
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Change of the kinetics of inclusion in cucurbit[7]uril upon hydrogenation and methylation of palmatine†
Zsombor
Miskolczy
 a,
Mónika
Megyesi
a,
Orsolya
Toke
b and
László
Biczók
*a
a,
Mónika
Megyesi
a,
Orsolya
Toke
b and
László
Biczók
*a
aInstitute of Materials and Environmental Chemistry, Research Centre for Natural Sciences, Hungarian Academy of Sciences, P.O. Box 286, 1519 Budapest, Hungary. E-mail: biczok.laszlo@ttk.mta.hu
bLaboratory for NMR Spectroscopy, Research Centre for Natural Sciences, Hungarian Academy of Sciences, P.O. Box 286, 1519 Budapest, Hungary
First published on 5th February 2019
Abstract
The inclusion of protonated (−)-tetrahydropalmatine (THP+) and dehydrocorydaline (DHC+), natural alkaloids, in the cavity of cucurbit[7]uril was monitored in real time by a spectrofluorimetric method in water at various temperatures. Both guests produced 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complexes in enthalpy controlled processes without any detectable intermediates. The tight entrance of CB7 imposed substantial steric hindrance for encapsulation making the entry into the host several orders of magnitude slower than diffusion. Despite the ∼6 kJ mol−1 lower activation enthalpy, the rate constant of THP+ ingression into CB7 was about 44-fold smaller at 298 K than that of DHC+ as a consequence of the considerably negative activation entropy of the former binding. The egression rates of the two studied alkaloids differed to a much lesser extent because the lower energy barrier of THP+ release was almost compensated by the unfavourable activation entropy. In comparison with the kinetics of the reversible confinement of the palmatine parent compound, the presence of the methyl substituent on the aromatic heterocyclic ring in DHC+ barely modified the rate constant of entry into CB7 but caused about 10-fold increase in the dissociation rate at 298 K.
:1 complexes in enthalpy controlled processes without any detectable intermediates. The tight entrance of CB7 imposed substantial steric hindrance for encapsulation making the entry into the host several orders of magnitude slower than diffusion. Despite the ∼6 kJ mol−1 lower activation enthalpy, the rate constant of THP+ ingression into CB7 was about 44-fold smaller at 298 K than that of DHC+ as a consequence of the considerably negative activation entropy of the former binding. The egression rates of the two studied alkaloids differed to a much lesser extent because the lower energy barrier of THP+ release was almost compensated by the unfavourable activation entropy. In comparison with the kinetics of the reversible confinement of the palmatine parent compound, the presence of the methyl substituent on the aromatic heterocyclic ring in DHC+ barely modified the rate constant of entry into CB7 but caused about 10-fold increase in the dissociation rate at 298 K.
Introduction
The non-covalent association of macrocyclic compounds has many potential biomedical applications in cellular imaging, photodynamic therapy, analyte sensing and drug and gene delivery.1,2 Inclusion complex formation is a versatile tool in supramolecular chemotherapy and in the creation of tailor-made functional nanostructures.3,4 The capability of synthetic host compounds to recognize amino acids, peptides, and protein surface elements can be used to modulate the interactions between proteins and to modify the activity of biomolecules.5,6 Among water-soluble biocompatible hosts, cucurbiturils (CBn) and their acyclic analogues have attracted particular attention due to their remarkable binding capability.7–10 The confinement in this type of molecular container improved the solubility, stability and bioactivity of guest molecules,11–14 which can be exploited to develop novel drug formulations.15–18 Because of the dynamic nature of complex formation, controlled release of the encapsulated molecule was achieved19 and CBn cavitands served as catalysts for 1,3-dipolar cycloaddition, hydrolysis, oxidation, and photoinduced reactions.20–23 The highly selective, strong binding of 1-adamantylamine-substituted substrates to cucurbit[7]uril-functionalized nanoparticles allowed facile non-covalent surface modification.24 The marked change of the fluorescent properties of guest molecules upon embedment in CBn25 was utilized for the development of indicator displacement assays and chemical sensors.26,27Understanding the effect of the major factors controlling the rate of entry into and exit from the CBn cavity is of pivotal importance for the rational design of functional supramolecular assemblies and for gaining insight into the details of the dynamics of inclusion. Time-resolved measurements provided valuable information on pseudorotaxane formation.28–30 In a recent excellent review, Masson summarized the current state of knowledge on the kinetics of CBn complex formation.31 Although activation parameters provide unique information on the transition state and the energy barrier of reversible host–guest binding, very few such data have been reported. Despite its moderate binding affinity, cyclohexanediammonium produced a kinetically extraordinarily stable complex with cucurbit[6]uril (CB6) because the sterically strongly hindered dissociation had an activation enthalpy32 as high as 125.5 kJ mol−1. The slow exchange of 4-methylbenzylammonium33 and cyclohexylmethylammonium34 cations in CB6 allowed kinetic measurements by NMR spectroscopy. In D2O:formic acid 1:1 mixture, an activation enthalpy of 56.1 and 78.7 kJ mol−1 was obtained for the ingression of the former and latter cations, respectively, indicating that a substantial widening of the tight CB6 portals was needed for the penetration of these guests.33,34 The inclusion of an uncharged amine in CB6 and CB7 was more rapid than its protonated form because the embedment of the ammonium cation was retarded by exclusion complex formation, whereas the unprotonated amine directly entered the host.35,36 The encapsulation in the more spacious cucurbit[7]uril (CB7) was much faster than in CB6 and depended on the relative size of the guest and the openings of the host macrocycle.36–41 Further increase of the cavity size from CB7 to CB8 accelerated berberine inclusion ∼7-fold but the rate of the process remained about 2 orders of magnitude slower than the diffusion-controlled limit.42 The study of the reversible binding between a dimethyl viologen−CB8 1:1 complex and electron-rich aromatic moieties tethered to a poly(ethylene glycol) chain demonstrated that the pressure and viscosity of the medium solely influence ingression rates whereas egression rates are sensitive mainly to the molecular structure of the electron-rich guest.43
Despite the key role of kinetic data in the rational design of CBn applications, limited knowledge has been gathered so far concerning the relationship between molecular structure and the rate constants of the inclusion in the CBn cavity. The main goal of the present study is to elucidate how the introduction of hydrogen atoms or a methyl substituent into palmatine influences the kinetic parameters of the embedment into and release from the CB7 cavitand. The chemical structures of the studied and parent compounds are shown in Scheme 1. The examined pharmacologically important natural alkaloids are the active ingredients of traditional Chinese herbal medicines. (−)-Tetrahydopalmatine is effective as an analgesic and sedative agent and has shown particular promise in the treatment of drug addiction.44,45 Dehydrocorydaline exerts antinociceptive, antiallergic, and antitumor effects and protects the cardiovascular system.46
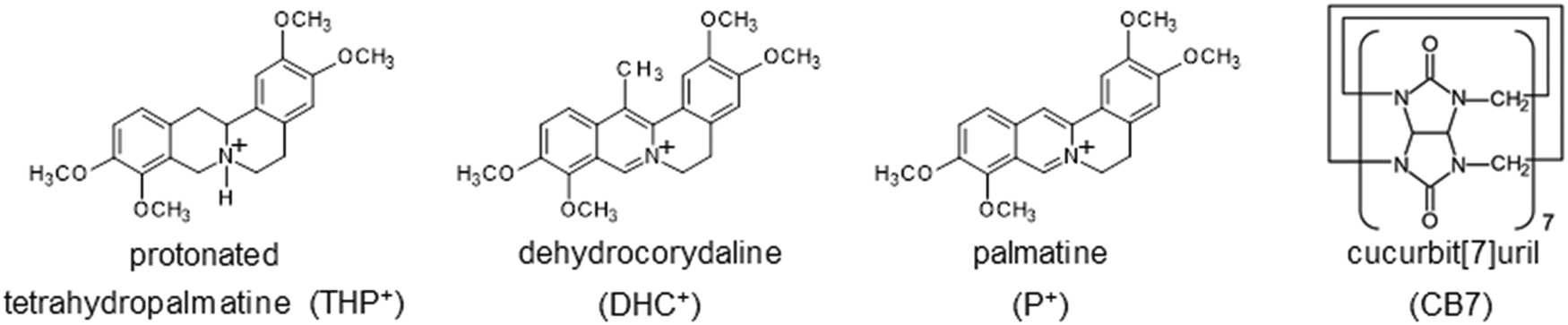 | ||
| Scheme 1 Chemical structures of the guest and host compounds. | ||
Experimental
(−)-Tetrahydopalmatine (THP+, Sigma-Aldrich) and dehydrocorydaline chloride (DHC+, BOC Sciences) were used as received. The alkaloid concentrations were determined spectrophotometrically using the molar absorption coefficient of 5369 M−1 cm−1 at 280 nm for protonated tetrahydopalmatine (THP+) in aqueous HCl solution of pH 4 and 22242 M−1 cm−1 at 333 nm for DHC+ in water. High-purity CB7 was kindly provided by Dr Anthony I. Day (University of New South Wales, Canberra, Australia). Experiments were performed in double distilled water whose pH was set to 4 with HCl in the case of THP+ solutions.
The absorption spectra were obtained on an Agilent Technologies Cary60 spectrophotometer. Fluorescence spectra were taken on a Jobin-Yvon Fluoromax-P spectrofluorometer, whereas stopped-flow experiments were performed with the same instrument using an Applied Photophysics RX2000 rapid mixing accessory and a pneumatic drive. For each measurement, 10–20 kinetic traces were averaged. The temperature of the samples was varied with a Julabo F25-ED thermostat. The results of spectrophotometric and fluorescence titrations as well as stopped-flow measurements were analysed with homemade programs written in MATLAB 7.9. Isothermal calorimetric titrations were performed with a VP-ITC (MicroCal) instrument at 298 K. All solutions were degassed prior to titration. The alkaloid solution (∼300–600 μM) was injected stepwise (10 μl each) from the computer-controlled microsyringe at an interval of 270 s into ∼20–40 μM CB7 solutions, while stirring at 300 rpm. The dilution heat, which was obtained by adding alkaloid solution into water under the same conditions as in the titration of CB7, was subtracted from the integrated heat evolved per injection. Data were analysed using the one-site binding model. The first data point was always removed. The measurements were repeated three times. NMR experiments were carried out on a 600 MHz Varian NMR system spectrometer equipped with a 5 mm indirect detection 1H/31P–15N/13C XYZ gradient probe. Measurements were performed in 0.1 mM DCl D2O solutions at 298 K. Solvent suppression was achieved by the PRESAT sequence. The 1H chemical shifts were referenced externally to 2,2-dimethylsilapentane-5-sulfonic acid.
Results
THP+ binding in CB7
Because of its nonaromatic heterocyclic ring, THP+ had entirely different absorption and fluorescence spectra (Fig. 1) than palmatine (P+).47Fig. 1 presents the spectral changes upon alteration of pH in aqueous solution. As the site of protonation is not directly connected to the aromatic ring, only slight absorbance variation was observed. In contrast, more than 2-fold emission intensity enhancement appeared when the pH was decreased from 9 to 4. A good match of the inflexion points of the sigmoid-shaped titration curves was obtained by spectrophotometric and fluorescence methods. The results were analysed by nonlinear least-squares fit of the following function: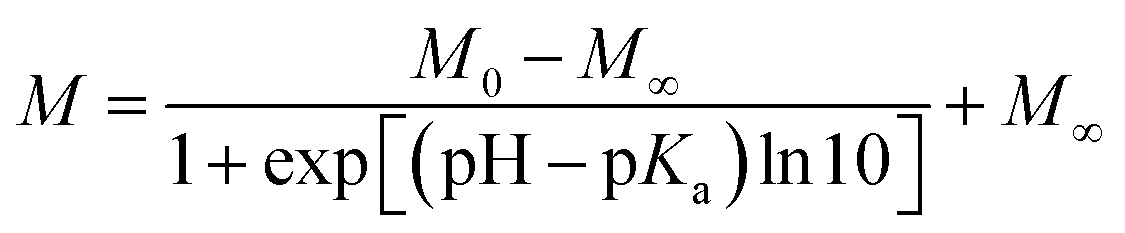 | (1) |
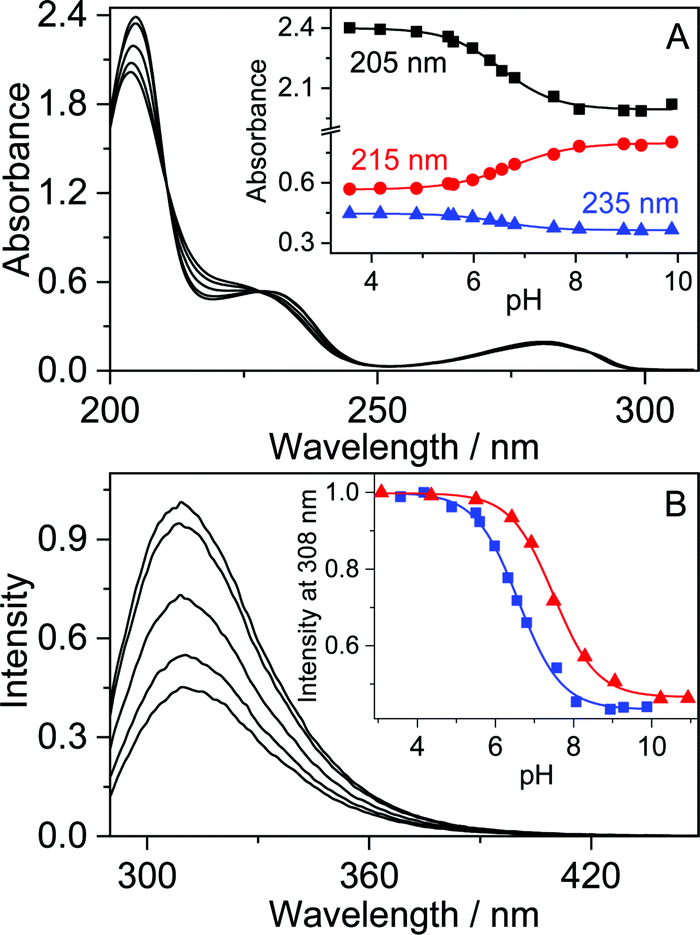 | ||
Fig. 1 Absorption (A) and fluorescence (B) spectra of 36 μM THP+ in water at pH 3.6, 5.5, 6.6, 7.6, and 9.3. Inset: (A) Absorbance at 205, 215, and 235 nm and (B) fluorescence intensity at 308 nm as a function of pH in water ( ) and in 300 μM CB7 aqueous solution ( ) and in 300 μM CB7 aqueous solution ( ) (excitation at 280 nm). ) (excitation at 280 nm). | ||
The absorption spectrum of THP+ barely varied upon addition of CB7 but the fluorescence titration curve shifted to larger pHs and pKa = 7.54 was obtained in the presence of 0.30 mM CB7 (inset to Fig. 1B). The acidity diminution of 0.89 pH units indicated the binding of THP+ in the nonpolar cavity of CB7. The hydrogen-bonding and ion–dipole interactions of the alkaloid N+–H bond with the carbonyl-laced portal of the host impeded proton release. The modification of the acidity of guests by inclusion complex formation has been reported for other amines, drugs, and dyes.48–51 As both free and CB7-encapsulated THP+s are completely protonated at pH 4, further experiments were performed under this condition.
The fluorescence quantum yield of THP+ (ΦF = 0.14) and the fluorescence maximum (λF = 310 nm) in the presence of 0.1 mM HCl barely changed when ethanol was used as the solvent instead of water. Fig. 2 demonstrates that addition of CB7 to THP+ aqueous solution at pH 4 brought about small fluorescence intensity diminution whereas the shape and maximum of the spectra did not alter. The scant variation of the fluorescent behaviour with the local environment arises from the lack of aromaticity of the fused heterocyclic ring in THP+. The steep initial emission intensity decline followed by the levelling off upon gradual increase of CB7 concentration (Fig. 2A) indicated complex formation. The fluorescence titration data could be fitted well assuming 1:1 binding. The global analysis of the results in the 290–388 nm domain provided K = (1.2 ± 0.2) × 105 M−1 for the equilibrium constant of THP+ confinement in CB7 at 298 K. The equimolar association was confirmed by Job's continuous variation method.52Fig. 2B shows the difference in fluorescence intensity recorded at 310 nm in the absence and presence of CB7 as a function of the mole fraction of THP+. The total concentration of reactants was kept constant (76 μM). The minimum appeared at [THP+]/([THP+] + [CB7]) = 0.5 implying 1:1 binding stoichiometry.
 | ||
| Fig. 2 Fluorescence spectra of 32 μM THP+ at pH 4 in water (thin line) and in 470 μM CB7 aqueous solution (thick line). Inset: (A) Fluorescence intensity at 308 nm as a function of CB7 concentration at 298 K (excitation at 280 nm). The line represents the results of the global analysis of the experimental data. (B) Job plot of the fluorescence intensity variation at 310 nm vs. the mole fraction of THP+. The total concentration [THP+] + [CB7] = 76 μM is constant. The line shows the fitted curve assuming K = 1.2 × 105 M−1 and 1:1 complexation. | ||
To gain insight into the structural characteristics of the THP+−CB7 complex, we turned to NMR spectroscopy. Because of the lower sensitivity of this method, much more concentrated (0.5 mM) alkaloid solutions had to be used than in absorption and fluorescence spectroscopic experiments. The THP+ signals in the absence and presence of CB7 were assigned on the basis of combined 1H–1H TOCSY, ROESY, and natural abundance 13C-HSQC measurements. The aromatic region of the 1H NMR spectra exhibited the most characteristic spectral differences between the free and bound THP+ (Fig. S1 in the ESI†). The two-dimensional 1H−13C-HSQC spectrum of unbound THP+ is depicted in Fig. S2 of the ESI.† The peak doubling observed in the free THP+ spectrum is related to a slow inversion at the cationic nitrogen giving rise to two THP+ species with slightly different chemical environments for the ring protons. The resonances corresponding to the aromatic ring protons of the tetrahydroisoquinolinium moiety (at positions 11 and 12) display substantial shifts to lower frequencies demonstrating the encapsulation of this segment of the guest molecule in CB7. In contrast, the resonance of the proton at position 1 is shifted downfield indicating the deshielding effect of the carbonyl groups of CB7 outside the cavity. The proton at position 4 resonates at nearly the same frequency as in free THP+ suggesting negligible interaction with the host. The most probable structure of the THP+−CB7 complex is presented in the upper panel of Scheme 2 (vide supra).
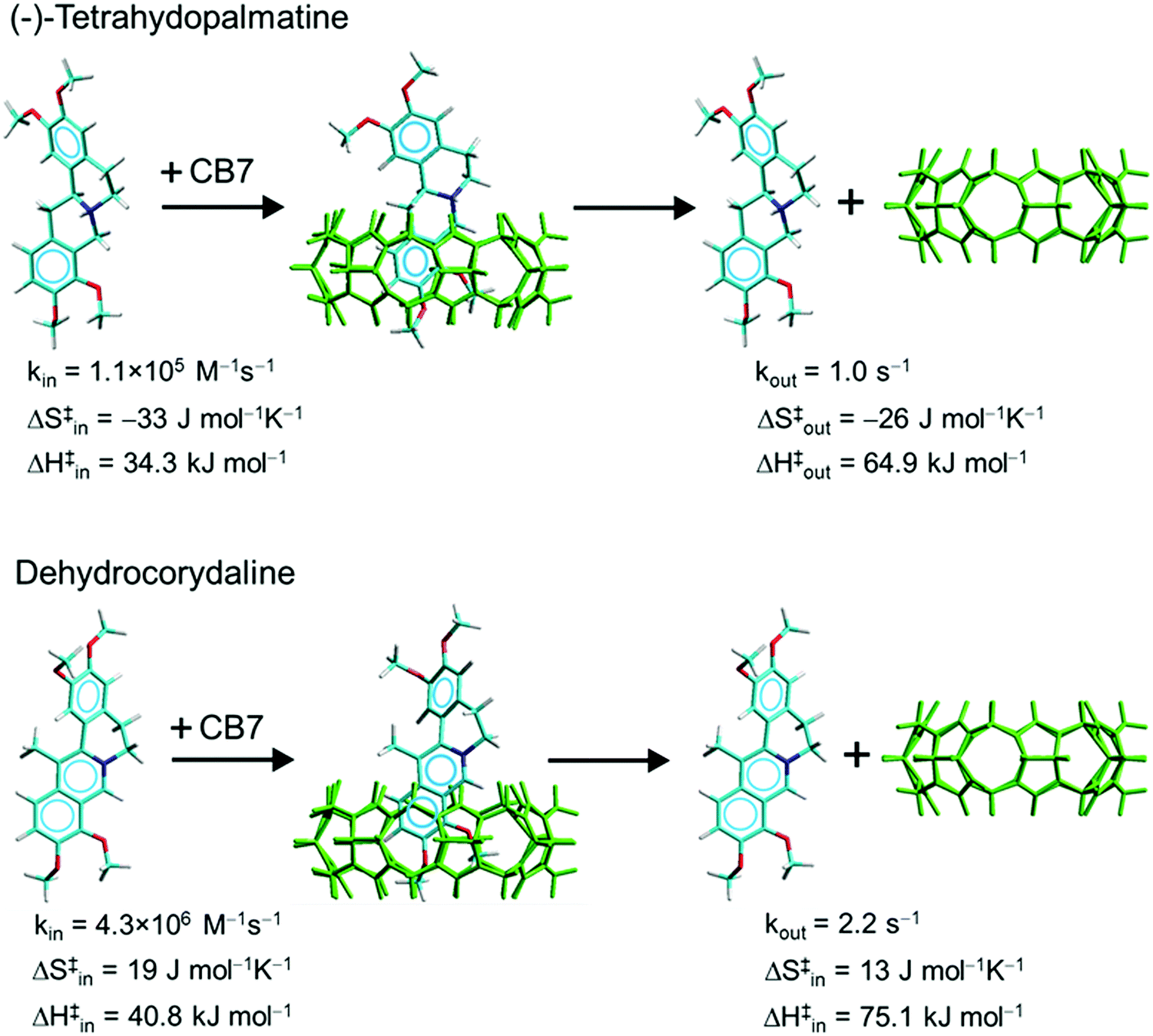 | ||
| Scheme 2 Rate constants at 298 K and activation parameters. | ||
To reveal the thermodynamics of association, isothermal calorimetric measurements were carried out. As a representative example, Fig. 3 shows the enthalpogram recorded for the titration of CB7 in water with THP+ solution at pH 4. Because of the decrease of the number of the free macrocycles, the evolved heat gradually decreased upon successive alkaloid additions. An inflexion point appeared at equimolar mixing ratio confirming 1:1 inclusion. Nonlinear least-squares fit of the titration data resulted in K = (1.1 ± 0.2) × 105 M−1 for the binding constant at temperature T = 298 K, in good agreement with the corresponding value derived from fluorescence measurements (vide supra). For the enthalpy change of complexation, ΔH = −30.6 ± 1.7 kJ mol−1 was calculated. Using these quantities, ΔS = −6 ± 5 J mol−1 K−1 was obtained for the entropy change of THP+−CB7 formation on the basis of the equation: ΔS = ΔH/T + RlnK, where R stands for the gas constant.
 | ||
| Fig. 3 Integrated heat released per injection for the titration of 38.3 μM CB7 by 610 μM THP+ solution at 298 K and pH 4. The line represents the best fit with a one-site binding model. | ||
The decrease of fluorescence quantum yield of THP+ upon confinement in CB7 was exploited to monitor the kinetics of the reversible host–guest binding in real time. Fig. 4A displays typical fluorescence intensity (I) decay at 310 nm after rapid mixing of the constituents. The signal reached a plateau when equilibrium was established. In the case of a simple 1:1 encapsulation, the kinetics of I alteration is defined as follows:
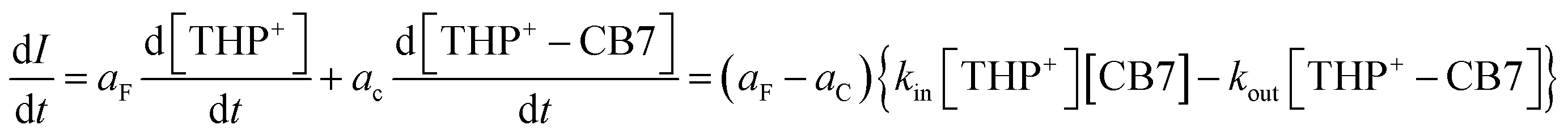 | (2) |
 | ||
| Fig. 4 (A) Stopped-flow signal at 310 nm after 1:1 mixing of 18 μM THP+ and 85 μM CB7 aqueous solutions at 298 K. Excitation at 280 nm. The line shows the result of the nonlinear least-squares analysis. (B) Arrhenius plots of the rate constants of inclusion in CB7 and the dissociation of the THP+−CB7 complex. | ||
To determine the activation parameters, the rate constants were measured by the stopped-flow technique at various temperatures. Fig. 4B presents the logarithm of kin and kout as a function of the reciprocal temperature. Nonlinear least-squares analysis of the data with the Arrhenius equation k = Aexp(−E/RT) provided the activation energies (E) and A factors. The excellent fit of the kinetic traces assuming simple 1:1 association indicates that no intermediate is produced in the course of THP++–CB7 complex formation within the time scale of the stopped-flow experiment (>10 ms). Hence, the transition state theory can be applied to understand the meaning of the Arrhenius parameters. From the Eyring–Polanyi equation, the following relationships can be derived for the standard entropy (ΔS‡) and enthalpy (ΔH‡) of activation:
| ΔH‡ = E − RT | (3) |
 | (4) |
| THP+ | DHC+ | Palmatinea | |
|---|---|---|---|
| a Ref. 53 b K = kin/kout. c ΔH = ΔH‡in − ΔH‡out. d ΔS = ΔS‡in − ΔS‡out. | |||
| k in at 298 K/M−1 s−1 | (1.1 ± 0.1) × 105 | (4.3 ± 0.4) × 106 | (5.4 ± 0.4) × 106 |
| A in/1013 M−1 s−1 | 0.032 ± 0.01 | 16 ± 4 | 6.3 ± 1.0 |
| E in/kJ mol−1 | 36.8 ± 1.0 | 43.2 ± 1.0 | 40.4 ± 2.0 |
| ΔH‡in/kJ mol−1 | 34.3 ± 1.0 | 40.8 ± 1.0 | 37.9 ± 2.0 |
| ΔS‡in/J mol−1 K−1 | −33 ± 3 | 19 ± 3 | 11 ± 3 |
| k out at 298 K/s−1 | 1.0 ± 0.1 | 2.2 ± 0.2 | 0.20 ± 0.04 |
| A out/1013 s−1 | 0.07 ± 0.02 | 8.6 ± 2.0 | 2.2 ± 0.3 |
| E out/kJ mol−1 | 67.4 ± 2.0 | 77.5 ± 2.0 | 79.3 ± 2.0 |
| ΔH‡out/kJ mol−1 | 64.9 ± 2.0 | 75.1 ± 2.0 | 76.8 ± 2.0 |
| ΔS‡out/J mol−1 K−1 | −26 ± 3 | 13 ± 2 | 2 ± 1 |
| K at 298 K/105 M−1 from kinetic datab | 1.1 ± 0.2 | 19 ± 3 | 270 ± 30 |
| K at 298 K/105 M−1 from fluorescence titration | 1.2 ± 0.2 | 21 ± 3 | 260 ± 30 |
| ΔH at 298 K/kJ mol−1 ITC measurement | −30.6 ± 1.7 | −34 ± 1 | −37 ± 2 |
| ΔH/kJ mol−1 from kinetic datac | −30.6 ± 3.0 | −34 ± 3 | −39 ± 3 |
| ΔS/J mol−1 K−1 ITC measurement | −6 ± 5 | 6 ± 2 | 14 ± 6 |
| ΔS/J mol−1 K−1 from kinetic datad | −7 ± 6 | 6 ± 3 | 9 ± 4 |
DHC+ inclusion in CB7
To unravel the effect of methylation in position 13 of palmatine on the kinetics of encapsulation in CB7, stopped-flow experiments were performed with DHC+. A previous study showed that the 1:1 embedment of this alkaloid in CB7 brought about substantial fluorescence intensity enhancement and the emission maximum of the complex appeared at 476 nm.54 The alteration of the absorption and fluorescence spectra upon gradual increase of CB7 concentration is presented in Fig. S3 in the ESI.† The sensitivity of the fluorescence properties to the local environment is characteristic of protoberberine alkaloids possessing an aromatic isoquinolinium moiety.53,55 We examined the inclusion in real time by monitoring the rise of the emission intensity after fast mixing of DHC+ and CB7 solutions. Low reactant concentrations (∼0.26 μM) were used to decelerate the bimolecular ingression into a conveniently measurable time range. Fig. 5A presents the normalized fluorescence intensity versus time traces at various temperatures. Since the free DHC+ emits negligible fluorescence, the intensity growth directly reflects the augmentation of DHC+−CB7 complex concentration. The substantial change of the initial slopes with temperature indicates that a considerable activation barrier hinders the DHC+ entry into CB7. The kinetic results were analysed as described for THP+−CB7 complex formation (vide supra). The Arrhenius plots of the derived kin and kout values are displayed in Fig. 5B, whereas the calculated activation parameters are given in Scheme 2 and Table 1. The ratio of the rate constants of ingression and egression provided K = kin/kout = (2.2 ± 0.3) × 106 M−1 for the binding constant of DHC+ in CB7. Practically the same value (K = (2.1 ± 0.3) × 106 M−1) was obtained from the results of spectrophotometric and fluorescence titrations plotted in Fig. S3 in the ESI.†
 | ||
| Fig. 5 (A) Rise of fluorescence intensity at 480 nm in 0.24 μM DHC+ and 0.27 μM CB7 solution after rapid mixing of the reactants at various temperatures. Excitation occurred at 335 nm. The signals were normalized at the intensity in the equilibrium. (B) Logarithm of the rate constants for the entry of DHC+ into CB7 and the dissociation of the 1:1 complex as a function of reciprocal temperature. The lines represent the results of the nonlinear fit of the Arrhenius equation on a logarithmic scale. | ||
Table 1 includes the thermodynamic parameters of host–guest complexation determined directly by isothermal titration calorimetry together with the values calculated as the difference of the corresponding activation parameters of ingression and egression. The integrated heat released per injection of 330 μM DHC+ into 22.8 μM CB7 solution as a function of reactant molar ratio is shown in Fig. 6. The location of the inflection point close to [DHC+]/[CB7] = 1 verifies the 1:1 association stoichiometry. The thermodynamic quantities calculated from the enthalpogram are shown in Table 1 and the obtained binding constant (K = (1.9 ± 0.3) × 106 M−1) is in accordance with those found by other methods (vide supra).
 | ||
| Fig. 6 Enthalpogram for the titration of 22.8 μM CB7 with 330 μM DHC+ aqueous solution at 298 K. | ||
Discussion
The kinetic and thermodynamic parameters of the host–guest binding of THP+ and DHC+ in CB7 are compared in Table 1 with those published for palmatine (P+) complexation.53 The introduction of a methyl substituent in position 13 slightly affects the rate constant of entry into CB7 because the increase of the activation barrier (ΔH‡in) is compensated by the activation entropy (ΔS‡in) enhancement. The presence of the methyl group in DHC+ alters the structure of the activated complex resulting in ∼3 kJ mol−1 higher transition state enthalpy than in the case of P+ ingression. The high ΔH‡in values for both alkaloids indicate constrictive binding. As the portals of CB7 are narrower than the cavity, an elliptical modification of the carbonyl rim of CB7 is the prerequisite for the inclusion. The positive ΔS‡in suggests that the activated complex resembles the free components more than the final host–guest associate. This is in agreement with the Hammond–Leffler postulate, which also predicts that the geometry of the transition state is more similar to the reactants than to the products in an exothermic process. The larger ΔS‡in for DHC+ compared with P+ probably originates from the looser binding of the former guest in the transition state. Positive ΔS‡in is found for DHC+ and P+ alike implying that the limitation of the degrees of freedom in the transition state is overcompensated by the entropy gain due to release of water from the hydrate shell of the reactants.The activation entropy of egression (ΔS‡out) is also positive for DHC+ but close to zero for P+. According to NMR spectra,54DHC+ is less deeply embedded in the CB7 cavity than P+ because of the steric hindrance caused by its methyl substituent. Therefore, more water remains in the host interior and the guest is more hydrated in DHC+−CB7 than in P+−CB7. As a consequence, the structural rearrangement of the former complex into the transition state is accompanied by lesser changes in solvation. Therefore, the entropy gain stemming from the partial release of the guest from CB7 upon reaching the weakly bound transition state is counterbalanced to a smaller extent by the entropy loss originating from the coordination of water molecules in the case of DHC+−CB7 dissociation.
The shallower inclusion of DHC+ in CB7 than its unmethylated analogue leads to less negative enthalpy change (ΔH) upon complex formation and more than one order of magnitude smaller binding constant (Table 1). The entropy term has similar and small contribution to the driving force of host–guest association of both alkaloids. The activation enthalpy of dissociation (ΔH‡out) also barely differs because the lower exothermicity of DHC+ confinement almost compensates the more substantial activation energy of the entry into CB7. The ∼10-fold faster egression for DHC+ than for P+ at 298 K originates from the combined effects of the larger ΔS‡out and slightly smaller ΔH‡out case.
The hydrogenation of P+ also significantly alters the kinetics of inclusion complex formation. Despite its lower activation enthalpy, THP+ enters into CB7 about 50-fold slower than its parent compound at 298 K and the process has ∼60000-fold lower rate constant than the diffusion-controlled limit56 (kdiff = 6.5 × 109 M−1 s−1). The surprisingly slow association is ascribed to the substantial negative activation entropy (ΔS‡in = −32 J mol−1 K−1). The negative ΔS‡in, which has not been observed for any other CB7 complexation, may indicate that the entropy gain arising from the change of hydration cannot overwhelm the reduction of the degrees of freedom within the tightly bound transition state. Hydrogen bonding of the N+−H moiety of THP+ to the oxygen(s) located at the opening of CB7 may strengthen the interaction between the components of the activated complex. In addition, the flexible character of free THP+ owing to its two fused nonaromatic rings allows the limitation of more degrees of freedom than in the activated complex of the much more rigid P+.
Kinetic results for 4-methylbenzylammonium ion assembly with cucurbit[6]uril (CB6) demonstrated the existence of both exclusion and inclusion complexes.57 Nau and coworkers suggested that primary ammonium cations first bind to the portal of CB6 producing an exclusion complex in which the organic moiety is still located in the bulk solvent. Then the hydrophobic part of the guest slips into the CB6 cavity in a “flip-flop” manner.34,35 When the more spacious CB7 served as a host, such an association pathway was found only for 2-aminoanthracenium embedment but the anthracene moiety of this guest could also directly enter into CB7.36 Exclusion complex formation seems to be possible when the ammonium group and the hydrophobic moiety are located at the two ends of the guest. No evidence was found for association into the exclusion complex in the case of THP+ binding to CB7.
The activation entropy of THP+ exit from CB7 (ΔS‡out) is also considerably negative because the transition to the tightly bound transition state allows relatively small entropy gain. Moreover, the coordination of water molecules upon partial release of THP+ also contributes to the unfavourable ΔS‡out. The remarkably negative ΔS‡out lessens the Arrhenius Aout factor but the almost 12 kJ mol−1 smaller activation enthalpy (ΔH‡out) for THP+ dissociation relative to that of P+ exit from CB7 exerts an opposite effect on the egression rate constant. As a consequence, THP+ is released from CB7 only 5 times faster than P+. The combined effects of the lower ΔH‡in and the less exothermic complexation causes the substantial ΔH‡out diminution going from P+−CB7 to THP+−CB7 because ΔH‡out is the difference of ΔH‡in and the enthalpy change upon binding (ΔH). The less negative ΔH for the latter complex is assigned to the lack of aromaticity in the heterocyclic ring of the guest. This unfavourably influences the binding enthalpy owing to the following: (i) because no charge delocalization can take place, the nitrogen of the guest becomes more hydrophilic and larger energy is needed for the reorganization of its hydrate shell, (ii) dipole−π interaction between CB7 and the heterocycle cannot stabilize THP+−CB7, and (iii) the nonplanar molecular structure and the increased number of hydrogens may make the host–guest binding sterically less advantageous. The more than two orders of magnitude decrease of the association constant upon the replacement of P+ with THP+ results not only from the diminution of the exothermicity of complexation but also from the unfavourable entropy changes.
Conclusions
Even minor variations in the molecular structure, such as addition of hydrogens or a methyl substituent considerably alter the formation and dissociation kinetics of inclusion complexes when constrictive binding takes place. The determination of activation parameters is necessary for a deeper understanding of the relationship between molecular structure and the rate constants of entry into and exit from the host because opposite changes in ΔH‡ and ΔS‡ often mask the trends. Such a phenomenon appears for THP+ ingression into CB7 whose rate constant is 50-fold lower than that of the corresponding process of P+ because the beneficial effect of ΔH‡in diminution is overwhelmed by the inclusion deceleration arising from the significantly negative ΔS‡in. A tightly bound transition state is produced in the course of entry into CB7 when the guest contains a nonaromatic heterocycle with localized charge, such as THP+. On the other hand, host–guest interaction is weak in the transition state when the positive charge of the nitrogen is distributed throughout the aromatic rings of the guests like in P+ and DHC+.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the National Research, Development and Innovation Office (NKFIH, Grant K123995), the BIONANO GINOP-2.3.2-15-2016-00017 project, and the János Bolyai Research Scholarship Program of the Hungarian Academy of Sciences (to ZM).References
- X. Ma and Y. Zhao, Chem. Rev., 2015, 115, 7794–7839 CrossRef CAS PubMed
.
- G. Ghale and W. M. Nau, Acc. Chem. Res., 2014, 47, 2150–2159 CrossRef CAS PubMed
- J. Zhou, G. Yu and F. Huang, Chem. Soc. Rev., 2017, 46, 7021–7053 RSC
- H. Zhu, L. Shangguan, B. Shi, G. Yu and F. Huang, Mater. Chem. Front., 2018, 2, 2152–2174 RSC
- S. van Dun, C. Ottmann, L.-G. Milroy and L. Brunsveld, J. Am. Chem. Soc., 2017, 139, 13960–13968 CrossRef CAS PubMed
- C. Hou, X. Zeng, Y. Gao, S. Qiao, X. Zhang, J. Xu and J. Liu, Isr. J. Chem., 2018, 58, 286–295 CrossRef CAS
-
E. Masson, in Comprehensive Supramolecular Chemistry II, ed. J. L. Atwood, Elsevier Inc., 2017, vol. 5, pp. 21–45 Search PubMed
- N. J. Wheate and C. Limantoro, Supramol. Chem., 2016, 28, 849–856 CrossRef CAS
- S. Ganapati and L. Isaacs, Isr. J. Chem., 2018, 58, 250–263 CrossRef CAS PubMed
- C. B. Rodell, J. E. Mealy and J. A. Burdick, Bioconjugate Chem., 2015, 26, 2279–2289 CrossRef CAS PubMed
- S. Li, J. Y.-W. Chan, Y. Li, D. Bardelang, J. Zheng, W. W. Yew, D. P.-C. Chan, S. M. Y. Lee and R. Wang, Org. Biomol. Chem., 2016, 14, 7563–7569 RSC
- Z. Miskolczy, M. Megyesi, G. Tárkányi, R. Mizsei and L. Biczók, Org. Biomol. Chem., 2011, 9, 1061–1070 RSC
- Z. Miskolczy and L. Biczók, J. Phys. Chem. B, 2011, 115, 12577–12583 CrossRef CAS PubMed
- H. S. El-Sheshtawy, S. Chatterjee, K. I. Assaf, M. N. Shinde, W. M. Nau and J. Mohanty, Sci. Rep., 2018, 8, 13925 CrossRef PubMed
- H. Yin and R. Wang, Isr. J. Chem., 2018, 58, 188–198 CrossRef CAS
-
D. H. Macartney, in Comprehensive Supramolecular Chemistry II, ed. J. L. Atwood, Elsevier, Oxford, 2017, vol. 1, pp. 479–494 Search PubMed
-
A. I. Day and J. G. Collins, in Supramolecular Chemistry: From Molecules to Nanomaterials, ed. P. A. Gale and J. W. Steed, John Wiley & Sons, Ltd, 2012 Search PubMed
- K. I. Kuok, S. Li, I. W. Wyman and R. Wang, Ann. N. Y. Acad. Sci., 2017, 1398, 108–119 CrossRef CAS PubMed
- L. Liu, J. Inclusion Phenom. Macrocyclic Chem., 2017, 87, 1–12 CrossRef CAS
- B. C. Pemberton, R. Raghunathan, S. Volla and J. Sivaguru, Chem. – Eur. J., 2012, 18, 12178–12190 CrossRef CAS PubMed
- K. I. Assaf and W. M. Nau, Chem. Soc. Rev., 2015, 44, 394–418 RSC
- L. Scorsin, J. A. Roehrs, R. R. Campedelli, G. F. Caramori, A. O. Ortolan, R. L. T. Parreira, H. D. Fiedler, A. Acuna, L. García-Río and F. Nome, ACS Catal., 2018, 8, 12067–12079 CrossRef CAS
- Y. Jiao, B. Tang, Y. Zhang, J.-F. Xu, Z. Wang and X. Zhang, Angew. Chem., Int. Ed., 2018, 57, 6077–6081 CrossRef CAS PubMed
- C. Sun, H. Zhang, S. Li, X. Zhang, Q. Cheng, Y. Ding, L.-H. Wang and R. Wang, ACS Appl. Mater. Interfaces, 2018, 10, 25090–25098 CrossRef CAS PubMed
- R. N. Dsouza, U. Pischel and W. M. Nau, Chem. Rev., 2011, 111, 7941–7980 CrossRef CAS PubMed
- S. Sinn and F. Biedermann, Isr. J. Chem., 2018, 58, 357–412 CrossRef CAS
- G. Ghale, A. G. Lanctôt, H. T. Kreissl, M. H. Jacob, H. Weingart, M. Winterhalter and W. M. Nau, Angew. Chem., Int. Ed., 2014, 53, 2762–2765 CrossRef CAS PubMed
- X. Ling, E. L. Samuel, D. L. Patchell and E. Masson, Org. Lett., 2010, 12, 2730–2733 CrossRef CAS PubMed
- V. Sindelar, S. Silvi and A. E. Kaifer, Chem. Commun., 2006, 2185–2187 RSC
- A. E. Kaifer, W. Li, S. Silvi and V. Sindelar, Chem. Commun., 2012, 48, 6693–6695 RSC
- E. Masson, M. Raeisi and K. Kotturi, Isr. J. Chem., 2018, 58, 413–434 CrossRef CAS
- P. Mukhopadhyay, P. Y. Zavalij and L. Isaacs, J. Am. Chem. Soc., 2006, 128, 14093–14102 CrossRef CAS PubMed
- W. L. Mock and N. Y. Shih, J. Org. Chem., 1986, 51, 4440–4446 CrossRef CAS
- C. Márquez, R. R. Hudgins and W. M. Nau, J. Am. Chem. Soc., 2004, 126, 5806–5816 CrossRef PubMed
- C. Marquez and W. M. Nau, Angew. Chem., Int. Ed., 2001, 40, 3155–3160 CrossRef CAS PubMed
- S. S. Thomas and C. Bohne, Faraday Discuss., 2015, 185, 381–398 RSC
- Z. Miskolczy, J. G. Harangozó, L. Biczók, V. Wintgens, C. Lorthioir and C. Amiel, Photochem. Photobiol. Sci., 2014, 13, 499–508 RSC
- H. Tang, D. Fuentealba, Y. H. Ko, N. Selvapalam, K. Kim and C. Bohne, J. Am. Chem. Soc., 2011, 133, 20623–20633 CrossRef CAS PubMed
- Z. Miskolczy and L. Biczók, J. Phys. Chem. B, 2014, 118, 2499–2505 CrossRef CAS PubMed
- Z. Miskolczy, L. Biczok and I. Jablonkai, Phys. Chem. Chem. Phys., 2017, 19, 766–773 RSC
- E. Mezzina, F. Cruciani, G. F. Pedulli and M. Lucarini, Chem. – Eur. J., 2007, 13, 7223–7233 CrossRef CAS PubMed
- Z. Miskolczy and L. Biczók, Phys. Chem. Chem. Phys., 2014, 16, 20147–20156 RSC
- E. A. Appel, F. Biedermann, D. Hoogland, J. del Barrio, M. D. Driscoll, S. Hay, D. J. Wales and O. A. Scherman, J. Am. Chem. Soc., 2017, 139, 12985–12993 CrossRef CAS PubMed
- H. E. Hassan, D. Kelly, M. Honick, S. Shukla, A. Ibrahim, D. A. Gorelick, M. Glassman, R. P. McMahon, H. J. Wehring, A. M. Kearns, S. Feldman, M. Yu, K. Bauer and J. B. Wang, J. Clin. Pharmacol., 2017, 57, 151–160 CrossRef CAS PubMed
- J. B. Wang and J. R. Mantsch, Future Med. Chem., 2012, 4, 177–186 CrossRef CAS PubMed
- Z.-Y. Yin, L. Li, S.-S. Chu, Q. Sun, Z.-L. Ma and X.-P. Gu, Sci. Rep., 2016, 6, 27129 CrossRef CAS PubMed
- K. Hirakawa, T. Hirano, Y. Nishimura, T. Arai and Y. Nosaka, J. Phys. Chem. B, 2012, 116, 3037–3044 CrossRef CAS PubMed
- D. H. Macartney, Isr. J. Chem., 2018, 58, 230–243 CrossRef CAS
- A. I. Lazar, J. Rohacova and W. M. Nau, J. Phys. Chem. B, 2017, 121, 11390–11398 CrossRef CAS PubMed
- I. Ghosh and W. M. Nau, Adv. Drug Delivery Rev., 2012, 64, 764–783 CrossRef CAS PubMed
- N. Barooah, J. Mohanty, H. Pal and A. C. Bhasikuttan, J. Phys. Chem. B, 2012, 116, 3683–3689 CrossRef CAS PubMed
- P. Job, Ann. Chim., 1928, 9, 113 CAS
- Z. Miskolczy, L. Biczók and G. Lendvay, Phys. Chem. Chem. Phys., 2018, 20, 15986–15994 RSC
- C. J. Li, J. Li and X. S. Jia, Org. Biomol. Chem., 2009, 7, 2699–2703 RSC
- K. Hirakawa and T. Hirano, Photochem. Photobiol., 2008, 84, 202–208 CAS
-
M. Montalti, A. Credi, L. Prodi and M. T. Gandolfi, Handbook of Photochemistry, CRC Press, Boca Raton, FL, 3rd edn, 2006 Search PubMed
- R. Hoffmann, W. Knoche, C. Fenn and H.-J. Buschmann, J. Chem. Soc., Faraday Trans., 1994, 90, 1507–1511 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cp07231k |
| This journal is © the Owner Societies 2019 |