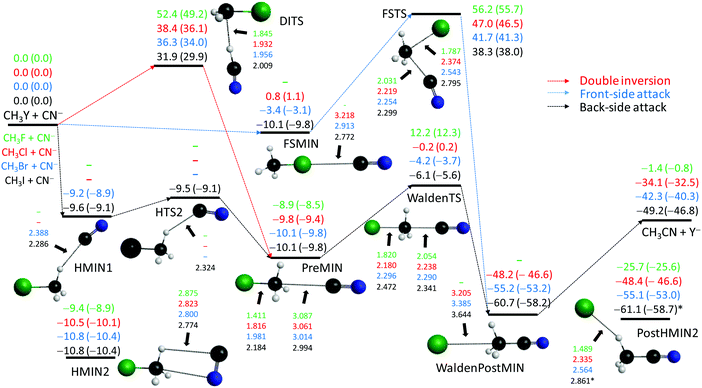 |
| | Fig. 3 Schematic potential energy surfaces of the CN− + CH3Y [Y = F, Cl, Br, I] SN2 reactions showing the classical (adiabatic) CCSD(T)-F12b/aug-cc-pVQZ (+ΔZPE[CCSD(T)-F12b/aug-cc-pVTZ]) relative energies, in kcal mol−1, and the most important CCSD(T)-F12b/aug-cc-pVTZ structural parameters, in Å, of the stationary points along the different reaction pathways. Results indexed by * correspond to MP2/aug-cc-pVDZ structures. | |
NH2− + CH3Y [Y = F, Cl, Br, I]
The SN2 reactions of NH2− with CH3Y are the most exothermic among the systems studied in this work; the enthalpies of the NH2− + CH3Y reactions are more negative by 15–20 kcal mol−1 than those of the corresponding, also highly exothermic OH− + CH3Y processes. Similar to the reactions of OH−, the global minima of the NH2− + CH3Y potential energy surfaces correspond to PostHMIN complexes, where Y− is connected to the NH2 group of CH3NH2 with a single hydrogen bond as seen in Fig. 4. The De (D0) dissociation energies of the CH3NH2⋯Y− complexes are 18.2 (17.9), 10.0 (9.5), 8.7 (8.3), and 7.3 (7.0) kcal mol−1 for Y = F, Cl, Br, and I, respectively, whereas the corresponding De (D0) values of CH3OH⋯Y− are 30.2 (30.5), 16.3 (15.9), 14.2 (13.6), and 11.7 (11.2) kcal mol−1, in order. The lower stability of the CH3NH2⋯Y− complexes is in accord with the longer NH⋯Y− bond lengths of 1.579 (F), 2.330 (Cl), 2.539 (Br), and 2.826 (I) Å with respect to the corresponding 1.345 (F), 2.097 (Cl), 2.303 (Br), and 2.584 (I) Å values of the OH systems. The back-side attack NH2− + CH3F SN2 reaction proceeds via the PreMIN → WaldenTS → PostHMIN inversion pathway featuring a slightly submerged transition state with a classical (adiabatic) energy of −3.0 (−1.9) kcal mol−1 relative to the reactants. For Y = Cl a hydrogen-bonded pre-reaction complex (HMIN) is also found with similar energy, around −14 kcal mol−1, to that of PreMIN and WaldenTS is at −12.8 (−12.1) kcal mol−1. In the case of Y = Br we could not find a WaldenTS, and HMIN is deeper than PreMIN by about 1 kcal mol−1. For Y = I neither a traditional PreMIN nor WaldenTS is found, but HMIN and a pre-reaction transition state (PreTS) exist as seen in Fig. 4. The submerged PreTS with a classical (adiabatic) energy of −15.6 (−15.2) kcal mol−1 relative to the reactants differs from WaldenTS in the orientation of the NH2 unit and the relative stretching of the C–Y distance, because C–F and C–Cl WaldenTS distances are stretched by 0.324 and 0.220 Å relative to the corresponding bond lengths of the CH3Y molecule, whereas the C–I PreTS distance is just longer by 0.107 Å than the same bond in CH3I. Therefore, PreTS may be viewed as an early WaldenTS. As always front-side complexes are found for Y = Cl, Br, and I with De (D0) dissociation energies of 1.7 (1.1), 12.5 (11.5), and 25.8 (24.4) kcal mol−1, respectively. Thus, the stability of the front-side complex is negligible for Y = F, comparable to that of HMIN in the case of Y = Br, and significantly stronger than that of HMIN for Y = I. Note that H3CI⋯NH2− is the most strongly bonded front-side complex investigated in the present study. The retention pathways of the title reactions are the most favored in the case of the NH2− nucleophile. All the DITS and FSTS barrier heights are below the corresponding, also favorable, OH− barriers by about 10 kcal mol−1. Furthermore, all the DITS barriers are below the corresponding FSTS ones by about 20 kcal mol−1; thus, double inversion opens a significantly lower energy retention pathway compared to the more traditional front-side attack. Moreover, for Y = Cl, Br, and I double inversion has negative classical (adiabatic) barrier heights of −3.6 (−2.6), −6.3 (−5.2), and −11.7 (−10.3) kcal mol−1, respectively, opening retention pathways which may become competitive with inversion especially for the iodine leaving group.
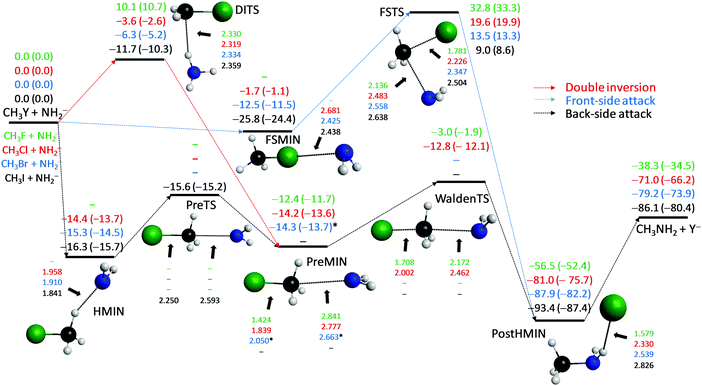 |
| | Fig. 4 Schematic potential energy surfaces of the NH2− + CH3Y [Y = F, Cl, Br, I] SN2 reactions showing the classical (adiabatic) CCSD(T)-F12b/aug-cc-pVQZ (+ΔZPE[CCSD(T)-F12b/aug-cc-pVTZ]) relative energies, in kcal mol−1, and the most important CCSD(T)-F12b/aug-cc-pVTZ structural parameters, in Å, of the stationary points along the different reaction pathways. Results indexed by * correspond to MP2/aug-cc-pVDZ structures. “Experimental” 0 K reaction enthalpies, with ±0.1 kcal mol−1 uncertainties, are −34.7, −66.1, −73.7, and −79.9 kcal mol−1 for Y = F, Cl, Br, and I, respectively.36 | |
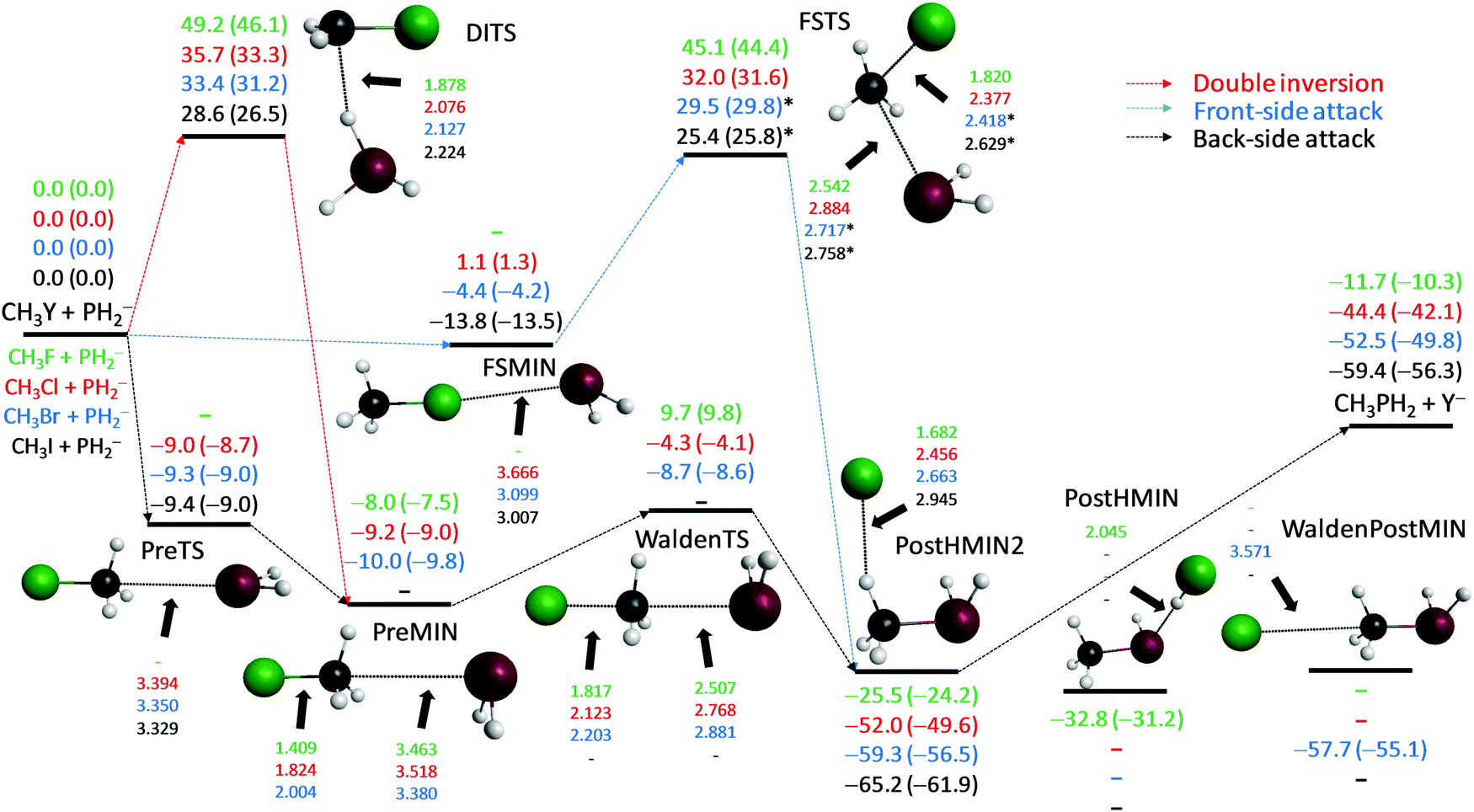
PH2− + CH3Y [Y = F, Cl, Br, I]
The PH2− + CH3Y reactions are less exothermic than the analogous NH2− + CH3Y systems due to the weaker C–P bonds compared to the binding energy of a C–N bond. For the reactions of PH2− we find a PostHMIN complex only for Y = F; however, its structure differs from the geometry of the PostHMIN complexes of the other nucleophiles. Here, the P⋯H distance is significantly stretched, whereas the F–H distance is close to the bond length in the HF molecule; thus, this PostHMIN is better viewed as a post-reaction proton-abstraction (proton transfer from the PH2 group to F−) complex. For all the four leaving groups we find hydrogen-bonded complexes in the exit channel (PostHMIN2), where Y− connects to one of the H atoms of the methyl group as seen in Fig. 5. Note that the proton-abstraction-like PostHMIN, found for Y = F, is a deeper minimum than PostHMIN2, but the enthalpy of the HF + CH3PH− channel (−8.4 kcal mol−1) is less exothermic than that corresponding to the F− + CH3PH2 SN2 products (−10.3 kcal mol−1). A traditional ion-dipole complex (WaldenPostMIN) is only found for Y = Br, but with slightly less stability than the corresponding PostHMIN2. In the entrance channel hydrogen-bonded complexes are not found unlike for the analogous NH2− reactions. For Y = F, Cl, and Br traditional ion-dipole complexes (PreMIN) and WaldenTSs are found as shown in Fig. 5. Note that in the Y = F case the Walden-inversion pathway has a positive classical (adiabatic) barrier of 9.7 (9.8) kcal mol−1, whereas WaldenTSs are submerged for Y = Cl and Br. For Y = Cl, Br, and I we find PreTSs similar to the Y = I case of the NH2− reactions. For Y = I PreTS is the only entrance-channel stationary point with a reactive orientation. Front-side complexes (FSMIN) are found for Y = Cl, Br, and I, but in the case of Y = Cl the stationary point has a slightly positive energy relative to the reactants. The H3CI⋯PH2− complex has De (D0) values of 13.8 (13.5) kcal mol−1; thus, this configuration is the global minimum of the entrance channel similarly to the OH− and NH2− + CH3I reactions. The retention pathways of the PH2− + CH3Y reactions proceed via high barriers and, unlike in the OH− and NH2− reactions, DITSs are slightly above the FSTSs similarly to the SH− systems.
 |
| | Fig. 5 Schematic potential energy surfaces of the PH2− + CH3Y [Y = F, Cl, Br, I] SN2 reactions showing the classical (adiabatic) CCSD(T)-F12b/aug-cc-pVQZ (+ΔZPE[CCSD(T)-F12b/aug-cc-pVTZ]) relative energies, in kcal mol−1, and the most important CCSD(T)-F12b/aug-cc-pVTZ structural parameters, in Å, of the stationary points along the different reaction pathways. Results indexed by * correspond to MP2/aug-cc-pVDZ structures. | |
IV. Summary and conclusions
Motivated by a recent perspective article entitled “Rethinking the SN2 reaction”3 and our previous findings about a novel double-inversion7 pathway and front-side complex formation,13 we have investigated the stationary points and possible reaction mechanisms of 20 different gas-phase SN2 reactions involving OH−, SH−, CN−, NH2−, and PH2− nucleophiles and methyl-halide molecules (CH3Y, Y = F, Cl, Br, I). Previous work usually reported three stationary points, i.e., pre- and post-reaction complexes and a central transition state, for some of these SN2 systems. In the present comprehensive high-level explicitly-correlated ab initio study we reveal that these reactions are much more complex and many stationary points play a role in the dynamics. In the entrance channel, besides the traditional ion-dipole complex and Walden-inversion transition state, hydrogen-bonded and front-side complexes are found. Pre-reaction hydrogen-bond formation usually occurs between OH−, CN−, and NH2− nucleophiles and CH3Y, where Y = Cl, Br, and I, and the hydrogen-bonded complex is more stable than the traditional ion-dipole complex for OH− and NH2−. Front-side complexes, where the nucleophile connects to the leaving group, are found in all cases except Y = F. For X = OH, NH2, and PH2, the H3CI⋯X− complex is the deepest region in the entrance channel suggesting indirect dynamics, because the deep front-side minimum may steer the reactants into a non-reactive orientation. In the exit channel a hydrogen-bonded H3CX⋯Y− complex may be formed for X = OH, SH, and NH2, which is the global minimum of the potential energy surface. In the case of X = CN and PH2, the Y− rather forms a hydrogen bond with one of the H atoms of the methyl group or in some cases a traditional ion-dipole complex is also found. Besides the Walden-inversion mechanisms, we report, in most cases for the first time, front-side attack and double-inversion retention pathways for the title reactions. Front-side attack always has a positive, usually large, barrier, whereas the double-inversion barrier heights are below the front-side attack ones in the case of the OH−, CN−, and NH2− nucleophiles. Moreover, for the OH− + CH3I and the NH2− + CH3Y [Y = Cl, Br, I] reactions the double-inversion transition state is submerged, thereby opening a barrier-less retention pathway for these SN2 reactions. We hope that the present study motivates future experimental and theoretical investigations on the dynamics of the title reactions and shapes the way how we think about SN2 reactions.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
This work was supported by the National Research, Development and Innovation Office – NKFIH, K-125317 and the Ministry of Human Capacities, Hungary grant 20391-3/2018/FEKUSTRAT. D. A. T. is thankful for support from the National Young Talent Scholarship (NTP-NFTÖ-18-B-0399).
References
- P. Walden, Ber. Dtsch. Chem. Ges., 1896, 29, 133 CrossRef.
- W. A. Cowdrey, E. D. Hughes, C. K. Ingold, S. Masterman and A. D. Scott, J. Chem. Soc., 1937, 1252 RSC.
- J. Xie and W. L. Hase, Science, 2016, 352, 32 CrossRef CAS PubMed.
- J. Mikosch, S. Trippel, C. Eichhorn, R. Otto, U. Louderaj, J.-X. Zhang, W. L. Hase, M. Weidemüller and R. Wester, Science, 2008, 319, 183 CrossRef CAS PubMed.
- J. Mikosch, J. Zhang, S. Trippel, C. Eichhorn, R. Otto, R. Sun, W. A. de Jong, M. Weidemüller, W. L. Hase and R. Wester, J. Am. Chem. Soc., 2013, 135, 4250 CrossRef CAS PubMed.
- J. Xie, R. Otto, J. Mikosch, J. Zhang, R. Wester and W. L. Hase, Acc. Chem. Res., 2014, 47, 2960 CrossRef CAS PubMed.
- I. Szabó and G. Czakó, Nat. Commun., 2015, 6, 5972 CrossRef PubMed.
- M. Stei, E. Carrascosa, M. A. Kainz, A. H. Kelkar, J. Meyer, I. Szabó, G. Czakó and R. Wester, Nat. Chem., 2016, 8, 151 CrossRef CAS PubMed.
- I. Szabó and G. Czakó, J. Phys. Chem. A, 2017, 121, 9005 CrossRef PubMed.
- I. Szabó and G. Czakó, J. Phys. Chem. A, 2015, 119, 3134 CrossRef PubMed.
- I. Szabó, H. Telekes and G. Czakó, J. Chem. Phys., 2015, 142, 244301 CrossRef PubMed.
- B. Olasz, I. Szabó and G. Czakó, Chem. Sci., 2017, 8, 3164 RSC.
- I. Szabó, B. Olasz and G. Czakó, J. Phys. Chem. Lett., 2017, 8, 2917 CrossRef PubMed.
- J. Zhang, J. Mikosch, S. Trippel, R. Otto, M. Weidemüller, R. Wester and W. L. Hase, J. Phys. Chem. Lett., 2010, 1, 2747 CrossRef CAS.
- I. Szabó, A. G. Császár and G. Czakó, Chem. Sci., 2013, 4, 4362 RSC.
- J. M. Gonzales, R. S. Cox, S. T. Brown, W. D. Allen and H. F. Schaefer, J. Phys. Chem. A, 2001, 105, 11327 CrossRef CAS.
- J. M. Gonzales, C. Pak, R. S. Cox, W. D. Allen, H. F. Schaefer III, A. G. Császár and G. Tarczay, Chem. – Eur. J., 2003, 9, 2173 CrossRef CAS PubMed.
- L. Sun, K. Song and W. L. Hase, Science, 2002, 296, 875 CrossRef CAS PubMed.
- L. Sun, K. Song, W. L. Hase, M. Sena and J. M. Riveros, Int. J. Mass Spectrom., 2003, 227, 315 CrossRef CAS.
- H. Tachikawa, M. Igarashi and T. Ishibashi, J. Phys. Chem. A, 2002, 106, 10977 CrossRef CAS.
- E. Carrascosa, M. Bawart, M. Stei, F. Linden, F. Carelli, J. Meyer, W. D. Geppert, F. A. Gianturco and R. Wester, J. Chem. Phys., 2015, 143, 184309 CrossRef CAS PubMed.
- Y. G. Proenza, M. A. F. de Souza and R. L. Longo, Chem. – Eur. J., 2016, 22, 16220 CrossRef CAS PubMed.
- J. Xie, S.
C. Kohale, W. L. Hase, S. G. Ard, J. J. Melko, N. S. Shuman and A. A. Viggiano, J. Phys. Chem. A, 2013, 117, 14019 CrossRef CAS PubMed.
- J. Xie, R. Sun, M. R. Siebert, R. Otto, R. Wester and W. L. Hase, J. Phys. Chem. A, 2013, 117, 7162 CrossRef CAS PubMed.
- J. Chen, Y. Xu and D. Wang, J. Comput. Chem., 2014, 35, 445 CrossRef CAS PubMed.
- H. Yin, D. Wang and M. Valiev, J. Phys. Chem. A, 2011, 115, 12047 CrossRef CAS PubMed.
- Y. Xu, T. Wang and D. Wang, J. Chem. Phys., 2012, 137, 184501 CrossRef PubMed.
- Y. Xu., J. Zhang and D. Wang, Phys. Chem. Chem. Phys., 2014, 16, 19993 RSC.
- Y. Xu., J. Zhang and D. Wang, J. Chem. Phys., 2015, 142, 244505 CrossRef PubMed.
- D. A. Tasi, Z. Fábián and G. Czakó, J. Phys. Chem. A, 2018, 122, 5773 CrossRef CAS PubMed.
- T. B. Adler, G. Knizia and H.-J. Werner, J. Chem. Phys., 2007, 127, 221106 CrossRef PubMed.
- C. Møller and M. S. Plesset, Phys. Rev., 1934, 46, 618 CrossRef.
- T. H. Dunning Jr., J. Chem. Phys., 1989, 90, 1007 CrossRef.
- K. A. Peterson, D. Figgen, E. Goll, H. Stoll and M. Dolg, J. Chem. Phys., 2003, 119, 11113 CrossRef CAS.
-
H.-J. Werner, P. J. Knowles, G. Knizia, F. R. Manby, M. Schützet al., Molpro, version 2015.1, a package of ab initio programs, see http://www.molpro.net Search PubMed.
-
B. Ruscic and D. H. Bross, Active Thermochemical Tables (ATcT) values based on ver. 1.122d of the ThermochemicalNetwork (2018); available at ATcT.anl.gov.
Footnote |
| † Electronic supplementary information (ESI) available: All the structural parameters, harmonic frequencies, absolute energies, and relative energies of the stationary points as well as Cartesian coordinates of the best geometries. See DOI: 10.1039/c8cp07850e |
|
| This journal is © the Owner Societies 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  ,
Zita
Fábián
and
Gábor
Czakó
,
Zita
Fábián
and
Gábor
Czakó