
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDNA-binding mechanism of spiropyran photoswitches: the role of electrostatics†
Davide
Avagliano
,
Pedro A.
Sánchez-Murcia
 * and
Leticia
González
*
* and
Leticia
González
*
Institute of Theoretical Chemistry, Faculty of Chemistry, University of Vienna, Währinger Strasse 17, 1090, Vienna, Austria. E-mail: leticia.gonzalez@univie.ac.at
First published on 15th February 2019
Abstract
The binding mechanism of the protonated open form of three spiropyran derivatives into a 12-mer (poly-dAT)2 has been unveiled by means of computational methods. It is found that the electrostatic term in the probe:DNA binding energy, modulates the binding mode, providing new guidelines for the design of spiropyran photoswitches with specific binding modes to DNA.
Molecular photoswitches – molecules that change their chemical configuration upon light absorption – are useful tools to monitor the conformational state of biomolecules, in both temporal and spatial scales.1 The advantages of these probes are that the implicated species in the photochemical equilibrium highly differ in the binding affinity to the macromolecule upon light absorption, that these species are interconvertible in a reversible manner, and most importantly, that the two species can be selectively accessed by illumination at different wavelengths and/or by thermal activation. Spiropyrans (SP) (Scheme 1) belong to a well-known class of photoswitches, which after light irradiation undergo heterocyclic cleavage to yield the trans-merocyanine form (MC).2–6
 | ||
| Scheme 1 Spirocyclic (SP) compounds 1a,71b,8 the proof of concept 1c and their corresponding unprotonated (MC, 2) and protonated (MCH, 3) merocyanine open forms. Compounds 3 are studied in this work. | ||
Spiropyrans have an indoline and a chromene moiety oriented perpendicular to each other and bound together via a Csp3 spiro-junction. Upon absorption of UV light, the sigma C–O bond on the spiranic carbon cleavages and builds the MC form. Thereafter, MC can reversibly turn to SP with heat or after irradiation with visible light. Whereas the SP species has moderate lipophilicity, the conjugated coplanar system MC is a zwitterionic species with increased polarity favouring intercalation to polyanions such as DNA via π–π stacking interactions. In contrast, the SP form shows no or very little affinity to DNA. The half-time of the MC form increases upon substitution on the phenyl ring with electron withdrawing groups, such as NO2.9–12 For instance, almost half of the population in solution of 1a (Scheme 1), which contains the scaffold 3′,3′-dimethyl-6-nitrospiro[2H-1-benzopyran-2,2′-indoline] (6-nitro-BIPS) with a trimethylammoniumpropyl group on the indole nitrogen, is converted to 2a after irradiation at 254 nm.7 Despite its efficiency, the binding mode of this family of photoswitches into DNA is still under debate. Experiments with flow-oriented linear dichroism (LD) spectroscopy strongly support that 2a intercalates into DNA given the dramatic blue-shift of the absorption spectrum of the drug in solution.7 A few years later, the same authors found13 that the maximum of the absorption spectrum of the protonated form 3a in buffered aqueous solution is blue-shifted compared to 2a, in analogy to what was previously observed after binding of 2a to DNA.7 Therefore, it is likely that 2a gets protonated to form 3a before binding to DNA.
The latter hypothesis was also supported by the measurement of the DNA binding constants of the amidinium-substituted derivatives 1b–3b.8 The protonated 3b form intercalates stronger into DNA by a factor of ca. 50 compared to the unprotonated 2b one. Again, and in analogy to 3a, the protonated form 3b was proposed to be the relevant DNA binder with a similar angle in the LD experiment (ca. 70°). Based on this experimental evidence, the authors ascribed the differences in the binding affinities of 3a and 3b to the total net positive charge on the photoprobe.8 However, the mechanism of the intercalation of 3a and 3b remains uncertain since before intercalation these species could bind to different grooves of the DNA.14 This is, for example, the case of the drug daunomycin (aka. doxorubicin) that can visit up to four states before intercalation, some of them pre-intercalative states,15 which were identified by molecular dynamics (MD) simulations as local minima bound in the minor groove of DNA.16,17
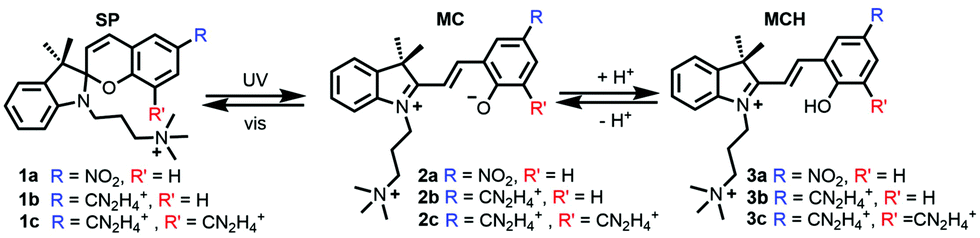
In this work, we unravel the intercalation modes of spiropyran derivatives from the solvent into DNA using MD and umbrella sampling simulations. To this aim, a 12-mer (poly-dAT)2 has been selected as these compounds are known to bind to AT.7 Upon light-driven ring opening and concomitant protonation of the phenoxy moiety (MCH in Scheme 1), 3a intercalates into (poly-dAT)2. However, before inserting into DNA, 3a has to travel from the bulk solvent into the nucleic acid. Potentially, the probe can visit a few binding modes before intercalation, i.e. entering DNA by the major (path M) or by the minor (path m) groove, as sketched in Fig. 1a. Experimentally, it is known7 from flow-oriented LD that 3a in the presence of DNA shows one negative peak around 420 nm. A single peak indicates that 3a has only one binding mode. Furthermore, it was determined that the photoprobe forms an angle of 80 ± 5° with the main Z-axis of DNA, meaning that the transition dipole moment of 3a is parallel to the plane of the sandwiched nucleobases, and thus, the molecule is intercalated in a sandwich-like way between the DNA base pairs. Unfortunately, and to the best of our knowledge, neither kinetics nor structural data of the intercalation process of any of the derivatives of the SP family have been reported to date. Therefore, we explore here the two possible binding pathways to the major (M) and minor (m) groove of 3a by means of umbrella sampling MD simulations.
 | ||
| Fig. 1 (a) Intercalation process of 3a into a 12-mer (poly-dAT)2 though path M (major groove) and path m (minor groove). (b) Free energy profiles (kcal mol−1) of path M (major) and m (minor) computed with umbrella sampling MD simulations. The bootstrap error values are shown over the plots. | ||
As the reaction coordinate (RC) for binding to both grooves, we define the distance between the center-of-mass of 3a and the center-of-mass of the four nucleobases in between 3a inserts. From the initial intercalated geometries (named M3 and m3 in Fig. 1a), we pull out the probe from the DNA (RC ∼ 2 Å) into the bulk solvent up to RC = 26 Å (M1 and m1). The whole RC was divided into 50 windows separated by 0.5 Å and the system was allowed to oscillate around the RC for 5 ns per window – the time at which the energy curves were converged (Sections S1 and S2, ESI†). The initial guesses for M3 and m3 were obtained by manual docking after structural superimposition of our system with the crystal structure of daunomycin intercalated into DNA (PDB id. 1VTH).18 Then, the two systems were minimized and equilibrated in explicit solvent. Due to the presence of the two methyl groups on the indoline ring of 3a (Scheme 1), there was only one possibility in each case for the relative orientation of these groups with respect to the methyl group of the two thymine units, in between the probe intercalates in the states M3 and m3.
Fig. 1b shows the obtained energy profiles for path M (blue line) and m (red line). In both cases, the chemical probe visits several pre-intercalative states before intercalation. However, our calculation clearly predicts that binding to the major groove (pathway M) is unfavorable. When 3a approaches to the DNA through the major groove it visits only metastable states since all the local minima between 25 and 3 Å are higher in energy compared to the minimum M1. Moreover, the intercalated state M3 is less stable than both the solvated state M1 (RC = 25 Å, ΔE > 3 kcal mol−1) and the local minimum M2 (at ∼10 Å). Inspection of the M3 geometry reveals that the ligand's rings are coplanar and inserted between four nucleobases parallel to them. In such a conformation, the binding site is highly distorted and the hydrogen bonds between the surrounding base pair are broken (A7:T6′ and A5:T8′, Fig. S1, ESI†). This explains the unfavorable energy for the state M3.
In contrast, the intercalated state m3 is the most thermodynamically stable binding mode. Along the minor groove binding pathway, we identified several metastable pre-intercalative states of 3a (between RC = 8 Å and RC = 23 Å) that end up in the local minimum m2 at RC = 9 Å. Since the energy of m2 is quite similar to m1 (ΔE = −1.5 kcal mol−1), the binding mode m2 is reversible and 3a could return to the bulk solvent. However, supplying only ca. 5 kcal mol−1, 3a can overcome the energy barrier between m2 and m3 to reach the latter intercalative state. This extra energy is used to open the space between the nucleobases.
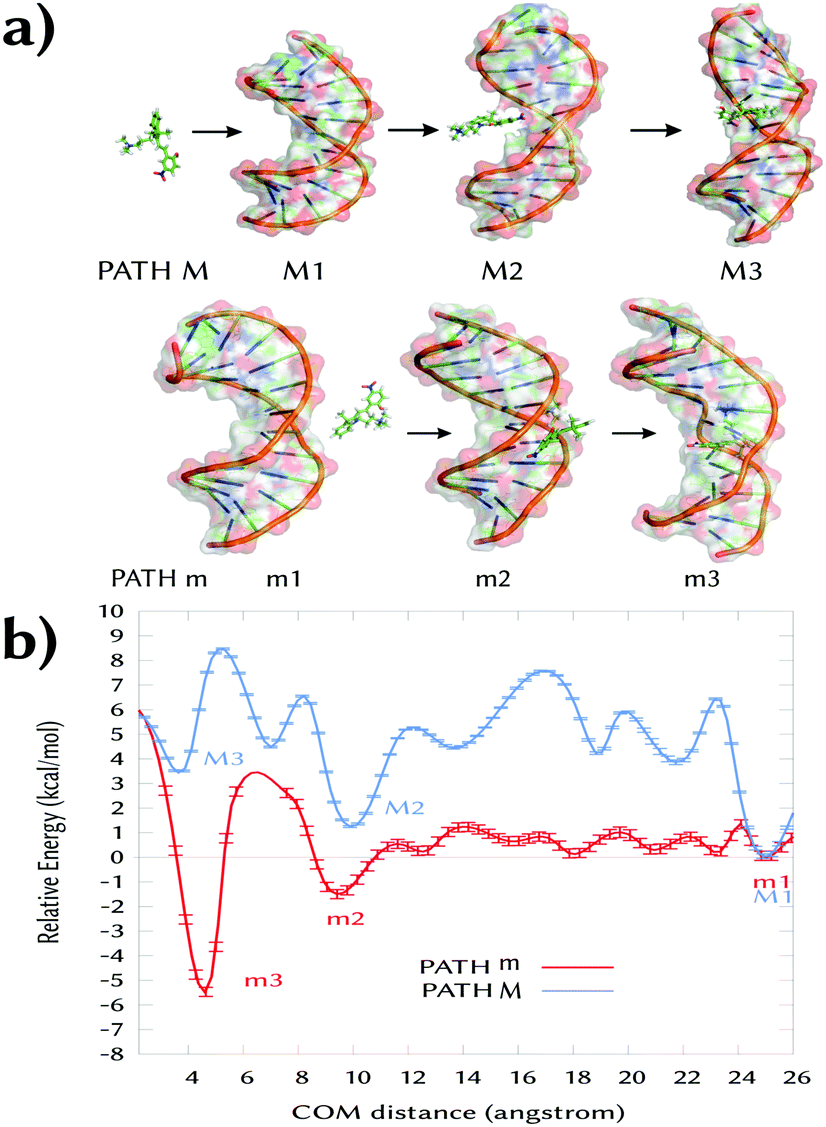
Fig. 2 illustrates the geometry of 3a in the m2 state. The indoline and the phenol rings are almost co-planar with a small rotation (ca. 15° with respect to each other, Fig. S2, ESI†) and the molecule is parallel to the DNA backbone. The nitro group is oriented to the same side of the ammoniumpropyl tail and projected outwards to the solvent. In such a disposition, the phenolic hydroxyl group establishes a hydrogen-bond with the carbonyl oxygen of a thymine T6′ (dashed line, Fig. 2a). This interaction remains when the system is further simulated during 100 ns without restraints. This observation, together with the orientation of the phenol of 3a in m2 and m3, could explain the higher experimental binding constant values for the MCH form (3a) compared to the MC one (2a).7 Indeed, as can be seen in Fig. 2b and 3a, the phenoxy group in 2a would face a negatively-charged region in both m3 and m2 states, respectively. In addition, the computed adaptive Poisson–Boltzmann solver (APBS) electrostatic potential on the surface of the DNA for m2 (Fig. 2b) shows that 3a binds to a region with high negative potential (red contour) and that the nitro group is oriented to a positive region (blue contour). Thus, 3a establishes a large number of contacts in the short range (ca. 243 in an inner sphere of 4 Å) along the MD simulation, including the interactions between the trimethylammoniumpropyl group of 3a and the phosphate groups in the DNA.
 | ||
| Fig. 2 (a) Representative binding geometry of 3a (sticks, carbon atoms colored in yellow) in m2 after 100 ns of unrestrained MD simulation. Only the closest water molecules (lines) are shown. (b) The APBS-generated electrostatic potential on the van der Waals surface of the DNA in m2 in the presence of 3a. | ||
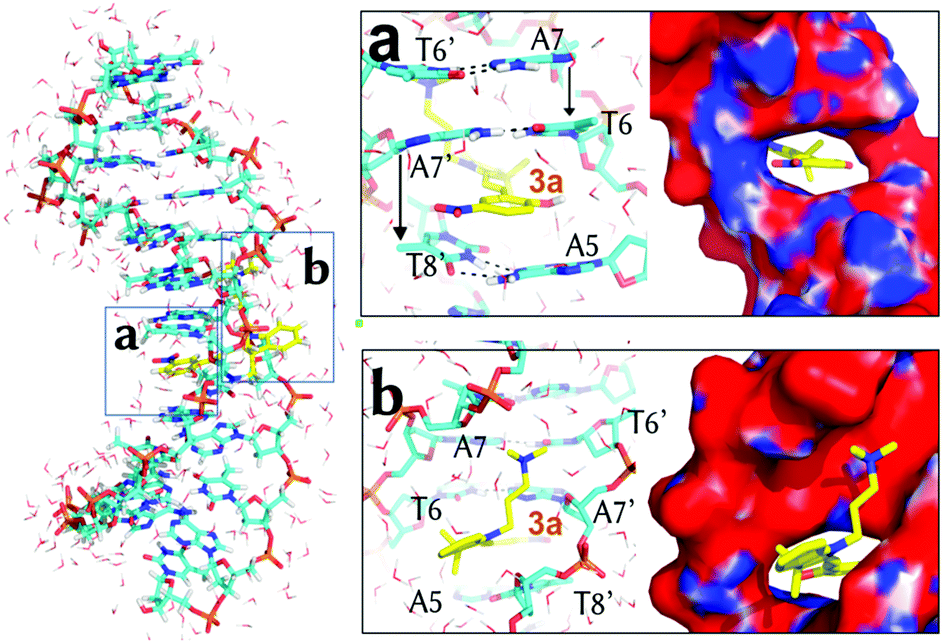 | ||
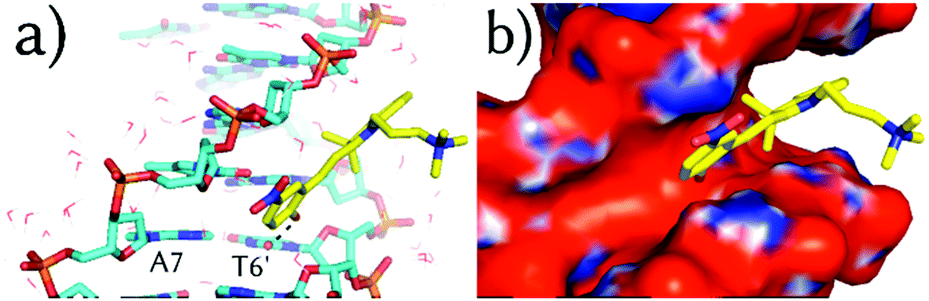
| Fig. 3 Representative binding geometry of 3a in m3 after 100 ns of unrestrained MD simulation. Boxes (a) and (b) zoom-in on the detailed orientation of the phenol and the indoline rings, respectively. Only the closest water molecules (lines) are shown. The APBS-generated electrostatic potential is also shown on the van der Waals surface of the DNA. | ||
Once 3a visits m2, the system requires ca. 5 kcal mol−1 to make room between the four stacking nucleobases A7:T6′ and A5:T8′ it binds to. Once in m3, only the phenol ring of 3a is parallel to the planes defined by the former four nucleobases and almost perpendicular to the main Z-axis of the DNA (Fig. 3a). In m3, 3a establishes face-to-face π–π stacking interactions between the phenol ring and the base pairs T6:A7′ and A5:T8′, as observed for other intercalative agents.19 The distances between the phenol ring and each of the former base pairs are, in fact, below 4 Å. The hydrogen-bonds of the Watson–Crick base pairs T6:A7′ and A5:T8′ and the surrounding A7:T6′ and T8:A5′ are kept in the presence of 3a but the distance between the nucleobases on the same strand T6 and A5 and A7′ and T8′ is increased from the non-intercalative crystallographic distance of 5 Å (like between A7 and T6) to 7.1 Å and 6.3 Å, respectively. Finally, the nitro group of 3a is again looking at a positive potential region (Fig. 3a) and the ammoniumpropyl tail interacts with a region with negative potential in between the phosphate groups (Fig. 3b).
The intercalation of 3a increases the angle between the indoline and the chromene moieties (Fig. S2, ESI†) and enlarges the amplitude of this hampered rotation between the two rings. This finding agrees with the larger experimental wavelength shift in the absorption spectrum upon intercalation7 than when going from water to chloroform in the absence of DNA, what may indicate a conformational change to a nonplanar structure of 3a in m3.
The substitution of the nitro group in 3a by an amidinium group (compound 3b)8 is known to increase the binding constant of the photoprobe to DNA. This effect could be due to the specific interactions of the amidinium group, the increment in the net positive charge in 3b, or a combination of both factors. In order to discern between these explanations, we simulated the binding states m2 and m3 of 3b with the (poly-dAT)2. We directly employed the already calculated binding states m2 and m3 of 3a, as there is no experimental evidence of different binding geometries or orientations between the nitro and the amidinium substituted MCH (3a and 3b), except for a slight difference in the measured angle in the flow-oriented linear dichroism (LD) spectra.8
Our computations show that 3b in both m2 and m3 states shares the main binding spots found in the nitro derivative (Fig. S3, ESI†) with the amidinium group projected to the solvent. In the m2 binding mode, the amidinium group turned out to have no specific interaction with the DNA backbone or with the nucleobases. Therefore, the first possibility highlighted above cannot explain why 3b binds stronger to DNA than 3a. Thus, we computed the binding energies of 3a and 3b to DNA using the molecular mechanics Poisson–Boltzmann surface area (MM-PBSA) method.20
We considered both m2 and m3 binding modes for each of the compounds. Table 1 collects the total solvated binding energies of 3a and 3b to DNA and their individual terms. In both cases, the intercalative state m3 is the most stable state (Fig. S4, ESI†). 3b shows the largest binding energy value due to a smaller positive (repulsive) electrostatic energy component (ΔEelec) than in 3a. This term has two contributions: the electrostatic component of the Poisson–Boltzmann solvation energy (ΔEPB) and the electrostatic term in the gas phase (ΔEEEL) (Table S1, ESI†). In the pre-intercalative state m2, ΔEelec strongly decreases between 3a and 3b (109%) so that the electrostatic component is so severely reduced in 3b that it turns negative (attractive by −1.02 kcal mol−1). Therefore, one is left to conclude that the increase of the positive charge on the probe strengthens the binding of 3 to DNA.
| Compound | Mode | ΔEelec | Non-electrostatic terms | TΔS | ΔGbind | |
|---|---|---|---|---|---|---|
| (ΔEEEL + ΔEPB) | ΔEvdW | ΔEnonpol | ||||
| 3a | m3 | 15.81 ± 0.14 | −55.17 ± 0.25 | −4.32 ± 0.01 | −21.73 ± 0.09 | −21.96 ± 0.24 |
| 3b | 10.10 ± 0.40 | −55.78 ± 0.23 | −4.62 ± 0.40 | −20.96 ± 0.17 | −29.35 ± 0.33 | |
| 3c | 11.09 ± 0.59 | −-50.71 ± 0.42 | −4.57 ± 0.03 | −26.42 ± 0.37 | −17.77 ± 0.74 | |
| 3a | m2 | 10.82 ± 0.22 | −37.72 ± 0.41 | −3.23 ± 0.01 | −20.97 ± 0.02 | −9.29 ± 0.47 |
| 3b | −1.02 ± 0.60 | −34.16 ± 0.26 | −3.22 ± 0.01 | −23.88 ± 0.13 | −14.52 ± 0.36 | |
| 3c | −12.07 ± 0.39 | −29.91 ± 0.49 | −3.19 ± 0.02 | −24.71 ± 0.26 | −20.46 ± 0.61 | |
With this conclusion in hand, and as a proof of concept, we designed the photoprobe 6,4-diamidinium-MCH (3c, Scheme 1), where an extra amidinium group was introduced in the ortho position to the hydroxyl group of the phenol ring. This compound – with an extra net charge – should validate our hypothesis that additional electrostatic contributions could then further increase the binding energy to DNA, and thereby, its binding constant. The ortho position was selected since it is chemically modifiable21 and the orientation of the group does not interfere with the proposed m2 and m3 binding modes. The values for the total binding energy are also shown in Table 1. Surprisingly, we observed that the extra amidinium group of 3c increases the binding energy only for the m2 mode, and the m3 state is less stable than the corresponding state in 3a or 3b. Note that the role of the entropy, although significant, is not the decisive term for the relative stability between states m2 and m3.
Fig. 4 depicts the binding energies for both the m2 and m3 binding modes of all three derivatives 3a–3c. Whereas in 3a and 3b the m2 state is always below in energy compared to m3, in 3c the pre-intercalative state m2 competes with m3 as the most stable binding mode. This means that, from the thermodynamical point of view, 3c would not intercalate, but rather bind in the minor groove, as other agents do.22 Our results are supported by the fact that 3c has similar size and charge compared to other minor groove binders (Fig. S5, ESI†) and also, like most of them, shows pronounced AT selectivity.
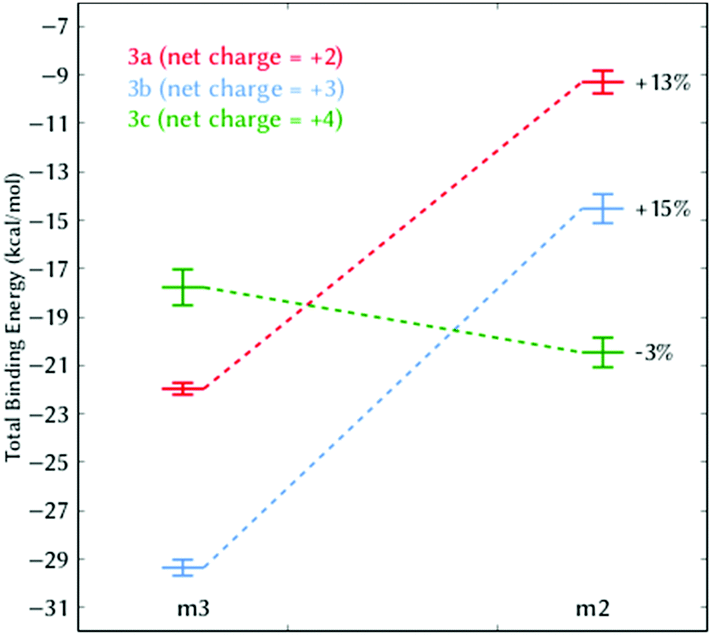 | ||
| Fig. 4 Total binding energy (kcal mol−1) of 3a–3c between the binding modes m2 and m3. The percentage indicates the relative energetic change of m2 compared to m3. | ||
These results indicate that care has to be taken in the modulation of the electrostatic component in photoactivatable DNA-targeting spiropyrans. Although experiments suggest that MCH species bind to DNA better than MC due to its increased electrostatics (e.g.2bversus3b),8 an excess of net positive charge on MCH can stabilize the pre-intercalative state m2 such that this binding site becomes competitive (e.g.3cversus3b); this is e.g. the case in polyamides or diamidines, which are species known to selectively bind AT pairs in the minor groove but not to intercalate.22
In summary, this work indicates that the modulation of the electrostatic potential in this family of spiropyrans can be used as a tool to control their binding mode to DNA (minor groove binder or intercalation). And most importantly, the current assumption that increasing the net positive charge on the spiropyranic photoswitcher always will favor the intercalation process is, at least, not generalizable and may require further experimentation.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
D. A. thanks the H2020-MSCA-ITN-2017 ETN programme LightDyNAmics, under grant agreement no. 765266. P. A. S.-M. thanks the Austrian FWF (Project M 2260). The authors would like to thank Dr J. J. Nogueira for preliminary discussions on spiropyrans. The Vienna Scientific Cluster (VSC) is acknowledged for kind allocation of computational resources.Notes and references
- W. Szymański, J. M. Beierle, H. A. V. Kistemaker, W. A. Velema and B. L. Feringa, Chem. Rev., 2013, 113, 6114–6178 CrossRef PubMed.
- A. Mustafa, Chem. Rev., 1948, 43, 509–523 CrossRef CAS PubMed.
- (a) R. Klajn, Chem. Soc. Rev., 2014, 43, 148–184 RSC; (b) C. L. Fleming, S. Li, M. Grøtli and J. Andréasson, J. Am. Chem. Soc., 2018, 140, 14069–14072 CrossRef CAS PubMed.
- V. I. Minkin, Chem. Rev., 2004, 104, 2751–2776 CrossRef CAS PubMed.
- S. Prager, I. Burghardt and A. Dreuw, J. Phys. Chem. A, 2014, 118, 1339–1349 CrossRef CAS PubMed.
- F. Liu and K. Morokuma, J. Am. Chem. Soc., 2013, 135, 10693–10702 CrossRef CAS PubMed.
- J. Andersson, S. Li, P. Lincoln and J. Andréasson, J. Am. Chem. Soc., 2008, 130, 11836–11837 CrossRef CAS PubMed.
- M. Hammarson, J. R. Nilsson, S. Li, P. Lincoln and J. Andréasson, Chem. – Eur. J., 2014, 20, 15855–15862 CrossRef CAS PubMed.
- C. Lenoble and R. S. Becker, J. Phys. Chem., 1986, 90, 62–65 CrossRef CAS.
- M. Gehrtz, C. Braeuchle and J. Voitlaender, J. Am. Chem. Soc., 1982, 104, 2094–2101 CrossRef CAS.
- Y. Sheng, J. Leszczynski, A. A. García, R. Rosario, D. Gust and J. Springer, J. Phys. Chem. B, 2004, 108, 16233–16243 CrossRef CAS.
- S. Aldridge and C. Jones, Chem. Soc. Rev., 2016, 45, 763–764 RSC.
- M. Hammarson, J. R. Nilsson, S. Li, T. Beke-Somfai and J. Andréasson, J. Phys. Chem. B, 2013, 117, 13561–13571 CrossRef CAS PubMed.
- A. A. Almaqwashi, T. Paramanathan, I. Rouzina and M. C. Williams, Nucleic Acids Res., 2016, 44, 3971–3988 CrossRef CAS PubMed.
- V. Rizzo, N. Sacchi and M. Menozzi, Biochemistry, 1989, 28, 274–282 CrossRef CAS PubMed.
- A. Mukherjee, R. Lavery, B. Bagchi and J. T. Hynes, J. Am. Chem. Soc., 2008, 130, 9747–9755 CrossRef CAS PubMed.
- M. Wilhelm, A. Mukherjee, B. Bouvier, K. Zakrzewska, J. T. Hynes and R. Lavery, J. Am. Chem. Soc., 2012, 134, 8588–8596 CrossRef CAS PubMed.
- C. M. Nunn, L. Van Meervelt, S. Zhang, M. H. Moore and O. Kennard, J. Mol. Biol., 1991, 222, 167–177 CrossRef CAS PubMed.
- G. Barone, C. F. Guerra, N. Gambino, A. Silvestri, A. Lauria, A. M. Almerico and F. M. Bickelhaupt, J. Biomol. Struct. Dyn., 2008, 26, 115–129 CrossRef CAS PubMed.
- N. Homeyer and H. Gohlke, Mol. Inf., 2012, 31, 114–122 CrossRef CAS PubMed.
- (a) C. Kaiser, T. Halbritter, A. Heckel and J. Wachtveitl, ChemistrySelect, 2017, 2, 4111–4123 CrossRef CAS; (b) H. Görner, Phys. Chem. Chem. Phys., 2001, 3, 416–423 RSC.
- (a) R. R. Tidwell and D. W. Boykin, in Dicationic DNA Minor Groove Binders as Antimicrobial Agents, in Small Molecule DNA and RNA Binders, ed. M. Demeunynck, C. Bailly, W. D. Wilson, John Wiley & Sons, Ltd, Chichester, 2002, pp. 414–460 Search PubMed; (b) B. Nguyen, D. Hamelberg, C. Bailly, P. Colson, J. Stanek, R. Brun, S. Neidle and W. David Wilson, Biophys. J., 2004, 86, 1028–1041 CrossRef CAS PubMed; (c) D. D. V. H. Xuemei Cai and P. J. Gray Jr., Cancer Treat. Rev., 2009, 35, 437–450 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cp07508e |
| This journal is © the Owner Societies 2019 |