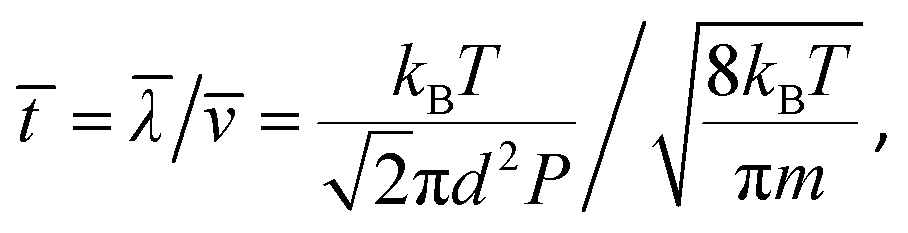Atomistic insights into the dynamics of binary collisions between gaseous molecules and polycyclic aromatic hydrocarbon dimers†
Received
15th November 2018
, Accepted 15th January 2019
First published on 16th January 2019
Abstract
Polycyclic aromatic hydrocarbon (PAH) dimers are important intermediates in combustion and soot formation. The scattering dynamics of gaseous molecules colliding with PAH dimers and the subsequent PAH dimer stability are investigated by performing molecular dynamics (MD) simulations. Effects of properties of the surrounding gaseous molecules and PAH dimers as well as temperature are investigated in this study. Depending on the residence time of N2 molecules trapped by the PAH dimers, two scattering types, that is, specular scattering and inelastic scattering, have been observed, which is correlated to the temperature and the type of the PAH dimer. Specifically, specular scattering preferentially takes place at high temperatures on small PAH dimers, while inelastic scattering tends to happen at low temperatures on large PAH dimers. During collision, energy transfer between the gaseous molecule and the PAH dimer changes the thermodynamic stability of the PAH dimer. Statistical analysis indicates that the decomposition rate of a PAH dimer to PAH monomers is sensitive to temperature and the PAH dimer type. Furthermore, effects of the gaseous molecule type on the PAH dimer stability are considered. The molecular mass of the colliding gaseous molecule is a key factor in determining the PAH dimer stability, as heavier gaseous molecules are more effective in promoting the PAH dimer decomposition. Results from this study indicate that collisions with gaseous molecules decrease the PAH dimer stability, while increasing the PAH dimer size and decreasing the collision temperature both decrease the decomposition rate of the PAH dimer.
1. Introduction
Soot particles formed from gaseous molecules under oxygen-starved conditions at high temperatures contribute to air pollution and climate change.1 Substantial understanding has been gained about the soot formation mechanism in the following stages: aromatic formation, nucleation, coagulation and surface growth.2–6 Among these stages, the nucleation stage that describes the process from gaseous PAH monomers to condensed nascent soot particles is the least understood. Conceptually, soot nucleation is typically assumed to stem from polycyclic aromatic hydrocarbon (PAH) dimerization.4,7 The irreversible dimerization of PAH molecules is widely used to represent soot nucleation in soot modeling.8 However, both experimental studies in low-temperature supersonic flows9 and theoretical studies by molecular dynamics simulations10–12 indicate that the physical dimerization of pyrene only happens at low temperatures. Later, studies by Thomson and coworkers13,14 proposed a reversible dimerization model by considering the chemical and physical dimerization of PAHs, which is reported to have a better prediction of the soot volume fraction, average primary particle diameter, etc. The above two widely accepted soot nucleation models both neglect the impact from gaseous molecules, which are inevitable in experiments and under real combustion conditions.4,15
Collisions between soot nuclei and surrounding gaseous moelcules are quite common in combustion, which are also of great importance to particle formation especially at high pressures.16,17 Serving as the soot nuclei, PAH dimers should survive the collision from surrounding gaseous molecules and be stable enough to grow with other PAH monomers or dimers. As the collision time between N2 molecules in engines (1600 K, 10 bar) is comparable to the PAH dimer lifetime, Schuetz and Frenklach18 stated that collisions with gaseous molecules is sufficient for PAH dimers to survive long enough for soot nucleation. Nonetheless, the effects of gaseous collisions on the PAH dimer stability have not yet been established with certainty. This results partly from the difficulty in taking experimental measurements with sufficient spatial and temporal resolutions to detect PAH dimers at low concentrations. From the aspect of the theoretical study or numerical modeling, the difficulties lie in the lack of a feasible force field description for interactions between gaseous molecules and PAHs in molecular dynamics (MD) simulations, and also in high computational costs of quantum chemistry (QC) calculations. Since higher peak pressures are becoming the norm in internal combustion engines, studying the interplay between the gaseous molecules and soot nuclei is ever more demanding.17,19
The objective of the present study is to provide a better understanding of the dynamics and kinetics of gaseous molecules colliding with PAH dimers. By performing a series of MD simulations, the influence of key factors, such as temperature, PAH dimer type, on the residence time and the scattering behavior of gaseous molecules is extensively examined. Furthermore, by quantifying the PAH dimer lifetime with and without gaseous molecule collisions, a kinetic model is developed with the aim to incorporate the key parameters that determine the PAH dimer stability.
2. Methodology
2.1 Simulation method
Binary collisions between gaseous molecules and PAH dimers are studied by MD simulations through tracing the movement of atoms. In the current study, a reactive force field (ReaxFF) is adopted, which includes most species in the periodic table.20 The ReaxFF MD can simulate both chemical reactions and physical dynamics in large systems with acceptable computational expense and accuracy.20 The computational cost for the single point energy calculation is several orders of magnitude lower than that of DFT calculations.9,11 In the ReaxFF, the energy calculation is composed of both bonded and non-bonded components based on the bond distance and bond order.21 Parameters in the force field are trained against a series of QC calculations.22,23 The interactions between gaseous molecules, such as N2, CO, CO2, and PAH dimers in the current study are described by the C/H/O/N force field given in the ESI.† This is integrated from the ReaxFF C-2013 carbon parameters22 and parameters of the C/H/O/N force field.23 This force field has been successfully used to study the thermodynamics and kinetics of PAHs and is reported to have a good description of the interactions between the gaseous molecules and PAHs.10,22,24–26
2.2 Computational setup
Before studying the binary collisions, the PAH dimer is firstly established. Our previous studies on PAH dimerization and soot inception have found that the PAH dimer in a stacked configuration is thermodynamically favored over the T-shaped one.10,24 Therefore, PAH dimers in stacked configurations are sampled evenly in time from binary collisions trajectories reported in our previous study of PAH dimerization.10Fig. 1(a) shows the initial configuration of the binary collision with the PAH dimer placed in the center of a cubic box (70 × 70 × 70 Å3). The initial distance between the PAH dimer and the gaseous molecule is Ld = 50 Å, which is much larger than the distance of effective intermolecular interaction. The impact parameter (b) is randomly selected from 0 to  , where ε is the enhancement factor,27ddimer is the effective diameter of the PAH dimer expressed as,
, where ε is the enhancement factor,27ddimer is the effective diameter of the PAH dimer expressed as,| |  | (1) |
where dA denotes the size of a single aromatic ring and equals  and nc is the number of carbon atoms in a PAH monomer.28 To achieve statistical significance, 1000 independent binary collision trajectories are studied at each temperature. The rotational energy (Erot) of the PAH dimer is randomly added in three dimensions according to the equipartition of energy. Relative translational velocities between the gaseous molecule and the PAH dimer are given to the gaseous molecules, which are in the Gaussian distribution of each temperature. In a real combustion system, collisions between gaseous molecules and PAH dimers happen at any orientation. Therefore, the collision angle (θ) is randomly sampled from the three dimensions. Gaseous molecules such as N2, CO, CO2 are diatomic and polyatomic. The bonds in these molecules are treated as a harmonic oscillator. Fig. 1(b) summarizes the initial configurations of the 1000 independent binary collisions between N2 molecules and pyrene dimers at 400 K. The arrows represent the collision orientations and relative collision velocities between gaseous molecules and the PAH dimer. The initial coordinate of the PAH dimer is the same for all the cases. MD simulations of the above binary collisions are performed adiabatically using a microcanonical ensemble (NVE) with a time step of 0.1 fs and last for 1 × 106 iterations, which accounts for a total simulation time of 100 ps. By solving the Newton's equations of motion with the ReaxFF, trajectories and velocities of the atom and molecules are given over time. MD simulations are performed using the large-scale atomic/molecular massively parallel simulator (LAMMPS) package29 implemented with reax/c. Snapshots and movies in this study are prepared by Ovito.30
and nc is the number of carbon atoms in a PAH monomer.28 To achieve statistical significance, 1000 independent binary collision trajectories are studied at each temperature. The rotational energy (Erot) of the PAH dimer is randomly added in three dimensions according to the equipartition of energy. Relative translational velocities between the gaseous molecule and the PAH dimer are given to the gaseous molecules, which are in the Gaussian distribution of each temperature. In a real combustion system, collisions between gaseous molecules and PAH dimers happen at any orientation. Therefore, the collision angle (θ) is randomly sampled from the three dimensions. Gaseous molecules such as N2, CO, CO2 are diatomic and polyatomic. The bonds in these molecules are treated as a harmonic oscillator. Fig. 1(b) summarizes the initial configurations of the 1000 independent binary collisions between N2 molecules and pyrene dimers at 400 K. The arrows represent the collision orientations and relative collision velocities between gaseous molecules and the PAH dimer. The initial coordinate of the PAH dimer is the same for all the cases. MD simulations of the above binary collisions are performed adiabatically using a microcanonical ensemble (NVE) with a time step of 0.1 fs and last for 1 × 106 iterations, which accounts for a total simulation time of 100 ps. By solving the Newton's equations of motion with the ReaxFF, trajectories and velocities of the atom and molecules are given over time. MD simulations are performed using the large-scale atomic/molecular massively parallel simulator (LAMMPS) package29 implemented with reax/c. Snapshots and movies in this study are prepared by Ovito.30
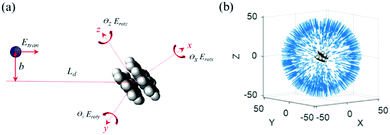 |
| | Fig. 1 (a) The initial configuration and setup for the binary collision between a gaseous molecule and a PAH dimer. (b) A summary of the initial configurations and the collision orientations of 1000 independent binary collisions between the gaseous molecule and the PAH dimer. | |
Experimental studies have indicated that the PAHs of naphthalene (A2), pyrene (A4) and coronene (A7) are typical precursors for soot inception.4 So PAH dimers formed from the above three types of PAH monomers are selected for the current study. The mean collision time (![[t with combining macron]](https://www.rsc.org/images/entities/i_char_0074_0304.gif) ) between a gaseous molecule and a PAH dimer is calculated from the mean free path (
) between a gaseous molecule and a PAH dimer is calculated from the mean free path (![[small lambda, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0cc.gif) ) and the mean velocity (
) and the mean velocity (![[small nu, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0ce.gif) ),31
),31
| |  | (2) |
where
kB is the Boltzmann constant,
T is the temperature,
P is the pressure,
m is the reduced mass expressed as
m =
mdimer·
mgas/(
mdimer +
mgas), and the collision diameter is
d = (
ddimer +
dgas)/2. Based on our previous study of PAH dimerization,
10 percentage of the PAH dimer with lifetime exceeding the mean collision time between the N
2 molecule and PAH dimer is presented in
Fig. 2, which decreases with increasing temperature and decreasing dimer size. At 1 atm, some coronene dimers survive longer than the mean collision time from 400 to 1600 K. While for naphthalene, only when the temperature decreases to 400 K, the dimer lifetime exceeds the mean collision time. Therefore, to reveal the scattering behavior of gas molecules and the PAH dimer stability, MD simulations are performed for PAH dimers whose lifetimes are longer than the mean collision time. Besides, effects of the combustion products, such as CO and CO
2, are additionally considered in the current study and listed in
Table 1.
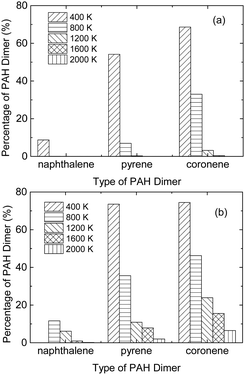 |
| | Fig. 2 Percentage of the PAH dimer with lifetime exceeding the mean collision time at (a) 1 atm, (b) 10 atm and temperatures from 400 to 2000 K. | |
Table 1 Overview of the initial settings for binary collisions
| Type |
PAH dimer |
Gaseous molecule |
Temperature (K) |
No. of binary collision (#) |
| 1 |
Naphthalene |
N2 |
400 |
1000 |
| 2 |
Naphthalene |
CO |
400 |
1000 |
| 3 |
Naphthalene |
CO2 |
400 |
1000 |
| 4 |
Pyrene |
N2 |
400 |
1000 |
| 5 |
Pyrene |
N2 |
600 |
1000 |
| 6 |
Coronene |
N2 |
400 |
1000 |
| 7 |
Coronene |
N2 |
600 |
1000 |
| 8 |
Coronene |
N2 |
800 |
1000 |
| 9 |
Coronene |
N2 |
1200 |
1000 |
| 10 |
Coronene |
N2 |
1600 |
1000 |
| 11 |
Coronene |
CO |
1600 |
1000 |
| 12 |
Coronene |
CO2 |
1600 |
1000 |
3. Results and discussion
Based on the above simulation method and setup, Section 3.1 presents the dynamic trajectories of the collisions between the gaseous molecule and the PAH dimer at different temperatures. A quantitative study of the residence time and the scattering behavior of gaseous molecules on PAH dimers is provided in Section 3.2. And finally, Section 3.3 focuses on the effects of temperature, gaseous molecule type on the PAH dimer stability.
3.1 Dynamics of binary collision
Dynamics of gaseous molecules colliding with the PAH dimer and the subsequent dimer behaviors are investigated at different temperatures. Firstly, spatio-temporal evolutions of the binary collision events are monitored according to the molecule trajectories. The collision trajectories of a N2 molecule colliding with the coronene dimer with different collision velocities and orientations at 400 K are shown in Fig. 3(a and b), while those at 1600 K are shown in Fig. 3(c and d), respectively. In Fig. 3, the movement directions and the mass center trajectories of the N2 molecules and PAH dimers are indicated in the snapshots. Moreover, Fig. 4 shows the changes in the translational, kinetic, potential and total energies of the N2 molecules during collisions with PAH dimers shown in Fig. 3(b and d).
 |
| | Fig. 3 Trajectories of the binary collision between the N2 molecule and the PAH dimer at varied temperatures ((a and b) T = 400 K; (c and d) T = 1600 K). The red and green lines represent the mass center trajectories of the N2 molecule and PAH dimer, respectively. Snapshots are recorded with the time evolution. Time and the position of N2 are recorded in blue and those of the PAH dimer or PAH monomers are in black. Blue, grey and white spheres represent nitrogen, carbon and hydrogen atoms. | |
 |
| | Fig. 4 Total, potential, kinetic and translational energy profiles of the colliding N2 molecules. The top and bottom panels show the results of the binary collisions of Fig. 3(b and d), respectively. | |
Firstly, the binary collision at a low temperature, e.g., 400 K, is discussed. According to the collision trajectories shown in Fig. 3(b) and the energy evolutions in the upper panel of Fig. 4, the translational energy of the coronene dimer is zero before collision and the N2 molecule moves towards the coronene dimer with its initial translational velocity. Afterwards, the translational, kinetic and total energies of the N2 molecule increase upon collision, while the potential energy decreases slightly because of the intermolecular interactions between the N2 molecule and the PAH dimer. And then, the N2 molecule diffuses randomly on the PAH dimer surface as indicated by the irregular curved mass-centered trajectories in Fig. 3(b). After adsorption, the N2 molecule continuously exchanges energy with the PAH dimer as indicated in the fluctuations of the energy profiles. The diffusion duration of the N2 molecule on the PAH dimer is named the residence time,32 which is evaluated from the adsorption and desorption moments as demonstrated in Fig. 4. Although the initial translational collision velocity for the event shown in Fig. 3(a) is similar to that in Fig. 3(b), difference in the collision orientation leads to greatly different residence time of N2 molecules. That is, the residence is about 3.25 ps for event (a) and as long as 21.25 ps for event (b). Subsequently, the N2 molecule desorbs back from the PAH dimer through gaining energies from the PAH dimer as indicated by the increase in the total energy as well as the translational and kinetic energies. Afterwards, the N2 molecule travels in the gas phase at a constant translational energy. The periodic fluctuations of the kinetic and potential energy profiles in Fig. 4 result from the harmonic vibration of the N atom in the N2 molecule, that is, the transfer between the kinetic component and the potential component in the N2 molecule. Due to the momentum and the energy transfer during binary collision, the translational velocity of the PAH dimer is initiated after N2 collisions and the dimer travels as indicated by the mass center trajectories shown in green colour.
Compared to the binary collision at 400 K, behaviors of the N2 molecule and PAH dimer at 1600 K are completely different as shown in Fig. 3(c and d). The energy profiles of the N2 molecule from binary collision shown in Fig. 3(d) are calculated and presented in the bottom panel of Fig. 4. After collision, the translational and kinetic energies of the N2 molecule increase, while the total and potential energies decrease. The residence time of the N2 molecule on the PAH dimer surface is much shorter compared with that at low temperatures as it reflects back simultaneously from the PAH dimer after collision. According to the energy profiles, the translational energy of the N2 molecule after reflection almost remains the same as that before collision. The behavior of the N2 molecule in this case can be termed as specular scattering as it looks like rigid body collisions.16 Due to energy transfer between the N2 molecule and the PAH dimer after collision, the total energy of the N2 dimer is smaller than that before collision. This consequently leads to the increased total energy of the PAH dimer as the binary collision is performed in the NVE ensemble. Another interesting phenomenon is that the PAH dimer stability decreases after N2 collision as the PAH dimer decomposes into its monomers, which is indicated by the PAH trajectories shown in Fig. 3(d). Compared to the binary collision event shown in Fig. 3(c), it seems that the PAH dimer is much less stable when the N2 molecule collides on the inter-layer space between the PAH dimer than on the planar surface of the PAH dimer even at a lower collision energy. Similar behaviors of adsorption, reflection and even dissociation are commonly present in collision systems, such as the Au atom and Au cluster reported by Yang et al.17
3.2 Gas-PAH dimer scattering
As discussed in Section 3.1, temperature is an important factor in determining the scattering behaviors of the N2 molecule. Therefore, quantitative study on its relation with the temperature is performed in this section. The scattering behavior is classified by the residence time of the N2 molecule trapped in the PAH dimer after collision. Firstly, the temperature effect on the residence time of a N2 molecule is examined. Fig. 5 shows the percentage distributions of the residence time of N2 molecules on the coronene dimer from 400 to 1600 K. Globally, the residence time distribution of the N2 molecule narrows with increasing temperature. That is, it ranges from 0 to more than 50 ps at 400 K, while it decreases to the range of 0–12.5 ps at an elevated temperature of 1600 K. According to Grančič et al.,32 the residence time of N2 molecules on the soot particle in the first bar is regarded as specular scattering, which is not strictly specular but close to it as the collision trajectories show in Fig. 3(d). It is noticeable that the number of N2 molecules within the first bar increases with increasing temperature. This demonstrates that the N2 molecules are more likely to take specular scattering during binary collision as temperature increases. However, few N2 molecules can permanently get absorbed in the coronene dimer based on our current study from 400 to 1600 K. The inelastic scattering is an intermediate state between the permanent absorption and the specular scattering as presented in Fig. 3(a and b). Thus the percentage of the inelastic scattering can be regarded as the sum of the rest of the grey bars. Opposite to the percentage of specular scattering, that of the inelastic scattering decreases with increasing temperature. The mean residence time (〈tres〉) of N2 molecules on the coronene dimer at each temperature is calculated and shown as the red line. Apparently, the mean residence time of N2 molecules on the coronene dimer is 4.381 ps at 400 K, and it decreases dramatically to 1.028 ps as the temperature increases to 1600 K. This means that the PAH dimer has higher adsorption ability at low temperatures, while it can hardly host the N2 molecules at high temperatures. Based on the energy profile shown in Fig. 4, the behavior of N2 molecules in the PAH dimers can be quantified by ΔEpot/kbT,16 where ΔEpot is the intermolecular interaction between the N2 molecule and the PAH dimer, and kb is the Boltzmann constant. Since the system of the PAH dimer and the N2 molecule has higher thermal fluctuations at high temperatures, it prevents the adsorption of gaseous molecules. The behavior of the N2 molecule on the PAH dimer is similar to that of N2 collision with Ag clusters16 as the absorption tendency gets weaker at a higher temperature.
 |
| | Fig. 5 Percentage distributions of the residence time of N2 molecules with the coronene dimer at temperatures from 400 to 1600 K. | |
Now, the characteristic effects of the PAH dimer on the scattering behavior of the N2 molecule are investigated. Fig. 6 shows the percentage distribution of the residence time of N2 molecules on the naphthalene and pyrene dimers at 400 K. Combined with the result of N2 molecules on the coronene dimer at 400 K in Fig. 5, it is found that the range of the residence time of N2 molecules is wider on the coronene dimer and it narrows with decreasing size of the PAH dimer. The mean residence time of N2 molecules on the PAH dimer decreases with increasing PAH dimer size. That is, the mean residence time decreases from 4.381 ps on the coronene dimer to 2.738 ps on the naphthalene dimer at 400 K. This illustrates that a larger PAH dimer has higher possibility to trap the gaseous N2 molecules. Moreover, this also indicates that the percentage of the inelastic scattering of N2 molecules increases with increasing PAH dimer size. The behavior of the N2 molecules on the PAH dimer agrees well with the N2 collision on Ag clusters as the specular-to-inelastic scattering transition shifts to larger particle sizes.16
 |
| | Fig. 6 Percentage distributions of the residence time of N2 molecule after colliding with naphthalene and pyrene dimers at the temperature of 400 K. | |
3.3 Stability of PAH dimers
As demonstrated in Fig. 3, binary collisions between the N2 molecule and the PAH dimer lead to the decrease in the PAH dimer stability as some of the dimers fall apart into PAH monomers after N2 collisions. To quantify this effect, the lifetime of the PAH dimer after the gaseous molecule collision is evaluated. Specifically, the rate of PAH dimer decomposition after collision with a gaseous molecule is expressed by:| |  | (3) |
| |  | (4) |
where ndimer is the number of the PAH dimers, k is the rate of dimer decomposition after gaseous molecule collision. Relation between ndimer and its lifetime t follows an exponential expression:| |  | (5) |
where n0 is the total number of PAH dimers before gaseous molecule collision. In order to explore the effects of gaseous molecule collision on the PAH dimer stability, the lifetime of the PAH dimer with and without gaseous molecule collision is compared by varying the gaseous molecule type, temperature and PAH dimer. Firstly, effects of temperature are investigated. Fig. 7 shows the ratio of the number of coronene dimers to the total number of gaseous molecule collisions (ndimer/n0) with respect to the dimer lifetime as well as the dimer decomposition rates at different temperatures. Apparently, the ratio of the coronene dimer after collisions with the N2 molecule at 400 K is mostly constant with its lifetime. This illustrates that no coronene dimer falls apart into monomers after N2 collisions within the simulation duration. As the temperature increases to 800 K, the ratio of the coronene dimer (n/n0) decreases slightly with the lifetime. Then, it decreases much quicker when the temperature increases to 1200 K and even 1600 K. The decomposition rate of the PAH dimer into PAH monomers is extracted from the slope of the curve in Fig. 7(a) and illustrated in Fig. 7(b), which indicates that temperature has a significant impact on the stability of the PAH dimer at high temperatures. In addition, effects of the gaseous molecules of CO and CO2 on the coronene dimer stability at 1600 K are also studied and compared against that of N2 molecules in Fig. 7. It is found that the profile of the ratio of the coronene dimer to its lifetime after the collision with CO molecules mostly coincides with that of collision with N2 molecules, while that of collision with CO2 molecules is much quicker. This correspondingly means that the heavier the colliding gaseous molecule, the quicker the decomposition of the PAH dimer into PAH monomers.
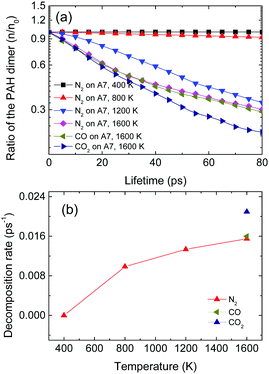 |
| | Fig. 7 (a) Ratio of the number of the coronene (A7) dimers to the total number of gaseous molecule collisions (n/n0), and (b) the decomposition rate of the coronene dimer under collisions with gaseous molecules (N2, CO and CO2) from 400 to 1600 K. | |
Finally, the effect of the PAH dimer characteristic is studied at 400 K and the results of the ratios of the number of PAH dimers to the total number of binary collisions with respect to the lifetime are presented in Fig. 8. It is noteworthy that the PAH dimer stability after collision with gaseous molecules is sensitive to the PAH dimer characteristic. Specifically, for N2 collision with different PAH dimers, the decomposition rate of the PAH dimer decreases dramatically with increasing PAH size. That is, the coronene dimer is relatively more stable compared to the pyrene and naphthalene dimers after N2 collisions at the same temperature, which results from the stronger binding energy between coronene monomers. In addition, the effect of the gaseous molecule type is considered. It is quite similar to the results for coronene PAHs at 1600 K in Fig. 7 as heavier colliding gaseous molecules are more likely to break the naphthalene dimer into naphthalene monomers. According to our previous studies on the nascent soot morphology,24 it is found that above 400, 600 and 800 K, PAHs of naphthalene, pyrene and coronene cannot physically stack into nascent soot particles, respectively. Therefore, by integrating the effects of gaseous molecules, it can be concluded that gaseous molecule collisions may further decrease the possibility of PAH dimerization and formation of nascent soot particles in stacked configurations.
 |
| | Fig. 8 (a) Ratio of the number of the surviving naphthalene, pyrene and coronene dimers to the total number of gaseous molecule collisions (n/n0), and (b) the decomposition rate of the PAH dimer, under collisions with gaseous molecules (N2, CO and CO2) at 400 K. | |
4. Conclusions
This paper presents a systematic study on the binary collision between the surrounding gaseous molecules and the PAH dimer by MD simulations. Key parameters, such as temperature, and properties of the PAH dimer and the gaseous molecule, are examined to clarify their influence on the scattering mechanism of the gaseous molecule on the PAH dimer and the PAH dimer stability. By analyzing the binary collision trajectory, the energy profile and the residence time of the gaseous molecule, it is found that the N2 molecules are more likely to result in specular scattering at elevated temperatures, while the possibility of inelastic scattering decreases as the temperature increases or the dimer size decreases. Moreover, few N2 molecules can become permanently absorbed on the coronene dimer even at 400 K. Because of the energy transfer during the binary collision, the increase in the energy of the PAH dimer results in decreasing the PAH dimer stability compared to the situation without gaseous molecule collisions. The decomposition rate of the PAH dimer due to gaseous molecule collision is calculated and found to increase with increasing temperature. Moreover, larger PAH dimers are much more stable than smaller PAH dimers during collisions with N2 molecules. In addition to N2 molecules, the effect of the gaseous molecule type on the PAH dimer stability is also considered by including other molecules such as CO and CO2, which indicates that the mass of the colliding gaseous molecule is important for the PAH dimer stability. That is, CO2 has a higher possibility to lead to PAH dimer decomposition, and the effect of CO is quite similar to that of N2. The present study demonstrates that collisions with N2, CO or CO2 molecules lead to decreased stability of the PAH dimer, while increasing the size of the PAH dimer or decreasing the collision temperature tends to decrease the decomposition rate of the PAH dimer.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Support from the National Natural Science Foundation of China (Grant No. 91441120 and 51390493) is gratefully acknowledged. Further support from the UK EPSRC under the project “UK Consortium on Mesoscale Engineering Sciences (UKCOMES)” (Grant No. EP/L00030X/1 and EP/R029598/1) is also acknowledged. The simulations were partly performed on the Tsinghua High-Performance Parallel Computer supported by the Tsinghua National Laboratory for Information Science and Technology.
References
- H. A. Michelsen, Probing soot formation, chemical and physical evolution, and oxidation: a review of in situ diagnostic techniques and needs, Proc. Combust. Inst., 2017, 36, 717–735 CrossRef CAS.
- J. H. Miller, W. G. Mallard and K. C. Smyth, Intermolecular potential calculations for polycyclic aromatic hydrocarbons, J. Phys. Chem., 1984, 88, 4963–4970 CrossRef CAS.
- M. Frenklach, Reaction mechanism of soot formation in flames, Phys. Chem. Chem. Phys., 2002, 4, 2028–2037 RSC.
- H. Wang, Formation of nascent soot and other condensed-phase materials in flames, Proc. Combust. Inst., 2011, 33, 41–67 CrossRef CAS.
- H. A. Michelsen, Probing soot formation, chemical and physical evolution, and oxidation: a review of in situ diagnostic techniques and needs, Proc. Combust. Inst., 2016, 000, 1–19 Search PubMed.
- K. Siegmann and K. Sattler, Formation mechanism for polycyclic aromatic hydrocarbons in methane flames, J. Chem. Phys., 2000, 112, 698–709 CrossRef CAS.
- B. Zhao, Z. Yang, M. V. Johnston, H. Wang, A. S. Wexler, M. Balthasar and M. Kraft, Measurement and numerical simulation of soot particle size distribution functions in a laminar premixed ethylene-oxygen-argon flame, Combust. Flame, 2003, 133, 173–188 CrossRef CAS.
- M. Frenklach and H. Wang, Detailed modeling of soot particle nucleation and growth, Symp. Combust., 1990, 23, 1559–1566 CrossRef.
- H. Sabbah, L. Biennier, S. J. Klippenstein, I. R. Sims and B. R. Rowe, Exploring the role of PAHs in the formation of soot: pyrene dimerization, J. Phys. Chem. Lett., 2010, 1, 2962–2967 CrossRef CAS.
- Q. Mao, Y. Ren, K. H. Luo and A. C. T. van Duin, Dynamics and Kinetics of Reversible Homo-molecular Dimerization of Polycyclic Aromatic Hydrocarbons, J. Chem. Phys., 2017, 147, 244305 CrossRef PubMed.
- S. H. Chung and A. Violi, Peri-condensed aromatics with aliphatic chains as key intermediates for the nucleation of aromatic hydrocarbons, Proc. Combust. Inst., 2011, 33, 693–700 CrossRef CAS.
- T. S. Totton, A. J. Misquitta and M. Kraft, A quantitative study of the clustering of polycyclic aromatic hydrocarbons at high temperatures, Phys. Chem. Chem. Phys., 2012, 14, 4081–4094 RSC.
- N. A. Eaves, S. B. Dworkin and M. J. Thomson, The importance of reversibility in modeling soot nucleation and condensation processes, Proc. Combust. Inst., 2015, 35, 1787–1794 CrossRef CAS.
- M. R. Kholghy, N. A. Eaves, A. Veshkini and M. J. Thomson, The role of reactive PAH dimerization in reducing soot nucleation reversibility, Proc. Combust. Inst., 2018, 000, 1–9 Search PubMed.
- A. D. Abid, J. Camacho, D. A. Sheen and H. Wang, Quantitative measurement of soot particle size distribution in premixed flames-The burner-stabilized stagnation flame approach, Combust. Flame, 2009, 156, 1862–1870 CrossRef CAS.
- Z. Li and H. Wang, Gas-nanoparticle scattering: a molecular view of momentum accommodation function, Phys. Rev. Lett., 2005, 95, 1–4 Search PubMed.
- H. Yang, E. Goudeli and C. J. Hogan, Condensation and dissociation rates for gas phase metal clusters from molecular dynamics trajectory calculations, J. Chem. Phys., 2018, 148, 164304 CrossRef PubMed.
- C. A. Schuetz and M. Frenklach, Nucleation of soot: molecular dynamics simulations of pyrene dimerization, Proc. Combust. Inst., 2002, 29, 2307–2313 CrossRef CAS.
- A. L. Miller, C. B. Stipe, M. C. Habjan and G. G. Ahlstrand, Role of lubrication oil in particulate emissions from a hydrogen-powered internal combustion engine, Environ. Sci. Technol., 2007, 41, 6828–6835 CrossRef CAS.
- T. Senftle, S. Hong, M. M. Islam, S. B. Kylasa, Y. Zheng, Y. K. Shin, C. Junkermeier, R. Engel-Herbert, M. J. Janik, H. M. Aktulga, T. Verstraelen, A. Grama and A. C. T. van Duin, The ReaxFF reactive force-field: development, applications, and future directions, npj Comput. Mater., 2016, 2, 15011 CrossRef CAS.
- A. C. T. van Duin, S. Dasgupta, F. Lorant and W. A. Goddard III, ReaxFF: a reactive force field for hydrocarbons, J. Phys. Chem. A, 2001, 105, 9396–9409 CrossRef CAS.
- S. Goverapet Srinivasan, A. C. T. van Duin and P. Ganesh, Development of a ReaxFF potential for carbon condensed phases and its application to the thermal fragmentation of a large fullerene, J. Phys. Chem. A, 2015, 119, 571–580 CrossRef PubMed.
- M. R. Weismiller, M. F. Russo, A. C. T. Van Duin and R. A. Yetter, Using molecular dynamics simulations with a ReaxFF reactive force field to develop a kinetic mechanism for ammonia borane oxidation, Proc. Combust. Inst., 2013, 34, 3489–3497 CrossRef CAS.
- Q. Mao, A. C. T. van Duin and K. H. Luo, Formation of incipient soot particles from polycyclic aromatic hydrocarbons: a ReaxFF molecular dynamics study, Carbon, 2017, 121, 380–388 CrossRef CAS.
- K. Yoon, A. Rahnamoun, J. L. Swett, V. Iberi, D. A. Cullen, I. V. Vlassiouk, A. Belianinov, S. Jesse, X. Sang, O. S. Ovchinnikova, A. J. Rondinone, R. R. Unocic and A. C. T. van Duin, Atomistic-scale simulations of defect formation in graphene under noble gas ion irradiation, ACS Nano, 2016, 10, 8376–8384 CrossRef CAS PubMed.
- Q. Mao and K. H. Luo, Trace metal assisted polycyclic aromatic hydrocarbons fragmentation, growth and soot nucleation, Proc. Combust. Inst., 2018, 000, 1–8 CAS.
- S. J. Harris and I. M. Kennedy, The coagulation of soot particles with van der Waals forces, Combust. Sci. Technol., 1988, 59, 443–454 CrossRef CAS.
- A. Raj, M. Sander, V. Janardhanan and M. Kraft, A study on the coagulation of polycyclic aromatic hydrocarbon clusters to determine their collision efficiency, Combust. Flame, 2010, 157, 523–534 CrossRef CAS.
- S. Plimpton, Fast parallel algorithms for short-range molecular dynamics, J. Comput. Phys., 1995, 117, 1–19 CrossRef CAS.
- A. Stukowski, Visualization and analysis of atomistic simulation data with OVITO–the Open Visualization Tool, Modell. Simul. Mater. Sci. Eng., 2010, 18, 015012 CrossRef.
-
C. Law, Combustion physics, Cambridge University Press, 2006 Search PubMed.
- P. Grančič, J. W. Martin, D. Chen, S. Mosbach and M. Kraft, Can nascent soot particles burn from the inside?, Carbon, 2016, 109, 608–615 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Parameter sets of ReaxFF force field for C/H/O/N descriptions. See DOI: 10.1039/c8cp07060a |
|
| This journal is © the Owner Societies 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *ab and
Adri C. T.
van Duin
c
*ab and
Adri C. T.
van Duin
c
 , where ε is the enhancement factor,27ddimer is the effective diameter of the PAH dimer expressed as,
, where ε is the enhancement factor,27ddimer is the effective diameter of the PAH dimer expressed as,
 and nc is the number of carbon atoms in a PAH monomer.28 To achieve statistical significance, 1000 independent binary collision trajectories are studied at each temperature. The rotational energy (Erot) of the PAH dimer is randomly added in three dimensions according to the equipartition of energy. Relative translational velocities between the gaseous molecule and the PAH dimer are given to the gaseous molecules, which are in the Gaussian distribution of each temperature. In a real combustion system, collisions between gaseous molecules and PAH dimers happen at any orientation. Therefore, the collision angle (θ) is randomly sampled from the three dimensions. Gaseous molecules such as N2, CO, CO2 are diatomic and polyatomic. The bonds in these molecules are treated as a harmonic oscillator. Fig. 1(b) summarizes the initial configurations of the 1000 independent binary collisions between N2 molecules and pyrene dimers at 400 K. The arrows represent the collision orientations and relative collision velocities between gaseous molecules and the PAH dimer. The initial coordinate of the PAH dimer is the same for all the cases. MD simulations of the above binary collisions are performed adiabatically using a microcanonical ensemble (NVE) with a time step of 0.1 fs and last for 1 × 106 iterations, which accounts for a total simulation time of 100 ps. By solving the Newton's equations of motion with the ReaxFF, trajectories and velocities of the atom and molecules are given over time. MD simulations are performed using the large-scale atomic/molecular massively parallel simulator (LAMMPS) package29 implemented with reax/c. Snapshots and movies in this study are prepared by Ovito.30
and nc is the number of carbon atoms in a PAH monomer.28 To achieve statistical significance, 1000 independent binary collision trajectories are studied at each temperature. The rotational energy (Erot) of the PAH dimer is randomly added in three dimensions according to the equipartition of energy. Relative translational velocities between the gaseous molecule and the PAH dimer are given to the gaseous molecules, which are in the Gaussian distribution of each temperature. In a real combustion system, collisions between gaseous molecules and PAH dimers happen at any orientation. Therefore, the collision angle (θ) is randomly sampled from the three dimensions. Gaseous molecules such as N2, CO, CO2 are diatomic and polyatomic. The bonds in these molecules are treated as a harmonic oscillator. Fig. 1(b) summarizes the initial configurations of the 1000 independent binary collisions between N2 molecules and pyrene dimers at 400 K. The arrows represent the collision orientations and relative collision velocities between gaseous molecules and the PAH dimer. The initial coordinate of the PAH dimer is the same for all the cases. MD simulations of the above binary collisions are performed adiabatically using a microcanonical ensemble (NVE) with a time step of 0.1 fs and last for 1 × 106 iterations, which accounts for a total simulation time of 100 ps. By solving the Newton's equations of motion with the ReaxFF, trajectories and velocities of the atom and molecules are given over time. MD simulations are performed using the large-scale atomic/molecular massively parallel simulator (LAMMPS) package29 implemented with reax/c. Snapshots and movies in this study are prepared by Ovito.30