
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The 3s Rydberg state as a doorway state in the ultrafast dynamics of 1,1-difluoroethylene†
Sandra
Gómez
,
Lea M.
Ibele
and
Leticia
González
 *
*
Institute for Theoretical Chemistry, Faculty of Chemistry, University of Vienna, Währingerstr. 17, 1090 Vienna, Austria. E-mail: leticia.gonzalez@univie.ac.at
First published on 13th February 2019
Abstract
The deactivation dynamics of 1,1-difluoroethylene after light excitation is studied within the surface hopping formalism in the presence of 3s and 3p Rydberg states using multi-state second order perturbation theory (MS-CASPT2). Due to the proximity of the Rydberg π-3s state with the ππ* state, the states are mixed favoring ultrafast exchange of population via a conical intersection that closely resembles the equilibrium structure. After excitation, it is found that the π-3s state acts as a doorway state, trapping the population and delaying internal conversion to the ππ* state, from which deactivation to the closed-shell ground state takes place. Besides the conical intersection between the π-3s and ππ* states, five additional conical intersections between the ππ* state and the ground state are found, indicating that after the system is excited, it stretches the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond before it twists and pyramidalizes at any of the carbon atoms, in the spirit of a hula-twist mechanism.
C bond before it twists and pyramidalizes at any of the carbon atoms, in the spirit of a hula-twist mechanism.
1 Introduction
Ethylene is a prototype for conjugated π systems in a large number of interesting systems, including polymers,1 organic dye sensitized solar cells,2,3 non linear optical chromophores,4 or organic light-emitting diodes using low molecular weight compounds with high fluorescence quantum yields.5 Additionally, its light-induced cis–trans isomerization plays an important role in biological processes, such as vision,6–8 and it can be exploited to operate molecular rotors such as those pioneered by Feringa based on overcrowded alkenes.9Given its many applications, it has been a recurrent topic of interest for theory10–28 as well as for experiment.29–38 As a result, the electronic states of ethylene are well characterized, with an intense ππ* or valence band dominating the absorption spectrum and surrounded by Rydberg series where the π-3s lies below the absorption maximum, well separated from the other π-3p states, and a mostly dark double excited or zwitterionic state.32,33 From the electronic ground state, an excitation to the bright ππ* state triggers an ultrafast deactivation in less than 50 fs,33,34,37,38 which has been interpreted as internal conversion to the electronic ground state through several conical intersections (CI), including twisted, twisted-pyramidalized, hydrogen-migration and ethylidene-like structures.14,20,22,23,39–41
An interesting question is whether the Rydberg states play an active role in the deactivation of ethylene. Theoretically, the 3s Rydberg orbital has been included in a number of stationary42,43 as well as dynamical calculations.40,41 The work by Mori et al.40 using multiple spawning dynamics showed that although the π-3s state lies below the bright ππ* state, little population arrives in this state during the first 10 fs and the excited state lifetime is only slightly longer (90 fs) than when the π-3s state is excluded (60 fs), thus concluding that the Rydberg state is merely a spectator. Another dynamical study by Sellner et al.,41 including the 3s orbital, also found the contribution of the π-3s state to be minor, although the details of the deactivation dynamics depend slightly on the method employed for the calculation of the excited states. Regardless of the methods employed, both studies agree that the inclusion of Rydberg states has a modest effect in ethylene and its photodynamics can be theoretically described including valence and zwitterionic states exclusively.
Adding halogen atoms can substantially alter the photochemical properties and the deactivation mechanism of ethylene. Experiments indicate that 1,1-difluoroethylene and fluoroethylene show a very similar absorption spectra,29,44,45 only slightly blue-shifted with respect to ethylene. However, while in ethylene, internal conversion from the ππ* to the ground state requires a CC stretching followed by a twisting around the double bond and a pyramidalization at one of the methylene units,46 in fluoroethylene, the torsion over the double bond directly drives to a CI between the ππ* state and the ground state.39 It is also curious that depending on which of the two H atoms are substituted by F in ethylene, CIs similar to those predicted in either ethylene or fluoroethylene can be located in 1,1- and 1,2-difluoroethylene,47 respectively. Also the CI corresponding to H-migration present in ethylene and 1,2-difluoroethylene was never found in 1,1-difluoroethylene.47
The aim of the present paper is to investigate the deactivation dynamics of 1,1-difluoroethylene (1,1-DFE hereafter) after light irradiation using non-adiabatic surface hopping simulations. Up to our knowledge, no excited state dynamics study is available in ethylene-fluoro-derivatives, beyond our previous one-dimension wave packet simulations in 1,1-DFE.48,49 Our intention is twofold. First, we aim at comparing the internal conversion mechanism of deactivation of 1,1-DFE against bare ethylene. Second, we want to investigate whether Rydberg orbitals play a role in the non-adiabatic dynamics of 1,1-DFE. Generally, we are interested to learn which main coordinates play a main role in the deactivation of 1,1-DFE. To this purpose, multiconfigurational calculations including the 3s and the three 3p orbitals have been carried out and used to perform non-adiabatic simulations at the multistate second order perturbation (MS-CASPT2) level of theory.
2 Computational details
The non-adiabatic dynamics simulations on 1,1-DFE were carried out using the Surface Hopping including ARbitrary Couplings (SHARC) approach.50–52 The trajectories were propagated during 200 fs with a nuclear time step of 0.5 fs, where the integration of the nuclear motion was done with the velocity-Verlet algorithm.53,54 Decoherence corrections were taken into account following the energy-based method of Granucci and Persico55 with a parameter of α = 0.1 Hartree. As a hopping scheme, the “Standard SHARC surface hopping probabilities” scheme was used.51Direct dynamical simulations including Rydberg and valence states are challenging as both type of states need to be described on the same footing with similar accuracy. The description of Rydberg states requires extensive diffuse functions and valence states profit from the inclusion of large amounts of dynamical correlation.56 A balanced description can be achieved using the multi-state complete active space second-order perturbation theory57,58 (MS-CASPT2) method and at least the aug-cc-pVDZ basis set.59 No IPEA shift60,61 and an imaginary level shift of 0.3 a.u. were employed. Accordingly, the electronic energies and gradients were calculated on-the-fly for each nuclear time step using MS-CASPT2, as implemented in MOLCAS8.0.62 To avoid the expensive calculation of non-adiabatic couplings at every time step, the propagation was done using the local diabatization scheme, that allows the use of (the full) wave function overlaps instead, as described in ref. 63.
Two different active spaces (see corresponding orbitals in Fig. 1) were employed in the underlying Complete Active Space Self-Consistent Field64 (CASSCF) calculation to investigate the effect of the Rydberg states on the deactivation dynamics. The smallest active space includes only the two electrons and the two active π and π* orbitals; accordingly, three roots were state-averaged (SA) with equal weights, resulting in the neutral (N) closed-shell ground state π2, the valence (V) ππ* state and the dark zwitterionic (Z) π*2 state – this calculation will be refereed as SA(3)-CAS(2,2)PT2. The larger active space includes the Rydberg 3s, 3px, 3py, 3pz orbitals besides the π and π* pair and it requires 6 or 11 roots to include all the possible single or single and double excitations from the π to the π* and Rydberg orbitals, respectively; these calculations are labelled SA(6)- or SA(11)-CAS(2,6)PT2.
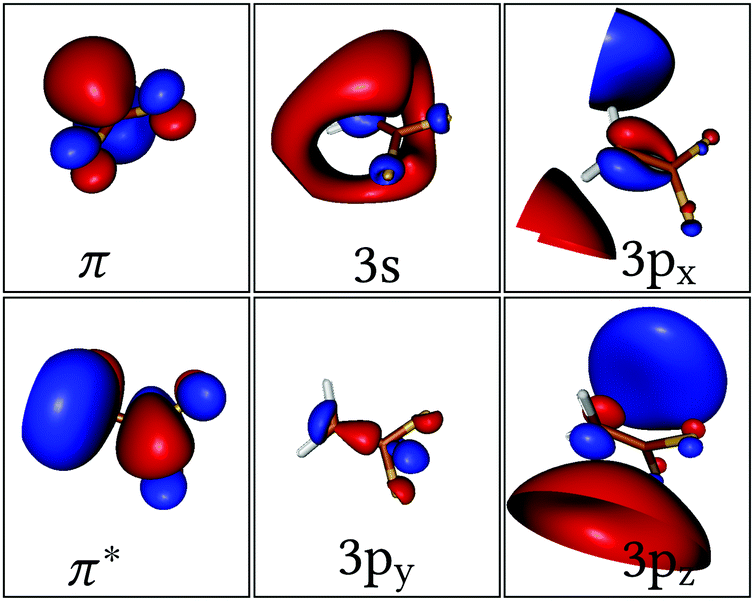 | ||
| Fig. 1 Orbitals included in the active space of 1,1-DFE. | ||
In order to generate initial conditions for the SHARC dynamical simulations, the ground state equilibrium geometry was optimized at both the MP2/aug-cc-pVDZ and SA(11)-CAS(2,6)PT2/aug-cc-pVDZ levels of theory. As it can be seen from Table 1, the geometrical parameters are very similar with both methods. Thus, the MP2 geometry was further employed to calculate harmonic frequencies that were used to generate a Wigner distribution of 1000 uncorrelated velocities and geometries. From each geometry, vertical excitations were calculated at MS-CASPT2 level of theory with both SA(3)-CAS(2,2) and SA(11)-CAS(2,6) active spaces to simulate an absorption spectrum from the oscillator strengths, as explained in ref. 65. The oscillator strengths served as a condition to select initial trajectories for the dynamics66 which were excited assuming an infinitely short δ-pulse. Since the dynamical results will be given in the adiabatic picture (i.e. governed by states strictly ordered by their energy that never cross), a transformation to the diabatic or spectroscopic picture51 was carried out a posteriori, directly analyzing the configurational interaction vectors resulting from subsequent MS-CASPT2 single point calculations carried out for the geometries taken every 2 fs of every trajectory. The state character was assigned to the largest reference weight. Note here that the diabatization performed does not consider the full configuration interaction vector, only the largest contribution to it. This however, allows us to follow in an approximate way the electronic character of the states, even though the unitary transformation diabatic–adiabatic is lost.
| Exp70 | MP2 | CASPT2 | |
|---|---|---|---|
| r(CC) | 1.315 | 1.326 | 1.341 |
| r(CF) | 1.323 | 1.322 | 1.339 |
| r(CH) | 1.078 | 1.079 | 1.089 |
| ∢(FCF) | 109.1 | 109.8 | 109.6 |
| ∢(HCH) | 121.8 | 121.2 | 121.1 |
| ∢(CCF) | 125.5 | 125.1 | 125.2 |
| ∢(HCC) | 119.1 | 119.4 | 119.4 |
To cluster the hopping geometries to a minimum energy CI (MECI), the root-mean-square deviation (RMSD) was minimized using quaternions.67 The structures were assigned according to their minimum value of the RMSD with respect to every MECI. The optimization of MECIs was done using MOLCAS in combination with the the external optimizer of ORCA,68 as described in ref. 69. The evolution of the relevant coordinates during the dynamics was performed using a Gaussian convolution of all trajectories. Trajectories were stopped after hopping to the lowest energy state (S0) and remaining there during ten femtoseconds.
3 Results and discussion
3.1 Vertical excitations and absorption spectra
The vertical excitations of 1,1-DFE computed at SA(3)-CAS(2,2)PT2, SA(6)- and SA(11)-CAS(2,6)PT2 levels of theory are collected in Table 2. The Rydberg states originate from excitations from the π orbital to every one of the Rydberg orbitals. As it can be observed, the inclusion of the Rydberg orbitals leads to closer agreement of the energy of the valence state V with the experimental value,44,45 but only when all single and double excitations are included in the calculation. If only single excitations are allowed (6 roots, SA(6)), the V state is mixed with the π-3pz state, deteriorating considerably the energy of the ππ* state. The most extensive SA(11)-CAS(2,6)PT2 calculation predicts that the spectrum of 1,1-DFE is composed of a main band due to the excitation to the bright V state surrounded by Rydberg series. The π-3s state lies slightly below the V state (≈0.5 eV) at the equilibrium geometry whilst the three π-3p states are more than 1 eV above.| State | ΔE | f | Ref. weight | Exp44 | Exp45 |
|---|---|---|---|---|---|
| SA(3)-CAS(2,2)PT2 | |||||
| N | 0.00 | 0.85 | |||
| V | 7.33 | 0.441 | 0.81 | 7.51 | 7.49 |
| Z | 13.93 | 0.025 | 0.82 | ||
| SA(6)-CAS(2,6)PT2 | |||||
| N | 0.00 | 0.85 | |||
| V | 8.02 | 0.165 | 0.62 | 7.51 | 7.49 |
| π-3s | 7.00 | 0.060 | 0.96 | 6.73 | 6.75 |
| π-3px | 8.06 | 0.000 | 0.99 | 7.92 | 7.91 |
| π-3py | 8.14 | 0.000 | 0.96 | ||
| π-3pz | 9.10 | 0.008 | 0.62 | ||
| SA(11)-CAS(2,6)PT2 | |||||
| N | 0.00 | 0.85 | |||
| V | 7.49 | 0.403 | 0.85 | 7.51 | 7.49 |
| π-3s | 7.05 | 0.072 | 0.85 | 6.73 | 6.75 |
| π-3px | 8.10 | 0.000 | 0.85 | 7.92 | 7.91 |
| π-3py | 8.20 | 0.008 | 0.85 | ||
| π-3pz | 8.93 | 0.111 | 0.81 | ||
| Z | 13.77 | 0.019 | 0.83 | ||
The absorption spectra calculated from a Wigner distribution including 1000 geometries at the SA(3)-CAS(2,2)PT2 and SA(11)-CAS(2,6)PT2 levels of theory are superimposed to the experimental spectrum in Fig. 2. Again, the spectrum obtained with the smallest active space is slightly red-shifted with respect to that calculated with the largest active space and the experimental one from Bélanger and coworkers.44 The overall width of the spectrum is well described with both methods, but it is noticeable that the fine structure of the main experimental band is very well reproduced with the (2,6) active space. The band at energies larger than 8 eV corresponds to excitations to 3d and higher order Rydberg orbitals that are not included in any of our calculations.
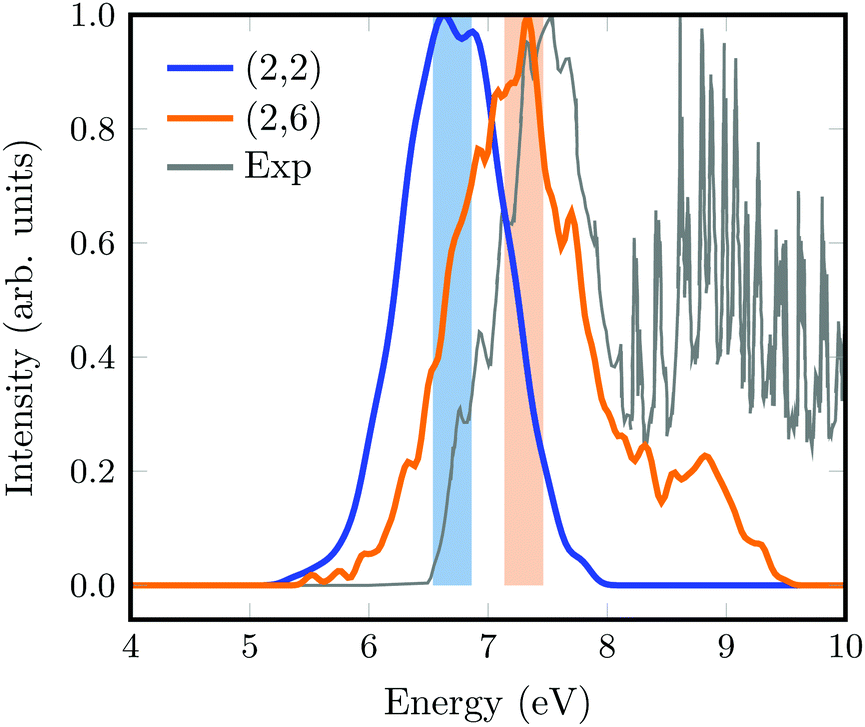 | ||
| Fig. 2 Absorption spectrum of 1,1-DFE computed from a Wigner distribution including 1000 geometries excited with SA(3)-CAS(2,2)PT2 (blue) and SA(11)-CAS(2,6)PT2 (orange) levels of theory. The experimental spectrum44 is shown in grey. The excitation windows used for the initial condition generation in SHARC are marked as light color boxes. | ||
3.2 Non-adiabatic SHARC dynamics
SHARC simulations were initiated from the initial geometries and velocities contained in an excitation window of 0.5 eV centered at the absorption maxima (color boxes in Fig. 2). For the SA(3)-CAS(2,2)PT2 calculation, this provides 152 geometries excited to the S1 state, which here is the ππ* state. Likewise, for the SA(11)-CAS(2,6)PT2 simulations, the same energetic window corresponds to 121 trajectories, from which 108 (89%) are excited to the S2, 11 (9%) to the S1 and 1 to the S3 as well as the S4 states. Note that in this case, the states are labelled by increasing energy from S0 to S10. Initially, besides the brightest ππ* state, also the π-3s state is excited. Note however, that during the dynamics the character of the states can change so that a one-to-one correlation with the energy ordering and character of the states at the Franck–Condon (FC) point is not straightforward.The evolution of the adiabatic populations for both sets of simulations is shown in Fig. 3a and b, together with exponential fits (thinner lines). For the small active space (Fig. 3a), after 20 fs the population of the S1 state decays very fast to the ground state, which then recovers to 93% at the end of the simulation. An exponential fit from the time interval between 20 and 200 fs returns a time scale of 30 fs for the decay from the S1 to the S0 state.
 | ||
| Fig. 3 Time evolution of SHARC adiabatic populations during 200 fs obtained at (a) SA(3)-CAS(2,2)PT2 and (b–d) SA(11)-CAS(2,6)PT2 levels of theory. In panel (c) the populations of the states S1–10 is plotted versus that of S0. Panel (d) shows the evolution of the N, V, Z and π-3s diabatic populations, where 'rest' (palatinate line) refers to the states not clearly assignable to one of the above. | ||
Interestingly, the dynamics including Rydberg states (Fig. 3b) shows during the first 20 fs that population is mainly transferred from the S2 to the S1 state, with some negligible amount of population in the S3 and S4 states. The ground state recovery starts after 26 fs, and at first glance one can see that the slope of the associated curve is smaller than when Rydberg states are excluded (compare with Fig. 3a). After 200 fs, 72% of the trajectories have decayed to the electronic ground state S0. In this case, the time constants for the exponential decay of the population of the respective states were fitted separately in two time intervals: from 0 to 26 fs and from 26.5 to 200 fs. Within the first 26 fs, population decays from the S2 to the S1 with a time constant of 36 fs. After 26 fs the S1 state decays with a time constant of 50 fs. The time constant for the decay from the S1 to the S0 was only fitted in the interval between 26 and 200 fs and was found to be 32 fs. This time constant appears to be the same as when Rydberg states are excluded (30 fs); however, one should notice that when Rydberg states are present, the dynamics starts with population mainly in the S2 state (which for some geometries is the V state and for others the Rydberg π-3s) and this population needs to be transferred first to the S1 before decaying to the S0; therefore, the overall effective decay is slower.
In order to compare the kinetic models obtained with both active spaces in the same way, another fit was then performed considering the sum of the population of all the electronic excited states versus that of S0. The evolution of these populations as well as the corresponding exponential fit is shown in Fig. 3c. In this case, a lifetime of 60 fs was obtained; this is the time that all the excited state population takes to decay to the electronic ground state when the Rydberg states are included. Thus, the population transfer to the ground state when Rydberg states are excluded occurs approximately twice as fast than when Rydberg orbitals were included.
For both sets of trajectories, the amount of hops between the two lowest energy states in time intervals of 5 fs was analyzed. Fig. 4 displays an histogram with the frequency of hops between S1 and S0 per time interval over all the trajectories. In the (2,2) active space the hopping frequency is more localized, with the majority of the hops occurring around 26 fs. The (2,6) active space produces a broader and generally slower behavior, mainly due to the transfer of population to the Rydberg states that at later times need to find their way back to the valence ππ* state before deactivating to the S0. The correspondence of the geometries where the above mentioned hops occurred and its relationship to the optimized MECIs will be discussed in the next section.
 | ||
| Fig. 4 Frequency of hops between S1 and S0 states during the dynamics obtained with SA(3)-CAS(2,2)PT2 (blue) and SA(11)-CAS(2,6)PT2 (orange) levels of theory. | ||
As it is clear that Rydberg states have an influence in the deactivation mechanism of 1,1-DFE, henceforth, only the ensemble of trajectories from the SA(11)-CAS(2,6)PT2 will be further analyzed and discussed. Fig. 5 shows the variation of the coordinates expected to play an important role before deactivation to the S0 takes place. Taking into account the initial Wigner distribution as a reference, during the first 10 fs of the dynamics the CC distance starts elongating while the other coordinates remain localized around the equilibrium geometry. This CC stretching oscillates between 1.2 and 1.8 Å for many trajectories, showing a coherent movement. Some trajectories, however, stretch until almost 2 Å and then recoil to shorter distances. The torsional angle shows a less coherent variation and the nuclear wavefunction spreads very fast. At some points (for example, at 30 fs) almost complete delocalization over all possible torsional angles can be appreciated. Both pyramidalizations show quite similar delocalized behaviors, including many oscillations in time.
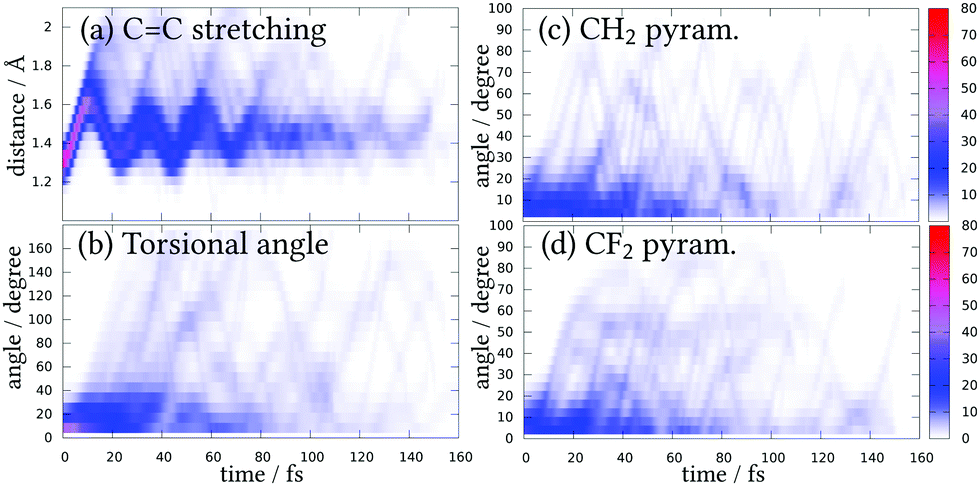 | ||
| Fig. 5 Evolution of most important coordinates before deactivation to the ground state. The colour bar indicates percentage of trajectories. (a) CC stretching. (b) Torsion. (c) CH2 pyramidalization. (d) CF2 pyramidalization. | ||
3.3 Minimum energy conical intersections
In order to understand better the internal conversion mechanism of 1,1-DFE to the S0, all the hopping geometries encountered during the SA(11)-CAS(2,6)PT2 dynamics were optimized as state-crossings. Starting from the S1/S0 hopping events, 78 geometries were optimized, which collapsed into the five MECIs, CI1–CI5, shown in Fig. 6 (see ESI† for Cartesian coordinates). All structures are substantially different from the equilibrium (FC) geometry, see Table 3, but have a similar torsional angle around 60–75°. According to their relative energies, after the ππ* state is excited, all MECIs are energetically accessible. The MECIs with the highest energy are CI1 and CI3, characterized by a very elongated CC bond and a very elongated C–F bond, respectively. While the CI1 and CI2 both show the large CF2 pyramidalization angle of about 80°, the latter is found at lower energy. In contrast, CI4 and CI5 have the lowest energy. The former is pyramidalized at the CH2 carbon whilst the latter is the most twisted one, showing also a small pyramidalization at the CH2 carbon of about 20°. In terms of the one-bond-flip (OBF) or hula-twist (HT) mechanisms,39 the CI1–4 allow for the HT deactivation pathway while the CI5 operates for the OBF mechanism. In the CI1–2, it is the CF2 which hula-hoops and the CH2 that twists; in CI3–4 the CF2 twists and the CH2 hula-hoops.
 | ||
| Fig. 6 Relative energy (in eV) with respect to the Franck–Condon (FC) ground state of the minimum energy conical intersections (CI) optimized in 1,1-DFE at the SA(11)-CAS(2,6)PT2 level of theory. Geometrical parameters are collected in Table 3. | ||
| Label | pyrCF2 | pyrCH2 | Torsion | r(CC) | r(CH) | r(CF) |
|---|---|---|---|---|---|---|
| FC | 0.00 | 0.00 | 0.00 | 1.33 | 1.08 | 1.32 |
| CIa | 0.14 | 0.92 | 6.33 | 1.44 | 1.10 | 1.29 |
| CI1 | 80.96 | 20.21 | 67.05 | 2.08 | 1.11 | 1.41 |
| CI2 | 76.07 | 2.80 | 75.57 | 1.55 | 1.08 | 1.61 |
| CI3 | 6.39 | 24.42 | 69.82 | 1.45 | 1.08 | 1.88 |
| CI4 | 1.98 | 48.84 | 59.13 | 1.37 | 1.08 | 1.44 |
| CI5 | 0.50 | 19.83 | 79.36 | 1.40 | 1.10 | 1.61 |
Starting from the S2/S1 hopping events, 26 geometries were optimized, all of which collapsed to the same geometry, the CIa. This structure enables population transfer between the π-3s Rydberg and the ππ* state. The geometry is very similar to that of the equilibrium, only with a slightly larger CC bond distance; accordingly, both structures are energetically very close, allowing for a very fast radiationless transfer between the ππ* state and the π-3s Rydberg state.
As all the MECIs are energetically accessible from the V state, their relative importance can only be assessed from the dynamical simulations. By minimizing the RMSD between the structures, it is possible to classify the hopping geometries to find that the majority of the trajectories went to the S0 through CI2 (44.9%) followed by CI5 and CI1 (21.8% and 20.5%). The less active MECIs are CI4 and CI3 with 9.0% and 3.8% of the trajectories, respectively. Considering their geometries, one can conclude that deactivation operates after the system elongates its CC bond, twists and pyramidalizes the CF2 carbon atom, the latter motion only being present in two out of the three predominant MECIs. This means that 65% of the CIs correspond to HT of the CF2, 13% to HT of the CH2 and 22% can be assigned to the OBF mechanism.
In comparison to the ethylene,40,41 no ethylidene-like CI was found here. Interestingly, in the dynamics of Sellner et al.41 on ethylene, the hops to the S0 required pyramidalized geometries, while in 1,1-DFE, 22% of the deactivation is possible without pyramidalization, i.e. following a OBF mechanism. Our optimized CIa between the ππ* and π-3s is energetically in agreement with the one found by Mori et al.40 for ethylene, close to the FC region. However, in 1,1-DFE, the CIa is closer to the equilibrium geometry, triggering a faster population transfer to the π-3s state than in ethylene. In contrast to the study of fluoroethylene done by Barbatti et al.,39 in 1,1-DFE neither H-migration CI or ethylidene-like were found within the dynamics.
3.4 Analysis of the dynamics in the diabatic representation
As during the dynamics the character of the states mix, it is convenient to transform the adiabatic populations into diabatic ones. Diabatic states can be described as those where the electronic character does not change with respect to the nuclear coordinates. A transformation from adiabatic to diabatic states is often a tedious task, as no general black-box recipe exists. Here, the diabatization is based on the configuration interaction vectors of the MS-CASPT2 method taken every 2 fs for each trajectory; the diabatic states are defined according to the state character of the largest squared coefficient of this vector. The resulting diabatic populations for the N, V, Z and π-3s states are depicted in Fig. 3d.Comparing to the adiabatic picture (Fig. 3a), only 60% of the trajectories deactivated to the ground state after 200 fs. This decrease evidences a mixing of the ππ* state and the closed-shell ground state during the dynamics, as expected for torsional angles close to 90°, where both orbitals π and π* are degenerated and thus indistinguishable. At the beginning of the dynamics, the population is well distributed between the valence state and the energetically close 3s-Rydberg in a ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Note that the initial distribution between both states is different from that obtained for the adiabatic S2 and S1 states (9:1), as state inversions take place for some geometries of the initial Wigner distribution. None of π-3p Rydberg states is populated during the propagation time, meaning that their contributions to the configuration interaction state vector are always minor. The population of the double excited π*2 state (Z) is always low, but not negligible, despite this state lies ca 7 eV above the V state at the FC region. This is the result of the ultrafast stabilization of the Z state, which after about 10 fs is already the adiabatic S2 state. In passing we note that this emphasizes the importance to include double excitations in the electronic wavefunction during the dynamics. As in Fig. 3a, the diabatic populations show a ground state recovery after a short time delay, in this case 24 fs. Thus, in this case the populations are fitted within two time intervals, one from 0 to 24 fs, where the V state transfers population to the π-3s and another from 24.5 fs onwards, in which the deactivation from V to N takes place. A scheme showing the kinetic constants obtained in Fig. 7. Note that the fact that the final populations on the ground state are different in the adiabatic and diabatic representations clearly evidences the importance of the diabatization, as it emphasizes the strong mixing between ππ* and ππ closed shell contributions.
:1. Note that the initial distribution between both states is different from that obtained for the adiabatic S2 and S1 states (9:1), as state inversions take place for some geometries of the initial Wigner distribution. None of π-3p Rydberg states is populated during the propagation time, meaning that their contributions to the configuration interaction state vector are always minor. The population of the double excited π*2 state (Z) is always low, but not negligible, despite this state lies ca 7 eV above the V state at the FC region. This is the result of the ultrafast stabilization of the Z state, which after about 10 fs is already the adiabatic S2 state. In passing we note that this emphasizes the importance to include double excitations in the electronic wavefunction during the dynamics. As in Fig. 3a, the diabatic populations show a ground state recovery after a short time delay, in this case 24 fs. Thus, in this case the populations are fitted within two time intervals, one from 0 to 24 fs, where the V state transfers population to the π-3s and another from 24.5 fs onwards, in which the deactivation from V to N takes place. A scheme showing the kinetic constants obtained in Fig. 7. Note that the fact that the final populations on the ground state are different in the adiabatic and diabatic representations clearly evidences the importance of the diabatization, as it emphasizes the strong mixing between ππ* and ππ closed shell contributions.
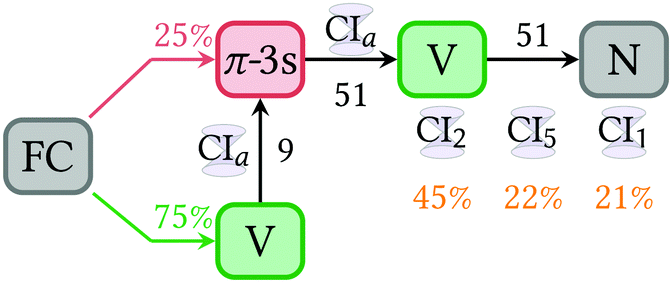 | ||
| Fig. 7 Kinetic model for the deactivation of 1,1-DFE after light irradiation. The black numbers are the kinetic constants in fs. Deactivation to the closed-shell N ground state occurs mainly via three conical intersections (CI) between the V and the N state. The Rydberg π-3s state slows down the deactivation as all the population excited to the V state makes a detour via the CIa to the π-3s state. | ||
From the FC structure, 75% of the population is vertically excited to the V state and 25% to the Rydberg π-3s state. The population transferred to the V state leaks non-adiabatically to the π-3s state with a time constant of 9 fs. After this initial ultrafast exchange to the π-3s state, the population is transferred back to the V state with a time constant of 51 fs, from which it deactivates to the close-shell N state, in another 51 fs. The population directly promoted to the π-3s state follows the same route, undergoing via the V state.
The initial ultrafast population inversion between the valence and the Rydberg state is in good agreement with the fact that the CIa structure found for this process is geometrically very close to the FC geometry. After the V state is populated, 1,1-DFE twists around the CC bond, stretches the CC bond and depending, on which of the CI is used for deactivation, pyramidalizes at any of the carbon atoms to undergo a radiationless transition to the N ground state.
Notably, the dynamics of 1,1-DFE is rather different from that reported by Sellner et al.41 for ethylene, where the Rydberg states, slightly populated at the beginning of the dynamics, decay very fast to the V state, becoming depopulated at already 10 fs. One should nevertheless note that in that study41 only three adiabatic states (S0, S1 and S2) were included in the dynamics and this might not be sufficient, in the light of the present work. In contrast, the population dynamics obtained here is similar to that obtained by Mori et al.40 for bare ethylene, where ca. 30% of the population goes initially from the valence state to the Rydberg 3s state, before deactivating to the ground state. However, in 1,1-DFE, almost all the population makes the detour via the Rydberg 3s; this makes the ground state recovery slower than in ethylene, as the system needs to find its way back to the V state to continue its deactivation pathway to the ground state.
4 Conclusions
In this paper, the non-adiabatic dynamics of 1,1-DFE including the 3s and 3p Rydberg manifold was investigated using surface-hopping simulations at the MS-CASPT2 level of theory. It is found that the inclusion of Rydberg states, (i) considerably improves the description of its absorption spectrum, and (ii) has an influence on the deactivation time scale, with respect to calculations that exclude Rydberg states. In particular, the π-3s state acts as a doorway state for the ultrafast deactivation of 1,1-DFE as it traps all the population before it gets transferred to the V state and deactivates through internal conversion to the closed-shell N electronic ground state.The π-3s and the V states are excited in a ratio 1:3, as they lie rather close in energy and there is intensity borrowing from the valence state. A conical intersection very similar to the equilibrium geometry allows for ultrafast exchange between these states within less than 10 fs. Once the population is finally in the V state, five structures have been located which enable radiationless decay from the V to the N state. From them, the most important three channels for internal conversion involve CC stretching, torsion, and pyramidalization on the CF2 carbon atom, following a HT mechanism. One of the other two less important pathways involves slight pyramidalization on the CH2 carbon atom and CC stretching, while the least important of these three conical intersections is strongly twisted.
A sequential kinetic fit indicates that the ultrafast deactivation of 1,1-DFE in the presence of the 3s Rydberg state is delayed due to the Rydberg detour experienced by the population excited to the V state. In either case, it takes about 50 fs to empty the π-3s state to the V state and another 50 fs to deactivate from the V to the N state. For comparison, when the Rydberg states are neglected, direct excitation to the V state deactivates to the N state within 30 fs.
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The authors thank the University of Vienna for funding, the Vienna Scientific Cluster for allocation of computer resources and the COST Action CM1405 (MOLIM) for inspiring discussions.References
- L. L. Böhm, Angew. Chem., Int. Ed., 2003, 42, 5010 CrossRef PubMed.
- B. O'Regan and M. Grätzel, Nature, 1991, 353, 737 CrossRef.
- K. Sun, Y. Ma, W. Zhang, Y. Wen, L. Wang and J. Zhang, Dyes Pigm., 2017, 139, 148 CrossRef CAS.
- P. N. Prasad and S. P. Karna, Int. J. Quantum Chem., 1994, 52, 395 CrossRef.
- S. Reineke, F. Lindner, G. Schwartz, N. Seidler, K. Walzer, B. Lüssem and K. Leo, Nature, 2009, 459, 234 CrossRef CAS PubMed.
- G. G. Kochendoerfer, S. W. Lin, T. P. Sakmar and R. A. Mathies, Trends Biochem. Sci., 1999, 24, 300 CrossRef CAS PubMed.
- K. Palczewski, Annu. Rev. Biochem., 2006, 75, 743 CrossRef CAS PubMed.
- C. Schnedermann, X. Yang, M. Liebel, K. M. Spillane, J. Lugtenburg, I. Fernández, A. Valentini, I. Schapiro, M. Olivucci, P. Kukura and R. A. Mathies, Nat. Chem., 2018, 10, 449 CrossRef CAS PubMed.
- T. van Leeuwen, A. Lubbe, P. Stacko, S. Wezenberg and B. Feringa, Nat. Rev. Chem., 2017, 1, 1 CrossRef.
- I. Ohmine, J. Chem. Phys., 1985, 83, 2348 CrossRef CAS.
- M. Ben-Nun and T. J. Martínez, Chem. Phys. Lett., 1998, 298, 57 CrossRef CAS.
- M. Ben-Nun and T. J. Martínez, J. Phys. Chem. A, 1999, 103, 10517 CrossRef CAS.
- M. Ben-Nun, J. Quenneville and T. J. Martínez, J. Phys. Chem. A, 2000, 104, 5161 CrossRef CAS.
- M. Ben-Nun and T. J. Martínez, Chem. Phys., 2000, 259, 237 CrossRef CAS.
- V. Molina, M. Merchan, B. O. Roos and P.-A. Malmqvist, Phys. Chem. Chem. Phys., 2000, 2, 2211 RSC.
- G. Granucci, M. Persico and A. Toniolo, J. Chem. Phys., 2001, 114, 10608 CrossRef CAS.
- A. Viel, R. P. Krawczyk, U. Manthe and W. Domcke, Angew. Chem., Int. Ed., 2003, 42, 3434 CrossRef CAS PubMed.
- J. Quenneville and T. J. Martínez, J. Phys. Chem. A, 2003, 107, 829 CrossRef CAS.
- R. P. Krawczyk, A. Viel, U. Manthe and W. Domcke, J. Chem. Phys., 2003, 119, 1397 CrossRef CAS.
- M. Barbatti, J. Paier and H. Lischka, J. Chem. Phys., 2004, 121, 11614 CrossRef CAS PubMed.
- A. Viel, R. P. Krawczyk, U. Manthe and W. Domcke, J. Chem. Phys., 2004, 120, 11000 CrossRef CAS PubMed.
- M. Barbatti, G. Granucci, M. Persico and H. Lischka, Chem. Phys. Lett., 2005, 401, 276 CrossRef CAS.
- B. G. Levine and T. J. Martínez, Annu. Rev. Phys. Chem., 2007, 58, 613 CrossRef CAS PubMed.
- M. Barbatti, M. Ruckenbauer, J. J. Szymczak, A. J. A. Aquino and H. Lischka, Phys. Chem. Chem. Phys., 2008, 10, 482 RSC.
- E. Fabiano, T. Keal and W. Thiel, Chem. Phys., 2008, 349, 334 CrossRef CAS.
- H. Tao, B. G. Levine and T. J. Martínez, J. Phys. Chem. A, 2009, 113, 13656 CrossRef CAS PubMed.
- K. Saita and D. V. Shalashilin, J. Chem. Phys., 2012, 137, 22A506 CrossRef PubMed.
- H. Tao, T. K. Allison, T. W. Wright, A. M. Stooke, C. Khurmi, J. van Tilborg, Y. Liu, R. W. Falcone, A. Belkacem and T. J. Martinez, J. Chem. Phys., 2011, 134, 244306 CrossRef CAS PubMed.
- B. A. Williams and T. A. Cool, J. Chem. Phys., 1991, 94, 6358 CrossRef CAS.
- B. A. Balko, J. Zhang and Y. T. Lee, J. Chem. Phys., 1992, 97, 935 CrossRef CAS.
- E. F. Cromwell, A. Stolow, M. J. J. Vrakking and Y. T. Lee, J. Chem. Phys., 1992, 97, 4029 CrossRef CAS.
- P. Farmanara, V. Stert and W. Radloff, Chem. Phys. Lett., 1998, 288, 518 CrossRef CAS.
- P. Farmanara, O. Steinkellner, M. T. Wick, M. Wittmann, G. Korn, V. Stert and W. Radloff, J. Chem. Phys., 1999, 111, 6264 CrossRef CAS.
- J. M. Mestdagh, J. P. Visticot, M. Elhanine and B. Soep, J. Chem. Phys., 2000, 113, 237 CrossRef CAS.
- J. J. Lin, C. C. Wang, Y. T. Lee and X. Yang, J. Chem. Phys., 2000, 113, 9668 CrossRef CAS.
- S.-H. Lee, Y. T. Lee and X. Yang, J. Chem. Phys., 2004, 120, 10983 CrossRef CAS PubMed.
- V. Stert, H. Lippert, H.-H. Ritze and W. Radloff, Chem. Phys. Lett., 2004, 388, 144 CrossRef CAS.
- K. Kosma, S. A. Trushin, W. Fuss and W. E. Schmid, J. Phys. Chem. A, 2008, 112, 7514 CrossRef CAS PubMed.
- M. Barbatti, A. J. A. Aquino and H. Lischka, J. Phys. Chem. A, 2005, 109, 5168 CrossRef CAS PubMed.
- T. Mori, W. Glover, M. S. Schuurman and T. J. Martínez, J. Phys. Chem. A, 2012, 116, 2808 CrossRef CAS PubMed.
- B. Sellner, M. Barbatti, T. Müller, W. Domcke and H. Lischka, Mol. Phys., 2013, 111, 2439 CrossRef CAS.
- C. Petrongolo, R. J. Buenker and S. D. Peyerimhoff, J. Chem. Phys., 1982, 76, 3655 CrossRef CAS.
- T. Müller, M. Dallos and H. Lischka, J. Chem. Phys., 1999, 110, 7176 CrossRef.
- G. Bélanger and C. Sandorfy, J. Chem. Phys., 1971, 55, 2055 CrossRef.
- P. Limao-Vieira, E. Vasekova, B. Sekhar, N. Mason and S. Hoffman, Phys. Chem. Chem. Phys., 2006, 8, 4766 RSC.
- J. Quenneville, M. Ben-Nun and T. J. Martínez, J. Photochem. Photobiol., A, 2001, 144, 229 CrossRef CAS.
- J. González-Vázquez and L. González, Chem. Phys., 2008, 349, 287 CrossRef.
- J. González-Vázquez, L. González, I. R. Sola and J. Santamaria, J. Chem. Phys., 2009, 131, 104302 CrossRef.
- S. Gómez, M. Oppel and L. González, Chem. Phys. Lett., 2017, 683, 205 CrossRef.
- M. Richter, P. Marquetand, J. González-Vázquez, I. Sola and L. González, J. Chem. Theory Comput., 2011, 7, 1253 CrossRef CAS PubMed.
- S. Mai, P. Marquetand and L. González, Int. J. Quantum Chem., 2015, 115, 1215 CrossRef.
- S. Mai, M. Richter, M. Heindl, M. F. S. J. Menger, A. Atkins, M. Ruckenbauer, F. Plasser, M. Oppel, P. Marquetand and L. González, SHARC2.0: Surface Hopping Including Arbitrary Couplings – Program Package for Non-Adiabatic Dynamics, http://sharc-md.org, 2018 Search PubMed.
- L. Verlet, Phys. Rev., 1967, 159, 98 CrossRef CAS.
- L. Verlet, Phys. Rev., 1968, 165, 201 CrossRef.
- G. Granucci and M. Persico, J. Chem. Phys., 2007, 126, 134114 CrossRef PubMed.
- L. González, D. Escudero and L. Serrano-Andrés, ChemPhysChem, 2012, 13, 28 CrossRef PubMed.
- B. Roos and K. Andersson, Chem. Phys. Lett., 1995, 245, 215 CrossRef CAS.
- B. Roos, K. Andersson, M. Fülscher, L. Serrano-Andrés, K. Pierloot, M. Merchán and V. Molina, THEOCHEM, 1996, 388, 257 CrossRef CAS.
- R. Kendall, T. Dunning, Jr. and R. Harrison, J. Chem. Phys., 1992, 96, 6796 CrossRef CAS.
- G. Ghigo, B. O. Roos and P.-Å. Malmqvist, Chem. Phys. Lett., 2004, 396, 142 CrossRef CAS.
- J. P. Zobel, J. J. Nogueira and L. González, Chem. Sci., 2017, 8, 1482 RSC.
- F. Aquilante, J. Autschbach, R. Carlson, L. Chibotaru, M. Delcey, L. De Vico, I. F. Galván, N. Ferré, L. Frutos, L. Gagliardi, M. Garavelli, A. Giussani, C. Hoyer, G. Li Manni, H. Lischka, D. Ma, P. Malmqvist, T. Müller, A. Nenov, M. Olivucci, T. Pedersen, D. Peng, F. Plasser, B. Pritchard, M. Reiher, I. Rivalta, I. Schapiro, J. Segarra-Martí, M. Stenrup, D. Truhlar, L. Ungur, A. Valentini, S. Vancoillie, V. Veryazov, V. Vysotskiy, O. Weingart, F. Zapata and R. Lindh, J. Comput. Chem., 2016, 37, 506 CrossRef CAS PubMed.
- F. Plasser, M. Ruckenbauer, S. Mai, M. Oppel, P. Marquetand and L. González, J. Chem. Theory Comput., 2016, 12, 1207 CrossRef CAS PubMed.
- B. O. Roos, P. R. Taylor and P. E. Sigbahn, Chem. Phys., 1980, 48, 157 CrossRef CAS.
- R. Crespo-Otero and M. Barbatti, Theor. Chem. Acc., 2012, 131, 1237 Search PubMed.
- M. Barbatti, M. Ruckenbauer, F. Plasser, J. Pittner, G. Granucci, M. Persico and H. Lischka, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 26 Search PubMed.
- E. A. Coutsias, C. Seok and K. A. Dill, J. Comput. Chem., 2004, 25, 1849 CrossRef CAS PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1327 Search PubMed.
- B. G. Levine, J. D. Coe and T. J. Martínez, J. Phys. Chem. B, 2008, 112, 405 CrossRef CAS PubMed.
- M. K. L. M. Sverdlov and E. Krainov, J. Mol. Struct., 1975, 26, 136 CrossRef.
- R. S. Mulliken, Phys. Rev., 1933, 43, 279 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Conical intersections optimized geometries. See DOI: 10.1039/c8cp07766e |
| This journal is © the Owner Societies 2019 |