
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMicrohydration of protonated 5-hydroxyindole revealed by infrared spectroscopy†
Johanna
Klyne
 and
Otto
Dopfer
*
and
Otto
Dopfer
*
Institut für Optik und Atomare Physik, Technische Universität Berlin, Hardenbergstr. 36, 10623 Berlin, Germany. E-mail: dopfer@physik.tu-berlin.de; Fax: +49-30-31423018
First published on 8th January 2019
Abstract
Controlled microsolvation of protonated aromatic biomolecules with water is fundamental to understand proton transfer reactions in aqueous environments. We measured infrared photodissociation (IRPD) spectra of mass-selected microhydrates of protonated 5-hydroxyindole (5HIH+–Wn, W = H2O, n = 1–3) in the OH and NH stretch ranges (2700–3800 cm−1), which are sensitive to the spectroscopic characteristics of interior solvation, water network formation, and proton transfer to solvent. Analysis of the IRPD spectra by dispersion-corrected density functional theory calculations (B3LYP-D3/aug-cc-pVTZ) reveals the coexistence of C3- and C4-protonated carbenium ions, 5HIH+(C3) and 5HIH+(C4), as well as the O-protonated oxonium ion, 5HIH+(O). Monohydrated 5HIH+–W clusters are formed by hydrogen-bonding (H-bonding) of the first water to the most acidic functional group, namely, the NH group in the case of 5HIH+(C3), the OH group for 5HIH+(C4), and the OH2 group for 5HIH+(O). The latter benefits from its twofold degeneracy and the outstandingly high binding energy of D0 ∼ 100 kJ mol−1. Larger 5HIH+–W2/3 clusters preferably grow (i) by H-bonding of the second water to the remaining vacant functional group and and/or (ii) by formation of W2 water chains at the respective most acidic functional group. Our IRPD spectra of 5HIH+–Wn do not indicate any proton transfer to the solvent up to n = 3, in line with the proton affinities of 5HI and Wn. Comparison of 5HIH+–Wn to neutral 5HI–W and cationic 5HI+–Wn clusters elucidates the impact of different charge states on the topology of the initial solvation shell. Furthermore, to access the influence of the size of the arene ion and a second functional group, we draw a comparison to microhydration of protonated phenol.
1. Introduction
The interaction of biomolecules, such as proteins, enzymes or hormones, with water (W) is crucial for their structure and function in living organisms. Controlled sequential microhydration of isolated biomolecules facilitates investigation of the interactions between solutes and solvents, which are blurred in the condensed phase. Infrared (IR) vibrational spectroscopy of size-selected hydrated clusters is particularly useful because it provides direct structural information. Combined with quantum chemistry, IR spectroscopy can elucidate the potential energy surface of hydration interactions.Aromatic heterocyclic molecules are ubiquitous biomolecular building blocks.1–3 For example, 5-hydroxyindole (5HI), the prototype chosen herein, consists of pyrrole fused to a phenol ring and occurs as a subunit in the neurotransmitter serotonin. The sequential microhydration of protonated aromatic molecules is particularly interesting because proton transfer from the biomolecule to the solvent may happen in these systems. Both, protonation and hydration are found to enhance the acidity of the functional groups (OH/NH), and hence their ability to donate a proton.4,5 Moreover, the proton affinity (PA) of W (PA = 691 kJ mol−1)6 and small Wn clusters (PA = 808–879 kJ mol−1 for n = 2–3)7,8 is in the same range as those of aromatic hydrocarbons (e.g., PA = 750 kJ mol−1 for benzene).6 Hence, formation of a hydronium ion (H3O+) or protonated water clusters H+(H2O)nvia proton transfer can eventually increase the interaction energy resulting in more stable hydrated clusters. For example, protonated benzene releases its excess proton already upon hydration by a single water molecule.9,10 Moreover, for the prototypical protonated arenes (A) naphthalene and benzaldehyde, proton transfer to water occurs at cluster sizes of n = 2 and 3, respectively, as revealed from IR photodissociation (IRPD) spectra of their microhydrated [A–Wn]H+ clusters.11,12 In general, the topology of the potential energy surface of hydrated clusters strongly depends on their charge or protonation state.5,13–15
The microhydration of the protonated phenol (PhH+) subunit of 5HI has been studied by quantum chemistry and IRPD spectroscopy.14,16 While the theoretical study focused on the electronic structure of the carbenium ion,16 the IRPD study indicates the coexistence of ortho/para C-protonated carbenium and O-protonated oxonium isomers, PhH+(o/p) and PhH+(O), in a molecular beam.14 For both types of protomers, small Wn clusters are formed at the OH group of phenol. However, proton transfer is observed at different critical sizes of the hydration shell (nc). While the Wn network accepts the excess proton already at nc = 3 for PhH+(O)–Wn, proton transfer is only possible for n ≥ 4 in PhH+(o/p)–Wn. This change in nc is explained by the geometry of the carbenium cluster. The protonated CH2 group is simply too far away from the Wn network at the OH group. Proton transfer is possible only when the Wn cluster forms a ring bridging the OH and CH2 groups at nc = 4. The role of binding energies (D0) of the PhH+–Wn clusters or differences in the acidity of the OH groups have not been discussed. However, our preceding IRPD study of the sequential microsolvation of protonated 5-hydroxyindole (5HIH+) by nonpolar Ar and quadrupolar N2 ligands (L) indicates a drastic difference in the ligand binding energies of the carbenium and oxonium protomers, 5HIH+(C) and 5HIH+(O).17 The 5HIH+–Ln spectra (n ≤ 3) reveal the coexistence of 5HIH+(C3)–Ln, 5HIH+(C4)–Ln, and 5HIH+(O)–Ln clusters with ion cores protonated at C3, C4, and O, respectively. This finding is surprising at first glance, because bare 5HIH+(O) is drastically less stable than the carbenium ions (ΔE0 > 100 kJ mol−1).5,17 The presence of clusters with an oxonium core can however be rationalized by the outstandingly large binding energies of 5HIH+(O)–Ln(OH) clusters with ligands attached to the OH2 group, the high barriers for isomerization from the oxonium to the carbenium ions (kinetic trapping), and the twofold degeneracy of these structures.14,17–20 Furthermore, the acidity of the NH and OH functional groups is found to strongly depend on the protonation site.17 Comparison with PhH+ indicates an increase in the acidity of the OH group from 5HIH+ to PhH+.17 Hence, we expect intracluster proton transfer in 5HIH+–Wn clusters for a larger nc value as compared to PhH+–Wn.
To the best of our knowledge, only one theoretical study describes the microhydration of protonated 5HIH+,5 and the protonation-induced change of the interaction potential compared to neutral hydrated clusters. This study reveals the preference of OH⋯W over NH⋯W H-bonds in the neutral ground state, in line with spectroscopic data.14 Both, OH⋯W and NH⋯W H-bonds are significantly strengthened upon protonation,5 whereas the increase in the acidity of the NH group is more pronounced than that of the OH group. As a result, the topology of the hydration shell is changed upon protonation and NH⋯W bonds are preferred. However, only C3-protonated 5HIH+(C3)–W clusters have been considered.5 Most likely, the topologies of 5HIH+(C4)–W and 5HIH+(O)–W clusters differ strongly.17 Thus, we address herein three main questions: (i) what is the protomer abundance within 5HIH+–Wn clusters and how is it changed compared to 5HIH+–Ln with L = Ar and N2; (ii) what is the structure of the initial solvation shell in 5HIH+–Wn; and (iii) do we observe proton transfer in the size range n ≤ 3 and a protomer dependence of nc?
To this end, we analyze IRPD spectra of mass-selected hydrated clusters of 5HIH+ with the aid of quantum chemical calculations. Vibrational spectroscopy in the NH and OH stretch range can determine the protonation site, discriminate interior ion solvation from water network formation, and signal potential proton transfer from 5HIH+ to the Wn solvent cluster. The comparison of our results on 5HIH+–W to those obtained for 5HI+–W and 5HI–W illustrates the influence of different charge states and protonation on the solvation of 5HI.
2. Experimental and computational techniques
The microsolvation of 5HIH+ is studied by IRPD spectroscopy of mass-selected 5HIH+–Wn (n = 1–3) clusters. Additional spectra of colder 5HIH+–W are obtained by Ar and N2 tagging. IRPD spectra are measured in the XH stretch range (X = N and O, 2600–3800 cm−1) with a quadrupole–octopole–quadrupole tandem mass spectrometer described elsewhere.18–26 Briefly, protonated clusters are generated in an electron ionization source coupled to a pulsed molecular beam expansion. Solid 5HI (Sigma-Aldrich, 97%) is heated to 145 °C and the resulting vapor is seeded in Ar or N2 carrier gas (8–9 bar) containing water. He/H2 gas (90/10) is added to pure Ar (N2) in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio to enhance the protonation efficiency. The gas mixture is expanded into vacuum through a pulsed nozzle. Close to the nozzle orifice, electron and/or chemical ionization of X (X = Ar, N2, H2, or Wn) forms XH+ ions, which subsequently protonate 5HI via proton transfer.18–20,24 Three-body collisions with W and/or carrier gas molecules lead to cluster formation. In the first quadrupole, the desired parent clusters are mass-selected. Pulsed IR radiation emitted from a tunable optical parametric oscillator (2–4 mJ pulse energy, 10 Hz repetition rate, 1 cm−1 bandwidth) is introduced into the adjacent octopole. Resonant vibrational excitation of the parent clusters leads to the loss of the least bonded ligand, i.e. single W molecules in the case of 5HIH+–Wn or the loosely bound Ar/N2 tag for 5HIH+–W–Ar/N2. The produced fragment ions are mass-selected with the second quadrupole and monitored by a Daly detector as a function of the IR laser frequency to derive the IRPD spectrum of the 5HIH+–Wn or 5HIH+–W–Ar/N2 parent clusters. The ion source is triggered at 20 Hz (twice the laser frequency) facilitating subtraction of the background signal (metastable decay). All IRPD spectra are normalized for laser intensity variations recorded with a pyroelectric detector. Collision-induced dissociation in the octopole confirms the composition of mass-selected parent clusters.22,23
:1 ratio to enhance the protonation efficiency. The gas mixture is expanded into vacuum through a pulsed nozzle. Close to the nozzle orifice, electron and/or chemical ionization of X (X = Ar, N2, H2, or Wn) forms XH+ ions, which subsequently protonate 5HI via proton transfer.18–20,24 Three-body collisions with W and/or carrier gas molecules lead to cluster formation. In the first quadrupole, the desired parent clusters are mass-selected. Pulsed IR radiation emitted from a tunable optical parametric oscillator (2–4 mJ pulse energy, 10 Hz repetition rate, 1 cm−1 bandwidth) is introduced into the adjacent octopole. Resonant vibrational excitation of the parent clusters leads to the loss of the least bonded ligand, i.e. single W molecules in the case of 5HIH+–Wn or the loosely bound Ar/N2 tag for 5HIH+–W–Ar/N2. The produced fragment ions are mass-selected with the second quadrupole and monitored by a Daly detector as a function of the IR laser frequency to derive the IRPD spectrum of the 5HIH+–Wn or 5HIH+–W–Ar/N2 parent clusters. The ion source is triggered at 20 Hz (twice the laser frequency) facilitating subtraction of the background signal (metastable decay). All IRPD spectra are normalized for laser intensity variations recorded with a pyroelectric detector. Collision-induced dissociation in the octopole confirms the composition of mass-selected parent clusters.22,23
The protonation sites in 5HIH+ have already been examined in a previous study.17 The 5HIH+ protomers offer two competing H-bonding sites for W molecules, namely, their NH and OH functional groups. The 5HIH+–Wn input structures for geometry optimization are constructed by hand, attaching W ligands successively to the NH and OH groups. Up to n = 3, this approach is still feasible to find all low-energy minima. For larger hydrates (and more flexible organic chromophores), it may be rather difficult to find geometries by hand and, as a consequence, systematic computational sampling techniques such as basin-hopping or molecular dynamics should be employed.28–30 Geometries, energies, and harmonic IR spectra of stable 5HIH+–Wn (n = 1–3) and 5HIH+–W–Ar/N2 (ESI†) structures are calculated at the B3LYP-D3/aug-cc-pVTZ level using GAUSSIAN09.27,31–34 This hybrid density functional with additive dispersion correction has proven to yield reliable results for related aromatic clusters.11,22,23,35–39 For example, the binding energies computed for the monohydrates of the benzene and naphthalene cations (D0 = 3209 and 2773 cm−1) compare favorably with the experimental values (D0 = 3290 ± 120 and 2800 ± 300 cm−1).40,41 Similarly, the calculated binding energy of W2 (D0 = 1108 cm−1) matches the measured value (D0 = 1105 ± 10 cm−1).42,43 We optimize selected structures also at the PBE0-D3/aug-cc-pVTZ level to yield reference data on structural and spectroscopic properties. The PBE0 functional is frequently used to compute properties of molecular clusters and is less empirical than B3LYP.28,30,44–46 Furthermore, single-point energy calculations of selected optimized structures are performed at the CC2/aug-cc-pVDZ level. Comparison to neutral and cationic s/a5HI–W clusters previously studied at the same level (B3LYP-D3/aug-cc-pVTZ) yields the effects of protonation.22,23 Total binding energies (D0) are derived by subtracting the zero-point corrected energies of the corresponding monomers from that of the cluster: D0 = E0(5HIH+–Wn) − E0(5HIH+) − n·E0(W). Binding energies of the Ar/N2 tag in 5HIH+–W–Ar/N2 are calculated in a similar way. For selected structures, we estimate the basis set superposition errors (BSSE) using the counterpoise method.47,48 Calculated harmonic vibrational frequencies are linearly scaled by a factor of 0.957 derived previously from fitting the free OH stretching frequency of cationic s/a5HI+.22 Yet, this scaling factor yields OH stretch frequencies of W (ν1/3 = 3635/3733 cm−1) systematically lower by ∼20 cm−1 than the experimental values (ν1/3 = 3657/3756 cm−1).49 For selected 5HIH+–W clusters, anharmonic spectra are calculated at the B3LYP-D3/aug-cc-pVDZ level as implemented in GAUSSIAN09.50
3. Results and discussion
3.1 IRPD spectra
An overview of the IRPD spectra of 5HIH+–Wn with n = 1–3 measured in the XH stretch range is given in Fig. 1. The positions, widths, and suggested vibrational and isomer assignments of the transitions observed (A–L, X) are listed in Table 1, along with computed frequencies and IR oscillator strengths. The spectra cover the antisymmetric and symmetric OH stretching modes of water (ν3 and ν1, bands A and B) and the OH and NH stretching modes of 5HIH+ (νOH and νNH). Dotted lines indicate the correspondence of related bands in the different spectra. Bands G and H emerging in the spectra of 5HIH+–Wn with n ≥ 2 already indicate the formation of a H-bonded water network. In the following, the IRPD spectra are disentangled by comparison with IR spectra calculated for the possible isomers and taking into account the previous interpretation of the 5HIH+–Ln spectra with L = Ar and N2.17 IRPD spectra of tagged 5HIH+–W–L clusters with L = Ar and N2 are shown in Fig. S1 in the ESI.†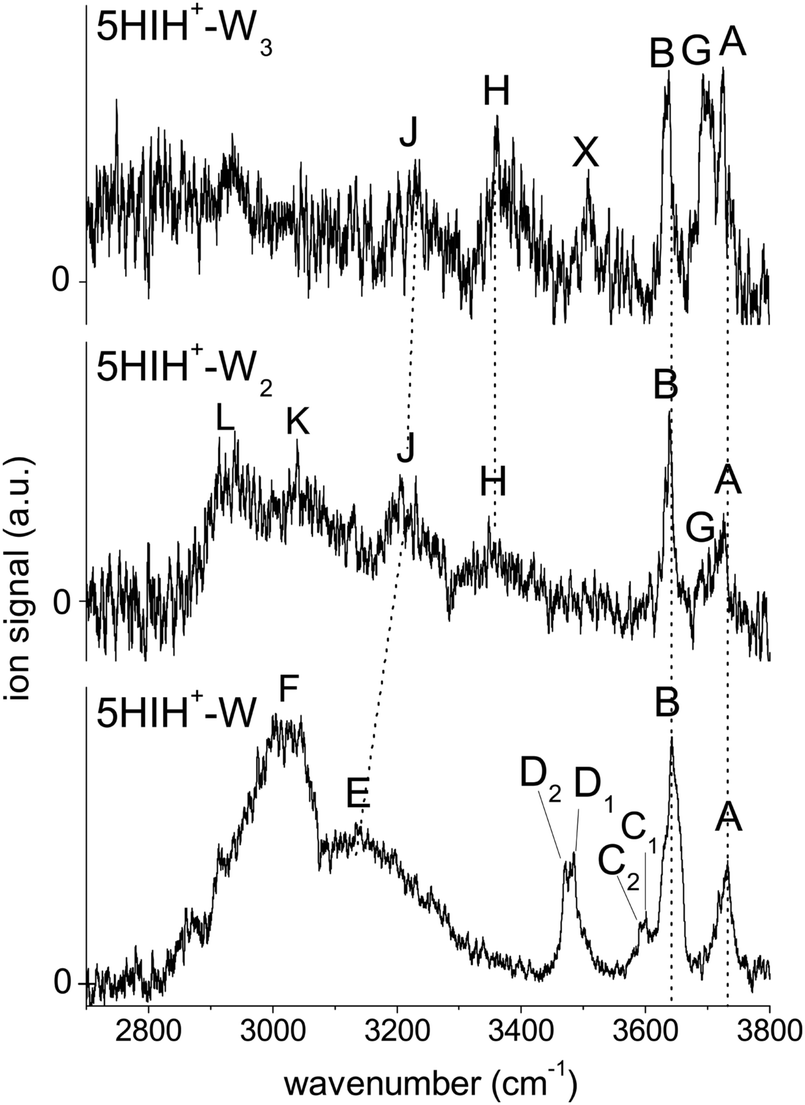 | ||
| Fig. 1 Overview of the IRPD spectra of 5HIH+–Wn (n = 1–3) measured in the XH stretch range (2700–3800 cm−1). The positions, widths, and vibrational and isomer assignments of the transitions are listed in Table 1. | ||
| Cluster | Exp. | Vibration | Calc. | Isomer |
|---|---|---|---|---|
| 5HIH+–W | A 3732 (30) | ν 3 | 3704 (119) | s5HIH+(C3)–W(NH) |
| ν 3 | 3708 (118) | s5HIH+(C4)–W(NH) | ||
| ν 3 | 3697 (134) | s5HIH+(C4)–W(OH) | ||
| ν 3 | 3685 (178) | s5HIH+(O)–W(OH) | ||
| B 3641 (25) | ν fOH | 3632 (164) | s5HIH+(C3)–W(NH) | |
| ν 1 | 3622 (54) | s5HIH+(C3)–W(NH) | ||
| ν 1 | 3612 (52) | s5HIH+(C4)–W(OH) | ||
| ν 1 | 3622 (40) | s5HIH+(C4)–W(NH) | ||
| ν 1 | 3600 (68) | s5HIH+(O)–W(OH) | ||
| C 1 3600 (5) | ν fOH | 3588 (219) | s5HIH+(C4)–W(NH) | |
| C 2 3591 (5) | ν fOH | 3558 (199) | s5HIH+(O)–W(OH) | |
| D 1 3485 (30) | ν fNH | 3494 (116) | s5HIH+(O)–W(OH) | |
| D 2 3470 (30) | ν fNH | 3478 (139) | s5HIH+(C4)–W(OH) | |
| E 3130 (100) | ν bNH | 3191 (1184) | s5HIH+(C4)–W(NH) | |
| F 3015 (70) | ν bOH | 3035 (2283) | s5HIH+(C4)–W(OH) | |
| ν bNH | 3011 (1399) | s5HIH+(C3)–W(NH) | ||
| 5HIH+–W2 | A 3725 (20) | ν 3 | 3711 (131)/3707 (113) | s5HIH+(C3)–W2(NH–OH) |
| ν 3 | 3717 (114) | s5HIH+(C3)–W2(NH–W) | ||
| ν 3 | 3711 (114)/3700 (128) | s5HIH+(C4)–W2(NH–OH) | ||
| G 3692 (10) | ν f(W)OH | 3690 (105) | s5HIH+(C3)–W2(NH–W) | |
| B 3638 (15) | ν 1 | 3624 (49)/3619 (33) | s5HIH+(C3)–W2(NH–OH) | |
| ν fOH | 3635 (156) | s5HIH+(C3)–W2(NH–W) | ||
| ν 1 | 3624 (37)/3615 (48) | s5HIH+(C4)–W2(NH–OH) | ||
| H 3348 (>50) | ν bOH | 3283 (1565) | s5HIH+(C3)–W2(NH–OH) | |
| ν b(W)OH | 3308 (921) | s5HIH+(C3)–W2(NH–W) | ||
| J 3205 (>50) | ν bNH | 3218 (1094) | s5HIH+(C4)–W2(NH–OH) | |
| K 3040 (>50) | ν bNH | 3041 (1331) | s5HIH+(C3)–W2(NH–OH) | |
| ν bOH | 3083 (2234) | s5HIH+(C4)–W2(NH–OH) | ||
| L 2940 (>50) | ν bNH | 2787 (2249) | s5HIH+(C3)–W2(NH–W) | |
| 5HIH+–W3 | A 3725 (20) | ν 3 | 3719 (112)/3708 (114) | s5HIH+(C3)–W3(OH–NH–W) |
| ν 3 | 3721 (111) | s5HIH+(C3)–W3(NH–W–W) | ||
| ν 3 | 3722 (208) | s5HIH+(C3)–W3(W–NH–W) | ||
| ν 3 | 3723 (106)/3702 (127) | s5HIH+(C4)–W3(OH–NH–W) | ||
| ν 3 | 3722 (115)/3712 (112) | s5HIH+(C4)–W3(OH–W–NH) | ||
| ν 3 | 3702 (147)/3695 (222) | s5HIH+(O)–W3(NH–OH–W1) | ||
| ν 3 | 3717 (128)/3704 (143) | s5HIH+(O)–W3(NH–OH–W2) | ||
| G 3692 (30) | ν fOH | 3692 (102) | s5HIH+(C3)–W3(OH–NH–W) | |
| ν f(W)OH | 3694 (70) | s5HIH+(C3)–W3(NH–W–W) | ||
| ν f(W)OH | 3693 (111) | s5HIH+(C3)–W3(NH–W–W) | ||
| ν f(W)OH | 3689 (92) | s5HIH+(C4)–W3(OH–NH–W) | ||
| ν f(W)OH | 3679 (100) | s5HIH+(C4)–W3(OH–W–NH) | ||
| ν f(W)OH | 3661 (140) | s5HIH+(O)–W3(NH–OH–W1) | ||
| ν f(W)OH | 3677 (97) | s5HIH+(O)–W3(NH–OH–W2) | ||
| B 3637 (20) | ν 1 | 3630 (27)/3620 (31) | s5HIH+(C3)–W3(OH–NH–W) | |
| ν f(W)OH | 3635 (151) | s5HIH+(C3)–W3(NH–W–W) | ||
| ν 1 | 3631 (23) | s5HIH+(C3)–W3(NH–W–W) | ||
| ν fOH | 3636 (147) | s5HIH+(C3)–W3(W–NH–W) | ||
| ν 1 | 3631 (29) | s5HIH+(C3)–W3(W–NH–W) | ||
| ν 1 | 3632 (24)/3616 (45) | s5HIH+(C4)–W3(OH–NH–W) | ||
| ν 1 | 3631 (29)/3624 (35) | s5HIH+(C4)–W3(OH–W–NH) | ||
| ν 1 | 3616 (54)/3586 (100) | s5HIH+(O)–W3(NH–OH–W1) | ||
| ν 1 | 3628 (36)/3617 (45) | s5HIH+(O)–W3(NH–OH–W2) | ||
| X 3508 (30) | ν fNH | 3497 (108) | s5HIH+(O)–W3(NH–OH–W1) | |
| ν fNH | 3500 (105) | s5HIH+(O)–W3(NH–OH–W2) | ||
| H 3360 (>50) | ν b(W)OH | 3375 (723) | s5HIH+(C4)–W3(OH–NH–W) | |
| ν b(W)OH | 3306 (891) | s5HIH+(C4)–W3(OH–W–NH) | ||
| ν b(W)OH | 3326 (867) | s5HIH+(C3)–W3(OH–NH–W) | ||
| ν bOH | 3301 (156) | s5HIH+(C3)–W3(OH–NH–W) | ||
| ν b(W)OH | 3378 (663) | s5HIH+(C3)–W3(NH–W–W) | ||
| ν b(as)OH | 3424 (380) | s5HIH+(C3)–W3(W–NH–W) | ||
| ν b(s)OH | 3381 (516) | s5HIH+(C3)–W3(W–NH–W) | ||
| J 3230 (>50) | ν bOH | 3110 (1927) | s5HIH+(C4)–W3(OH–NH–W) | |
| ν bNH | 3078 (1906) | s5HIH+(C4)–W3(OH–NH–W) | ||
| ν bNH | 3236 (1063) | s5HIH+(C4)–W3(OH–W–NH) | ||
| ν b(W)OH | 3126 (1404) | s5HIH+(C3)–W3(NH–W–W) | ||
| ν bOH | 3183 (1263) | s5HIH+(O)–W3(NH–OH–W2) | ||
| ν bOH | 3183 (1263) | s5HIH+(O)–W3(NH–OH–W2) | ||
3.2 5HIH+–W
Internal OH rotation yields syn and anti rotamers of 5HIH+, denoted by s5HIH+ and a5HIH+, respectively. Our previous study of 5HIH+–Ln demonstrates the coexistence of the most stable s/a5HIH+(C3) protomers (ΔE0 = 0/1.8 kJ mol−1), as well as the s/a5HIH+(C4) and s/a5HIH+(O) isomers (ΔE0 = 5.1/11.1 and 117.1/117.7 kJ mol−1).17 With the exception of 5HIH+(C4), the syn and anti rotamers of the 5HIH+ protomers have similar energies and IR spectra and thus cannot be distinguished at the current spectral resolution. A deep potential well (ΔE > 150 kJ mol−1) prevents the interconversion of the drastically less stable 5HIH+(O) isomers into 5HIH+(C4).17 We compute proton affinities (PAs) of 905/902 kJ mol−1 for s/a5HIH+(C3), 900/892 kJ mol−1 for s/a5HIH+(C4), and 786/785 kJ mol−1 for s/a5HIH+(O).17 On the other hand, for Wn clusters we calculate PAs of 681, 818, and 898 kJ mol−1 for n = 1–3, respectively. Hence, the predicted PA of s/a5HIH+(O) is smaller than that of the Wn cluster already at n = 2, and the proton may be transferred from this 5HIH+ protomer to the Wn solvent cluster. The PAs of s/a5HIH+(C3/C4) are in the same range as that of the W3 cluster. Yet, proton transfer may still occur if the solvation energy of the proton-transferred structure is higher. Due to the possible proton transfer to the Wn moiety, the notation [5HI–Wn]H+ is more precise. However, for consistency, we keep the 5HIH+–Wn notation for all clusters throughout this paper. Based on our previous results,17 we calculate herein hydrated clusters of s/a5HIH+(C3), s/a5HIH+(C4), and s/a5HIH+(O) with W attached to the acidic functional OH(2) and NH groups, the π-electron cloud, or the protonated CH2 group. In general, the binding motifs are very similar for the syn and anti rotamers of the 5HIH+–Wn clusters, with s5HIH+–Wn being systematically more stable (Table S1, ESI†). Therefore, in the main text and figures, we concentrate on s5HIH+–Wn. Corresponding data for a5HIH+–Wn are available in the ESI.†Fig. 2 summarizes the calculated s5HIH+–W clusters relevant for the present study, along with their binding and relative energies (D0, E0) and intermolecular H-bond lengths (R). Additional geometric and spectroscopic properties are listed in Table S1 (ESI†). Further stable binding motifs of s5HIH+–W are shown in Fig. S2, and corresponding structures of a5HIH+–W are given in Fig. S3 (Table S1, ESI†).
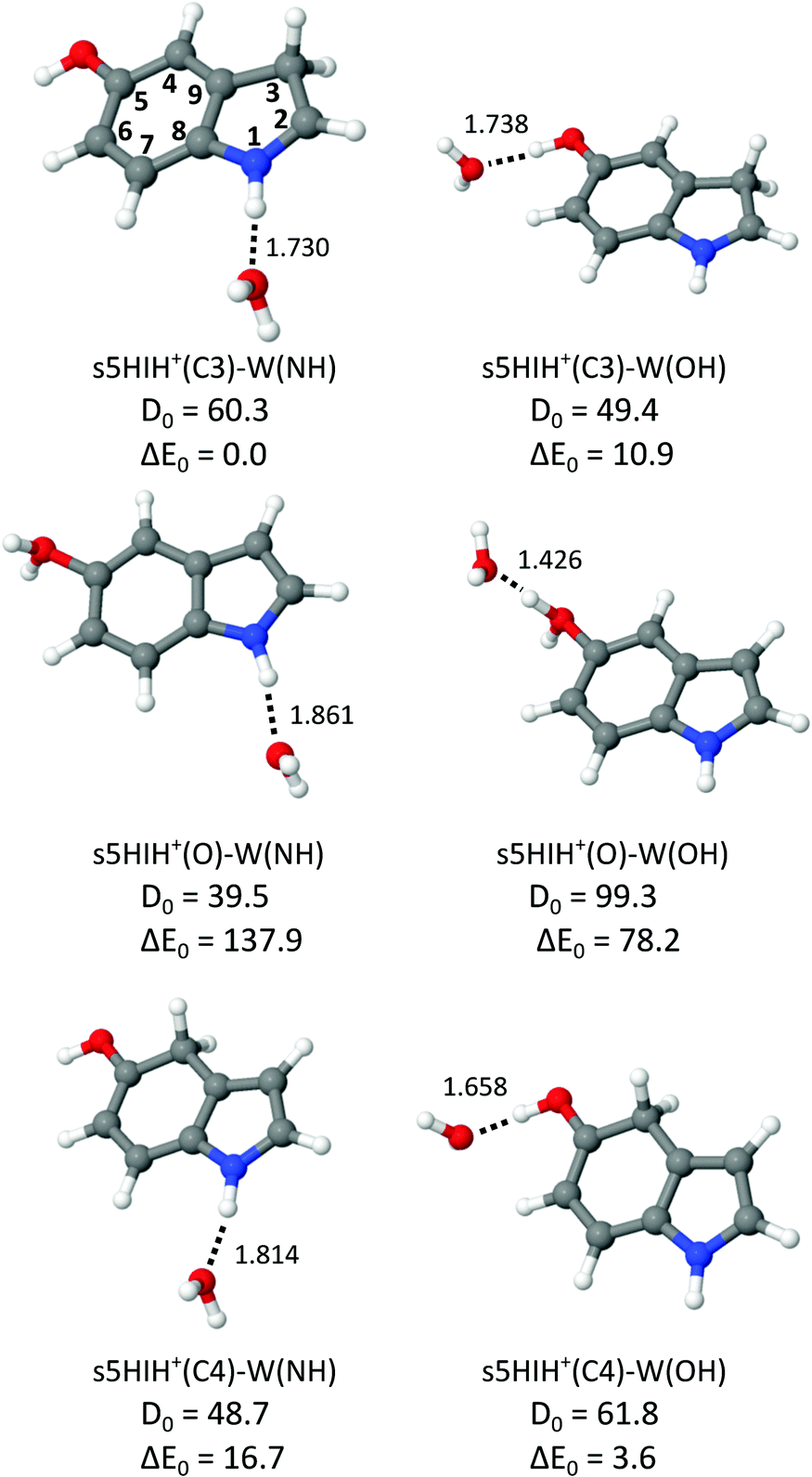 | ||
| Fig. 2 Structures of selected s5HIH+–W clusters calculated at the B3LYP-D3/aug-cc-pVTZ level (Table S1, ESI†) along with binding energies and relative energies (D0 and E0 in kJ mol−1) and intermolecular bond lengths (R in Å). | ||
All 5HIH+ protomers exhibit competing H-bonding sites, and most important are the acidic functional OH(2) and NH groups acting as proton donors in OH⋯O and NH⋯O H-bonds to W. As already observed for related heterocyclic arene cations,23,36,41,51–53 π-stacking of W is rather unfavorable, e.g., D0 = 33.3 kJ mol−1 for s5HIH+(C3)–W(π), and thus not considered further. Only one isomer with W attached to the protonated CH2 group could be located, namely, s5HIH+(C4)–W(CH) with D0 = 28.6 kJ mol−1. Because of the low binding energy of the CH⋯O H-bonds, this binding motif is also not considered further. Depending on the protonation site, the acidity of the OH and NH functional groups is changed,17 and the H-bond strengths vary in the same way. For example, the NH group is the strongest proton donor in s5HIH+(C3)–W(NH) (ΔE0 = 0 and D0 = 60.3 kJ mol−1, R = 1.730 Å for NH⋯O), while the OH group is preferred in s5HIH+(C4)–W(OH) (ΔE0 = 3.6 and D0 = 61.8 kJ mol−1, R = 1.658 Å for OH⋯O). The OH⋯O H-bond of the protonated OH2 oxonium group is outstandingly strong, with D0 = 99.3 kJ mol−1 and R = 1.426 Å for s5HIH+(O)–W(OH). This strong H-bond and the twofold degeneracy may thus again somewhat compensate for the large energy gap of ΔE0 = 78.2 kJ mol−1 between s5HIH+(O)–W(OH) and the s5HIH+(C3)–W(NH) global minimum. The NH group of the oxonium protomer is a far less acidic than the OH2 group, with D0 = 39.5 kJ mol−1 and R = 1.861 Å for the NH⋯O H-bond in s5HIH+(O)–W(NH). All considered s5HIH+–W isomers are readily distinguishable by their IR spectra in the investigated spectral range (Fig. 3 and Fig. S4, ESI†). Our calculated binding energies of s5HIH+(C3)–W(NH) and s5HIH+(C3)–W(OH), D0 = 60.3 and 49.4 kJ mol−1, are substantially but systematically smaller than the corresponding H-bond energies reported earlier at the RI-MP2/aug-cc-pVDZ level, De = 88.6 and 76.8 kJ mol−1.5 Similar to the MP2 results,5 the single-point energies computed at the CC2/aug-cc-pVDZ level yield the same energy hierarchy as predicted at the B3LYP-D3/aug-cc-pVTZ level, but the spread of relative energies is somewhat larger (Table S1, ESI†). In general, BSSE corrections of the computed binding energies evaluated at the B3LYP-D3/aug-cc-pVTZ level are small (on the order of 1% or less) because the aug-cc-pVTZ basis set is rather large (Table S1, ESI†). Reference calculations at the PBE0-D3/aug-cc-pVTZ level yield energies, structural and spectroscopic properties comparable to those obtained at the B3LYP-D3/aug-cc-pVTZ level (Table S1, ESI†). Indeed, the IR spectra computed at the B3LYP-D3/aug-cc-pVTZ level match somewhat better the experimental spectra (Fig. S5, ESI†).
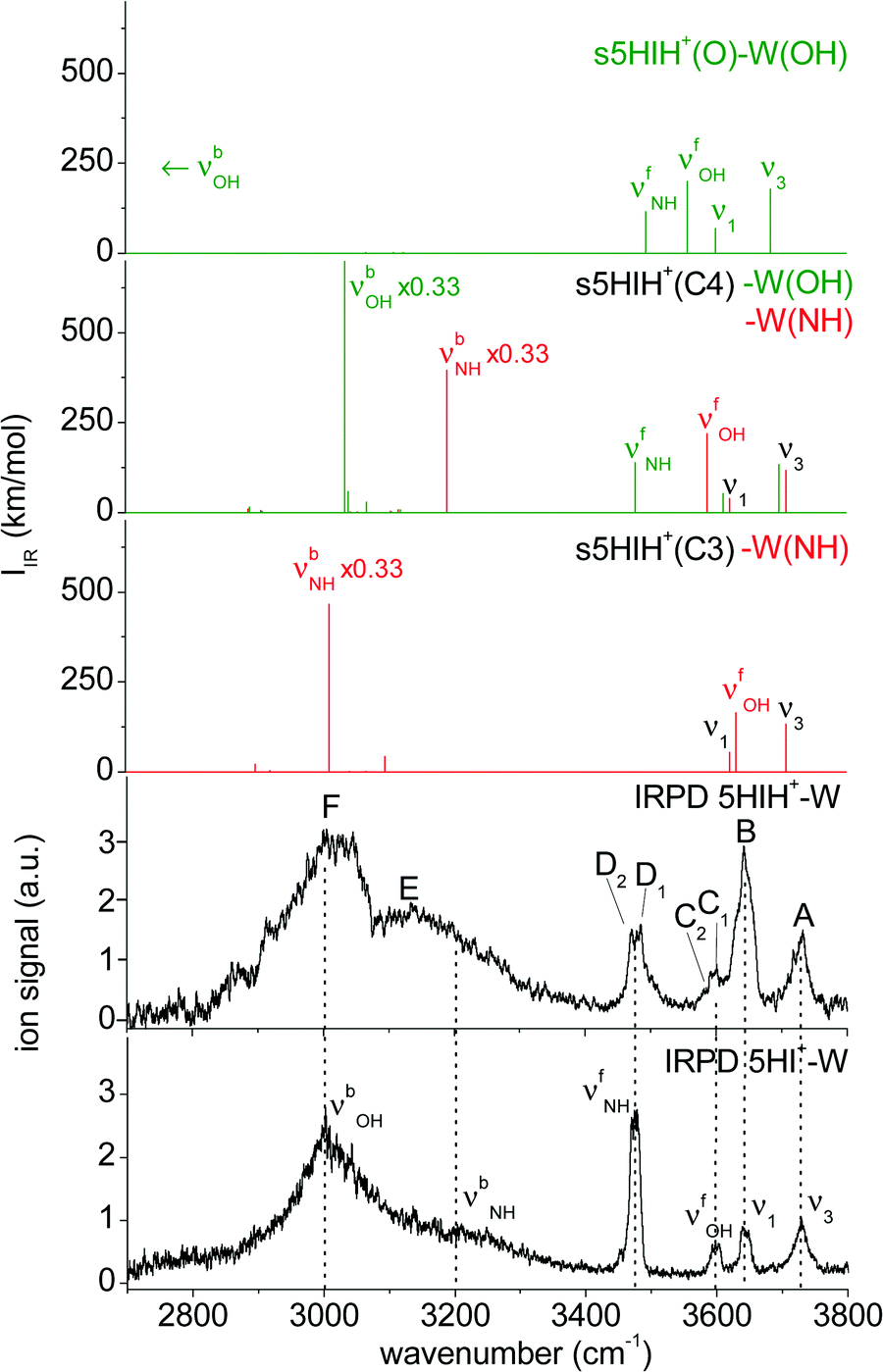 | ||
| Fig. 3 Comparison of the IRPD spectrum of 5HIH+–W to linear IR spectra calculated for the most stable isomers at the B3LYP-D3/aug-cc-pVTZ level. For comparison, the IRPD spectrum of 5HI+–W is also shown.23 | ||
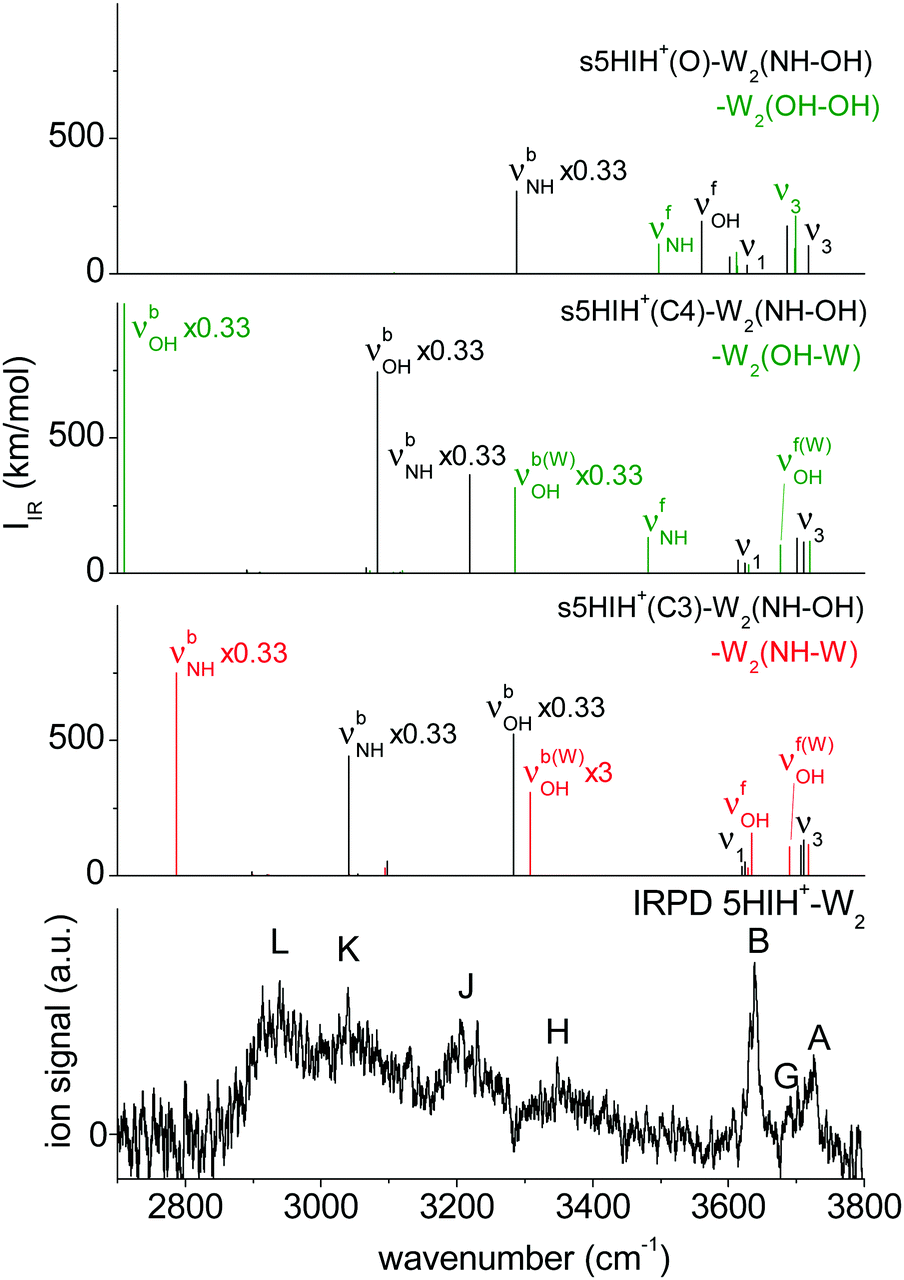
In Fig. 3 the IRPD spectrum of 5HIH+–W is compared to that of cationic 5HI+–W and to linear IR absorption spectra calculated for relevant s5HIH+–W isomers (Table 1). The vibrational assignment is facilitated by comparing the spectra of 5HIH+–W and 5HI+–W. Bands A and B at 3732 and 3641 cm−1 are readily assigned to the antisymmetric and symmetric free OH stretching modes of W (ν3 and ν1). Band A may also contain the red-shifted ν3 mode of s5HIH+(O)–W(OH) and s5HIH+(C4)–W(OH) predicted at 3685 and 3697 cm−1, respectively. The relative intensity of band B is substantially enhanced for 5HIH+–W as compared to 5HI+–W, while this is not the case for band A. Some contribution of the free phenolic OH stretching mode (νfOH) of s5HIH+(C3)–W(NH) predicted at 3632 cm−1 rationalizes the higher intensity of band B. Indeed, comparison of the IRPD spectra of 5HIH+–L with L = Ar, N2, and W further strengthens this assignment.17 The νfOH band of s5HIH+(C3)–Ar(π) is observed at 3635 cm−1, that is only 6 cm−1 red-shifted from the maximum of peak B. By comparison to 5HI+–W, bands C1/C2 are clearly assigned to phenolic νfOH modes. We attribute C1 at 3600 cm−1 to νfOH of s5HIH+(C4)–W(NH) predicted at 3588 cm−1, and C2 at 3591 cm−1 to νfOH of s5HIH+(O)–W(OH) predicted at 3558 cm−1. Another indication for the production of s5HIH+(O)–W(OH) is the doublet D1/D2 at 3485/3470 cm−1 (Δν = 15 cm−1). By analogy to 5HI+–W, it is assigned to the free NH stretching mode (νfNH). Here, D1 is attributed to s5HIH+(O)–W(OH) and D2 to s5HIH+(C4)–W(OH) predicted at 3494 and 3478 cm−1 (Δν = 16 cm−1), respectively. The broad band E centered at 3130 cm−1 is interpreted as H-bonded NH stretch (νbNH) of s5HIH+(C4)–W(NH) predicted at 3191 cm−1. Band E is somewhat red-shifted compared to νbNH in the IRPD spectrum of cationic 5HI+–W, indicating a destabilization of the NH bond induced by C4-protonation. The broad transition F at 3015 cm−1 contains both νbOH of s5HIH+(C4)–W(OH) and νbNH of s5HIH+(C3)–W(NH) predicted at 3035 and 3011 cm−1. The IRPD spectrum shows no clear signature of the s5HIH+(C3)–W(OH) isomer (Fig. S4, ESI†). This finding is somewhat surprising because this isomer is predicted to be slightly more stable than s5HIH+(C4)–W(NH) (ΔE0 = 10.9 and D0 = 49.4 kJ mol−1vs. ΔE0 = 16.7 and D0 = 48.7 kJ mol−1). We also rule out the presence of the s5HIH+(O)–W(NH) isomer, mostly on the basis of stability and spectroscopy (Fig. S4, ESI†).
In conclusion, we assign the measured IRPD spectrum of 5HIH+–W to C3-protonated s5HIH+(C3)–W(NH) with NH-bonded W, C4-protonated s5HIH+(C4)–W(OH) and s5HIH+(C4)–W(NH) with OH- and NH-bonded W, and the OH-bonded s5HIH+(O)–W(OH) oxonium ion. This interpretation is strengthened by a comparison of the IRPD spectrum to exemplary anharmonic spectra computed for the monohydrates (Fig. S6 and Table S1, ESI†). Obviously, within the harmonic approximation, the red-shifts of the H-bond donor stretching vibrations (νbOH and νbNH) are underestimated by 60–110 cm−1, but both the anharmonic and scaled harmonic spectra yield the same assignment of the experimental transitions. At the current experimental resolution, we cannot discriminate syn and anti rotamers but assume that both are present in significant abundance for the assigned isomers. The estimation of population ratios for the three protomers (C3, C4, O) based on the comparison of measured and computed band intensities is not straightforward due to overlapping transitions. Furthermore, the binding energies of the 5HIH+–W clusters are higher than the IR photon energies in the XH stretch range, such that absorption of a single photon will not lead to fragmentation from cold clusters. Hence, multiple-photon effects or ions with high internal energy must be considered. Moreover, due to the very different binding energies of the individual 5HIH+–W clusters, the photodissociation cross sections may be different, too. Still, assuming similar fragmentation cross sections for the assigned isomers, a rough estimate of populations is possible. In contrast to the predictions for s5HIH+(O)–W(OH), band C (C1 + C2, νfOH) is significantly less intense than D (D1 + D2, νfNH). This result suggests that we probe only a few oxonium ions (∼10%), while the main contribution to band D arises from s5HIH+(C4)–W(OH). The comparison of bands C and D also indicates that significantly fewer s5HIH+(C4)–W(NH) than s5HIH+(C4)–W(OH) isomers are probed. Band B is roughly twice as intense as band A, while ν1 is predicted to be half as intense as ν3 in any of the calculated IR spectra. Thus, we conclude that most signals of band B stem from νfOH of s5HIH+(C3)–W(NH). Hence, we suggest that the major contribution (roughly 70%) to the IRPD spectrum arises from the most stable s5HIH+(C3)–W(NH) and s5HIH+(C4)–W(OH) isomers.
3.3 5HIH+–W–Ar/N2
Tagging with Ar or N2 reduces the peak widths in the IRPD spectra of 5HIH+–W because the binding energy of the least bonded ligand limits the internal temperature of the cluster. Fig. S7 and S8 (ESI†) compare the IRPD spectra of 5HIH+–W–Ar and 5HIH+–W–N2 to the IR spectra calculated for relevant tagged isomers. The IRPD spectra of 5HIH+–W–Ar/N2 do not add any new information about the monohydrated clusters but confirm the assignment of s5HIH+(C3)–W(NH), s5HIH+(C4)–W(OH), s5HIH+(C4)–W(NH) and s5HIH+(O)–W(OH) being the predominant monohydrates in the molecular beam. The Ar tag is mainly attached to the W moiety or π-stacked. The N2 ligand is H-bonded to either W or to the remaining functional group not occupied by W. Selected structures of s5HIH+–W–Ar and s5HIH+–W–N2 are depicted in Fig. S9 and S10 (ESI†), respectively, and Table S1 (ESI†) contains structural and spectroscopic information for all calculated 5HIH+–W–Ar/N2 isomers. For a detailed discussion of the 5HIH+–W–L spectra, the interested reader is referred to the ESI.†3.4 5HIH+–W2
Fig. 4 shows the structures of selected s5HIH+–W2 clusters along with binding energies and H-bond lengths. Additional stable s5HIH+–W2 isomers are shown in Fig. S11 (ESI†). Corresponding a5HIH+–W2 structures are depicted in Fig. S12 (ESI†). Selected structural and spectroscopic properties of all calculated s/a5HIH+–W2 isomers are listed in Table S1 (ESI†).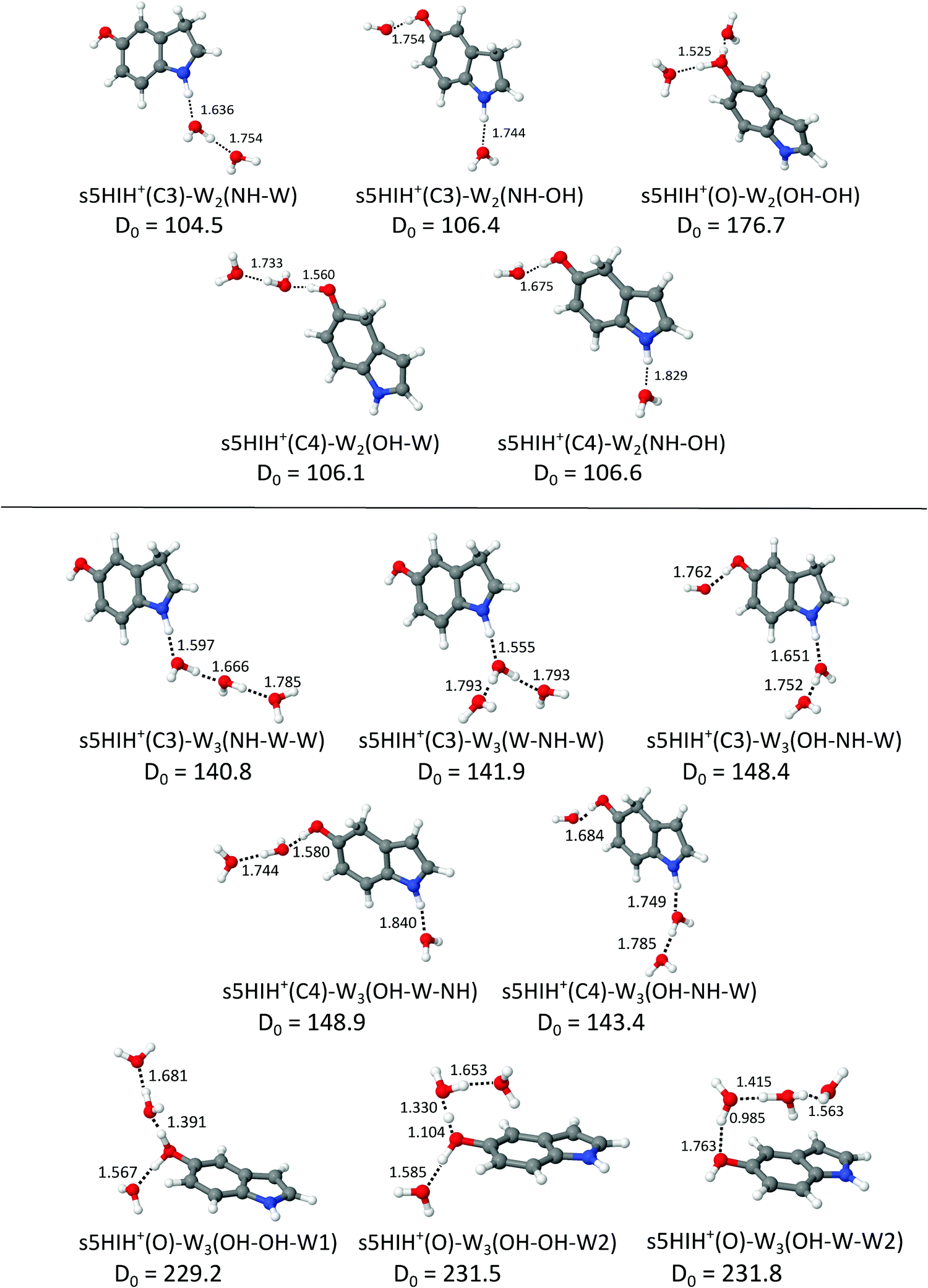 | ||
| Fig. 4 Structures of selected s5HIH+–W2/3 clusters calculated at the B3LYP-D3/aug-cc-pVTZ level (Table S1, ESI†) along with binding energies (D0 in kJ mol−1) and intermolecular distances (R in Å). | ||
For 5HIH+(C3/C4)–W2, we consider three binding motifs. H-bonding of two individual W ligands to both functional groups yields the most stable s5HIH+(C3/C4)–W2(NH–OH) with D0 = 106.4/106.6 kJ mol−1. BSSE corrections are again on the order of 1% of the computed binding energies (Table S1, ESI†). Noncooperative three-body effects weaken the individual H-bonds compared to the monohydrates, as revealed by comparison of binding energies and H-bond lengths. For example, corresponding H-bonds elongate from R = 1.730 and 1.738 Å in s5HIH+(C3)–W(NH) and s5HIH+(C3)–W(OH) to R = 1.744 and 1.754 Å in s5HIH+(C3)–W2(NH–OH). The same trend holds for s5HIH+(C4)–W2(NH–OH) with R = 1.814 and 1.658 Å compared to R = 1.829 and 1.675 Å. This weakening of the H-bonds directly translates into reduced complexation-induced red-shifts of the corresponding νbNH and νbOH modes (Table S1, ESI†). For example, νbNH of s5HIH+(C3)–W(NH) is predicted at 3011 cm−1 and that of s5HIH+(C3)–W2(NH–OH) at 3041 cm−1. For the C3-protonated ion, formation of a H-bonded water network (i.e., attachment of a H-bonded W2) at the NH group is more favorable than at the OH group, with D0 = 104.5 and 86.8 kJ mol−1 for s5HIH+(C3)–W2(NH–W) and s5HIH+(C3)–W2(OH–W), respectively. Due to cooperativity, the initial H-bond is shortened from R = 1.730 Å in s5HIH+(C3)–W(NH) to 1.636 Å in s5HIH+(C3)–W2(NH–W). In line with the altered acidity of the functional groups for the C4-protonated ion,17s5HIH+(C4)–W2(OH–W) is more stable than s5HIH+(C4)–W2(NH–W), with D0 = 106.1 and 87.6 kJ mol−1, respectively. Again, network formation strengthens the first H-bond with R = 1.658 Å in s5HIH+(C4)–W(OH) vs. 1.560 Å in s5HIH+(C4)–W2(OH–W). Formation of water networks yields characteristic spectroscopic features, most prominently, νf(W)OH and νb(W)OH of the solvated water in the W2 unit, predicted at 3690 and 3307 cm−1 for s5HIH+(C3)–W2(NH–W). The corresponding ν3 and ν1 of the terminal free water are blue-shifted from 3708 and 3622 cm−1 in s5HIH+(C3)–W(NH) to 3717 and 3628 cm−1 in s5HIH+(C3)–W2(NH–W). The impact on the respective functional group by solvation with W2 is largely enhanced. For example, νbOH of s5HIH+(C4)–W2(OH) is predicted at 3035 cm−1 and shifts down to 2710 cm−1 for s5HIH+(C4)–W2(OH–W).
The largest binding energy of D0 = 176.7 kJ mol−1 is calculated for s5HIH+(O)–W2(OH–OH), i.e. the oxonium ion monohydrated at both OH groups. Yet, due to noncooperativity, this value is significantly lower than twice the binding energy of s5HIH+(O)–W(OH), D0 = 99.3 kJ mol−1. Formation of a water network at the OH2 group yields s5HIH+(O)–W2(OH–W1) and s5HIH+(O)–W2(OH–W2) with D0 = 167.3 and 169.7 kJ mol−1 (Fig. S11, ESI†). The NH group of the oxonium ion is far less attractive, leading to s5HIH+(O)–W2(NH–OH) and s5HIH+(O)–W2(NH–W) with only D0 = 135.5 and 73.4 kJ mol−1. Again, BSSE corrections are on the order of only 1% of the computed binding energies (Table S1, ESI†). In s5HIH+(O)–W2(OH–W1), the water chain points away from the phenol ring. Proton transfer to the W2 chain is indicated, because the OH bond of s5HIH+(O) is already longer (rOH = 1.262 Å) than the OH⋯W bond (R = 1.147 Å). Thus, the ion may be better described by s5HI–H5O2+. Yet, for consistency, we keep the introduced notation for the structures. On the other hand, in s5HIH+(O)–W2(OH–W2) the water chain is bent toward the aromatic ring facilitating an additional OH⋯π interaction with the aromatic π-electron cloud. In this structure, proton transfer is more pronounced, with rOH = 1.434 and R = 1.056 Å.
The assignment of the IRPD spectrum of 5HIH+–W2 (Table 1) is based on the comparison to the corresponding calculated spectra (Fig. 5 and Fig. S13, ESI†). Exemplary anharmonic spectra depicted in Fig. S14 (ESI†) are in line with the following assignments based on the harmonic spectra. Bands A and B at 3725 and 3638 cm−1 are readily assigned to ν3 and ν1 and are well reproduced by essentially all considered isomers. Band G at 3692 cm−1 is characteristic of an H-bonded W2 network and appears at 3690 and 3676 cm−1 in the calculated spectra of s5HIH+(C3)–W2(NH–W) and s5HIH+(C4)–W2(OH–W), respectively. In line with the assignments above, νfOH of s5HIH+(C3)–W2(NH–W) predicted at 3635 cm−1 contributes to band B, which is again significantly more intense than band A. The νfNH mode characteristic of s5HIH+(C4)–W2(OH–W) at 3481 cm−1 is however not observed. Hence, we assign only s5HIH+(C3)–W2(NH–W). Consequently, band H centered at 3348 cm−1 is attributed to a superposition of νbOH of s5HIH+(C3)–W2(NH–OH) predicted at 3283 cm−1 and νbOH of the W2 unit in s5HIH+(C3)–W2(NH–W), which is predicted at 3308 cm−1 (νb(W)OH). Band J at 3205 cm−1 arises from νb(W)NH of s5HIH+(C4)–W2(NH–OH) predicted at 3218 cm−1. Its νbOH mode at 3083 cm−1 and νbNH of s5HIH+(C3)–W2(NH–OH) predicted at 3041 cm−1 account for band K observed at 3040 cm−1. Finally, band L at 2940 cm−1 may be assigned to νbNH of s5HIH+(C3)–W2(NH–W), which is predicted to be rather low (2787 cm−1). However, below 2800 cm−1, the laser power is very low such that this mode may also not be detected. We cannot safely assign any 5HIH+(O)–W2 oxonium isomer because there is no strong signal in the IRPD spectrum around 3500 cm−1. In this range, the νfNH and νfOH modes of s5HIH+(O)–W2(OH–OH) and s5HIH+(O)–W2(NH–OH) are predicted (3498 and 3560 cm−1, Fig. 5). Furthermore, the νfNH modes of the two proton-transferred structures s5HIH+(O)–W2(OH–W1) and s5HIH+(O)–W2(OH–W2) and their intense νbOH modes of the H-bonded W2 are expected around 3500 cm−1 (Fig. S13, ESI†). For the 5HIH+–W cluster, the population of the OH-bound oxonium isomer s5HIH+(O)–W(OH) was most likely enhanced by its high binding energy of D0 = 99.3 kJ mol−1 and the twofold degeneracy. However, degeneracy effects no longer promote the most stable s5HIH+(O)–W2(OH–OH) cluster or the formation of W2 chains in s5HIH+(O)–W2(OH–W1/W2). Thus, our spectra do not indicate proton transfer for any of the 5HIH+–Wn clusters at n = 2.
 | ||
| Fig. 5 Comparison of the IRPD spectrum of 5HIH+–W2 to calculated IR spectra of the relevant isomers at the B3LYP-D3/aug-cc-pVTZ level. | ||
3.5 5HIH+–W3
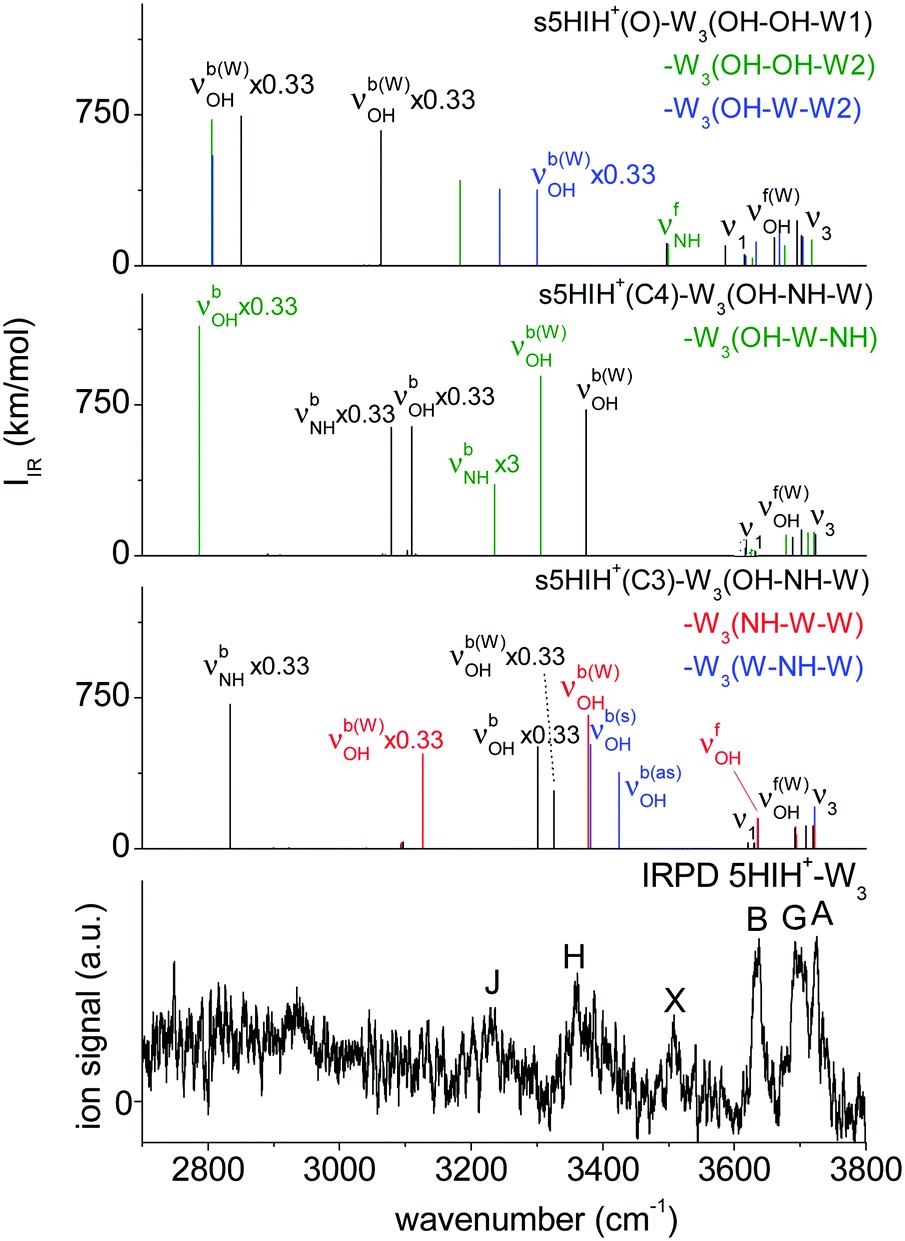
In s/a5HIH+–W3, interior ion solvation competes with water network formation. W3 chains at either one of the functional groups (NH–W–W and OH–W–W) are comparably strong as bifurcated W3 H-bonds (W–NH–W and W–OH–W). Selected structures of s5HIH+–W3 are shown in Fig. 4. Additional s5HIH+–W3 and selected a5HIH+–W3 isomers are depicted in Fig. S15 and S16 (ESI†). Corresponding structural and spectroscopic data are listed in Table S1 (ESI†). Again, we observe a strong modulation in the acidity of the functional groups depending on the protonation site. As the NH group is more acidic in s5HIH+(C3), formation of W3 networks at the NH site (D0 ≈ 141 kJ mol−1) is favored over the OH site (D0 ≈ 119 kJ mol−1). The bifurcated W–NH–W H-bond (D0 = 141.9 kJ mol−1) is slightly favored over the linear NH–W–W chain (D0 = 140.8 kJ mol−1). However, interior ion solvation with one water molecule attached to the OH group and two to the NH group yields the most stable s5HIH+(C3)–W3(OH–NH–W) isomer with D0 = 148.4 kJ mol−1. In contrast, W3 networks at the OH site are preferred for s5HIH+(C4) with D0 ≈ 142–145 kJ mol−1 (Table S1, ESI†). Solvation of both functional groups yields s5HIH+(C4)–W3(OH–W–NH) and s5HIH+(C4)–W3(OH–NH–W) with D0 = 148.9 and 143.4 kJ mol−1 (Fig. 4). Noncooperative effects destabilize the individual W2 chains. For example, comparing s5HIH+(C4)–W2(OH–W) and s5HIH+(C4)–W3(OH–W–NH), the H-bonds within the W2 chain at the OH group elongate from R = 1.560/1.733 to 1.580/1.744 Å. The same trend is observed for all four interior solvated s5HIH+–W3 isomers (Fig. 4 and Table S1, ESI†). In line with the weakening of the individual H-bonds, for n = 3 (interior solvation) the impact on the s5HIH+ core is also weaker than for n = 2 (water chain). This directly translates into blue-shifts of the affected proton donor XH stretches. For instance, νbNH of s5HIH+(C3)–W2(NH–W) and s5HIH+(C3)–W3(OH–NH–W) are predicted at 2787 and 2834 cm−1, respectively.Solvation of both OH groups of the oxonium ion yields s5HIH+(O)–W3(OH–OH–W1) and s5HIH+(O)–W3(OH–OH–W2) isomers with D0 = 229.2 and 231.5 kJ mol−1, respectively. The latter is again stabilized by its additional interaction with the aromatic π-electron cloud. A W2 chain is attached to one OH site, significantly elongating the affected OH bond (rOH = 1.068 and 1.104 Å). The OH⋯W bond is still longer (R = 1.391 and 1.330 Å). Hence, in contrast to what has been predicted for the corresponding s5HIH+(O)–W2(OH–W1) and s5HIH+(O)–W2(OH–W2) clusters, the proton is not transferred due to the noncooperative character of this W3 binding motif. However, proton-transferred structures cannot be neglected for n = 3. We find four proton-transferred structures, namely, s5HIH+(O)–W3(NH–OH–W1/W2) with a W2 chain at one OH group and a single W at the NH group as well as s5HIH+(O)–W3(OH–W–W1/W2) with a W3 chain at its OH group (Fig. 4 and Fig. S15, ESI†). Interestingly, s5HIH+(O)–W3(OH–W–W2) is the most stable structure found with D0 = 231.8 kJ mol−1. The W3 chain is entirely detached from the OH group (rOH = 1.763 and ROH–W = 0.985 Å), and is arranged over the phenol ring of neutral 5HI (Fig. 4). This isomer is distinguished by its νfOH and νfNH predicted at 3633 and 3497 cm−1, and three intense νb(W)OH at 3300, 3243, and 2806 cm−1. The other three structures are significantly less stable with D0 = 201.9, 208.8, and 216.5 kJ mol−1, respectively.
Fig. 6 compares the IRPD spectrum of 5HIH+–W3 to the calculated IR spectra of selected s5HIH+–W3 isomers. Spectra calculated for some additional s/a5HIH+–W3 conformers are given in Fig. S17 (ESI†). The triplet A, G, and B at 3725, 3692, and 3637 cm−1 unambiguously reveals the predominance of chainlike W2 and/or W3 solvation structures. Band A is thus assigned to ν3, band B to ν1, and band G to νf(W)OH of the single-donor water molecules. Candidates to explain this pattern are at least the isomers considered in Fig. 6, except for s5HIH+(C3)–W3(W–NH–W) with a bifurcated H-bond. However, a clear isomer assignment is challenging, because the IRPD spectrum is not well resolved below 3600 cm−1, possibly due to overlapping transitions of several isomers. Still, we can draw some conclusions from the comparison with the IRPD spectra of 5HIH+–W1/2 (Fig. 1). First, band X at 3508 cm−1 is considered to be a contamination band. It is not convincingly rationalized by any calculated mode and it occurs at the same position as band X in the spectrum of 5HIH+–W–N2 (ESI†). Yet, one may argue that νfNH of s5HIH+(O)–W3(NH–OH–W1/W2) predicted at 3497/3500 cm−1 gives rise to band X. However, already for 5HIH+–W2 it remains unclear whether oxonium clusters are probed. Any other modes of s5HIH+(O)–W3(NH–OH–W1/W2) are also not clearly observed. Second, bands H and J at 3360 and 3230 cm−1 can be related to corresponding transitions in the IRPD spectra of 5HIH+–W1/2. In analogy to the spectrum of 5HIH+–W2, band H may be assigned to νb(W)OH of the solvated water and/or νbOH of the 5HIH+(C3) protomer. Corresponding transitions are predicted at 3225 and 3301 cm−1 for s5HIH+(C3)–W3(OH–NH–W). In the case of 5HIH+–W2, band J is assigned to νbNH of the 5HIH+(C4) core. The corresponding intense νbNH mode of s5HIH+(C4)–W3(OH–W–NH) is predicted at 3236 cm−1. Bands J and H may also be explained by the two intense νb(W)OH of the very stable proton-transferred s5HIH+(O)–W3(OH–W–W2) oxonium isomer, which are predicted at 3300 and 3243 cm−1. Still, the IRPD spectrum of 5HIH+–W3 is already well reproduced by the two most stable carbenium ions s5HIH+(C3)–W3(OH–NH–W) and s5HIH+(C4)–W3(OH–W–NH). Finally, probably due to overlapping bands of several isomers, the spectral resolution is not sufficient to definitely exclude any of the isomers considered in Fig. 6 based on their IR spectra. A suggested, detailed assignment of the observed bands to vibrational modes and isomers is listed in Table 1. We most likely do not observe proton-transferred structures for n = 3, or at most only at low percentage. To unambiguously evidence whether proton transfer happens at n = 3 or not, the isomer contribution must be disentangled more accurately by double-resonance spectroscopy.
 | ||
| Fig. 6 Comparison of the IRPD spectrum of 5HIH+–W3 to calculated IR spectra of the relevant isomers at the B3LYP-D3/aug-cc-pVTZ level. | ||
3.6 Comparison to 5HIH+–Ln (L = Ar, N2)
Recently, we studied the sequential microsolvation of 5HIH+ with nonpolar (L = Ar) and quadrupolar (L = N2) solvents.17 The IRPD spectra of the 5HIH+–L clusters with L = Ar, N2, and W are compared in Fig. S18 (ESI†).The same protomers have been identified in the 5HIH+–Ln clusters, namely, C3, C4 and O. C4-Protonation significantly affects the adjacent OH group. As a result, s- and a5HIH+(C4) rotamers are distinguishable by their well-resolved νfOH bands appearing in the spectra of 5HIH+–Ar and 5HIH+–N2. In contrast, the current 5HIH+–Wn spectra are not sufficiently well resolved to distinguish s- and a5HIH+ rotamers. O-Protomers have unambiguously been identified by the IRPD spectra of larger 5HIH+–(N2)2/3 clusters recorded in different fragmentation channels.17 While the IRPD spectra measured in the n → 0 loss channel correspond to the superposition of all three protomers, 2 → 1 and 3 → 2 loss channels provide the isomer-pure spectrum of O-protonated 5HIH+(O)–(N2)2/3 clusters.
In general, the growth of 5HIH+–Ln clusters (L = Ar, N2, W) follows similar trends. Our previous study related the acidity of the functional groups to the charge distribution in the individual protomers.17 In 5HIH+(C3) the NH group is most acidic, whereas in 5HIH+(C4) it is the OH group. For the oxonium, exclusively H-bonding to its OH2 group is observed due to its high binding energies of D0 = 15.2 (Ar), 29.8 (N2), and 99.3 (W) kJ mol−1 compared to only D0 = 5.5 (Ar), 9.3 (N2), and 39.5 (W) kJ mol−1 for the NH-bound minimum. H-bonding and π-stacking compete in the growth of 5HIH+–Arn clusters. In contrast, the microsolvation of 5HIH+ with N2 is dominated by H-bonding to the functional groups instead of π-stacking, and the same is true for microhydration with W. The L–L interaction is rather small for both Ar and N2 (∼100 cm−1),54,55 because it relies mainly on dispersion. Hence, their 5HIH+–Ln clusters strongly prefer interior ion solvation over the formation of solvent networks. In contrast, the permanent dipole moment of W promotes the formation of water networks as the W–W interaction is rather strong (∼1000 cm−1).42,51,52 Upon network formation, strong cooperative effects strengthen preexisting H-bonds, in particular in the presence of a positive charge. Indeed, the formation of W2 and W3 chains is indicated by the characteristic triplet of ν3, νf(W)OH, and ν1 in the IRPD spectra of 5HIH+–W2/3. Complexation-induced frequency red-shifts (ΔνXH) of H-bonded proton donor stretching modes are a convenient measure of intermolecular H-bond strengths. Therefore, we evaluate the ΔνbOH and ΔνbNH red-shifts (Table 2) observed in 5HIH+–L dimers as a function of the ligand (L = Ar, N2, W). Fig. S19 (ESI†) illustrates the dependence of the ΔνbOH and ΔνbNH red-shifts on the PAs of the ligands.6 Generally, the impact on the proton donor group increases monotonically in the order Ar < N2 < W. The ΔνbOH red-shifts are larger than the ΔνbNH ones. However, one must be careful with their direct comparison because only for s/a5HIH+(C4) both ΔνbOH and ΔνbNH are observed (Table 2). Our previous IRPD study of 5HIH+–Ar/N2 already revealed an increase of the acidity of the OH group in the order s5HIH+(C3) < a5HIH+(C4) < s5HIH+(C4) < s/a5HIH+(O).17 Indeed, complexation has the largest impact on the OH group of s/a5HIH+(O)–L, which is the strongest H-bond donor with D0 = 15.2, 29.8, and 61.8 kJ mol−1 for Ar, N2, and W, respectively.
| Isomer | ν XH | ΔνXH | ||
|---|---|---|---|---|
| L = Ar | L = N2 | L = W | ||
| a Values correspond to Ne-tagged PhH+(O)–Ne(OH),20 which closely approximate those of bare PhH+(O). Complexation-induced red-shifts are calculated relative to PhH+(O)–Ne(OH). b Value corresponds to π-bonded PhH+(o/p)–Ar(π),20 which closely approximates that of bare PhH+(o/p). c Values calculated at the B3LYP-D3/aug-cc-pVTZ level. d Values taken from ref. 14. | ||||
| PhH+(O) | 3552 (X = O)a | −18a | −9a | +33d |
| 3477 (X = O)a | −148a | −440a | >−877d | |
| PhH+(o/p) | 3554 (X = O)b | −61 | −146 | −654 |
| a/s5HIH+(O) | 3555 (X = O)c | −20 | −15 | +36 |
| 3480 (X = O)c | −115 | −335 | −1304c | |
| 3503 (X = N) | Not observed | Not observed | Not observed | |
| s5HIH+(C3) | 3635 (X = O) | Not observed | Not observed | Not observed |
| 3405 (X = N) | −40 | −65 | −390 | |
| a5HIH+(C4) | 3598 (X = O) | −63 | −118 | −583 |
| 3478 (X = N) | −24 | −68 | −348 | |
| s5HIH+(C4) | 3584 (X = O) | −49 | −131 | −569 |
| 3478 (X = N) | −13 | −68 | −348 | |
3.7 Comparison to neutral 5HI–W and cationic 5HI+–Wn
A direct comparison of the (structural) properties of neutral 5HI–W and cationic 5HI+–W to the protonated 5HIH+–W clusters is challenging because protonation strongly affects the chemical structure. While only two isomers, namely, syn and anti rotamers, exist in the S0 and D0 states of 5HI(+), we observe (at least) six protomers for 5HIH+, namely, s/a5HIH+(C3), s/a5HIH+(C4), and s/a5HIH+(O).In the neutral ground state (S0), NH- and OH-bound hydrates of both s- and a5HI rotamers could be identified by isomer-selective IR spectroscopy.56 While neutral a5HI–W clusters are more stable by ΔE0 > 1.1 kJ mol−1, protonation reverses this trend, such that protonated s5HIH+–W are more stable by ΔE0 > 1.5 kJ mol−1. In the S0 state, OH⋯W bonds are stronger than NH⋯W bonds. Upon C3-protonation the neutral NH⋯W bond is strengthened from De = 40.2 to 88.6 kJ mol−1.5 The strengthening of the OH⋯W bond is less pronounced with De = 43.3 vs. 76.8 kJ mol−1.5 These results were rationalized by the large charge density on the pyrrole ring of 5HIH+(C3), which affects the acidity of the OH and NH groups. Our recent analysis of the NBO charge distribution in 5HIH+(C3), 5HIH+(C4), and 5HIH+(O) quantifies this qualitative argument.17
The IRPD spectrum of 5HI+–W is depicted in Fig. 3. Number and positions of the bands (A–F) are similar to those observed for 5HIH+–W, indicating comparable structures of the 5HI core and similar microhydration motifs (OH⋯W and NH⋯W H-bonds). However, band widths and intensities differ significantly. The spectrum of 5HI+–W was explained by the coexistence of 5HI+–W(OH) and 5HI+–W(NH).23 Also in the cationic ground state (D0), OH⋯W bonds are stronger than NH⋯W bonds, yet the interaction strengths are significantly enhanced compared to the neutral clusters. Hence, OH⋯W bonds are strongly preferred over NH⋯W bonds by a factor of 10.23 A very rough estimate of the population of the 5HIH+–W protomers yields that 5HIH+(C3)–W(NH) and 5HIH+(C4)–W(OH) contribute ≈70%, 5HIH+(C4)–W(NH) ≈20%, and 5HIH+(O)–W(OH) ≈10%. Thus, due to the strong variation of the acidity of the NH and OH groups upon protonation, there is no longer any preference for OH⋯W or NH⋯W bonds. In the D0 state, clusters of s5HI+ are more stable than those of a5HI+, and the same is true for the protonated species.
IRPD spectroscopy of 5HI+–Wn clusters reveals the competition between interior ion solvation and the formation of H-bonded water networks.23 For 5HI+–W2, interior ion solvation at both acidic groups (OH/NH) is strongly preferred (≥90%). For 5HI+–W3, two isomers coexist which bear one single W and one W2 dimer H-bonded to either of the functional groups. The IRPD spectrum of 5HI+–W3 suggests a strong preference for attachment of the W2 dimer at the OH group, leading to an estimated population ratio of 10:1 for OH/W/NH:OH/NH/W.23 Proton transfer from 5HI+–Wn to the solvent was not observed for n ≤ 3. These results are in line with our current findings on the 5HIH+–Wn clusters.
3.8 Comparison to PhH+–Wn
It is instructive to compare our results for 5HIH+–Wn to those obtained by IRPD spectroscopy of the related PhH+–Wn clusters because (i) similar protonation mechanisms have been evidenced for PhH+, resulting in carbenium PhH+(p/o) and oxonium PhH+(O) ions,14,18–20,24 and (ii) as a consequence, the microhydration is expected to be similar. In our previous study,17 we compared the protonation of 5HIH+ and PhH+ and their microsolvation by Ar and N2, revealing that the acidity of the OH group increases as s5HIH+(C3) < a5HIH+(C4) < s5HIH+(C4) < PhH+(p/o) < s5HIH+(O) < a5HIH+(O) < PhH+(O).17 The acidity of the functional group(s) correlates with the H-bond strength and is crucial for proton transfer.The IRPD spectra of PhH+–Wn with n ≤ 5 were interpreted by PhH+(p/o)–Wn and PhH+(O)–Wn clusters, for which proton transfer occurs at different critical sizes of the hydration shell (nc). In the case of PhH+(O)–Wn, the critical size is determined as nc = 3, and for PhH+(o/p)–Wn as nc = 4.14 Most likely, the transferred proton is the excess proton, coming from the OH2 group of PhH+(O) and the CH2 group of PhH+(p/o). However, for all PhH+–Wn exclusively the OH group is solvated. Hence, the Wn network has to bridge the OH group and the protonated CH2 group in PhH+(p/o) which is only possible for Wn≥4 chains. Our current IRPD spectra of 5HIH+–Wn do not indicate proton transfer at n ≤ 3 for any of the assigned protomers. This finding is interesting as we also observe hydrated oxonium ions, 5HIH+(O)–Wn. To elucidate the microsolvation mechanism, we compare the acidity of the OH groups of PhH+(o/p) and PhH+(O) to those of the observed 5HIH+(C3), 5HIH+(C4), and 5HIH+(O) clusters with the aid of the measured complexation-induced red-shifts of the OH stretch (ΔνOH) (Table 2). Most obviously, the acidity of the OH group of 5HIH+(C3) is the smallest, because its νOH has the highest measured frequency (3635 cm−1),17 and we do not observe OH⋯W bonds for 5HIH+(C3)–W. Preferentially, s/a5HIH+(C4) are solvated at the OH group, yet the observed ΔνOH shifts are of medium size (ΔνOH = −569/−583 cm−1). The OH⋯W H-bond in PhH+(o/p)–W is comparably strong with ΔνOH = −654 cm−1. Unfortunately, ΔνOH has not been measured for PhH+(O)–W,14 but is estimated to be larger than −877 cm−1. Hence, we determine the largest value of ΔνOH = −1304 cm−1 for a/s5HIH+(O)–W. Considering the red-shifts induced by attachment of Ar and N2 at the OH group of PhH+(O) and 5HIH+(O), we again infer a somewhat larger acidity of the OH group of PhH+(O).17 Hence, proton transfer may occur at nc ≥ 4 for 5HIH+(O)–Wn.
4. Conclusions
Herein, we investigate the initial microhydration of a prototypical protonated heteroaromatic biomolecule using IRPD spectroscopy of size-selected 5HIH+–Wn (W = H2O, n = 1–3) clusters in the XH stretching range and calculations at the B3LYP-D3/aug-cc-pVTZ level. Our results may be summarized as follows.We observe clusters of the C3- and C4-protonated carbenium ions, 5HIH+(C3) and 5HIH+(C4), and the oxonium ion, 5HIH+(O). Detection of 5HIH+(O)–Wn clusters is surprising at first glance because they are significantly less stable (ΔE0 > 75 kJ mol−1). However, the H-bonds to the protonated OH2 group are very strong (D0 > 99 kJ mol−1). In line with our previous results obtained for 5HIH+–L with L = Ar and N2,17 5HIH+(O)–Wn clusters benefit from the strong OH⋯W bond and the twofold degeneracy of the corresponding minimum. At the current spectral resolution, syn and anti rotamers (s- and a5HIH+) are not distinguishable. As the energy differences between their clusters are rather small (ΔE0 = 1–5 kJ mol−1), we assume the production of both s- and a5HIH+–Wn clusters.
5HIH+–Wn grow by H-bonding of the first W ligand to the acidic NH and OH groups, and π-stacking of W is unlikely. The absolute and relative strengths of the NH⋯W and OH⋯W H-bonds strongly depend on the 5HIH+ protomer. The strongest H-bond is found in 5HIH+(O)–W(OH) with W attached to one of its OH groups with an outstandingly high binding energy of D0 = 99.3 kJ mol−1. The acidity of the OH group (NH group) increases (decreases) in the order 5HIH+(C3) < 5HIH+(C4) < 5HIH+(O).17 Thus, we predominantly observe 5HIH+(C3)–W(NH), both 5HIH+(C4)–W(NH) and 5HIH+(C4)–W(OH), and 5HIH+(O)–W(OH) clusters. IRPD spectra of tagged 5HIH+–W–Ar/N2 clusters confirm this assignment. Interior ion solvation and formation of water networks compete for 5HIH+–W2. We assign carbenium clusters with both functional groups solvated, s5HIH+(C3)–W2(NH–OH) and s5HIH+(C4)–W2(NH–OH), and those with W2 water chains at their respective most acidic functional group, s5HIH+(C3)–W2(NH–W) and s5HIH+(C4)–W2(OH–W). The IRPD spectrum of 5HIH+–W2 does not clearly show features of any 5HIH+(O)–W2 clusters. The spectrum of 5HIH+–W3 clearly indicates W2 water chains at the NH and OH groups. It can readily be explained by the two most stable carbenium ions, s5HIH+(C3)–W3(OH–NH–W) and s5HIH+(C4)–W3(OH–W–NH). Isomer s5HIH+(C3)–W3(W–NH–W) with a bifurcated H-bond may also contribute to the measured spectrum. Again, we exclude the oxonium 5HIH+(O)–W3 clusters. Future IR-UV or IR-IR double resonance spectroscopy may facilitate disentangling the isomer contribution to the IRPD spectrum of 5HIH+–Wn.
Compared to the growth of 5HIH+–Ln clusters (L = Ar, N2), H-bonding is strongly preferred for L = W, and Wn networks compete with interior ion solvation, which is not the case for L = Ar/N2. The strength of individual H-bonds increases in the order Ar < N2 < W as shown by comparison of the respective complexation-induced frequency red-shifts (ΔνXH) of the corresponding H-bonded proton donor stretching modes.
Protonation significantly strengthens the OH⋯W and NH⋯W H-bonds observed in neutral 5HI–W due to the excess charge.5 The distribution of the excess positive charge is very different in the individual protomers directly affecting the acidity of the OH and NH groups. In the neutral S0 ground state, a5HI–W clusters are more stable than s5HI–W.56 In contrast, ionization into the D0 state and protonation reverse the relative stability of the rotamers.22,23 The mechanism of the growth of the initial solvation shell (n < 4) is very similar for cationic and protonated hydrates.
IRPD spectra of the PhH+ subunit of 5HIH+ revealed proton transferred at critical sizes nc = 3 and 4 in the case of PhH+(O)–Wn and PhH+(o/p)–Wn, respectively.14 In contrast, we do not observe any clear characteristics of proton-transferred structures in the 5HIH+–Wn clusters up to n = 3. Yet, we observe an increase in the acidity of the OH group in the order 5HIH+(C3) < 5HIH+(C4) < PhH+(p/o) < 5HIH+(O) < PhH+(O). Hence, proton transfer may occur at nc ≥ 4 for 5HIH+(O)–Wn.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by Deutsche Forschungsgemeinschaft (DO 729/3). J. K. is grateful for a fellowship of the Studienstiftung des deutschen Volkes.References
- A. T. Balaban, D. C. Oniciu and A. R. Katritzky, Chem. Rev., 2004, 104, 2777–2812 CrossRef CAS PubMed.
- J. M. Berg, J. L. Tymoczko and L. Stryer, Biochemistry, Freeman, New York, 2002 Search PubMed.
- J. P. Schermann, Spectroscopy and modeling of biomolecular building blocks, Elsevier, 2007 Search PubMed.
- H.-S. Andrei, N. Solcà and O. Dopfer, ChemPhysChem, 2006, 7, 107–110 CrossRef CAS PubMed.
- R. Omidyan, M. Omidyan and A. Mohammadzadeh, RSC Adv., 2016, 6, 33148–33158 RSC.
- E. P. L. Hunter and S. G. Lias, J. Phys. Chem., 1998, 27, 413–656 CAS.
- D. J. Goebbert and P. G. Wenthold, Eur. J. Mass Spectrom., 2004, 10, 837–845 CrossRef CAS PubMed.
- M. Miyazaki, A. Fujii, T. Ebata and N. Mikami, Chem. Phys. Lett., 2004, 399, 412–416 CrossRef CAS.
- E. S. Kryachko and M. T. Nguyen, J. Phys. Chem. A, 2001, 105, 153–155 CrossRef CAS.
- T. C. Cheng, B. Bandyopadhyay, J. D. Mosley and M. A. Duncan, J. Am. Chem. Soc., 2012, 134, 13046–13055 CrossRef CAS PubMed.
- I. Alata, M. Broquier, C. Dedonder-Lardeux, C. Jouvet, M. Kim, W. Y. Sohn, S. Kim, H. Kang, M. Schütz and A. Patzer, et al. , J. Chem. Phys., 2011, 134, 74307 CrossRef PubMed.
- O. Dopfer, A. Patzer, S. Chakraborty, I. Alata, R. Omidyan, M. Broquier, C. Dedonder and C. Jouvet, J. Chem. Phys., 2014, 140, 124314 CrossRef PubMed.
- O. Dopfer and M. Fujii, Chem. Rev., 2016, 116, 5432–5463 CrossRef CAS PubMed.
- M. Katada and A. Fujii, J. Phys. Chem. A, 2018, 122, 5822–5831 CrossRef CAS PubMed.
- K. Tanabe, M. Miyazaki, M. Schmies, A. Patzer, M. Schütz, H. Sekiya, M. Sakai, O. Dopfer and M. Fujii, Angew. Chem., Int. Ed., 2012, 51, 6604–6607 CrossRef PubMed.
- M. Ataelahi and R. Omidyan, J. Phys. Chem. A, 2013, 117, 12842–12850 CrossRef CAS PubMed.
- J. Klyne and O. Dopfer, J. Phys. Chem. B, 2018, 122, 10700–10713 CrossRef CAS PubMed.
- N. Solcà and O. Dopfer, Chem. Phys. Lett., 2001, 342, 191–199 CrossRef.
- N. Solcà and O. Dopfer, J. Chem. Phys., 2004, 120, 10470–10482 CrossRef PubMed.
- N. Solcà and O. Dopfer, J. Am. Chem. Soc., 2004, 126, 1716–1725 CrossRef PubMed.
- S. A. Nizkorodov, O. Dopfer, T. Ruchti, M. Meuwly, J. P. Maier and E. J. Bieske, J. Phys. Chem., 1995, 99, 17118–17129 CrossRef CAS.
- J. Klyne and O. Dopfer, J. Mol. Spectrosc., 2017, 337, 124–136 CrossRef CAS.
- J. Klyne, M. Miyazaki, M. Fujii and O. Dopfer, Phys. Chem. Chem. Phys., 2018, 20, 3092–3108 RSC.
- N. Solcà and O. Dopfer, J. Chem. Phys., 2004, 121, 769–772 CrossRef PubMed.
- O. Dopfer, Int. Rev. Phys. Chem., 2003, 22, 437–495 Search PubMed.
- O. Dopfer, Z. Phys. Chem., 2005, 219, 125–168 CrossRef CAS.
- M. J. Frisch, et al., GAUSSIAN09, Rev. D.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- J. Klyne, A. Bouchet, S. Ishiuchi, M. Fujii, M. Schneider, C. Baldauf and O. Dopfer, Phys. Chem. Chem. Phys., 2018, 20, 28452–28464 RSC.
- W. Fu and W. S. Hopkins, J. Phys. Chem. A, 2017, 122, 167–171 CrossRef PubMed.
- M. Rossi and C. Baldauf, J. Phys.: Condens. Matter, 2015, 27, 493002 CrossRef PubMed.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS.
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS.
- M. Miyazaki, A. Naito, T. Ikeda, J. Klyne, K. Sakota, H. Sekiya, O. Dopfer and M. Fujii, Phys. Chem. Chem. Phys., 2018, 20, 3079–3091 RSC.
- M. Schütz, Y. Matsumoto, A. Bouchet, M. Öztürk and O. Dopfer, Phys. Chem. Chem. Phys., 2017, 19, 3970–3986 RSC.
- Q. Zhang and L. Du, Comput. Theor. Chem., 2016, 1078, 123–128 CrossRef CAS.
- H. Zhao, J. Chang and L. Du, Comput. Theor. Chem., 2016, 1084, 126–132 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- M. Miyazaki, A. Fujii and N. Mikami, J. Phys. Chem. A, 2004, 108, 8269–8272 CrossRef CAS.
- K. Chatterjee and O. Dopfer, Phys. Chem. Chem. Phys., 2017, 19, 32262–32271 RSC.
- B. E. Rocher-Casterline, L. C. Ch'ng, A. K. Mollner and H. Reisler, J. Chem. Phys., 2011, 134, 211101 CrossRef PubMed.
- K. Chatterjee and O. Dopfer, Chem. Sci., 2018, 9, 2301–2318 RSC.
- C. Baldauf, K. Pagel, S. Warnke, G. von Helden, B. Koksch, V. Blum and M. Scheffler, Chem. – Eur. J., 2013, 19, 11224–11234 CrossRef CAS PubMed.
- V. Scutelnic, M. A. S. Perez, M. Marianski, S. Warnke, A. Gregor, U. Rothlisberger, M. T. Bowers, C. Baldauf, G. von Helden and T. R. Rizzo, et al. , J. Am. Chem. Soc., 2018, 140, 7554–7560 CrossRef CAS PubMed.
- M. Ropo, M. Schneider, C. Baldauf and V. Blum, Sci. Data, 2016, 3, 160009 CrossRef CAS PubMed.
- S. Simon, M. Duran and J. J. Dannenberg, J. Chem. Phys., 1996, 105, 11024–11031 CrossRef CAS.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
- G. Herzberg, Molecular spectra and molecular structure, Read Books Ltd, 2013 Search PubMed.
- V. Barone, J. Chem. Phys., 2004, 122, 14108 CrossRef PubMed.
- J. Klyne, M. Schmies, M. Miyazaki, M. Fujii and O. Dopfer, Phys. Chem. Chem. Phys., 2018, 20, 3148–3164 RSC.
- J. Klyne, M. Schmies, M. Fujii and O. Dopfer, J. Phys. Chem. B, 2015, 119, 1388–1406 CrossRef CAS PubMed.
- N. Solcà and O. Dopfer, Chem. Phys. Lett., 2001, 347, 59–64 CrossRef.
- O. Couronne and A. Ellinger, Chem. Phys. Lett., 1999, 306, 71–77 CrossRef CAS.
- E. A. Colbourn and A. E. Douglas, J. Chem. Phys., 1976, 65, 1741–1745 CrossRef CAS.
- T. Ikeda, K. Sakota and H. Sekiya, J. Phys. Chem. A, 2016, 120, 1825–1832 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cp06950f |
| This journal is © the Owner Societies 2019 |