
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The role of symmetric functionalisation on photoisomerisation of a UV commercial chemical filter†
Jack M.
Woolley
a,
Jack S.
Peters
a,
Matthew A. P.
Turner
ab,
Guy J.
Clarkson
a,
Michael D.
Horbury
*a and
Vasilios G.
Stavros
 *a
*a
aDepartment of Chemistry, University of Warwick, Gibbet Hill Road, Coventry, CV4 7AL, UK. E-mail: v.stavros@warwick.ac.uk; m.horbury@warwick.ac.uk
bDepartment of Physics, University of Warwick, Gibbet Hill Road, Coventry, CV4 7AL, UK
First published on 31st January 2019
Abstract
Photoisomerisation has been shown to be an efficient excited-state relaxation mechanism for a variety of nature-based and artificial-based molecular systems. Here we report on the excited-state relaxation dynamics and consequent photostability of a symmetrically functionalised cinnamate by transient electronic absorption spectroscopy, along with complementary computational and steady-state spectroscopy methods. The findings are then discussed in comparison to 2-ethylhexyl-E-4-methoxycinnamate, a structurally related ‘off the shelf’ chemical filter present in commercial sunscreens with a similar absorption profile. The present study allows for a like-for-like comparison beween 2-ethylhexyl-E-4-methoxycinnamate and the functionalised cinnamate, driven by the need to enhance solar protection across both the UVA and UVB regions of the electromagnetic spectrum.
Introduction
Overexposure to ultraviolet radiation (UVR) is an increasing concern in today's Society.1–5 Chemical filters, found in commercial formulations, have been designed to allay such overexposure.6 Chemical filters based on cinnamates7,8 are currently being used as a class of UVR absorbing molecules in commercial formulations due to their strong absorption of UVR and potent antioxidant properties.9–12 Investigations into their photochemistry has shown a photoisomerisation pathway as a main route of deactivation, irrespective of solvent environment,13–17 and starting geometric isomer (either E or Z);14,18 although a change in lifetimes is noted. One such cinnamate-derivative contained in sunscreen formulations is the FDA and EU approved 2-ethylhexyl-E-4-methoxycinnamate (E-EHMC).6,19 Studies have shown that, following UVR excitation to the first electronic excited (S1) state, E-EHMC undergoes ultrafast relaxation, repopulating the electronic ground (S0) state through an energetically accessible S1/S0 conical intersection, with a percentage forming Z-EHMC.20–23 The formation of Z-EHMC is cause for concern as recent studies suggest its adverse effects on human liver cells.24 These reports follow a growing trend of chemical filters with harmful photoproducts, both to humans and the environment, triggered by photoexcitation.24–27 As such, there is mounting impetus to develop ‘safer’ chemical filters for use in formulations, such as derivatives of commercial chemical filters.The present work takes our current knowledge on photoisomerisation in these cinnamate systems one step further. It assesses the influence of symmetric functionalisation of cinnamates through the addition of a second methyl acrylate moiety onto the base cinnamate, methyl cinnamate (highlighted in red, Fig. 1a). This gives rise to (potentially) three distinct geometric isomers: dimethyl 3,3′-(1,4-phenylene)(2E,2′E)-diacrylate, dimethyl 3,3′-(1,4-phenylene)(2Z,2′E)-diacrylate and dimethyl 3,3′-(1,4-phenylene)(2Z,2′Z)-diacrylate, henceforth termed E,E-DPD, Z,E-DPD and Z,Z-DPD respectively and all shown in Fig. 1a. Drawing inspiration from the recent gas-phase experiments reported by Kinoshita et al. on the influence of hydroxy substitution on the photochemistry of methyl hydroxycinnamates,28 we study the photochemistry of the geometric isomer E,E-DPD and track its possible evolution into the remaining geometric isomers (vide supra). In doing so we attempt to grasp a handle, on the influence of multiple geometric structures on dynamics, and how these may ultimately influence function.
 | ||
| Fig. 1 (a) The three geometric isomers for DPD, with the methyl cinnamate base unit highlighted in red. (b) Steady-state absorption spectra for E,E-DPD: at ∼10 μM concentration in ethanol (blue line) and acetonitrile (red line), and (owing to poorer solubility) <10 μM for E,E-DPD in cyclohexane (black). | ||
To facilitate these studies, we use a combination of synthesis, spectroscopy and theory. E,E-DPD was synthesised following a literature procedure.29 We subsequently used femtosecond transient electronic (UV/visible) absorption spectroscopy (TEAS) to probe (E,E-DPD) in three solvents of varying polarity and ability to hydrogen bond: cyclohexane, acetonitrile and ethanol. Complementary time dependent density functional theory (TD-DFT) was also carried out to elucidate the relative energies of the different geometric isomers and provide insight into their associated electronic structure, ultimately assisting in the assignment of the relaxation dynamics of these systems.
Experimental
Synthesis
DPD was prepared using a method adapted from the literature.29 Briefly, 5 drops of dimethylformamide (99.8% purity, Sigma-Aldrich) were added to a mixture of 1 g p-phenylenediacrylic acid (97% purity, Sigma-Aldrich) and 10 ml thionyl chloride (97% purity, Sigma-Aldrich) and was refluxed overnight. The mixture was allowed to cool and the excess of thionyl chloride was removed at reduced pressure to give a yellow powder. 50 ml of methanol (99% purity, Alfa-Aesar) was added gradually to the powder, which started to reflux due to the exothermic nature of the reaction. The mixture was refluxed for 4 hours and then the solid was isolated via filtration. The reaction mixture was analysed using thin layer chromatography (TLC) with dichloromethane (DCM, 99.8% purity, Sigma-Aldrich) as an eluent, which showed that the starting material had been consumed and that multiple new spots were visualised under UV irradiation. The mixture was recrystallized using a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 solution of DCM and petrol (Sigma-Aldrich), to form a light-yellow crystalline product. The resulting compound was analysed using 1H, 13C NMR along with X-ray crystallography (see ESI,† Fig. S1–S4).
:1 solution of DCM and petrol (Sigma-Aldrich), to form a light-yellow crystalline product. The resulting compound was analysed using 1H, 13C NMR along with X-ray crystallography (see ESI,† Fig. S1–S4).
Transient electronic absorption spectroscopy (TEAS)
TEAS measurements were executed within the Warwick Centre for Ultrafast Spectroscopy (WCUS; www.go.warwick.ac.uk/WCUS), the capabilities of which have been previously detailed and thus only summarised here.30 800 nm pulses (12 W, 1 kHz, 40 fs) were generated by a commercially available Ti-Sapphire regenerative amplified laser system (Spectra-Physics, Dual Ascend Pumped Spitfire Ace) seeded by a Mai Tai (Spectra-Physics). The beam was split into four fractions, each having an independent recompression grating. One of these beams (3.5 W) was used to produce the pump and probe pulses. The pump beam (2.5 W) seeds an optical parametric amplifier (Topas-Prime with UV extension, Light Conversion) allowing variability in pump wavelength. The wavelengths of excitation were: cyclohexane 315 nm, ethanol 318 nm, and acetonitrile 317 nm, corresponding to the peak maxima of E,E-DPD for each solvent as shown in Fig. 1. The pump beam was focussed beyond the sample holder, to give a beam diameter of 500 μm at the sample. 5% of the remaining 1 W of fundamental 800 nm was further attenuated and irised (<0.05 W), before being focussed into a vertically translated 2 mm thick CaF2 window to generate a white light supercontinuum from 330 to 680 nm. The relative polarisation between the pump and probe pulses was held at magic angle (54.7°) to negate dynamics from ensemble molecular reorientation. Pump–probe time-delays (Δt) were achieved out to the maximum temporal delay of 2.5 ns using a motorized optical delay line in the probe beam. Changes in optical density (ΔOD) were measured through the change in probe intensities using a fibre-coupled spectrometer (Avantes, AvaSpec-ULS1650F). Collated spectra were subsequently chirp corrected using the KOALA package.31 Samples were prepared in 1 mM concentrations and delivered to the interaction region through a demountable liquid cell (Harrick Scientific Products Inc.) with a sample thickness of 100 μm placed between two CaF2 windows. The sample was flowed through the cell by a diaphragm pump (SIMDOS 02) at a rate to ensure that fresh sample was interrogated with each laser pulse.Steady-state spectroscopy
Steady-state UV/Vis absorption measurements were taken on a Cary 60 spectrophotometer with sample concentrations of ∼10 μM. Steady-state irradiation (of varying time-delay) measurements of ∼10 μM samples (ethanol and acetonitrile solvents, >10 μM in cyclohexane) were also taken using a Horiba FluroLog-3, with an 8 nm slit width centred on the TEAS excitation wavelength and irradiation power equal to that of solar irradiance. Samples (each 10 mM, >10 mM in cyclohexane) for 1H NMR analysis, were irradiated for 1 h using the full available power of the Xenon lamp at the TEAS excitation wavelength using an 8 nm slit width. Samples were then dried of solvent and subsequently dissolved in deuterated chloroform for 1H NMR analysis. Fluorescence measurements were performed on the FluroLog-3, at excitation wavelengths matching those for the TEAS measurements. A 2.5 nm slit width was applied for both excitation and emission measurements. A quartz cuvette with a 1 cm path length was used for UV/Vis, fluorescence, steady-state irradiation and 1H NMR irradiation measurements.Computational
The structures of DPD were generated for all three isomers (E,E, E,Z, Z,Z), using visual molecular dynamics,32 with the molefacture plugin. Each of these structures underwent a geometry optimization within density functional theory using a cc-pVTZ basis set and PBE0 functional,33,34 using the NWChem software.35 Following this, time-dependant density functional theory calculations were conducted on the optimized geometries of the E,E, E,Z and Z,Z forms. Again, the level of theory was cc-pVTZ/PBE0 and the NWChem software was employed, generating the vertical excitation energies along with the oscillator strengths for each transition. For all calculations, a conductor-like screening model (COSMO) was used to replicate the effect of the solvent.36,37 The default COSMO implicit solvent model for each environment within NWChem was used, the descriptors of which are based on the Minnesota Solvent Descriptor Database.38 Computational output files, for the discussed calculations are available at: DOI: 10.5281/zenodo.1305150.Results
The dominant product of the synthesis was found to be E,E-DPD, determined through X-ray crystallography, which showed half the molecule in the asymmetric unit with an inversion centre in the middle of the phenyl ring, and E stereochemistry at each double bond.391H NMR spectra also confirms the E,E-isomer as the major reaction product (see ESI,† Fig. S1). Computed structures predict that the lowest energy structure is the E,E-isomer, showing good agreement with the experimental structures (see ESI,† Fig. S5). Table 1 shows the calculated energy difference between the three isomers, along with the first singlet vertical excitations of each isomer in each solvent.| Solvent | Isomer | E,E | E,Z | Z,Z |
|---|---|---|---|---|
| Acetonitrile | Ground state difference relative to E,E-DPD (eV) | 0 | 0.27 | 0.54 |
| Vertical excitation energies (eV, nm) | 3.79, 327 | 3.80, 326 | 3.81, 325 | |
| Ethanol | Ground state difference relative to E,E-DPD (eV) | 0 | 0.27 | 0.55 |
| Vertical excitation energies (eV, nm) | 3.76, 330 | 3.80, 326 | 3.81, 325 | |
| Cyclohexane | Ground state difference relative to E,E-DPD (eV) | 0 | 0.35 | 0.58 |
| Vertical excitation energies (eV, nm) | 3.80, 326 | 3.81, 325 | 3.81, 325 |
Fig. 2 shows the transient absorption spectra (TAS) resulting from photoexcitation of E,E-DPD, at the absorption maximum: 317 nm (3.91 eV) in acetonitrile and 318 nm (3.90 eV) in ethanol (cf.Fig. 1). We note the good agreement between the calculated vertical and experimentally measured excitation energies for E,E-DPD with differences being between 10–12 nm across all solvents. From the TAS presented as false colour heat maps in Fig. 2a and b, two main features are clearly visible: a stimulated emission centred at 375 nm and a strong excited state absorption centred at 525 nm. We add here that in Fig. 2b, we see evidence of a doublet around 350 nm, due to the ground state bleach and stimulated emission being spectrally offset in ethanol (cf. acetonitrile this is a single feature). This is also evidenced in the evolutionary associated difference spectra (EADS) presented in Fig. 2c and d (discussed below). The excited state absorption and stimulated emission features return to baseline within 50 ps leaving a small excited state absorption (not immediately apparent, highlighted in Fig. 2e and f) centred around 410 nm, which persists to the maximum temporal delay in our measurements (Δt = 2.5 ns). Fig. 2e and f also show the presence of a ground state bleach centred at 340 nm. We discuss this, as well as the aforementioned excited state absorption, below. We note that further studies were carried out in cyclohexane and additional excitation wavelengths, showing similar spectral features. This data is presented in the ESI,† Fig. S7–S9.
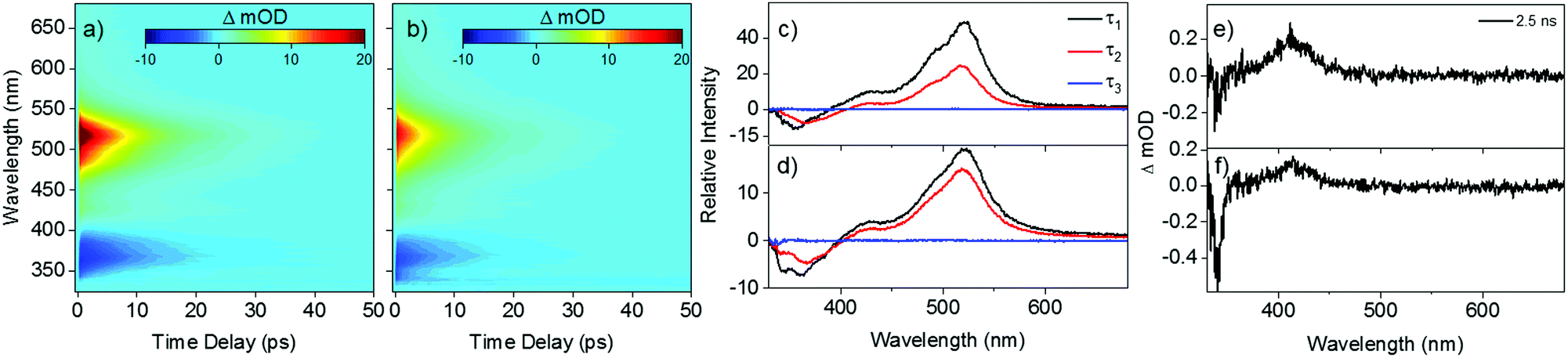 | ||
| Fig. 2 False colour heat maps of E,E-DPD following photoexcitation at (a) 317 nm in acetonitrile and (b) 318 nm in ethanol. Evolutionary associated difference spectra (EADS) for the TAS of E,E-DPD in (c) acetonitrile and (d) ethanol. TAS at Δt = 2.5 ns of E,E-DPD in (e) acetonitrile and (f) ethanol. Note the intensity scale in (a) is cropped as a visual aid to the dynamical features beyond the first 200 fs. An expanded intensity scale of (a) is presented in the ESI,† Fig. S6. | ||
To quantitatively assign lifetimes to the reported TAS features, a global fit across all wavelengths (330–680 nm), using a sequential model 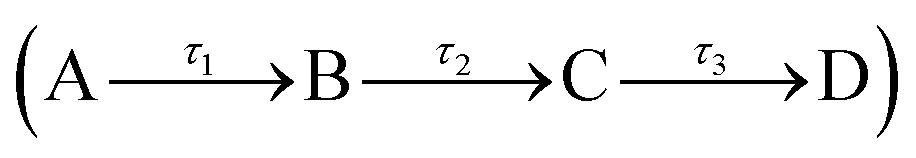 , was performed using the software package: Glotaran.40,41 The TAS were fitted with a three-step sequential model, with three lifetimes returned; these incorporate a convolution with a Gaussian to model our instrument response (FWHM = 80 fs). The extracted lifetimes are shown in Table 2, along with the associated errors. Transient slices are also provided in the ESI,† (Fig. S10 and S11) to illustrate the quality of the fit using this model. We note that where the error returned was less than 50% of our instrument response, we elected to quote an error of 40 fs (i.e. 50% of our instrument response). The corresponding EADS are also shown in Fig. 2c (acetonitrile) and d (ethanol). The associated residuals for each TAS (difference between experimentally measured and calculated TAS) are provided in the ESI,† (Fig. S12 and S13).
, was performed using the software package: Glotaran.40,41 The TAS were fitted with a three-step sequential model, with three lifetimes returned; these incorporate a convolution with a Gaussian to model our instrument response (FWHM = 80 fs). The extracted lifetimes are shown in Table 2, along with the associated errors. Transient slices are also provided in the ESI,† (Fig. S10 and S11) to illustrate the quality of the fit using this model. We note that where the error returned was less than 50% of our instrument response, we elected to quote an error of 40 fs (i.e. 50% of our instrument response). The corresponding EADS are also shown in Fig. 2c (acetonitrile) and d (ethanol). The associated residuals for each TAS (difference between experimentally measured and calculated TAS) are provided in the ESI,† (Fig. S12 and S13).
Discussion
From the reported lifetimes given in Table 2 and associated EADS presented in Fig. 2c and d, we can now discuss the reported TAS, with reference to the schematic shown in Fig. 3. We note that we draw considerable insight from comparative studies (with associated lifetimes; also shown in Table 2) of E-EHMC.20 Photoexcitation to the S1 (1ππ*) state leads to population traversing out of the Franck–Condon region described by τ1. Notably τ1 for acetonitrile is significantly shorter than that of either ethanol or cyclohexane, as has previously been reported for similar systems.42,43 This also (likely) encompasses any geometry rearrangement of solute and surrounding solvent. As with similar systems, this geometry rearrangement is not thought to involve a change in the electronic state.16,44τ2 then describes the evolution of excited-state population, as it moves towards an S1/S0 conical intersection along the E → Z isomerisation coordinate and then funnels through this conical intersection. This latter step leads to effective repopulation of the ground (S0) state. Previous reports of photoisomerisation,7,15,18,20,42,43 have identified the presence of an absorption feature at long time delays, attributed to the formation of the corresponding geometric isomer; in the present case, this would correspond to E,Z-DPD (or Z,Z-DPD). This may be the case here, evidenced through the incomplete ground state bleach recovery of E,E-DPD (negative feature <350 nm; see Fig. 2e and f). However, the positive feature extending >400 nm points towards a long-lived excited state, which would also restrict ground state bleach recovery. We attribute this long-lived excited state to either an 1nπ* or triplet state. We draw confidence that this is due to an excited state absorption, given that neither E,Z-DPD or Z,Z-DPD isomer is likely to absorb beyond 400 nm (see Fig. 4). Both these features in the TAS, persist to Δt = 2.5 ns and are assigned a lifetime of τ3. We discuss these features and the origin of τ3 in further detail below.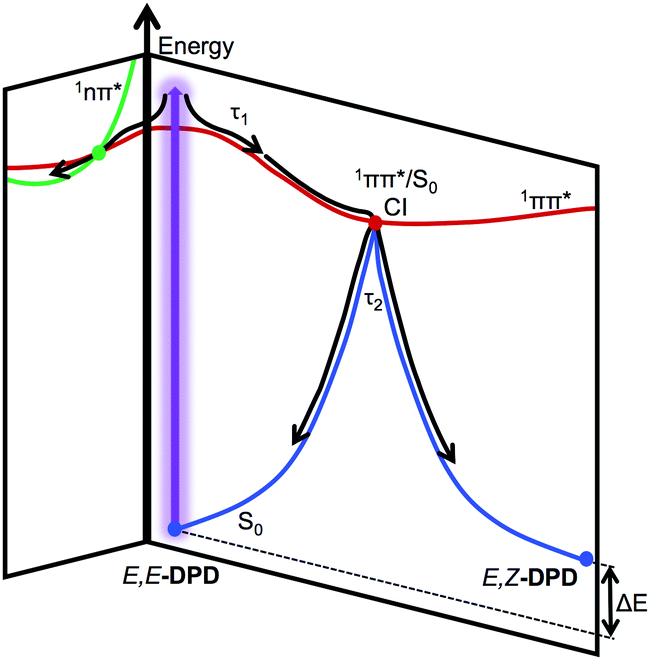 | ||
| Fig. 3 Schematic of relaxation for photoexcited E,E-DPD, showing the progression of τ1 and τ2. We add that τ3 (not shown) would correspond to population trapped in the E,Z-DPD and 1nπ* state. | ||
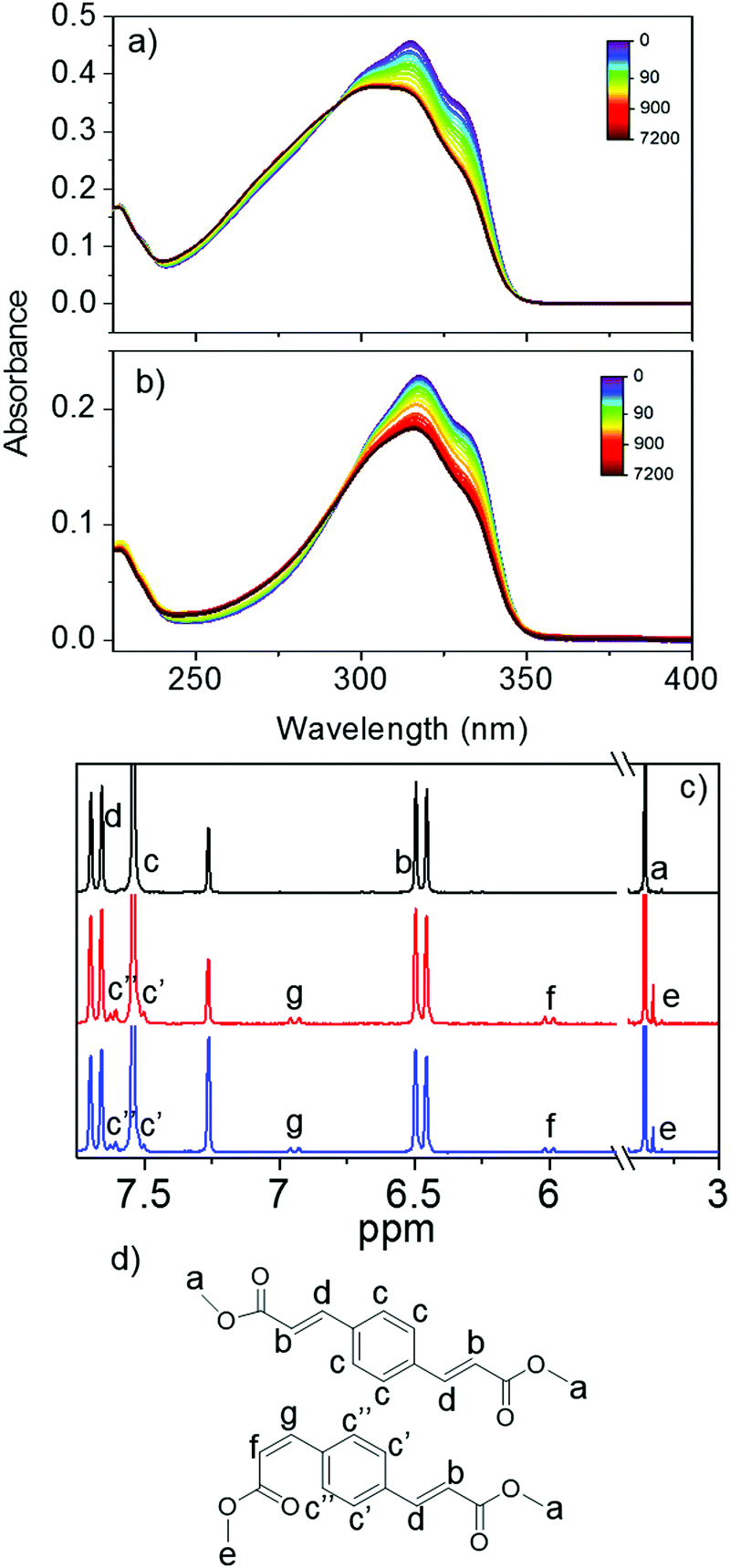 | ||
| Fig. 4 Steady-state UV/Vis absorption of 10 μM E,E-DPD in (a) acetonitrile and (b) ethanol, following irradiation at solar fluences over the peak absorption (λmax = 317 nm (acetonitrile) and 318 nm (ethanol)). The colour bar indicates length of irradiation in seconds. Selected region of (c) pre- (black) and post-irradiation 1H NMR of E,E-DPD irradiated at 10 mM concentration in acetonitrile and ethanol (red and blue respectively). (d) Peak assignment of chemical shifts in E,E and E,Z isomers. | ||
Steady-state irradiation at solar irradiance intensity was conducted to explore the long term photostability of E,E-DPD upon exposure to UVR. The results for both acetonitrile and ethanol are shown in Fig. 4; steady-state irradiation studies for E,E-DPD in cyclohexane are shown in the ESI.† All absorption spectra show a depletion of the initial main peak before plateauing following 20 minutes (1200 seconds) of irradiation. Accompanying this depletion is a small shift in the absorption to higher energy. The depletion of absorbance, is assigned to the generation of a photoequilibrium between E,E-DPD and E,Z-DPD; see Fig. 1 for associated structures. This depletion is consistent with the calculated oscillator strengths (S1 ← S0, at the calculated absorption maximum) between the two geometric isomers; e.g. 1.30 and 1.17 (arb. units) for E,E-DPD and E,Z-DPD respectively in acetonitrile. To further prove the existence of this photoequilibrium, and to exclude formation of the Z,Z-isomer, 1H NMR of the irradiated samples was performed. The samples were irradiated for 5 h using the full power of the Xenon lamp (approximately 30 mW) at the absorption maximum (λmax = 317 and 318 nm, for acetonitrile and ethanol respectively), to obtain sufficient conversion to the E,Z-DPD for 1H NMR identification; the sample concentration was also increased to 10 mM to facilitate this. The 1H NMR spectra in deuterated chloroform for the region of interest for both pre- and post-irradiation are shown in Fig. 4c.
From the analysis of the 1H NMR spectra, we can conclude that E → Z isomerisation occurs, with peaks f and g showing a 13 Hz splitting characteristic of the Z-isomer, along with the appearance of peak e indicating a change in environment of the terminating methyl. This is in contrast to peak b and d which have a splitting of 16 Hz (see ESI,† Fig. S3), attributed to E-isomer. One would anticipate a progression towards Z,Z-DPD to be a stepwise multiple-photon process, with the first conversion to E,Z-DPD then subsequently conversion to Z,Z-DPD. From the NMR we can deduce that no detectable presence of Z,Z-DPD is observed, confirmed through the splitting of the hydrogens on the aromatic ring (see signals c′ and c′′) indicating unsymmetrical substitution.
To conclude our discussion, we shall return to the nature of the long-lived features in the TAS (Fig. 2e and f) and the origin of τ3. It is evident from steady-state irradiation measurements that the geometric isomer E,Z-DPD has a weaker absorbance <350 nm than the E,E-DPD (cf.Fig. 4a and b, 0 and 7200 seconds irradiation). Given the TAS represents a difference spectrum (post-irradiated sample compared to pre-irradiated sample), it is unsurprising that we see a negative feature in the TAS (along with an incomplete, and overlapping, ground state bleach recovery) and is further evidenced, via1H NMR data, of isomerisation from E,E-DPD → E,Z-DPD. However, there is no absorption of either isomer at wavelengths greater than 350 nm, pointing to a different origin of the positive feature in the TAS >400 nm. This relatively small excited state absorption is likely due to either a 1nπ* or a triplet state. Several reports have suggested the presence of 1nπ* states playing a role within relaxation of similar systems.8,28,45 Fluorescence has also been reported for similar systems7,20,46 and was again observed here with the quantum yield reported as 0.039 ± 0.002 in cyclohexane (see ESI,† Fig. S17), hence a relatively minor pathway. Also, no phosphorescence was observed. The possibility of the absorption feature being due to a radical cation species, generated via a step-wise two-photon ionisation, was considered (as seen for similar species),9,17,42 but dismissed due to the linear power dependence of the feature (see ESI,† Fig. S19). Taken together, we assign τ3 to a combination of a long-lived 1nπ* state and presence of E,Z-DPD. We conclude our discussion of the long lived feature in the TAS by noting that a quantitative analysis of the conversion of E,E-DPD to E,Z-DPD is not possible from the TAS given the convoluted nature of the features both at early times (from the stimulated emission) and at Δt = 2.5 ns (from the presence of the 1nπ* state).
It is evident from the data thus far that E,E-DPD may possess suitable properties for applications as a chemical filter. Notably, its absorption spectrum is similar to that of E-EHMC,19,20 but with a slight red-shift (∼10 nm), meaning it extends further into the UVA which is a sought-after quality for a chemical filter. Coupled to this is the rapid relaxation of the excited state through an accessible conical intersection, consistent with the previous literature of similar species.7,42,43 Furthermore, the apparent degradation upon constant UVR is attributed to interconversion to the E,Z-DPD isomer. This finding is, once again, in good agreement with previous reports on E-EHMCversus Z-EHMC.20,23
Whilst there are similarities to E-EHMC, there are evident differences. Firstly, the apparent interconversion between the two isomers is longer for E,E-DPD in comparison with E-EHMC; e.g. in cyclohexane, τ2 ∼ 10 ps for E,E-DPDversus ∼2 ps for E-EHMC (see Table 2).20 This indicates that photoisomerisation is impinged by the different substituent in the para position. In this case, the addition of a second methyl acrylate alters the excited state dynamics by increasing the time for ground state repopulation (be it in E,Z-DPD (more likely) or E,E-DPD (less likely), Fig. 3). Whilst the underlying reason for this is not known, this inevitably points to alterations to the excited state landscape along the photoisomerisation coordinate. We add here that the increase in excited state lifetime could promote competing pathways; the ns component (τ3) of the E-EHMC can entirely be attributed to occurrence of the Z-isomer whilst for E,E-DPD, this appears to be a multicomponent, with the front runners being the E,Z-DPD isomer and a long-lived nπ* state. Additionally, compared to E-EHMC, the time taken to reach a photoequilibrium is increased to 20 minutes (cf. 2 minutes for E-EHMC).23 We finally note that no detectable spectroscopic evidence for Z,Z-DPD was observed in any experiments, leading to the hypothesis that a photoequilibrium is only established between E,E-DPD and E,Z-DPD.
Conclusions
This study highlights the effect of chemical substitution on an established photoisomerisation pathway. In this instance, we compare E,E-DPD with the commercial chemical filter E-EHMC. Following photoexcitation in solvents of varying polarity, the excited state population in E,E-DPD evolves out of the Franck–Condon region within 500 fs, along with geometry rearrangement of the solute and surrounding solvent, enabling access to an energetically accessible S1/S0 conical intersection. As the population transfers to the electronic ground state, the first difference between E,E-DPD and E-EHMC is in the relaxation times; ∼10 ps for E,E-DPD and ∼2 ps E-EHMC (both in cyclohexane). Ultimately this change is assigned to effects of the additional methyl acrylate on the excited state landscape. The second difference relates to the assignment of τ3. Here τ3 describes the presence of the corresponding E,Z-DPD isomer, along with a small percentage of the excited state population trapped in an 1nπ* (or triplet) state, which persists beyond the time-window of the present measurements (<ns). This ns component is absent in E-EHMC. Furthermore no evidence for Z,Z-DPD was observed, leading to the conclusion that its formation is unfavoured and the photoequilibrium is generated between E,E-DPD, and E,Z-DPD.As a final comparison, the steady-state absorption spectra of E,E-DPD (in solvents of varying polarity) are red-shifted relative to the absorption spectra of E-EHMC, allowing for stronger absorption within the UV-A region of the spectrum. This is a key quality, which is expected for new chemical filters, given the growing need for photoprotection across the UVB and UVA regions of the solar spectrum.1 Taken together the results shown here provide a potential blue-print into how a current chemical filter can be adapted and improved for use in future cosmetic products.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Dr Michael Staniforth at the Warwick Centre for Ultrafast Spectroscopy (WCUS; www.go.warwick.ac.uk/WCUS), for experimental aid. The authors would also like to thank Dr Nicholas Hine for valuable discussions regarding the theory. J. M. W. is grateful to EPSRC and Newport Spectra-Physics Ltd for a joint studentship. M. A. P. T. thanks EPSRC for a doctoral studentship through the EPSRC Centre for Doctoral Training in Molecular Analytical Science, grant number EP/L015307/1. M. D. H. thanks the Leverhulme Trust for postdoctoral funding. V. G. S. is grateful to the EPSRC for an equipment grant (EP/N010825) and the Royal Society and the Leverhulme Trust for a Royal Society Leverhulme Trust Senior Research Fellowship. Computing facilities were provided by the Scientific Computing Research Technology Platform of the University of Warwick.Notes and references
- R. P. Sinha and D.-P. Häder, Photochem. Photobiol. Sci., 2002, 1, 225–236 RSC.
- G. P. Pfeifer, Y. H. You and A. Besaratinia, Mutat. Res., Fundam. Mol. Mech. Mutagen., 2005, 571, 19–31 CrossRef CAS PubMed.
- F. R. de Gruijl, Eur. J. Cancer, 1999, 35, 2003–2009 CrossRef CAS PubMed.
- M. Brenner and V. J. Hearing, Photochem. Photobiol., 2008, 84, 539–549 CrossRef CAS PubMed.
- M. Krause, A. Klit, M. Blomberg Jensen, T. Søeborg, H. Frederiksen, M. Schlumpf, W. Lichtensteiger, N. E. Skakkebaek and K. T. Drzewiecki, Int. J. Androl., 2012, 35, 424–436 CrossRef CAS PubMed.
- N. A. Shaath, Photochem. Photobiol. Sci., 2010, 9, 464–469 RSC.
- M. D. Horbury, L. A. Baker, N. D. N. Rodrigues, W.-D. Quan and V. G. Stavros, Chem. Phys. Lett., 2017, 673, 62–67 CrossRef CAS.
- N. D. N. Rodrigues, M. Staniforth, J. D. Young, Y. Peperstraete, N. C. Cole-Filipiak, J. R. Gord, P. S. Walsh, D. M. Hewett, T. S. Zwier and V. G. Stavros, Faraday Discuss., 2016, 194, 709–729 RSC.
- S. Foley, S. Navaratnam, D. J. McGarvey, E. J. Land, T. G. Truscott and C. A. Rice-Evans, Free Radical Biol. Med., 1999, 26, 1202–1208 CrossRef CAS PubMed.
- A. Gaspar, M. Martins, P. Silva, E. M. Garrido, J. Garrido, O. Firuzi, R. Miri, L. Saso and F. Borges, J. Agric. Food Chem., 2010, 58, 11273–11280 CrossRef CAS PubMed.
- J. C. Murray, J. A. Burch, R. D. Streilein, M. A. Iannacchione, R. P. Hall and S. R. Pinnell, J. Am. Acad. Dermatol., 2008, 59, 418–425 CrossRef PubMed.
- K. Sevgi, B. Tepe and C. Sarikurkcu, Food Chem. Toxicol., 2015, 77, 12–21 CrossRef CAS PubMed.
- J. Luo, Y. Liu, S. Yang, A. L. Flourat, F. Allais and K. Han, J. Phys. Chem. Lett., 2017, 8, 1025–1030 CrossRef CAS PubMed.
- S. Wang, S. Schatz, M. C. Stuhldreier, H. Böhnke, J. Wiese, C. Schröder, T. Raeker, B. Hartke, J. K. Keppler, K. Schwarz, F. Renth and F. Temps, Phys. Chem. Chem. Phys., 2017, 19, 30683–30694 RSC.
- M. D. Horbury, W.-D. Quan, A. L. Flourat, F. Allais and V. G. Stavros, Phys. Chem. Chem. Phys., 2017, 19, 21127–21131 RSC.
- A. Espagne, D. H. Paik, P. Changenet-Barret, M. M. Martin and A. H. Zewail, ChemPhysChem, 2006, 7, 1717–1726 CrossRef CAS PubMed.
- M. Vengris, D. S. Larsen, M. A. van der Horst, O. F. A. Larsen, K. J. Hellingwerf and R. van Grondelle, J. Phys. Chem. B, 2005, 109, 4197–4208 CrossRef CAS PubMed.
- M. D. Horbury, A. L. Flourat, S. E. Greenough, F. Allais and V. G. Stavros, Chem. Commun., 2018, 54, 936–939 RSC.
- U. Osterwalder and B. Herzog, Photochem. Photobiol. Sci., 2010, 9, 470–481 RSC.
- Y. Peperstraete, M. Staniforth, L. A. Baker, N. D. N. Rodrigues, N. C. Cole-Filipiak, W.-D. Quan and V. G. Stavros, Phys. Chem. Chem. Phys., 2016, 18, 28140–28149 RSC.
- T. M. Karpkird, S. Wanichweacharungruang and B. Albinsson, Photochem. Photobiol. Sci., 2009, 8, 1455–1460 RSC.
- S. Pattanaargson, T. Munhapol, P. Hirunsupachot and P. Luangthongaram, J. Photochem. Photobiol., A, 2004, 161, 269–274 CrossRef CAS.
- K. M. Hanson, S. Narayanan, V. M. Nichols and C. J. Bardeen, Photochem. Photobiol. Sci., 2015, 14, 1607–1616 RSC.
- A. Sharma, K. Bányiová, P. Babica, N. El Yamani, A. R. Collins and P. Čupr, Sci. Total Environ., 2017, 593–594, 18–26 CrossRef CAS PubMed.
- N. Blüthgen, S. Zucchi and K. Fent, Toxicol. Appl. Pharmacol., 2012, 263, 184–194 CrossRef PubMed.
- M. Coronado, H. De Haro, X. Deng, M. A. Rempel, R. Lavado and D. Schlenk, Aquat. Toxicol., 2008, 90, 182–187 CrossRef CAS.
- M. Ghazipura, R. McGowan, A. Arslan and T. Hossain, Reprod. Toxicol., 2017, 73, 175–183 CrossRef CAS PubMed.
- S. Kinoshita, Y. Miyazaki, M. Sumida, Y. Onitsuka, H. Kohguchi, Y. Inokuchi, N. Akai, T. Shiraogawa, M. Ehara, K. Yamazaki, Y. Harabuchi, S. Maeda, T. Taketsugu and T. Ebata, Phys. Chem. Chem. Phys., 2018, 20, 17583–17598 RSC.
- H. Miura, S. Takahashi, H. Kasai, S. Okada, K. Yase, H. Oikawa and H. Nakanishi, Cryst. Growth Des., 2010, 10, 510–517 CrossRef CAS.
- J. M. Woolley, M. Staniforth, M. D. Horbury, G. W. Richings, M. Wills and V. G. Stavros, J. Phys. Chem. Lett., 2018, 9, 3043–3048 CrossRef CAS PubMed.
- M. P. Grubb, A. J. Orr-Ewing and M. N. R. Ashfold, Rev. Sci. Instrum., 2014, 85, 64104 CrossRef PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- M. Valiev, E. J. Bylaska, N. Govind, K. Kowalski, T. P. Straatsma, H. J. J. Van Dam, D. Wang, J. Nieplocha, E. Apra, T. L. Windus and W. A. D. Jong, Comput. Phys. Commun., 2010, 181, 1477–1489 CrossRef CAS.
- A. Klamt and G. Schüürmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799–805 RSC.
- D. M. York and M. Karplus, J. Phys. Chem. A, 1999, 103, 11060–11079 CrossRef CAS.
- P. Winget, D. M. Dolney, D. J. Giesen, C. J. Cramer and D. G. Truhlar, Minnesota solvent descriptor database, http://comp.chem.umn.edu/solvation/mnsddb.pdf.
- K. Ueno, H. Nakanishi and M. Hasegawa, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 1978, 34, 2034–2035 CrossRef.
- J. J. Snellenburg, S. P. Laptenok, R. Seger, K. M. Mullen and I. H. M. van Stokkum, J. Stat. Softw., 2012, 49, 1–22 Search PubMed.
- K. M. Mullen and I. H. M. van Stokkum, J. Stat. Softw., 2007, 18, 1–46 CrossRef.
- M. D. Horbury, L. A. Baker, W.-D. Quan, S. E. Greenough and V. G. Stavros, Phys. Chem. Chem. Phys., 2016, 18, 17691–17697 RSC.
- L. A. Baker, M. D. Horbury, S. E. Greenough, F. Allais, P. S. Walsh, S. Habershon and V. G. Stavros, J. Phys. Chem. Lett., 2016, 7, 56–61 CrossRef CAS PubMed.
- H. Kuramochi, S. Takeuchi and T. Tahara, J. Phys. Chem. Lett., 2012, 3, 2025–2029 CrossRef CAS.
- X. P. Chang, C. X. Li, B. Bin Xie and G. Cui, J. Phys. Chem. A, 2015, 119, 11488–11497 CrossRef CAS PubMed.
- M. Promkatkaew, S. Suramitr, T. Karpkird, S. Wanichwecharungruang, M. Ehara and S. Hannongbua, Photochem. Photobiol. Sci., 2014, 13, 583 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra, crystallographic data, computed structures, additional transient absorption, and steady-state spectra, fitting residuals, fluorescence quantum yields and power dependency measurements. See DOI: 10.1039/c8cp06536e |
| This journal is © the Owner Societies 2019 |