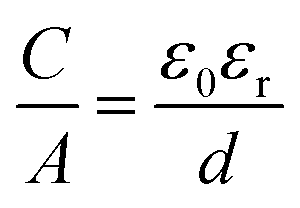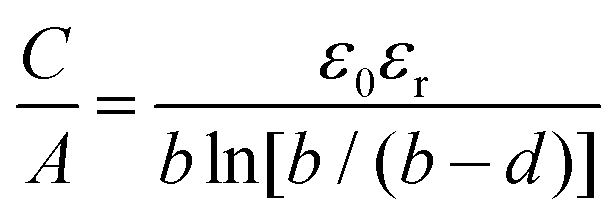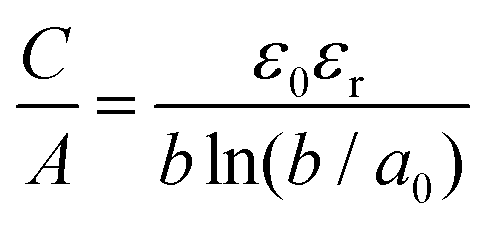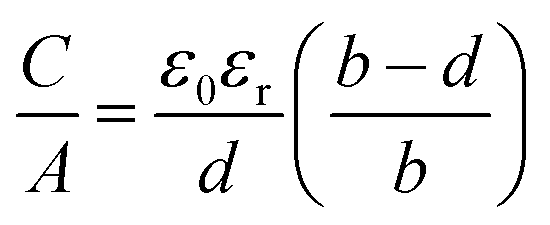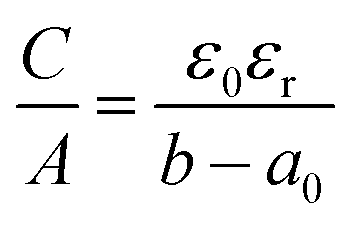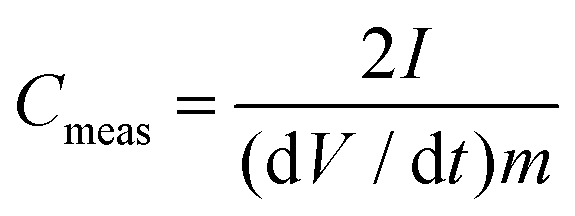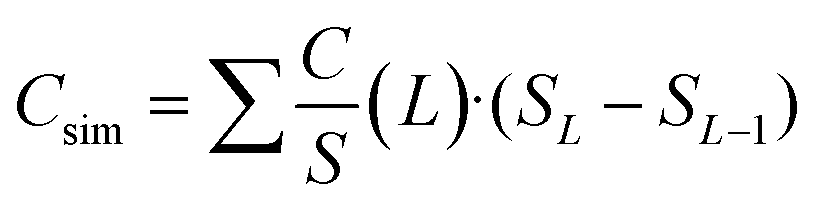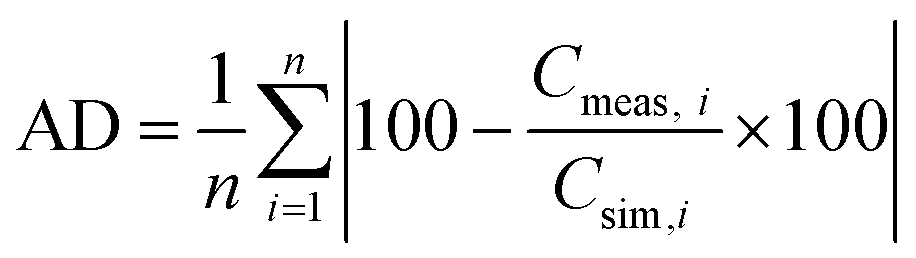Insights into the influence of the pore size and surface area of activated carbons on the energy storage of electric double layer capacitors with a new potentially universally applicable capacitor model†
Received
16th October 2018
, Accepted 15th January 2019
First published on 15th January 2019
Abstract
The electric double layer formation of supercapacitors is governed by ion electrosorption at the electrode surface. Large surface areas are beneficial for the energy storage process, typically achieved by carbon electrode materials. It is a matter of debate whether pores provide the same contribution to the capacitance regardless of the size, or if subnanometer pores lead to an anomalous increase of capacitance. In our work, we developed a new model for normalized capacitance depending on pore sizes, using a combination of a sandwich type capacitor for micropores and double-cylinder capacitor model for larger pores. Modification factors for each capacitance value were calculated using the nonlinear generalized reduced gradient method to obtain a modified electric sandwich double-cylinder capacitor (ESDCC) model. The model was validated by comparing the measured capacitance values of a set of prepared activated carbons in organic electrolytes with simulated values according to the modified ESDCC model, using combined physisorption data of carbon dioxide and nitrogen. We concluded a non-constant capacitive contribution, with pores having the size of bare cations contributing to the capacitance to a larger extent and mesopores with the size of three solvated ions providing an unusual low contribution to the overall capacitance.
1. Introduction
Electrochemical double layer capacitors (EDLCs), which belong to the supercapacitors, are emerging energy storage devices that offer the benefits of high power density, long cycle life, rapid charging rates and moderate energy density.1–4 Supercapacitors can be divided into pseudocapacitors and asymmetric hybrid capacitors, combining both double layer and pseudocapacitive energy storage mechanisms. Here, we will focus on symmetric EDLCs, which store and release energy by physical ion adsorption and desorption on the electrode surface in the electric double layer.5–14 The double layer is formed, when a charged electrode is placed in an electrolyte, which causes the electrolyte ions to migrate towards the charged surface in order to balance the applied potential.1,2 Helmholtz proposed originally the concept of a double layer on a charged solid surface, which consists of a rigid single layer of adsorbed counter ions on the electrode surface.15,16 The surface normalized capacitance of the electric double layer on a flat surface is described by means of the following formula:| | 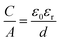 | (1) |
where the capacitance C normalized by the accessible electrode surface area A depends on the vacuum permittivity ε0, the relative permittivity of the electrolyte εr and the double-layer thickness d.16
Regarding EDLC electrode materials, a lot of research was dedicated to porous carbons such as activated carbons, which have a good electrical conductivity, chemical stability, are of low cost, easily available and possess a high specific surface area.17,18 Therefore, most commercial supercapacitors use biomass derived activated carbons like carbonized and activated coconut shell residues.8,19 We recently demonstrated the importance of the pre-carbonization temperature used prior to the chemical activation with KOH.20 A lower pre-carbonization temperature leads to an enlargement of pores, resulting in carbon materials with larger pore volume and specific surface area. Therefore, it is possible to tailor the pore size, total pore volume, micropore volume and surface area by varying the pre-carbonization temperature and the amount of activation agent. Besides a high specific surface area, the pore size and pore geometry of the carbon materials are important factors, as the latter has a direct influence on the electric double layer. It was observed that micropores (<2 nm) can increase the specific capacitance, when their size matches the size of electrolyte ions. By distorting or removing the solvation shell of the electrolyte ions the thickness of the double layer is reduced, which increases the specific capacitance.21,22 The validity of this confinement effect has been a subject of discussion over the recent years. While some authors assume that pore sizes are independent of capacitance, which is described by a “regular pattern”,23–25 it was experimentally proven that partially or fully desolvated ions participate in the formation of the electric double layer.26 The Helmholtz model of the rigid double layer can be used for flat plate configurations and can describe the pore size-dependent thickness of the double layer in perfectly ordered slit pores, but is inaccurate for nanopores with different degrees of curvature, caused by different pore geometries. Therefore, new models of capacitors have been proposed to describe the double layer formation in a curved and highly nanoporous carbon network in porous carbons. Depending on the synthesis, nanoporous carbons can have various pore shapes, such as slit, cylindrical and sometimes spherical types.27 Huang et al. adapted the classic Helmholtz model to develop a concept based on cylindrical mesopores (2 to 50 nm) and micropores (<2 nm). In the mesopore regime, solvated ions enter the pores and approach the pore walls to form electric double-cylinder capacitors (Fig. 1d).28,29 The surface area normalized double-cylinder capacitance is given by the following formula:
| | 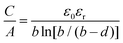 | (2) |
where
b is the radius of the cylinder pore and
d the double-layer thickness. In such a case, the effect of the pore size and pore curvature becomes prominent compared to the double-layer thickness
d. In the micropore regime, partially or fully desolvated ions enter the pores and line up to form electric wire-in-cylinder capacitors the capacitance of which is given by the formula:
| | 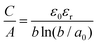 | (3) |
where the key quantity is
a0, the effective size of the ion (the extent of electron density around the ions).
28,29 These models can be combined to simulate the total capacitance of a carbon electrode material with a multimodal pore size distribution using formula
(3) for micropores, formula
(2) for mesopores and formula
(1) for macropores with their respective surface areas. The electric double cylinder-capacitor and electric wire-in-cylinder capacitor models assume a cylindrical pore shape for micropores and mesopores and alternative models have been proposed for different pore geometries. If the mesopores have a spherical shape, the geometric configuration of the adsorbed electrolyte ions would differ leading to the following formula derived by Wang
et al.:
30| | 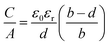 | (4) |
Although the electric double-cylinder capacitor model assumes a cylindrical pore geometry for micropores, it is supposed that micropores in carbon materials are rather slit pores, which can also show a curvature of the pore walls.
31 Feng
et al. suggested that micropores are too narrow for electrolyte ions to experience the effect of pore wall curvature on the electric double layer. They adjusted the classic flat plate model, assuming slit-shaped micropores, to account for the confined double layers leading to a sandwich-type capacitor model, as described in formula
(5) and shown in
Fig. 1a.
32 Furthermore, the effective double-layer thickness
deff was introduced, which is the distance between the pore wall and the outer electron shell of the electrolyte ion.
| | 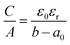 | (5) |
With the effective double-layer thickness, it is possible to describe an anomalous increase of capacitance for subnanometer pores very well. Hsieh
et al. used the new capacitor model for slit pores (
Fig. 1a) in combination with the electric double-cylinder capacitor model for mesopores (
Fig. 1d) to simulate the capacitance of activated carbons based on data derived from physisorption measurements.
33 In addition, they proposed a pore size dependent dielectric permittivity, which increases linearly up to a pore size of 2 nm. According to formula
(5), the capacitance increases with increasing pore size, considering the relative permittivity. However, since the term (
b −
a0) becomes larger with increasing pore size, the capacitance is reduced. These two counteracting effects lead to a constant capacitance for the entire range of micropores. Despite this approach, which contradicts the anomalous increase of capacitance from subnanometer pores, experimental and simulated capacitance values matched reasonably for organic and aqueous electrolytes. Just recently, Zuliani
et al. evaluated different combinations of double layer capacitor models. They used the electrical wire and slit pore model for micropores and the cylinder and spherical model for mesopores for various activated carbons, and compared the simulated capacitance with the measured capacitance.
34 Using high resolution scanning electron microscopy (SEM) for investigation of the pore shape it was found that the activated carbon materials had curved mesopores. Furthermore, they observed the best match of simulated and measured capacitances for a combined model of slit-shaped pores for micropores and cylindrical shaped pores for mesopores, confirming the approach of Hsieh
et al.
 |
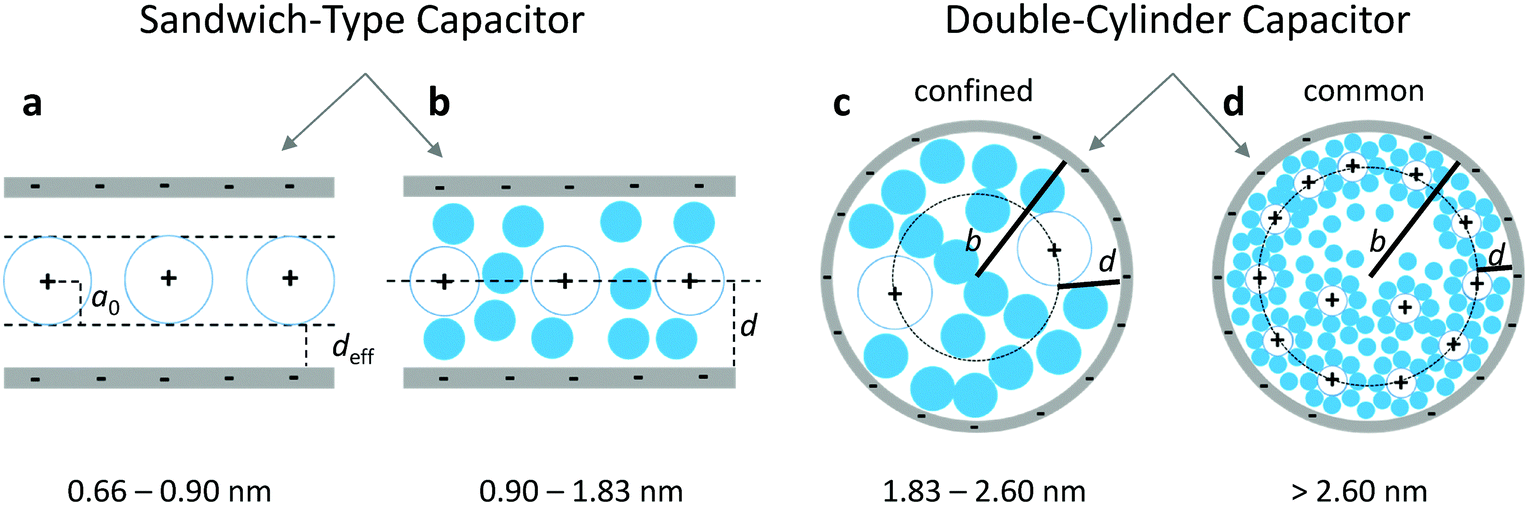
| | Fig. 1 Scheme of the ESDCC model. (a) The sandwich double layer capacitor with deff and the average ion radius a0 = 0.278 nm for pore diameters between 0.66 and 0.90 nm, (b) the sandwich double layer capacitor for pores with diameters between 0.90 and 1.83 nm and the double layer thickness d equals to the pore radius, (c) the confined double-cylinder capacitor with restricted access for solvated ions and thinner double layer thickness d = 0.820 nm for pores with radius b and a diameter between 1.83 and 2.60 nm and (d) the common double cylinder-capacitor model with the double layer thickness d = 1.050 nm for pores with radius b and diameters larger than 2.60 nm. | |
The evaluation of the experimentally derived data from gas physisorption is an important factor for describing the effects of porosity on electrochemical performance. Nitrogen physisorption is one of the most widely used methods to investigate the porosity of porous materials and the BET method is a standard method for determining the specific surface area.35 However, as mentioned by Centeno and Stoeckli and their co-workers, the use of the BET method to determine the specific surface area can be misleading, because it easily overestimates the surface area of microporous activated carbons.23,25,36,37 The DFT method is widely used and is currently the most advanced methodology to extract porosity data from gas sorption isotherms and effectively avoids the limitations of BET theory.38–40 The results of the DFT method are the pore size related cumulative pore volume and the cumulative specific surface area. Therefore, the electrochemical active surface can be determined, by subtracting the surface area of pore sizes, the area of which is inaccessible to the electrolyte ions, from the total surface area. Taking for example the ion diameter of tetraethylammonium (TEA+, 0.67 nm) and tetrafluoroborate (BF4−, 0.45 nm) into account, pores smaller than these values are inaccessible to the ions, which results in the reduction of the electrochemically active specific surface area which can be calculated using the results of the DFT method, as demonstrated by Jäckel et al.41,42 Although nitrogen physisorption is a powerful tool to investigate the pore size and specific surface areas of porous materials, it has its limitations for pore sizes below 1 nm, due to its slow diffusion kinetics at low pressures.38 Carbon dioxide physisorption was suggested in order to overcome the diffusion limitations of nitrogen in micropores due to the faster equilibration at a higher temperature and smaller molecular cross section. On the other hand, carbon dioxide loses its sensitivity for pores larger than 1 nm due to the high vapour pressure at the commonly used temperature.43 For the optimal characterization of the porous properties of activated carbons, the method of combining carbon dioxide and nitrogen physisorption has recently been demonstrated as a legitimate method of evaluating gas physisorption data.34,41,42,44,45
In this study, we have developed a new model that describes the dependence of capacitance on pore size. The pore sizes are divided into four ranges, based on the ion size of the electrolyte system used. Each of these pore ranges is described with a capacitor model to eventually obtain a combined model that describes the dependence of the capacitance on the pore sizes in the range from 0.66 to 10.7 nm. So far, capacitor models were divided into the respective pore sizes strictly according to the definition of IUPAC, namely the micropore range of up to 2 nm and the mesopore range.40,46 An electrolyte specific classification of the pore size ranges and the respective capacitor models, as well as a possible extension of these, has not yet been carried out and is necessary to achieve an extended understanding of the dependence of the capacitance on pore sizes. The four individual models of the combined model are based on the sandwich-type- and the double-cylinder capacitor model. Taking into account the size of the electrolyte ions, models were expanded, which entails a possible change in the thickness of the double layer, as well as a pore size-dependent change in relative permittivity. To evaluate the combined model, simulated capacitance values of synthesized activated carbons were compared with experimentally measured capacitance values. The carbon electrode materials based on phenol-formaldehyde (PF) resins differ in their porosity, from being purely microporous to having additional mesopores up to 5 nm.
2. Materials and methods
2.1 Material synthesis
The typical synthesis of high surface area carbons (HSACs) was already presented in ref. 20. In short, 2.2 g phenol (ABCR) were dissolved in 30 g ethanol. While stirring, 4.4 mL of formaldehyde (Grüssing, 37 wt% in water) solution and 10 mL of hydrochloric acid (HCl, 37 wt%) were added. The mixture was heated to 95 °C for 24 h. The bright brown PF resin was filtered, washed with ethanol and dried for 12 h at 100 °C. The obtained powdery dark red polymer was then heated in a quartz tube furnace at temperatures ranging from 300 °C to 900 °C for 3 h under an argon inert gas atmosphere with a heating rate of 5 °C min−1. The pre-carbonized material was mixed with potassium hydroxide in a ratio of 5 to 1 and dissolved in water and ethanol. The mixture was then dried at 100 °C for 24 h to ensure a good amalgamation of the pre-carbonized carbon and potassium hydroxide. The carbon/KOH mixture was activated at 900 °C with no holding time under a nitrogen gas atmosphere. The resulting product was washed with dilute HCl and distilled water to remove any impurities until the filtrate was pH neutral. The product was dried for 24 h at 100 °C. The prepared samples are named HSAC-300-5, HSAC-400-5, HSAC-600-5, HSAC-700-5 and HSAC-800-5 depending on the pre-carbonization temperature and the mass ratio of KOH to carbon. The additionally presented samples HSAC-500-5, HSAC-500-2.5, HSAC-500-8 and HSAC-900-5 were previously reported.20
2.2 Porosity analysis and SEM images
Nitrogen adsorption–desorption isotherms were obtained on a Quadrasorb SI-MP analyser from Quantachrome at 77 K. The pore size distributions, pore volumes and surface areas were calculated by the QSDFT method for nitrogen using the slit pore equilibrium kernel for nitrogen at carbon. For the evaluation of the pore range from 1 to 10.7 nm nitrogen sorption was used, because no larger pores were observed for any HSAC. Carbon dioxide sorption was used to study the range of pores below 1 nm. Carbon dioxide adsorption–desorption isotherms were obtained on an Autosorb-iQ-MP analyser from Quantachrome at 273 K. Pore size distributions, pore volumes and surface areas were calculated by the NLDFT method using the slit pore equilibrium kernel for carbon dioxide at carbon. SEM images were taken on NTS LEO-1525 from Carl Zeiss Company.
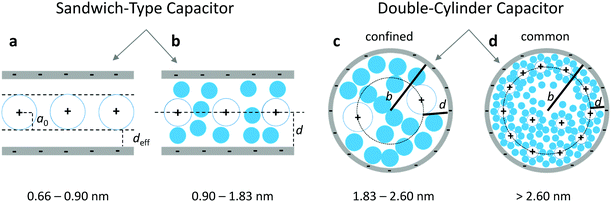
2.3 Cell preparation and electrochemical measurements
Electrodes were prepared by mixing carbon active materials with a polyvinylidene fluoride (PVDF) binder (Sigma-Aldrich, 10% suspension in N-methyl-pyrrolidone, NMP) and VULCAN XC72R (CABOT) as conductive additives in a ratio of 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1 in a slurry with NMP. The slurry was coated on aluminium foil (Custom Cells, 20 μm) and dried at 100 °C in vacuo. The thickness of the electrodes of each sample is about 70 μm (including Al foil). Electrodes were cut out with a diameter of 1.8 cm with mass loadings from 0.50 to 1.05 mg cm−2 of the total active carbon material. The electrochemical measurements were performed in a two-electrode test cell setup (El-Cell) using stainless steel electrodes (1.8 cm diameter). Symmetrical electrochemical capacitors were built using two carbon electrodes of identical mass, electrically isolated by a glassy fibrous separator (Whatman, 50 μm). A 1 M tetraethylammonium tetrafluoroborate (TEABF4, electrochemical grade, Sigma-Aldrich) solution in acetonitrile (AN, 99.999%, Sigma-Aldrich) was used as an electrolyte. The electrochemical characterization was performed using a computer-controlled potentiostat (Biologic VMP3). Before electrochemical tests were carried out, each test cell was cycled at a scan rate of 75 mV s−1 25 times to ensure the wettability of the electrode and the equilibrium state of electrolyte ions on the carbon electrode surface. The specific capacitance was calculated from galvanostatic charge/discharge cycling, which was performed in the 0 to 2.5 V range at a low current density of 1 A g−1 based on the total electrode mass of both electrodes, which is the most reliable current density to determine the largest possible specific capacitance.47 The specific capacitance Cmeas of both electrodes determined from galvanostatic cycles was calculated by means of the formula:
:2:1 in a slurry with NMP. The slurry was coated on aluminium foil (Custom Cells, 20 μm) and dried at 100 °C in vacuo. The thickness of the electrodes of each sample is about 70 μm (including Al foil). Electrodes were cut out with a diameter of 1.8 cm with mass loadings from 0.50 to 1.05 mg cm−2 of the total active carbon material. The electrochemical measurements were performed in a two-electrode test cell setup (El-Cell) using stainless steel electrodes (1.8 cm diameter). Symmetrical electrochemical capacitors were built using two carbon electrodes of identical mass, electrically isolated by a glassy fibrous separator (Whatman, 50 μm). A 1 M tetraethylammonium tetrafluoroborate (TEABF4, electrochemical grade, Sigma-Aldrich) solution in acetonitrile (AN, 99.999%, Sigma-Aldrich) was used as an electrolyte. The electrochemical characterization was performed using a computer-controlled potentiostat (Biologic VMP3). Before electrochemical tests were carried out, each test cell was cycled at a scan rate of 75 mV s−1 25 times to ensure the wettability of the electrode and the equilibrium state of electrolyte ions on the carbon electrode surface. The specific capacitance was calculated from galvanostatic charge/discharge cycling, which was performed in the 0 to 2.5 V range at a low current density of 1 A g−1 based on the total electrode mass of both electrodes, which is the most reliable current density to determine the largest possible specific capacitance.47 The specific capacitance Cmeas of both electrodes determined from galvanostatic cycles was calculated by means of the formula:| |  | (6) |
where I is the current (mA), m the mass (g) of the total active carbon material of both electrodes and dV/dt is the slope of the discharge curve (V s−1).
2.4 EDLC model description and capacitance simulation
For our combined model, we have divided the pore widths obtained from gas physisorption into four ranges and described each range with a different capacitor model as shown in Fig. 1. We only consider pore sizes larger than 0.66 nm, because the bare TEA+ ion cannot penetrate pores smaller than its ion diameter and therefore do not contribute to the cell capacitance.1,2,6,8,48 The sandwich double layer capacitor model as shown in Fig. 1a is used for pore sizes between 0.66 nm and 0.90 nm and the capacitance can be calculated according to formula (3). Considering the size of the fully solvated ions (1.3 nm for TEA+ and 1.1 nm for BF4− in acetonitrile),49 they need to be fully or partial desolvated to enter small pores between 0.66 and 0.90 nm. For larger pores between 0.90 and 1.83 nm, solvent molecules are able to enter the pore either free or in a solvation shell and become involved in the double layer formation. Therefore, we assume that the screening length of the double layer changes from the effective double-layer thickness deff (Fig. 1a) to the classic double-layer thickness d. We also assume that the model can still be described as a sandwich-type double layer capacitor as shown in Fig. 1b and the capacitance can be calculated according to formula (1) developed by Helmholtz, while the double-layer thickness is the pore radius of the carbon material. For pores larger than 1.83 nm, we suspect an influence of the pore wall curvature on the double layer formation, which is best described with a cylindrical capacitor model as shown in Fig. 1c and d.28,29,33,34 Although our synthesized carbon materials probably possess mainly slit shaped pores regardless of the pore diameter, the curvature of pore walls for larger pores plays a significant role in the double layer formation. Since it is not possible for two or more fully solvated ions to enter pores between 1.83 and 2.60 nm, we presume a restriction of the double layer formation, resulting in a smaller assumed double-layer thickness of 0.820 nm compared to 1.015 nm for the unrestricted double layer formation for pores larger than 2.60 nm. These values related to the double layer thickness are taken from the literature, whereby the first refers to higher concentrated electrolyte systems and the latter refers to the electrolyte concentration from this work (1 M).29,33 Pores in the range of 1.83 to 2.60 nm can be described with a confined electric double-cylinder capacitor model as shown in Fig. 1c and pores larger than 2.60 nm can be described with the common electric double-cylinder capacitor model as shown in Fig. 1d. For both ranges, the theoretical capacitance can be calculated according to formula (2) for the electric double-cylinder-capacitor model with the respective double-layer thickness d. This new combination of the four models shown in Fig. 1a–d can be referred to as an electric sandwich double-cylinder capacitor (ESDCC) model. The relative permittivity was also taken into account more explicitly, since it has been shown that in confined spaces, such as in nanopores of porous carbons, the permittivity is significantly lower in comparison to bulk space. We have considered two pore size ranges, between 0.45 and 2.60 nm and between 2.60 and 10.7 nm. The space in the latter pore size range is still considered restricted, which is why we postulated a constant value of 9.73 there. For the relative permittivity in the range of 0.45 to 2.60 nm, a pore size dependent progression is assumed, as described by Hsieh et al.33 In their work they assumed that the capacitance is constant for the entire micropore range, which means a linear increase of the relative permittivity with the pore diameter. In this work we do not assume a constant capacitance in the micropore range, but a dependence of the relative permittivity on the pore diameter. Therefore, a linear increase of the relative permittivity in pore diameters from 0.47 to 2.60 nm is assumed. The initial value of the relative permittivity is assumed to be 1 at a pore size of 0.47 nm, at which the bare BF4− ion can penetrate the pore without a solvent molecule being involved in the formation of the double layer. The pore range of 0.47 to 0.67 nm is taken into account, although this only makes a negligibly small contribution to the cell capacitance. All values for ion sizes, relative permittivity and double-layer thicknesses are taken from the literature.29,33 The simulated gravimetric capacitance Csim for the synthesized activated carbons was calculated by means of the formula:| | 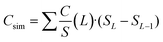 | (7) |
where L is the pore width, SL the pore size associated cumulative specific surface area and SL − SL−1 the associated surface area of two consecutive pore sizes. Pore sizes, their theoretical normalized capacitance values C/S (L) and the pore size dependent relative permittivity values are listed in Table S1 (ESI†).
3. Results and discussion
3.1 Material characterization
The prepared HSACs were characterized by scanning electron microscopy and the images of HSAC-800-5, HSAC-700-5 and HSAC-600-5 are shown in Fig. 2a–c. It can be seen that HSAC-800-5, HSAC-700-5 and HSAC-600-5 possess a network structure which is composed of spherical carbon particles with sizes around 3 μm, which is typical for carbons based on formaldehyde resins.50 The sintering of the spherical carbon nanoparticles leads to the formation of cavities within the network, which are 2 to 20 μm in diameter. The network structures of HSAC-400-5 and HSAC-300-5 differ significantly from the other HSACs, as can be seen in Fig. 2d and e. The carbon surface is smooth without sintered spherical carbon nanoparticles, while they still possess cavities within the network with similar diameters. The spherical nanoparticles agglomerated strongly during the activation process due to the low pre-carbonization temperature prior to the chemical activation, which led to a less rigid network as demonstrated previously.20,51
 |
| | Fig. 2 SEM images of the activated carbons (a) HSAC-800-5, (b) HSAC-700-5, (c) HSAC-600-5, (d) HSAC-400-5 and (e) HSAC-300-5. | |
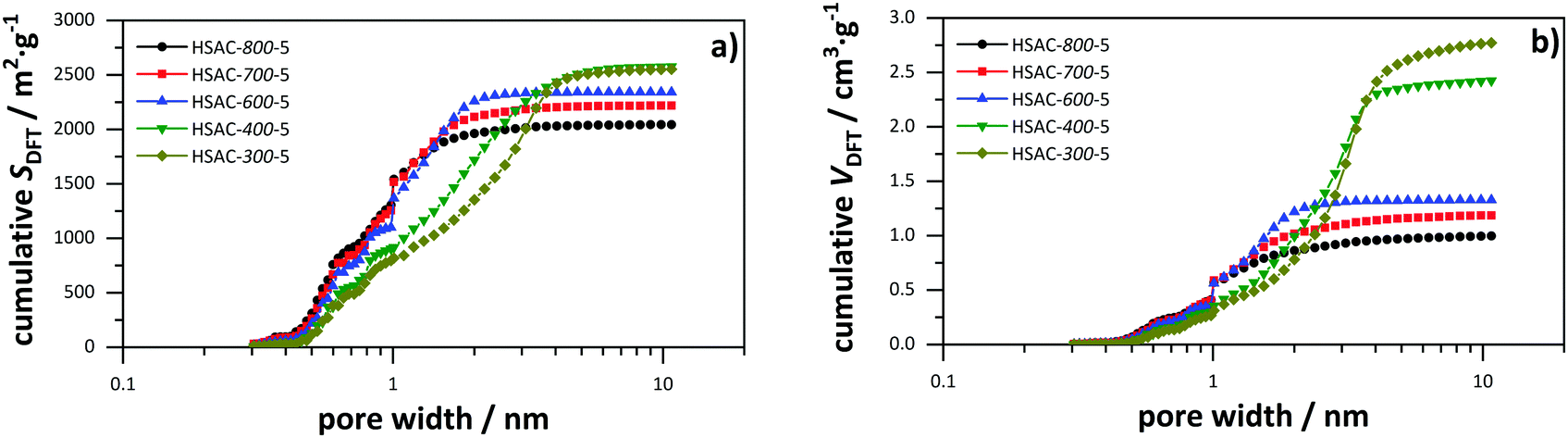
The prepared HSAC samples were characterized with carbon dioxide physisorption (273 K) for a better assessment of the subnanometer pores down to 0.3 nm in addition to nitrogen physisorption (77 K). The combined results of both methods contain the data of carbon dioxide desorption in the range of 0.3 to 1 nm and nitrogen desorption in the range of 1 to 10.7 nm. The cumulative surface area and pore volume for the HSAC samples are shown in Fig. 3, with the full isotherms and pore size distributions provided in the ESI† (Fig. S1–S3). The physisorption-derived data are summarized in Table 1. The corresponding isotherms of the nitrogen physisorption of HSAC-800-5, HSAC-700-5 and HSAC-600-5 are typical type I isotherms, indicating carbon materials with predominantly micropores. The isotherms of HSAC-400-5 and HSAC-300-5 show a slope at relative pressures from 0.1 to 0.4 resulting from broad pore size distributions of large micropores and mesopores.40 Besides pore sizes below 1 nm, all HSACs contain pore sizes up to 2 nm originating from the carbonization process and from the etching of carbon during the KOH activation. Additionally, HSAC-400-5 and HSAC-300-5 have also mesopores up to 5 nm, while HSAC-300-5 has slightly larger pore sizes than HSAC-400-5. All HSAC samples have high DFT surface areas, ranging from 2045 to 2606 m2 g−1, with HSAC-400-5 having the largest specific surface area. The specific surface area of the carbon samples increases with decreasing pre-carbonization temperatures. The reason for this is the more effective chemical activation of the less rigid carbon network caused by lower pre-carbonisation temperatures, resulting in pore size enlargements and larger surface areas. Assuming that pores smaller than 0.66 nm cannot contribute to the total capacitance for a symmetric capacitor in a two electrode setup due to the size of the bare TEA+ cation, the accessible surface areas Sacc were calculated and are listed in Table 2. Caused by the pore enlargement, the surface area of HSAC-300-5 is mostly generated by pores larger than 0.7 nm leading to a large accessible surface area of 2104 m2 g−1. Considering the previously presented and discussed samples HSAC-500-5 and HSAC-900-5,20 a direct correlation of an increasing accessible surface area with a decreasing pre-carbonization temperature is observable. Compared to the differences in the total surface areas, the differences of the accessible surface areas are significant (2104 m2 g−1 for HSAC-300-5 to 972 m2 g−1 for HSAC-900-5), which is supposed to have a major impact on the total gravimetric capacitance.23,25 Similar to the surface area, the total pore volume (Vt) correlates with the pre-carbonization temperature.
 |
| | Fig. 3 Combined cumulative surface area (a) and cumulative pore volume (b) derived from carbon dioxide and nitrogen physisorption data of the prepared HSACs with different pre-carbonization temperatures. | |
Table 1 Nitrogen and carbon dioxide physisorption derived data of the HSAC samples
| Sample |
S
DFT/m2 g−1 |
V
t/cm3 g−1 |
Ref. |
|
S
DFT: DFT surface area; Vt: total pore volume. |
| HSAC-300-5 |
2558 |
2.82 |
This work |
| HSAC-400-5 |
2606 |
2.45 |
This work |
| HSAC-500-5 |
2480 |
1.96 |
20
|
| HSAC-600-5 |
2342 |
1.33 |
This work |
| HSAC-700-5 |
2220 |
1.20 |
This work |
| HSAC-800-5 |
2045 |
1.00 |
This work |
| HSAC-900-5 |
1810 |
0.86 |
20
|
| HSAC-500-2.5 |
2108 |
0.93 |
20
|
| HSAC-500-8 |
2529 |
2.62 |
20
|
Table 2 Specific capacitance calculated from charge/discharge tests of EDLCs prepared from the HSACs in 1 M TEABF4/AN with the accessible surface area
| Sample |
S
acc/m2 g−1 |
C
meas/F g−1 |
Ref. |
|
S
acc: accessible DFT surface area of pores >0.66 nm; Cmeas: specific capacitance (galvanostatic discharge) at 1 A g−1. |
| HSAC-300-5 |
2104 |
143.3 |
This work |
| HSAC-400-5 |
2074 |
157.9 |
This work |
| HSAC-500-5 |
1954 |
162.2 |
20
|
| HSAC-600-5 |
1656 |
151.5 |
This work |
| HSAC-700-5 |
1445 |
135.4 |
This work |
| HSAC-800-5 |
1181 |
126.7 |
This work |
| HSAC-900-5 |
972 |
118.5 |
20
|
| HSAC-500-2.5 |
1217 |
133.8 |
20
|
| HSAC-500-8 |
2039 |
165.9 |
20
|
3.2 Electrochemical characterization
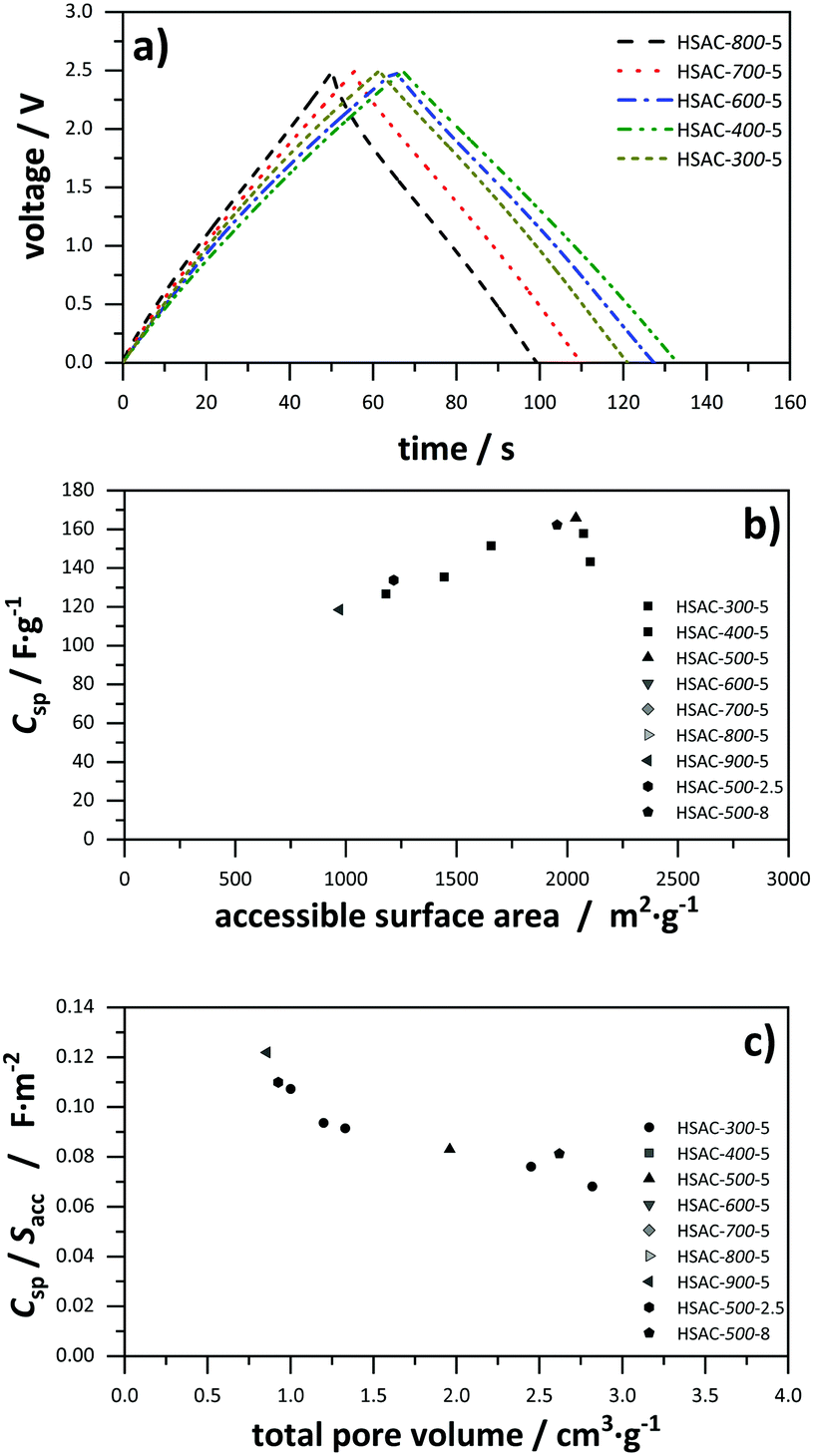
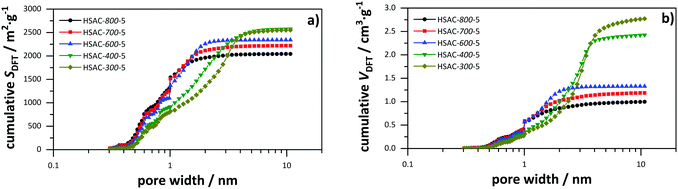
The prepared symmetric EDLCs were investigated with galvanostatic charge/discharge tests at a low current density of 1 A g−1 to obtain the highest possible capacitance and the results are listed in Table 2. The charge/discharge profiles of all HSACs with different pre-carbonization temperatures show symmetrical and linear shapes indicating well developed electrochemical double-layer capacitance properties as shown in Fig. 4a. From the presented HSAC samples prepared with different pre-carbonization temperatures, HSAC-400-5 has the highest specific capacitance of 157.9 F g−1. The specific capacitance values for HSAC-300-5, HSAC-700-5 and HSAC-800-5 are lower, but still as high as 143.3, 135.4 and 126.7 F g−1 respectively. HSAC-500-8 has a similar high specific capacitance to HSAC-400-5 of 165.9 F g−1. These high values compare very well with those of various state-of-the-art porous carbon materials and phenolic resin based carbon materials used as electrode materials for capacitors.52–59 The high capacitance values in general can be explained with the large accessible surface areas ranging from 1000 to 2100 m2 g−1 (Table 2). A general correlation of a higher specific capacitance with a larger accessible surface area is observable, as shown in Fig. 4b. However, HSAC-400-5 and especially HSAC-300-5 have comparatively lower capacitance values than HSAC-500-5 and HSAC-500-8 despite having similar large accessible surface areas. That is an indication of a more complex correlation between the porosity of carbon electrode materials and their respective capacitance than a simple linear surface area–specific capacitance correlation. The total pore volume of the HSACs correlates quite well with the average pore size (see Table 1 and Fig. S1, S2, ESI†) and is directly dependent on the surface area normalized capacitance, as can be seen in the application of the capacitance against the total pore volume in Fig. 4c. To be more precise, the normalized surface area decreases with increasing total pore volume, which indicates different influences of different pore sizes on the total capacitance. Therefore, a complex model of normalized capacitance and pore sizes was developed and compared with the experimentally derived data.
 |
| | Fig. 4 (a) Galvanostatic charge/discharge profile of all HSACs with different pre-carbonization temperatures at a current density of 1 A g−1; (b) specific capacitance calculated from charge/discharge tests plotted against the accessible surface area (Sacc for pores >0.66 nm) of all presented HSACs (including ref. 20); (c) accessible surface area normalized specific capacitance plotted against the total pore volume (Vt) of all presented HSACs (including ref. 20). | |
3.3 Evaluation of model applicability
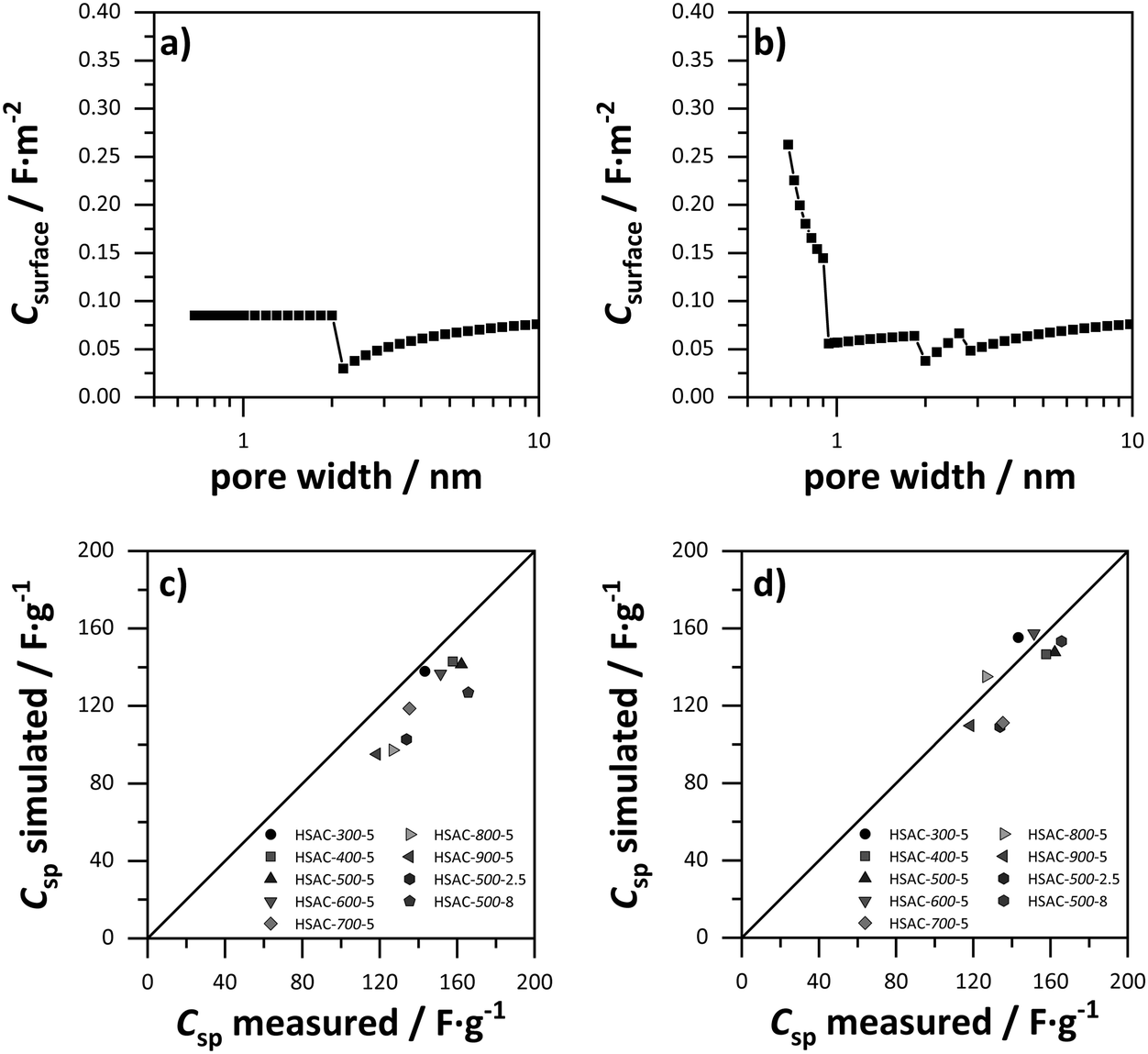
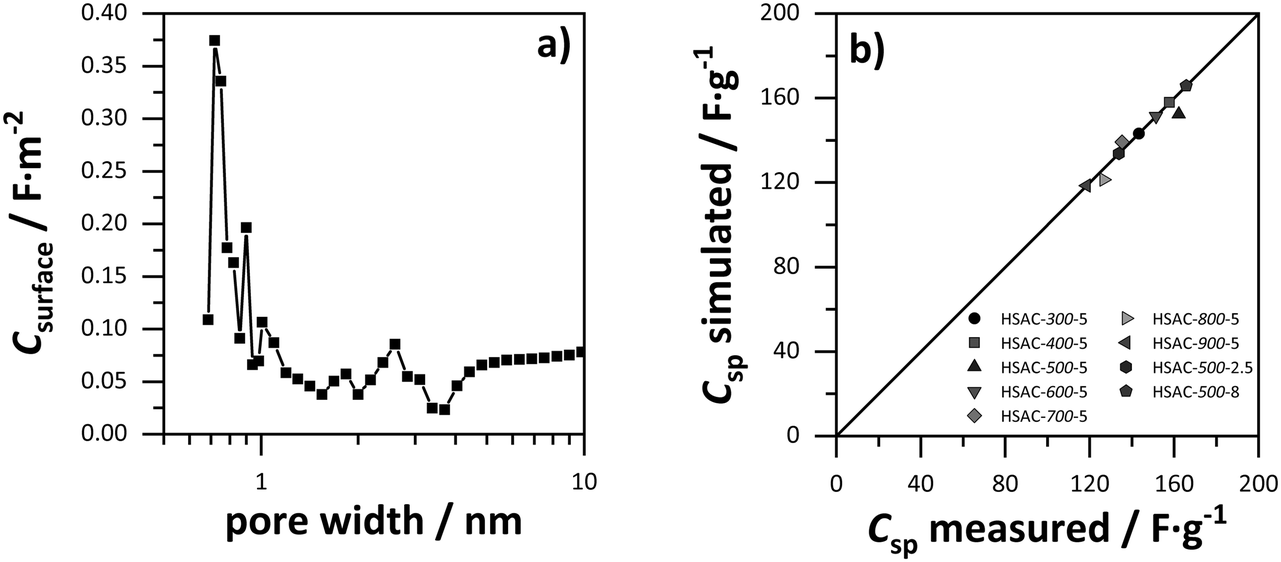
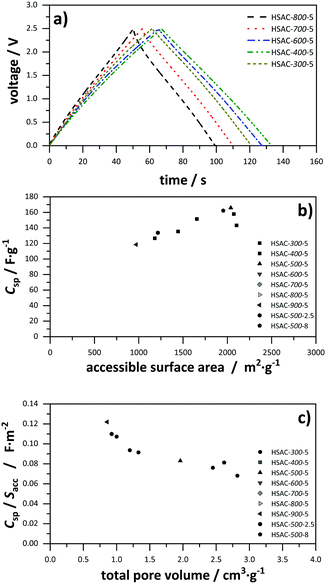
In order to elucidate the relation of pore sizes and capacitance, values for the gravimetric capacitance of all HSACs were simulated using a combination of sandwich-type- and double-cylinder capacitor model (Fig. 1a and d) and the combined ESDCC model (Fig. 1a–d). The simulated capacitance values were compared with the measured values to verify the applicability of the models. At first, values for gravimetric capacitance were calculated according to the approach developed by Hsieh et al.33 Two capacitor models were used for two pore size ranges, which were divided into micro- and mesopores according to the IUPAC definition. For the micropore region (<2 nm) the sandwich capacitor model was assumed (Fig. 1a), where the relative permittivity increases linearly with the pore size, resulting in a constant surface area normalized capacitance for the entire micropore region which was calculated using the classic Helmholtz equation (formula (1)). The mesopore region (>2 nm) is described with the double-cylinder capacitor model (Fig. 1d), using formula (2) for the calculation of the normalized capacitance. The capacitance–pore size relation is displayed in Fig. 5a and the corresponding values are listed in Table S1 (ESI†). The simulated gravimetric capacitance values for all HSACs are displayed in Fig. 5c and listed in Table 3. It is noteworthy that all simulated capacitance values were lower than the measured ones, indicating a systematic incorrect assumption. Furthermore, the simulated values of the HSACs deviate significantly from the measured values and the deviation of the value of HSAC-500-8 is even as high as −31%. The inaccurate simulation of capacitance values can be attributed to three different reasons: (1) the assumption of a constant capacitance for subnanometer pores (<2 nm) contradicts many observations and is too simplistic. The confinement effect of electrolyte ions in pores below 1 nm has been observed on numerous occasions.21,22,28,29,32,41,42,60 (2) Only two models were used to describe the pore ranges, which is not sufficient for the complicated network structures of nanopores in carbons and confinement effects of electrolyte ions. (3) The pores were divided into ranges according to the definition of IUPAC and not into ranges, which take into account the ion sizes of the respective electrolyte ions. Therefore, a more sophisticated model was developed which considers the previous mentioned aspects.
 |
| | Fig. 5 Surface area normalized capacitance depended on the pore size, based on (a) the sandwich-type capacitor for micropores and the cylindrical capacitor model for mesopores (values for ε0 = 9.73 and d = 1.015 nm were obtained from ref. 29). (b) The ESDCC model (values for a0 = 0.278 nm and d = 0.820 nm were obtained from ref. 33). Comparison of simulated and measured capacitance values of the HSACs for (c) the sandwich-type capacitor for micropores and the cylindrical capacitor model for mesopores and (d) the ESDCC model. | |
Table 3 Specific capacitance calculated from charge/discharge tests of EDLCs prepared from the HSACs in 1 M TEABF4/AN and the simulated specific capacitance based on different theoretical models
|
|
C
sp/F g−1 measured |
Sandwich + cylindrical model |
ESDCC |
ESDCC modified |
|
C
sp/F g−1 simulated |
Percentage difference (%) |
C
sp/F g−1 simulated |
Percentage difference (%) |
C
sp/F g−1 simulated |
Percentage difference (%) |
|
C
sp: specific capacitance; value of a0 = 0.278 in the pore range of 0.66–0.90 nm taken from ref. 33; value of d = 0.820 nm in the pore range of 1.83–2.6 nm taken from ref. 33; value of d = 1.015 nm for pores larger than 2.6 nm taken from ref. 29; values for εr = 9.73 and for the ESDCC models given in Table S1 (ESI) and adapted from ref. 29. |
| HSAC-300-5 |
143.3 |
138.0 |
−4 |
155.3 |
+7 |
143.3 |
±0 |
| HSAC-400-5 |
157.9 |
142.9 |
−11 |
146.6 |
−8 |
157.9 |
±0 |
| HSAC-500-5 |
162.2 |
141.5 |
−14 |
147.7 |
−10 |
152.2 |
−7 |
| HSAC-600-5 |
151.5 |
136.7 |
−11 |
157.5 |
+4 |
151.5 |
±0 |
| HSAC-700-5 |
135.4 |
118.7 |
−14 |
111.2 |
−21 |
139.3 |
+3 |
| HSAC-800-5 |
126.7 |
97.2 |
−30 |
135.0 |
+6 |
121.5 |
−4 |
| HSAC-900-5 |
118.5 |
95.1 |
−25 |
109.8 |
−8 |
118.5 |
±0 |
| HSAC-500-2.5 |
133.8 |
102.8 |
−30 |
109.1 |
−23 |
133.8 |
±0 |
| HSAC-500-8 |
165.8 |
126.8 |
−31 |
153.4 |
−8 |
165.8 |
±0 |
As mentioned in Section 2.4, we developed a new combined model of the individual models shown in Fig. 1a–d, which can be described as an electric sandwich double-cylinder capacitor model (ESDCC). As shown in Fig. 5b the correlation between the normalized capacitance and pore sizes became more differentiated. Pores with a size of 0.66 nm deliver the largest contribution to the normalized capacitance and with an increase of pore size, the capacitance decreases exponentially. Pores larger than 0.90 nm have significantly smaller values for the normalized capacitance and differ to a smaller extent, except for the pores ranging from 1.83 to 2.6 nm (Fig. 5b). Comparing the simulated gravimetric capacitance values of the HSACs employing the ESDCC model with the measured values, a better match is observable, as shown in Fig. 5d. With the exception of HSAC-700-5 and HSAC-500-2.5, all simulated values match with the measured values with a maximum difference of 10% (Table 3). Considering the higher impact on the capacitance of subnanometer pores below 0.90 nm, the ESDCC model confirms the anomalous increase of capacitance for pores around 0.7 nm. It also shows that the more sophisticated division of pore sizes depending on the electrolyte ions that are used and the extension of capacitor models (Fig. 1b and c) is important for the determination of capacitance values of carbon materials with different pore sizes. However, the percentage difference of the simulated values, compared to the measured values for HSAC-700-5 and HSAC-500-2.5 is still as high as −21% and −23% respectively. Although the ESDCC model is a more sophisticated approach to describe the influence of the pore size on capacitance, the assumed pore geometries (slit-shaped pores and cylindrical pores) are rather simple considering the complex structure of activated carbons. As a result, some simulated values can deviate from the measured values.
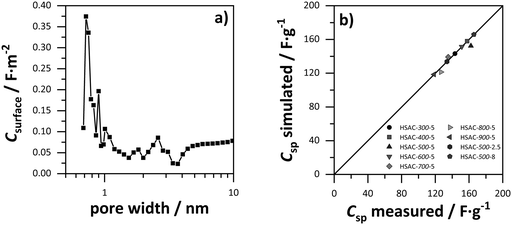
Therefore, a modification for the ESDCC model was necessary, to adapt the simulated gravimetric capacitance values to the experimentally measured values for all HSAC samples. For this purpose, a factor modification of the capacitance values of each individual pore size was performed to minimize the average deviation (AD) of the overall measured and simulated capacitance values. To determine the factors, we used the nonlinear generalized reduced gradient (GRG) method to solve for the smallest possible average deviation of all presented HSAC samples. The GRG method is a generalization of the reduced gradient method by allowing nonlinear constraints and arbitrary bound on the variables. The method is based on the Davidon–Fletcher–Powell algorithm.61 The average deviation was calculated using the following formula:
| | 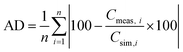 | (8) |
This was used to develop the modified ESDCC model. All factors for the ESDCC model and theoretical normalized capacitance values for the modified ESDCC model are listed in Table S1 (ESI
†). It is worth mentioning that the factor modification with the nonlinear GRG method only performed well for the ESDCC model. Neither the factor modification for the first discussed combined sandwich/double-cylinder capacitor model (
Fig. 1a and d), nor a factor modification of a constant normalized capacitance for all pore sizes provided satisfactory results in terms of a low average deviation and reasonable physical correlation of the pore size dependent capacitance values. As shown in
Fig. 6a, the correlation of capacitance and pore size of the modified ESDCC model became more complex, but the simulated capacitance values are in good agreement with the measured values, which is displayed in
Fig. 6b and
Table 3. The difference is as small as 7% for HSAC-
500-5 and the simulated capacitance values for HSAC-
300-5, HSAC-
400-5, HSAC-
600-5, HSAC-
900-5, HSAC-
500-2.5 and HSAC-
500-8 are even identical to the measured capacitance values. This emphasizes the mathematical validity of the modified ESDCC model. In the following, the physical meaning of the correlation between capacitance and pore sizes will be discussed. As can be seen in
Fig. 6a pore sizes between 0.72 and 0.75 nm exhibit the largest values for the normalized capacitance, which indicates the strong influence of partially or fully desolvated ions on the capacitance of EDLCs as proposed in the theory of the anomalous increase of capacitance for subnanometer pores.
21,22 For increasing pore sizes the capacitance decreases, which is in accordance to the sandwich capacitor model displayed in
Fig. 1a and described with formula
(5). Interestingly, the normalized capacitance increases again for pores with a size of 0.90 nm and again for pores around 1.0 nm, though the capacitance of the former pore size is larger than the capacitance of the latter. Electrolyte ions that penetrate subnanometer ultramicropores contribute more to the capacitance, but the number of electrolyte ions that can enter the pores is still limited due to steric impairment. This may change for pores with a size of 0.9 nm. Electrolyte ions can enter the pores without removing the entire solvent shell, which leads to an increase of the surface capacitance. For pores around 1 nm, BF
4− ions can enter the pores almost fully solvated, which can result in a minor increase of the normalized capacitance. For pores in the range of 1.0 to 1.3 nm the capacitance decreases again. This can be explained with the sandwich capacitor model as shown in
Fig. 1b. The electrolyte ion is centred between the pore walls and with increasing pore size the distance to the pore walls and thus the thickness of the double layer increases, which leads to a lower capacitance. Full accessibility of solvated TEA
+ ions may lead to a slight increase for pores from 1.30 to 1.83 nm. A larger increase of capacitance is observed for pores ranging from 2.0 to 2.6 nm. This can be attributed to the fact that more solvated ions are able to enter the pores, which participate in the electric double layer formation for both ions. Additionally, a smaller thickness of the double layer is assumed due to the previously discussed confinement effect (see
Fig. 1c). Despite the Coulomb repulsion of two equal polarized ions, we assume that two ions of equal charge are able to enter pores between 2.3 and 2.6 nm at their respective electrodes, which results in smaller double layer thickness leading to a higher normalized capacitance. It is noteworthy that the capacitance decreases again and reaches an overall minimum for pore sizes between 3.4 and 3.7 nm. A destructive interference of the electric double layer from opposing pore walls may be the cause, as Jiang
et al.62 and Feng
et al.,
60 respectively, found an oscillatory behaviour for capacitance with the increase of the pore size. However, this has only been proven for ionic liquids and is yet to be validated for organic electrolytes. A confinement effect is more likely to be the reason than an interference of electric double layers. For these pore sizes, a maximum of three fully – or partially – desolvated ions are able to enter a pore at the same time due to steric impairment caused by the pore geometry and Coulomb repulsion of the electrolyte ions. Therefore, the degree of adsorbed ions on the pore surface is rather low similar to pores in the range of 1.8 to 2.6 nm (
Fig. 1c). Nevertheless, compared to the pore size range of 1.8 to 2.6 nm, the double-layer thickness for pores between 3.4 and 3.7 nm is estimated to be thicker with 1.015 nm compared to 0.820 nm, which results in a lower capacitance for these pore sizes. The capacitance increases with pores larger than 3.7 nm and shows no significant deviations from the ESDCC model described in
Fig. 1. More solvated ions are able to enter the pores and occupy the pore surface, thus increasing the capacitance until confinement effects and the influence of the pore wall curvature becomes less significant. Having rationalized the pore-size dependent contribution to the overall capacitance in this way, the comparatively low specific capacitance of HSAC-
400-5 and especially HSAC-
300-5 can be explained, despite their large accessible surface area of ∼2000 m
2 g
−1. HSAC-
300-5 and HSAC-
400-5 have a larger number of pores between 3.4 and 3.7 nm, which contribute to a very low extent to the overall capacitance. The capacitance values simulated with the modified ESDCC method matched with a very good accuracy the measured values of nine activated carbons with different pore sizes. Therefore, we believe that the modified ESDCC model is applicable to a variety of activated carbons to simulate an accurate total gravimetric capacitance with TEABF
4 in acetonitrile as electrolytes. However, a thorough investigation of the porosity is mandatory, preferentially by a combination of carbon dioxide and nitrogen physisorption derived data.
 |
| | Fig. 6 (a) Surface area normalized capacitance depended on the pore size, based on the modified ESDCC model. (b) Comparison of simulated and measured capacitance. | |
4. Conclusions
Understanding the influence of the pore sizes of carbon electrode materials on the capacitance of EDLC is desirable, since the amount of stored energy should be as large as possible. In this study, we developed a new combined model based on sandwich-type capacitors for micropores and double-cylinder capacitors for mesopores, respectively, which includes confinement effects and is named the ESDCC model. Factors for the capacitance values of each pore size were optimized with the nonlinear GRG method to obtain a refined relation of pore sizes and capacitance, which were validated by comparing simulated capacitance values with experimentally measured capacitance values. Our results show that pores below 1 nm provide the largest contribution to the capacitance, especially pores around 0.74 nm and 0.90 nm, confirming the effect of an anomalous increase of capacitance for pores with the size of bare TEA+ ions, while pores between 3.4 and 3.7 nm provide the lowest contribution to the capacitance. This is supported by the fact that the surface capacitance decreases with increasing pore volume of the carbon samples generated by larger pores. However, the accessible surface area remains a key factor regarding the capacitance. Although specific surface area and average pore sizes are not linearly dependent on each other, it has been observed that synthesis parameters leading to carbons with high specific surface areas also lead to larger pores. Therefore, maximizing the capacitance is difficult, as an increase in specific surface area is associated with a simultaneous enlargement of pore sizes, which neutralize their effects on the capacitance. Thus, the capacitance decreases which is demonstrated with the carbon materials HSAC-300-5 and HSAC-400-5. With our developed modified ESDCC model, it is possible to simulate the gravimetric capacitance values for activated carbon materials on the basis of physisorption-derived data from carbon dioxide and nitrogen in combination. The model is applicable for EDLCs with TEABF4 in acetonitrile. However, we expect that the factor modification approach of the ESDCC model can also be applied to other carbon/electrolyte systems for the optimization of EDLCs with regard to energy performance for the following reasons: (1) the modified ESDCC model was developed and applied for various carbons with different ranges of pore sizes. The capacitance of both purely microporous carbons as well as carbons with additional mesopores up to 5 nm was simulated adequately. (2) It was possible to assign any feature of capacitance dependency on pore size a physical meaning, with the ion size being an important factor. The basic assumptions of the ESDCC model can be applied to other electrolyte systems. (3) The pore size dependence of the relative permittivity was also considered in the ESDCC model, which is an important factor for the capacitance and can be adjusted to other solvents.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors thank Renate Walter and Dr Frank Friedrich from the University of Hamburg for taking SEM images of the samples. The study is financed by the city of Hamburg as a part of the graduate school Keytechnologies for Sustainable Energy Systems and Smart Grids.
References
- A. Burke, J. Power Sources, 2000, 91, 37–50 CrossRef CAS.
- R. Kötz and M. Carlen, Electrochim. Acta, 2000, 45, 2483–2498 CrossRef.
- A. González, E. Goikolea, J. A. Barrena and R. Mysyk, Renewable Sustainable Energy Rev., 2016, 58, 1189–1206 CrossRef.
- W. Gu and G. Yushin, Wiley Interdiscip. Rev.: Energy Environ., 2014, 3, 424–473 CAS.
- J. Liu, J. Wang, C. Xu, H. Jiang, C. Li, L. Zhang, J. Lin and Z. X. Shen, Adv. Sci., 2018, 5, 1700322 CrossRef PubMed.
- Y. Zhang, S. Yu, G. Lou, Y. Shen, H. Chen, Z. Shen, S. Zhao, J. Zhang, S. Chai and Q. Zou, J. Mater. Sci., 2017, 52, 11201–11228 CAS.
- F. Wang, X. Wu, X. Yuan, Z. Liu, Y. Zhang, L. Fu, Y. Zhu, Q. Zhou, Y. Wu and W. Huang, Chem. Soc. Rev., 2017, 46, 6816–6854 RSC.
- E. Frackowiak, Q. Abbas and F. Béguin, J. Energy Chem., 2013, 22, 226–240 CrossRef CAS.
- M. Sevilla and R. Mokaya, Energy Environ. Sci., 2014, 7, 1250–1280 RSC.
- W. Gu and G. Yushin, Wiley Interdiscip. Rev.: Energy Environ., 2013, 3, 424–473 Search PubMed.
- E. Frackowiak, Phys. Chem. Chem. Phys., 2007, 9, 1774–1785 CAS.
- A. Burke, Electrochim. Acta, 2007, 53, 1083–1091 CrossRef CAS.
- W. G. Pell and B. E. Conway, J. Power Sources, 2001, 96, 57–67 CrossRef CAS.
- W. Li, J. Liu and D. Zhao, Nat. Rev. Mater., 2016, 1, 16023 CAS.
- H. Helmholtz, Ann. Phys. Chem., 1853, 165, 211–233 CrossRef.
- H. Helmholtz, Ann. Phys. Chem., 1879, 243, 337–382 CrossRef.
- C. Li, X. Zhang, K. Wang, X. Sun and Y. Ma, J. Power Sources, 2018, 400, 468–477 CAS.
- C. Li, X. Zhang, K. Wang, X. Sun, G. Liu, J. Li, H. Tian, J. Li and Y. Ma, Adv. Mater., 2017, 29, 1604690 CrossRef PubMed.
- M. Inagaki, H. Konno and O. Tanaike, J. Power Sources, 2010, 195, 7880–7903 CrossRef CAS.
- R. Heimböckel, S. Kraas, F. Hoffmann and M. Fröba, Appl. Surf. Sci., 2018, 427, 1055–1064 CrossRef.
- J. Chmiola, G. Yushin, Y. Gogotsi, C. Portet, P. Simon and P. L. Taberna, Science, 2006, 313, 1760–1763 CrossRef CAS PubMed.
- J. Chmiola, C. Largeot, P.-L. Taberna, P. Simon and Y. Gogotsi, Angew. Chem., Int. Ed., 2008, 47, 3392–3395 CrossRef CAS PubMed.
- T. A. Centeno, O. Sereda and F. Stoeckli, Phys. Chem. Chem. Phys., 2011, 13, 12403 RSC.
- F. Stoeckli and T. A. Centeno, J. Mater. Chem. A, 2013, 1, 6865 RSC.
- B. Lobato, L. Suárez, L. Guardia and T. A. Centeno, Carbon, 2017, 122, 434–445 CrossRef CAS.
- W.-Y. Tsai, P.-L. Taberna and P. Simon, J. Am. Chem. Soc., 2014, 136, 8722–8728 CrossRef CAS PubMed.
- Y. Meng, D. Gu, F. Zhang, Y. Shi, H. Yang, Z. Li, C. Yu, B. Tu and D. Zhao, Angew. Chem., Int. Ed., 2005, 44, 7053–7059 CrossRef CAS PubMed.
- J. Huang, B. G. Sumpter and V. Meunier, Angew. Chem., Int. Ed., 2008, 47, 520–524 CrossRef CAS PubMed.
- J. Huang, B. G. Sumpter and V. Meunier, Chem. – Eur. J., 2008, 14, 6614–6626 CrossRef CAS PubMed.
- H. Wang and L. Pilon, J. Power Sources, 2013, 221, 252–260 CrossRef CAS.
- K. Urita, C. Urita, K. Fujita, K. Horio, M. Yoshida and I. Moriguchi, Nanoscale, 2017, 9, 15643–15649 RSC.
- G. Feng, R. Qiao, J. Huang, B. G. Sumpter and V. Meunier, ACS Nano, 2010, 4, 2382–2390 CrossRef CAS PubMed.
- W. Hsieh, T.-L. A. Horng, H.-C. Huang and H. Teng, J. Mater. Chem. A, 2015, 3, 16535–16543 RSC.
- J. E. Zuliani, C. Q. Jia and D. W. Kirk, J. Phys. Chem. C, 2017, 121, 20555–20566 CrossRef CAS.
- S. Brunauer, P. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 407, 309–319 CrossRef.
- T. A. Centeno and F. Stoeckli, Carbon, 2010, 48, 2478–2486 CrossRef CAS.
- F. Stoeckli and T. A. Centeno, Phys. Chem. Chem. Phys., 2012, 14, 11589–11591 RSC.
-
M. Thommes, K. A. Cychosz and A. V. Neimark, Novel Carbon Adsorbents, Elsevier, 2012, pp. 107–145 Search PubMed.
- G. Y. Gor, M. Thommes, K. a. Cychosz and A. V. Neimark, Carbon, 2012, 50, 1583–1590 CrossRef CAS.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CAS.
- N. Jäckel, P. Simon, Y. Gogotsi and V. Presser, ACS Energy Lett., 2016, 1, 1262–1265 CrossRef.
- N. Jäckel, M. Rodner, A. Schreiber, J. Jeongwook, M. Zeiger, M. Aslan, D. Weingarth and V. Presser, J. Power Sources, 2016, 326, 660–671 CrossRef.
- S. K. Bhatia, Langmuir, 2017, 33, 831–847 CrossRef CAS PubMed.
- J. Jagiello, C. Ania, J. B. Parra and C. Cook, Carbon, 2015, 91, 330–337 CrossRef CAS.
- J. N. Caguiat, D. W. Kirk and C. Q. Jia, Carbon, 2014, 72, 47–56 CrossRef CAS.
- K. S. W. Sing, Pure Appl. Chem., 1985, 57, 603–619 CAS.
-
C. Breitkopf and K. Swider-Lyons, Springer Handbook of Electrochemical Energy, Springer Berlin Heidelberg, Berlin, Heidelberg, 1st edn, 2017 Search PubMed.
- Y. Zhai, Y. Dou, D. Zhao, P. F. Fulvio, R. T. Mayes and S. Dai, Adv. Mater., 2011, 23, 4828–4850 CrossRef CAS PubMed.
- Y.-J. Kim, Y. Masutzawa, S. Ozaki, M. Endo and M. S. Dresselhaus, J. Electrochem. Soc., 2004, 151, E199 CrossRef CAS.
- L. Zhang, H. Liu, M. Wang and L. Chen, Carbon, 2007, 45, 1439–1445 CrossRef CAS.
- J. Górka, A. Zawislak, J. Choma and M. Jaroniec, Carbon, 2008, 46, 1159–1161 Search PubMed.
- Z. Wang, M. Zhou, H. Chen, J. Jiang and S. Guan, Chem. – Asian J., 2014, 9, 2789–2797 CrossRef CAS PubMed.
- C. Lei, N. Amini, F. Markoulidis, P. Wilson, S. Tennison and C. Lekakou, J. Mater. Chem. A, 2013, 1, 6037 RSC.
- Y. Xu, Z. Lin, X. Zhong, X. Huang, N. O. Weiss, Y. Huang and X. Duan, Nat. Commun., 2014, 5, 4554 CrossRef CAS PubMed.
- G. A. Ferrero, M. Sevilla and A. B. Fuertes, Carbon, 2015, 88, 239–251 CrossRef CAS.
- H. Zhang, K. Wang, X. Zhang, H. Lin, X. Sun, C. Li and Y. Ma, J. Mater. Chem. A, 2015, 3, 11277–11286 RSC.
- M. Härmas, T. Thomberg, H. Kurig, T. Romann, A. Jänes and E. Lust, J. Power Sources, 2016, 326, 624–634 CrossRef.
- H. Chen, F. Wang, S. Tong, S. Guo and X. Pan, Appl. Surf. Sci., 2012, 258, 6097–6102 CAS.
- X. Zhang, J. Wang, Z. Yu, R. Wang and H. Xie, Mater. Lett., 2009, 63, 2523–2525 CrossRef CAS.
- G. Feng and P. T. Cummings, J. Phys. Chem. Lett., 2011, 2, 2859–2864 CrossRef CAS.
- L. S. Lasdon, R. L. Fox and M. W. Ratner, Rev. Fr. d’Automatique, Inform. Rech. Opérationnelle, 1974, 3, 73–104 Search PubMed.
- D. Jiang, Z. Jin and J. Wu, Nano Lett., 2011, 11, 5373–5377 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cp06443a |
|
| This journal is © the Owner Societies 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence and
Michael
Fröba
and
Michael
Fröba