
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cations brought together by hydrogen bonds: the protonated pyridine–boronic acid dimer explained†
Íñigo
Iribarren
a,
M. Merced
Montero-Campillo
 b,
Ibon
Alkorta
*a,
José
Elguero
a and
David
Quiñonero
c
b,
Ibon
Alkorta
*a,
José
Elguero
a and
David
Quiñonero
c
aInstituto de Química Médica (CSIC), Juan de la Cierva, 3, E-28006 Madrid, Spain. E-mail: ibon@iqm.csic.es
bDep. de Química, Módulo 13, Facultad de Ciencias, Universidad Autónoma de Madrid, Campus de Excelencia UAM-CSIC, Cantoblanco, 28049-Madrid, Spain
cDepartament de Química, Universitat de les Illes Balears, Crta. de Valldemossa km 7.5, E-07122 Palma de Mallorca, Spain
First published on 18th February 2019
Abstract
According to the Cambridge Structural Database, protonated pyridine–boronic acid dimers exist in the solid phase, apparently defying repulsive coulombic forces. In order to understand why these cation–cation systems are stable, we carried out M06-2X/6-311++G(3df,2pd) electronic structure calculations and used a set of computational tools (energy partitioning, topology of the electron density and electric field maps). The behavior of the charged dimers was compared with the corresponding neutral systems, and the effect of counterions (Br− and BF4−) and the solvent (PCM model) on the binding energies has been considered. In the gas-phase, the charged dimers present positive binding energies but are local minima, with a barrier (16–19 kJ mol−1) preventing dissociation. Once the environment is included via solvent effects or counterions, the binding energies become negative; remarkably, the strength of the interaction is very similar in both neutral and charged systems when a polar solvent is considered. Essentially, all methods used evidence that the intermolecular region where the HBs take place is very similar for both neutral and charged dimers. The energy partitioning explains that repulsion and electrostatic terms are compensated by the desolvation and exchange terms in polar solvents, thus giving stability to the charged dimer.
1. Introduction
The chemical and biochemical mechanisms are often very impressively efficient thanks to the establishment of noncovalent interactions.1 Therefore, a deep understanding and a correct and precise description of the interactions that take place between organic molecules are needed to fully comprehend the basis of highly specific recognition, transport and regulation mechanisms.All these intermolecular interactions are due to forces of diverse nature and, on the whole, can be hierarchically classified according to the strength with which molecules are attracted to each other. Among them, those possessing greater strength of attraction are the ones based purely on electrostatic interactions. Thus, the strongest interaction is established between ions of opposite charge.
The electrostatic interactions coming from hydrogen bonding interactions and salt bridges are ubiquitous in nature. They play a critical role in the determination of protein structures and affect a wide range of biochemical processes such as ion transport channels, and DNA–DNA, RNA–RNA and antibody–antigen interactions.2–6 For instance, in proteins the association of charged amino acids leads to the generation of electrostatic fields that play a vital role in enzymatic transformations, specific interaction with the ligand, allosteric control, and folding and stability of proteins.7–9 The recognition of the interactions between charges is also important in the design of site-directed mutagenesis studies, whereas the interactions of amino acids with protein surfaces influence protein signaling and recognition processes.10–13
In contrast, if we put together two like charges without environment stabilization effects, we would expect such an intermolecular interaction to be highly repulsive, resulting in both ions being infinitely separated instead of getting closer until an equilibrium distance is reached. Consequently, for a long time it has been assumed that it was not possible to find minima between ions with charges of identical signs in the gas phase. However, in 2005 Kass showed computationally that, despite their repulsive interaction energies, dianion complexes formed by carboxylates (derived from oxalic, malonic, terephthalic and glycine among others) could be stable in the gas phase, due to the existence of an energy barrier that prevents the complexes from dissociating.14
Nonetheless, it was not until 2012 that this work inspired researchers to explore the formation of such electrostatically-defying complexes in the gas phase. In this regard, Espinosa and coworkers have been active in this field: they have studied the formation of phosphate–phosphate and other oxoanion-based hydrogen-bonded complexes.15–19 In addition, other authors have reported computational studies on the stability of other complexes formed, thanks to hydrogen-bonding interactions.20–23
From the experimental point of view, the detection of these cation–cation and anion–anion complexes in the gas phase has remained elusive, unless they are stabilized by receptors that can accommodate such dimeric species through noncovalent interactions, as reported for sulfamic acid clusters,24 bisulphate dimers,25 organophosphate dimers,26 phosphate dimers27 and oligomers,28,29 and pyrophosphate dimers.28
Very recently, halogen bonding interactions have also been the subject of theoretical studies of ion like-charge interactions in both dianionic30–32 and dicationic complexes.31–33
In this article, we have explored the Cambridge Structural Database (CSD) for aromatic boronic acid dimers [R–B(OH)2] in neutral (R = phenyl, pyridinyl and pirymidyl) and dicationic forms (R = pyridinium), all summarized in Fig. 1. The reasons behind the stability of the latter charged dimers are studied and the systems confronted by bonding in the neutral systems. For this purpose, DFT calculations are used to find and rationalize the existence of those minima, together with a set of computational methods (energy partitioning, topology of the electron density and electric field maps) to analyze the nature of the interaction between monomers. The potential effect of the environment modeled using the PCM model and counterions (Br− and BF4−) has also been taken into account. All in all, we will try to explain whether charged systems interacting through hydrogen bonds are essentially different from their neutral counterparts and, if so, to what extent.
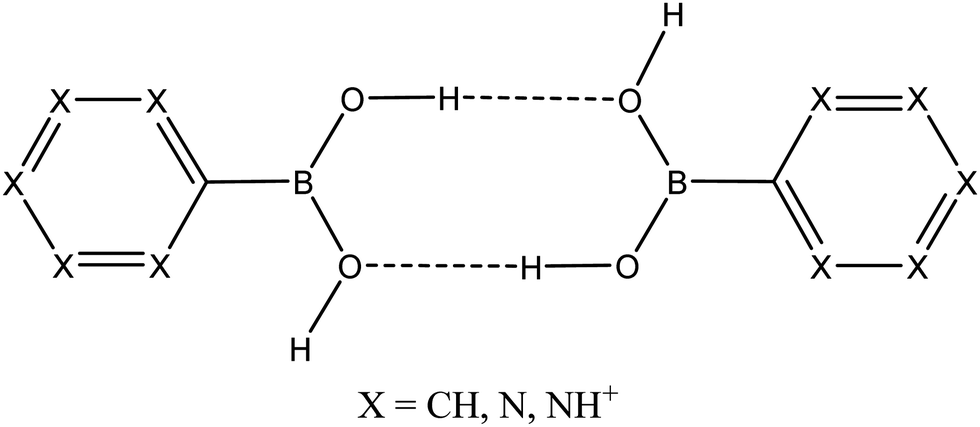 | ||
| Fig. 1 Schematic representation of aromatic boronic acid dimers [R–B(OH)2]2 in neutral (R = phenyl, pyridinyl and pirymidyl) and dicationic forms (R = pyridinium) considered in this work. | ||
2. Computational methods
A CSD search of aromatic-boronic acid dimers has been carried out in version 5.39 (Nov. 17) with three updates (Feb-2018, May-2018 and Aug-2018).34 Only those structures with atomic coordinates have been considered. The geometrical parameters of the structures have been obtained using the programs available in the CSD program package.DFT calculations of the selected systems have been carried out at the M06-2X/6-311++G(d,p) computational level35,36 using the Gaussian09/16 software.37 After optimization, frequency calculations have been performed to confirm that the structures obtained correspond to energetic minima. In order to improve the energy description, single point M06-2X/6-311++G(3df,2pd)//M06-2X/6-311++G(d,p) calculations have been performed. These latter values are used to calculate the binding energy of the systems.
The dissociation profile of the cationic dimers has been studied at the M06-2X/6-311++G(d,p) computational level. To scan the relationship between the energy and distance between monomers, the geometry of the sytems has been optimized at fixed distances in steps of 0.1 Å up to 4 Å from the initial minima. In addition, a few points at shorter distances than the minima have been considered to properly visualize the location of the minima.
The environment of the cationic dimers has been taken into account in two different ways. In the first one, the solvent effect has been implicitly considered by means of the PCM model38 with the n-hexane (ε = 1.882), chloroform (ε = 4.71), acetone (ε = 20.49) and water (ε = 78.36) parameters. In the second one, the effect of the counterion in protonated pyridines has been taken into account by placing a Br− atom or BF4− group close to the N–H groups in the gas phase.
The electron density of the systems has been analyzed within the quantum theory of atoms in molecules (QTAIM)39,40 using the AIMAll program.41 The presence of a bond critical point between two centers has been associated to an attractive bonding interaction. Electric field lines have been calculated from the electrostatic potential, as calculated from AIMAll, and represented with an in-house python program using the Matplotlib library.42
An energy decomposition analysis based on the generalized Kohn–Sham (GKS) and the localized molecular orbital energy decomposition analysis (LMO-EDA) scheme, named GKS-EDA,43 has been carried out at the M06-2X/6-311++G(d,p) computational level in different environments. The interaction energy is obtained as a sum of different energetic terms, as shown in eqn (1):
| Eint = Eelec + Eexc + Erep + Epol + Edisp + Edesolv | (1) |
3. Results and discussion
This section considers firstly the CSD analysis of aromatic boronic acids and later the theoretical study on the dimers of the neutral and cationic structures. We start this second part discussing the optimized structures obtained in the gas phase, followed by the analysis of the effect of the environment (solvent and counterions). After that, an energy partition analysis with explicit consideration of the solvation allows the physics underlying the interaction between neutral and charged species to be explained. As electrostatics plays the main role in these systems, the last sub-section analyzes the electronic characteristics of the systems.3.1. CSD search
The CSD search provided 175 neutral aromatic boronic acid dimers (162 phenyl; 1, 4-pyridyl; 10, 3-pyridyl and 2, 5-pyrimidyl derivatives) and 4 protonated pyridine–boronic acid dimers (3,4-pyridyl and 1,3-pyridyl derivatives) involving two hydrogen bonds (HB) responsible for the interaction between monomers. The list of refcodes of all the structures and intermolecular distances are tabulated in Table S1 of the ESI.† The structure of the phenylboronic acid and protonated 4-pyridylboronic acid dimer (refcodes PHBORA and DUKJUQ), taken as suitable examples, is shown in Fig. 2, whereas those of the four dimers of protonated pyridine derivatives are shown in Fig. S1 (ESI†). | ||
| Fig. 2 X-ray structure of the phenylboronic acid and protonated 4-pyridylboronic acid dimer (CSD refcodes PHBORA and DUKJUQ). | ||
The distribution of the 402 O⋯O distances in the neutral dimers is shown in Fig. 3. Most of the distances are between 2.7 and 2.8 Å (83% of the total), 2.76 Å being the average value of all the distances. The distances found between the protonated pyridine derivatives for the four dimers (Table 1) are between 2.72 and 2.80 Å, with an average value of 2.77 Å. Thus, a first important observation is that no significant differences are found between the neutral and protonated complexes with respect to the intermolecular distances. Among the geometrical characteristics of the dimers, it is interesting to note that, in general, the aromatic rings and boronic acid forming the HB interactions are in the same plane (Fig. S2, ESI†).
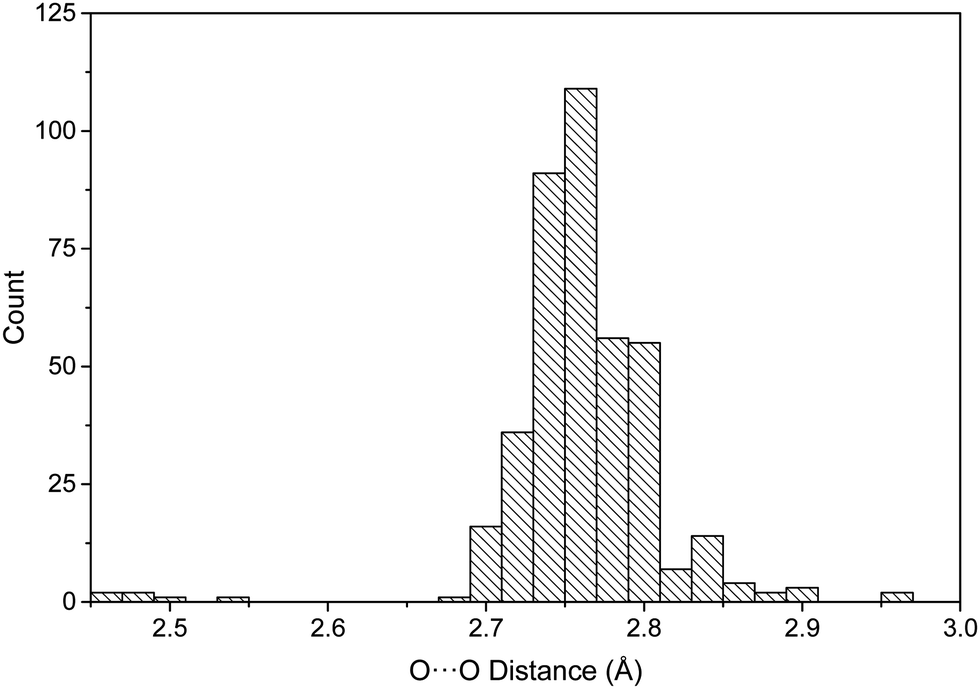 | ||
| Fig. 3 Histogram of the O⋯O distances (Å) in the neutral complexes found in the CSD database for dimers with a double hydrogen bond interaction. | ||
| Refcode | O⋯O distance | Aromatic ring | |
|---|---|---|---|
| DUKJOK | 2.769 | 2.769 | 4-Pyridyl |
| DUKKAX | 2.781 | 2.771 | 4-Pyridyl |
| DUKKEB | 2.725 | 2.761 | 4-Pyridyl |
| DUKKIF | 2.780 | 2.804 | 3-Pyridyl |
3.2. Theoretical calculations in the gas phase
From all the information extracted from the CSD, we will focus on the cationic systems derived from the 2-, 3- and 4-pyridyl boronic acid and their corresponding neutral structures. For each studied complex in the gas phase, we found a minimum with a double HB interaction belonging to the C2 or C2h symmetry groups. The neutral [R–B(OH)2]2 and cationic [(RH–B(OH)2)2]2+ dimers are labeled in Table 2 according to the ring R, where the main geometrical features and the corresponding binding energies are summarized. The geometries of these complexes are given in Table S2 (ESI†).| Aromatic substituent (R) | E b (kJ mol−1) | O⋯H (Å) | O⋯O (Å) | O–H⋯O (°) |
|---|---|---|---|---|
| Ph | −42.67 | 1.843 | 2.815 | 178.0 |
| 4-Py | −42.06 | 1.843 | 2.815 | 177.5 |
| 3-Py | −42.35 | 1.842 | 2.814 | 178.3 |
| 2-Py | −49.89 | 1.813 | 2.784 | 174.0 |
| 4-Py(H+) | 101.43 | 1.896 | 2.867 | 176.6 |
| 3-Py(H+) | 103.90 | 1.896 | 2.867 | 176.6 |
| 2-Py(H+) | 120.70 | 1.900 | 2.869 | 174.5 |
The first look at Table 2 shows negative (attractive) binding energies for the neutral molecules, ranging between −42 and −50 kJ mol−1, which is in clear contrast with positive (repulsive) binding energies for the cations, all above a hundred kJ mol−1 (101 [4-Py(H+)] and 121 kJ mol−1 [2-Py(H+)]). This is not surprising, as we are trying to approach two charged moeities in the gas phase. However, even with the disparity of energies obtained in these two sets, very small differences are found in the geometries of the HBs. The neutral complexes show O⋯H intermolecular distances between 1.84 and 1.81 Å, compared to nearly 1.90 Å in the protonated structures. In all cases, the HB is very linear, corresponding to O–H⋯O angles close to 180° (always larger than 174°). The O⋯O distances are, as expected, slighly larger than the average found in the CSD search (between 2.78 and 2.87 Å).
The fact that the protonated dimers show a stable minimum with positive binding energy is an indication that a barrier should be present somewhere between this minimum configuration and both monomers taken apart at infinite distance, as has been shown in other similar cases.16–19,31 In fact, the dissociation profile of the three protonated dimers exhibits a maximum lying between 16 and 19 kJ mol−1 above the energy of the dimer, as shown in Fig. 4a. It is interesting to note that the intermolecular distance decreases from 4-Py(H+) to 2-Py(H+), i.e., the more distant the NH moiety is from the HB region, the more both monomers are able to approach each other. This is fully consistent with the ranking of the Eb values for the three complexes 4-Py(H+) < 3-Py(H+) < 2-Py(H+) along all the intermolecular distances.
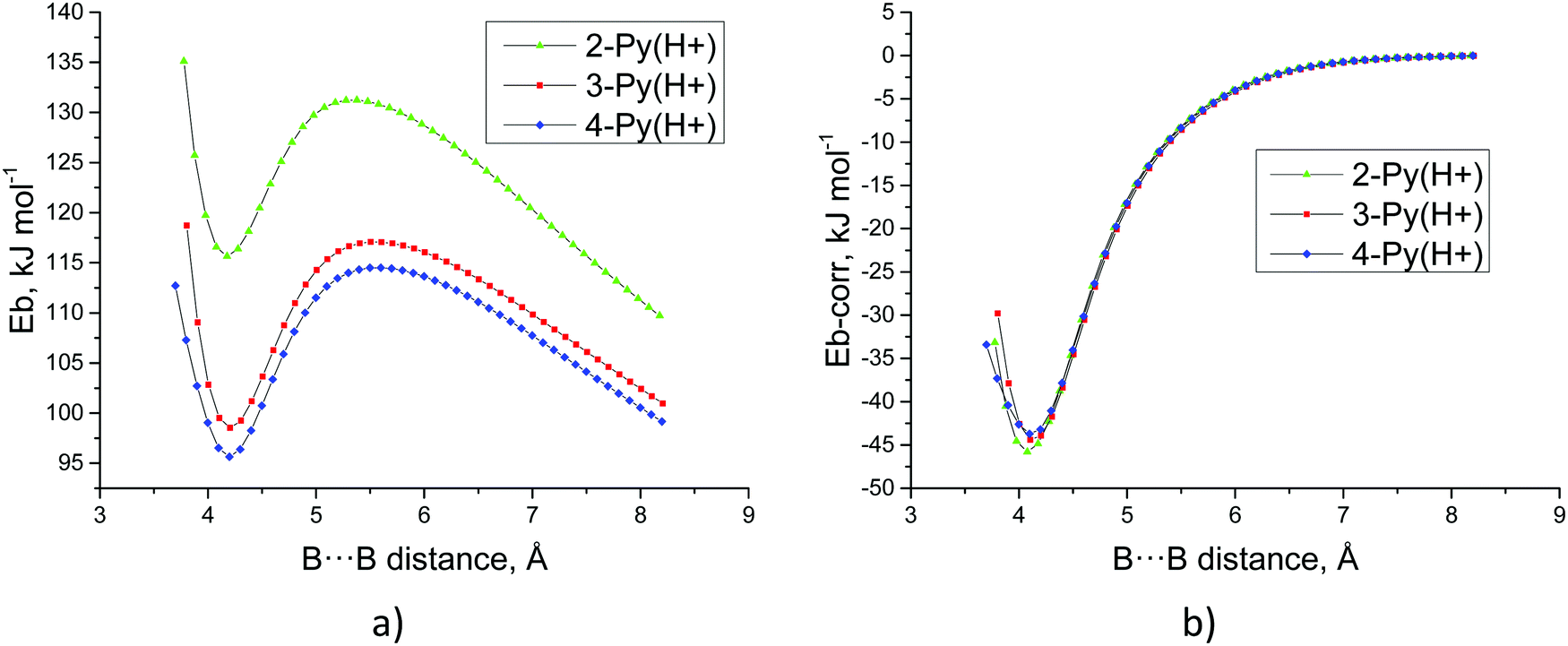 | ||
| Fig. 4 Energy profiles (kJ mol−1) of the dissociation scan (distances in Å): (a) binding energy vs. B⋯B distance and (b) corrected binding energy vs. B⋯B distance. | ||
Using the energetic values for the longest intermolecular distances in the scan, where the only important contribution to the energy corresponds to the electrostatic repulsion, we can obtain an estimation of the distance between the centers of charge of the two molecules. They correspond to 5.81, 5.55 and 4.48 Å plus the B–B distance in each structure along the dissociation profile for the dimers of 4-Py(H+), 3-Py(H+) and 2-Py(H+), respectively. Using such values, the charge–charge electrostatic repulsion energy on each point of the dissociation scan can be calculated and used to correct the binding energy (Eb-corr, see Fig. 4b). These corrected binding energies exhibit their minima at around −45 kJ mol−1. Note that these results are very similar to the binding energies obtained for the neutral dimers (Table 2).
3.3. Solvent effects
Four solvents covering a wide range of polarity [dielectric constants: 1.9 (n-hexane), 4.71 (chloroform), 20.49 (acetone) and 78.4 (water)] have been considered using the PCM method to estimate the effect of the environment on the complexes with respect to the vacuum situation.The binding energies of the neutral complexes (Table 3) decrease in absolute value steadily as the dielectric constant of the solvent increases, since the solvation of the isolated monomers is larger than the one in the corresponding complex, save for R = Ph and 4-Py in water where they show a slight increment. The opposite trend is observed in the charged complexes. Only in n-hexane are the binding energies positive, while the use of chloroform, acetone and water results in more and more negative binding energies. Remarkably, the binding energies of the neutral and charged complexes show small differences in the water–PCM model. This latter result is quite important in terms of energy, showing that if the environment is polar enough, two positively charged systems can attract each other similar to the corresponding neutral partners. Based on this, we found linear correlations between the inverse of the dielectric constant of the solvent and the binding energy in both neutral (R2 between 0.98 and 0.99) and charged dimers (R2 > 0.998, Fig. 5). We used 1/ε following the Coulomb's law, FC = q1q2/4πε0r2. The significant variation of the binding energy of the charged complexes with the solvent and the linear correlation with the inverse of the dielectric constant are clear indications of the importance of the shielding effect of the solvent on the electrostatic component of the energy in these complexes.
| Aromatic substituent (R) | Gas | n-Hexane | CHCl3 | Acetone | Water |
|---|---|---|---|---|---|
| Ph | −42.67 | −39.72 | −36.53 | −34.56 | −34.81 |
| 4-Py | −42.06 | −38.95 | −35.85 | −34.16 | −34.28 |
| 3-Py | −42.35 | −39.39 | −36.60 | −34.84 | −34.40 |
| 2-Py | −49.89 | −45.71 | −41.54 | −38.92 | −38.23 |
| 4-Py(H+) | 101.43 | 34.28 | −7.98 | −27.42 | −31.49 |
| 3-Py(H+) | 103.90 | 35.53 | −7.32 | −27.19 | −31.33 |
| 2-Py(H+) | 120.70 | 45.71 | −2.48 | −25.71 | −30.66 |
 | ||
| Fig. 5 Binding energy (kJ mol−1) vs. 1/ε for the charged dimers in different solvent enviroments. | ||
The inclusion of the solvent reinforces the HBs and produces a shortening of the H⋯O intermolecular distances in all the dimers (Table S3, ESI†). The larger the dielectric constant is, the stronger the HBs become, this effect being more pronounced in the protonated dimers (up to 0.078 Å) than in the neutral ones (up to 0.013 Å).
3.4. Counterion effect
Two model counterions have been considered in this study, Br− and BF4−, placed close to the N–H group of the pyridine in all the complexes. Minima similar to those found in the gas phase or considering the solvent model has been obtained for all cases, save for the case of the 2-Py(H+) dimer with BF4− where the systems is deformed and will not be considered here. The binding energies of these complexes are always negative (between −33 and −63 kJ mol−1), considering as a unit the corresponding protonated pyridine with its counterion. The intermolecular distances (Table 4) are similar to those found in the neutral complexes in the gas phase (Table 2). It is interesting to note that the stabilization of the systems is pretty similar to the one obtained by surrounding the dimer by a polar solvent except for the 2-Py(H+) system, where the counterion effect is more notable.| Aromatic ring | Br− | BF4− | ||
|---|---|---|---|---|
| E b | O⋯H | E b | O⋯H | |
| 4-Py(H+) | −34.88 | 1.846 | −37.04 | 1.847 |
| 3-Py(H+) | −37.76 | 1.845 | −32.56 | 1.849 |
| 2-Py(H+) | −62.59 | 1.834 | — | — |
3.5. Energy partition analysis
We carried out an energy decomposition analysis on the protonated dimers to interpret the contribution of the different kinds of energies to the interaction. The LMOEDA partition (Table 5) shows that the largest term in absolute value corresponds to the repulsion term, which ranges between 130 and 186 kJ mol−1. Notably, the other term that shows a repulsive contribution is the electrostatic one, ranging between 92 and 50 kJ mol−1. In general, it decreases as the dielectric constant of the solvent increases, even though the two monomers are close together. In addition, for a given solvent this term is ranked as follows: 2-Py(H+) > 3-Py(H+) > 4-Py(H+). The most important attractive term is the desolvation energy, increasing in absolute value with the polarity of the solvent except in n-hexane and vacuum, where the exchange is more important. The desolvation and exchange terms can reach values of −165 and −103 kJ mol−1 in water. The stabilization contribution of the polarization term is more modest ranging between −44 and −22 kJ mol−1, being more important in the less polar environments, but not enough to overcome repulsion and electrostatics.| Energy term | Vacuum | n-Hexane | Chloroform | Acetone | Water | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 2-Py(H+) | 3-Py(H+) | 4-Py(H+) | 2-Py(H+) | 3-Py(H+) | 4-Py(H+) | 2-Py(H+) | 3-Py(H+) | 4-Py(H+) | 2-Py(H+) | 3-Py(H+) | 4-Py(H+) | 2-Py(H+) | 3-Py(H+) | 4-Py(H+) | |
| Electrostatic | 85.9 | 82.6 | 79.4 | 91.5 | 68.7 | 63.7 | 59.6 | 83.1 | 56.8 | 78.2 | 54.9 | 51.3 | 77.0 | 53.3 | 49.8 |
| Exchange | −91.4 | −71.1 | −71.5 | −81.7 | −87.0 | −91.6 | −96.6 | −91.8 | −95.2 | −98.5 | −101.9 | −101.9 | −100.3 | −102.7 | −102.2 |
| Repulsion | 170.6 | 131.4 | 131.9 | 150.5 | 159.1 | 156.1 | 175.2 | 168.0 | 172.8 | 179.5 | 183.8 | 184.0 | 183.0 | 185.9 | 185.0 |
| Polarization | −44.3 | −36.2 | −35.4 | −34.6 | −31.3 | −35.9 | −26.6 | −31.3 | −25.1 | −30.0 | −24.4 | −23.3 | −30.0 | −23.2 | −22.1 |
| Desolvation | — | — | — | −75.6 | −68.4 | −72.2 | −115.5 | −128.7 | −113.5 | −153.7 | −137.0 | −134.8 | −165.6 | −146.8 | −144.4 |
| DFT correlation | −13.6 | −11.5 | −12.1 | −13.9 | −14.2 | −18.4 | −16.1 | −15.7 | −16.3 | −16.7 | −16.9 | −17.0 | −17.1 | −17.7 | −17.5 |
| Total interaction energy | 107.2 | 95.2 | 92.2 | 36.2 | 26.9 | 21.8 | −20.0 | −16.5 | −20.7 | −41.3 | −41.3 | −41.8 | −53.2 | −51.2 | −51.3 |
3.6. Electrostatic and electron density
The existence of minima dimers for the neutral and corresponding protonated monomers allows a comparison of the electric field maps. The ones corresponding to the 4-pyridine complexes (neutral and protonated) are shown in Fig. 6 as a suitable example (the rest are tabulated in Fig. S3, ESI†). The atomic basins of the B(OH)2 group of one of the monomers are shaded in grey. What is more remarkable in Fig. 6 is that the intermolecular region where the HBs take place is very similar for both neutral and charged dimers. It can be seen that the region of electric field gradient associated with the hydrogen atoms involved in the HB includes part of the electron density basin of the oxygen that forms the HB. Thus, in both cases there is a very similar electrostatic attraction between these two atoms involved in the HB, independent of the overall charge of the system. In contrast, the two maps look very different out of the HB region. In the neutral system, the hydrogen atom bonded to the oxygen atoms and not involved in the HB can interact with the oxygen of the other molecule. This feature is absent in the protonated dimers since all the rest of the electric field gradient lines not involved in the HB no longer connect the two molecules. | ||
| Fig. 6 Electric field maps of the neutral and protonated 4-pyridine dimers. The region in grey corresponds to the volume of the B(OH)2 group in one of the molecules. The electric field lines are colored based on the atom of origin (grey for carbon, black for hydrogen, red for oxygen, blue for nitrogen and orange for boron). | ||
Complementing this picture, the electron density analysis shows two BCPs associated with the HBs formed in all the dimers, independent of the environment considered (gas, solvent or counterion). The molecular graphs of the dimers in the gas phase are tabulated in Table S2 (ESI†). The values of the electron density at the BCP range between 0.033 and 0.025 a.u., the Laplacian between 0.128 and 0.098 a.u. and the total energy density between 0.002 and 0.001 a.u. These values are typical of hydrogen bonds in the pure closed shell region.45 Importantly, the characteristics of the BCPs at the HBs are independent of the charge or environment of the complexes, and show very good correlations with the interatomic distances (Fig. S4, ESI†).
4. Conclusions
A search in the CSD database shows that both neutral and protonated pyridine–boronic acid dimers exist, indicating that two cationic moieties can be held together through hydrogen bonds in the solid phase. The analysis of the geometrical properties of all the complexes shows no significant differences between the neutral cases and the cation–cation complexes. A theoretical analysis of the cation–cation complexes in the gas phase helped to find a metastable minimum for each protonated 2-, 3- and 4-pyridine–boronic acid dimer. The dissociation profile shows a barrier that avoids spontaneous dissociation of the cation–cation complexes, which increases in the order 2- < 3- < 4-pyridine. If the dissociation profiles are corrected with the charge–charge repulsion, they resemble the ones expected for a neutral system.The effect of the environment by means of implicit PCM solvent models and explicit counterions has been considered. In both cases, the electrostatic repulsion is significantly reduced producing stable complexes. The analysis of the electronic and electrostatic properties of the neutral and cation–cation complexes evidenced that the intermolecular region where the hydrogen bond interactions are formed is very similar in both cases.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Thanks are given to Dr Ignasi Mata (ICMAB-CSIC) for providing a copy of the program to plot the electric field. This work was carried out with financial support from the Ministerio de Economía, Industria y Competitividad (Projects No. CTQ2015-63997-C2-2-P, CTQ2014-57393-C2-1-P and CTQ2017-85821-R FEDER funds) and Comunidad Autónoma de Madrid (S2013/MIT2841, Fotocarbon).References
- H. J. Schneider, Angew. Chem., Int. Ed., 2009, 48, 3924 CrossRef CAS PubMed.
- A. Warshel, Acc. Chem. Res., 1981, 14, 284 CrossRef CAS.
- M. F. Perutz, Science, 1978, 201, 1187 CrossRef CAS PubMed.
- P. G. Schultz, Acc. Chem. Res., 1989, 22, 287 CrossRef CAS.
- A. A. Kornyshev and S. Leikin, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 13579 CrossRef CAS.
- B. H. Honig, W. L. Hubbell and R. F. Flewelling, Annu. Rev. Biophys. Biophys. Chem., 1986, 15, 163 CrossRef CAS PubMed.
- J. M. Baldwin, Prog. Biophys. Mol. Biol., 1975, 29, 225 CrossRef CAS PubMed.
- D. Van Belle, I. Couplet, M. Prevost and S. J. Wodak, J. Mol. Biol., 1987, 198, 721 CrossRef CAS PubMed.
- K. A. Sharp and B. Honig, Annu. Rev. Biophys. Biophys. Chem., 1990, 19, 301 CrossRef CAS PubMed.
- G. von Heijne, J. Mol. Biol., 1992, 225, 487 CrossRef CAS PubMed.
- G. von Heijne, Eur. J. Biochem., 1983, 133, 17 CrossRef CAS PubMed.
- M. J. Sternberg, F. R. Hayes, A. J. Russell, P. G. Thomas and A. R. Fersht, Nature, 1987, 330, 86 CrossRef CAS PubMed.
- M. del Carmen Fernandez-Alonso, D. Diaz, M. Alvaro Berbis, F. Marcelo, J. Canada and J. Jimenez-Barbero, Curr. Protein Pept. Sci., 2012, 13, 816 CrossRef PubMed.
- S. R. Kass, J. Am. Chem. Soc., 2005, 127, 13098 CrossRef CAS PubMed.
- I. Mata, I. Alkorta, E. Molins and E. Espinosa, ChemPhysChem, 2012, 13, 1421 CrossRef CAS PubMed.
- I. Mata, I. Alkorta, E. Molins and E. Espinosa, Chem. Phys. Lett., 2013, 555, 106 CrossRef CAS.
- I. Mata, E. Molins, I. Alkorta and E. Espinosa, J. Phys. Chem. A, 2015, 119, 183 CrossRef CAS PubMed.
- I. Alkorta, I. Mata, E. Molins and E. Espinosa, Chem. – Eur. J., 2016, 22, 9226 CrossRef CAS PubMed.
- I. Alkorta, I. Mata, E. Molins and E. Espinosa, ChemPhysChem, 2019, 20, 148 CrossRef CAS PubMed.
- A. Shokri, M. Ramezani, A. Fattahi and S. R. Kass, J. Phys. Chem. A, 2013, 117, 9252 CrossRef CAS PubMed.
- F. Weinhold and R. A. Klein, Angew. Chem., Int. Ed. Engl., 2014, 53, 11214 CrossRef CAS PubMed.
- G. Frenking and G. F. Caramori, Angew. Chem., Int. Ed., 2015, 54, 2596 CrossRef CAS PubMed.
- F. Weinhold, Inorg. Chem., 2018, 57, 2035 CrossRef CAS PubMed.
- M. Pszona, K. Haupa, A. Bil, K. Mierzwicki, Z. Szewczuk and Z. Mielke, J. Mass Spectrom., 2015, 50, 127 CrossRef CAS PubMed.
- E. M. Fatila, E. B. Twum, A. Sengupta, M. Pink, J. A. Karty, K. Raghavachari and A. H. Flood, Angew. Chem., Int. Ed., 2016, 55, 14057 CrossRef CAS PubMed.
- W. Zhao, B. Qiao, C. H. Chen and A. H. Flood, Angew. Chem., Int. Ed., 2017, 56, 13083 CrossRef CAS PubMed.
- Q. He, M. Kelliher, S. Bahring, V. M. Lynch and J. L. Sessler, J. Am. Chem. Soc., 2017, 139, 7140 CrossRef CAS PubMed.
- D. Mungalpara, A. Valkonen, K. Rissanen and S. Kubik, Chem. Sci., 2017, 8, 6005 RSC.
- E. M. Fatila, M. Pink, E. B. Twum, J. A. Karty and A. H. Flood, Chem. Sci., 2018, 9, 2863 RSC.
- G. Wang, Z. Chen, Z. Xu, J. Wang, Y. Yang, T. Cai, J. Shi and W. Zhu, J. Phys. Chem. B, 2016, 120, 610 CrossRef CAS PubMed.
- D. Quinonero, I. Alkorta and J. Elguero, Phys. Chem. Chem. Phys., 2016, 18, 27939 RSC.
- C. Wang, Y. Fu, L. Zhang, D. Danovich, S. Shaik and Y. Mo, J. Comput. Chem., 2018, 39, 481 CrossRef CAS PubMed.
- S. M. Chalanchi, I. Alkorta, J. Elguero and D. Quinonero, ChemPhysChem, 2017, 18, 3462 CrossRef CAS PubMed.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171 CrossRef CAS PubMed.
- Y. Zhao and D. Truhlar, Theor. Chem. Acc., 2008, 120, 215 Search PubMed.
- M. J. Frisch, J. A. Pople and J. S. Binkley, J. Chem. Phys., 1984, 80, 3265 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, 2009 Search PubMed.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999 CrossRef CAS PubMed.
- P. L. A. Popelier, Atoms In Molecules. An introduction, Prentice Hall, Harlow, England, 2000 Search PubMed.
- R. F. W. Bader, Atoms in Molecules: A Quantum Theory, Clarendon Press, Oxford, 1990 Search PubMed.
- T. A. Keith, AIMAll, 2017 Version 17.11.14.
- J. D. Hunter, Comput. Sci. Eng., 2007, 9, 90 Search PubMed.
- P. Su, Z. Jiang, Z. Chen and W. Wu, J. Phys. Chem. A, 2014, 118, 2531 CrossRef CAS PubMed.
- M. W. Schmidt, K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. H. Jensen, S. Koseki, N. Matsunaga, K. A. Nguyen, S. Su, T. L. Windus, M. Dupuis and J. A. Montgomery, J. Comput. Chem., 1993, 14, 1347 CrossRef CAS.
- E. Espinosa, I. Alkorta, J. Elguero and E. Molins, J. Chem. Phys., 2002, 117, 5529 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cp07542e |
| This journal is © the Owner Societies 2019 |